

Abstract
Background
Volatile anesthetic preconditioning confers delayed cardioprotection against ischemia/reperfusion injury (I/R). AMP-activated protein kinase (AMPK) takes part in autophagy activation. Furthermore, autophagic flux is thought to be impaired after I/R. We hypothesized that delayed cardioprotection can restore autophagic flux by activating AMPK.
Material/Methods
All male rat hearts underwent 30-min ischemia and 120-min reperfusion with or without sevoflurane exposure. AMPK inhibitor compound C (250 μg/kg, iv) was given at the reperfusion period. Autophagic flux blocker chloroquine (10 mg/kg, ip) was administrated 1 h before the experiment. Myocardial infarction, nicotinamide adenine dinucleotide (NAD+) content, and cytochrome c were measured. To evaluate autophagic flux, the markers of microtubule-associated protein 1 light chain 3 (LC3) I and II, P62 and Beclin 1, and lysosome-associated membrane protein-2 (LAMP 2) were analyzed.
Results
The delayed cardioprotection enhanced post-ischemic AMPK activation, reduced infarction, CK-MB level, NAD+ content loss and cytochrome c release, and compound C blocked these effects. Sevoflurane restored impaired autophagic flux through a lower ratio of LC3II/LC3I, downregulation of P62 and Beclin 1, and higher expression in LAMP 2. Consistently, compound C inhibited these changes of autophagy flux. Moreover, chloroquine pretreatment abolished sevoflurane-induced infarct size reduction, CK-MB level, NAD+ content loss, and cytochrome c release, with concomitant increase the ratios of LC3II/LC3I and levels of P62 and Beclin 1, but p-AMPK expression was not downregulated by chloroquine.
Conclusions
Sevoflurane exerts a delayed cardioprotective effects against myocardial injury in rats by activation of AMPK and restoration of I/R-impaired autophagic flux.
MeSH Keywords: AMP-Activated Protein Kinases; Autophagy; Ischemic Preconditioning, Myocardial; Oxidative Stress; Reperfusion Injury
Background
Perioperative myocardial ischemia/reperfusion (I/R) is a serious challenge in patients undergoing cardiac surgery, and they are subjected to a high risk of myocardial infarction within 24 h after surgery [1]. Therefore, there is a need to develop feasible and effective solutions for alleviating myocardial I/R injury. Volatile anesthetics confer cardioprotective effects against I/R injury, even when administered shortly before ischemia, and this phenomenon includes acute and delayed cardioprotective phases [2]. This delayed cardioprotection, referred to as the second window of protection (SWOP), arises starting 12 h after volatile anesthetics administration and can last up to 72 h [3]. SWOP is extremely important in perioperative patients with a coronary artery bypass graft when cardiac arrest occurs.
We previously showed that sevoflurane exposure upregulates autophagy before ischemia and provides a beneficial cardioprotection against I/R injury [4]. Autophagy is a mainly degradative pathway that eliminates intracellular pathogens and aggregates proteins and damaged organelles under pathological conditions such as stress, hypoxia, and ischemia [5,6]. Autophagy is rapidly activated after myocardium I/R. Therefore, AMP-induced protein kinase (AMPK) pathway activation is pivotal in the initiation of autophagy by inhibiting mammalian target of rapamycin (mTOR). It is a protein kinase involved in regulating cell growth, proliferation, and survival [7,8]. It was reported that induction of autophagy with AMPK activation is protective in the myocardial ischemic phase. However, reperfusion does not enhance AMPK actives; instead, autophagosome formation by myocardial I/R is dependent on Beclin 1 upregulation, but overabundance of autophagosomes is detrimental to cardiomyocytes survival [9]. Ma et al. [10] reported reperfusion impaired the function of autophagosome clearance, which is involved in cardiac myocyte death following I/R injury, the rate of autophagosome formation, and clearance determines the function of “autophagic flux” through the autophagic pathway. Moreover, lysosomes are pivotal to maintaining cellular metabolism by degrading excessive extra- and intracellular organelles [11]. Lysosomal enzymes, including lysosome-associated membrane protein 2 (LAMP 2), fuse abundantly with autophagosomes in dying cells, reflecting adaptive autophagy upregulation. However, the dynamic lysosomal degradative process is impaired, with reduction of autophagosomes clearance, inducing subsequent programmed cell death [12]. In addition, mitochondrial dysfunction is accompanied by spectacular autophagosomes induction during myocardial I/R, which is important in maintaining mitochondrial function by attenuating autophagosome over-accumulation [13]. Sevoflurane-activated Akt/mTOR signaling improves mitochondrial function and ameliorates oxidative stress, but the precise mechanism remains unclear [14].
Our study investigated the SWOP mechanism underlying sevoflurane-induced delayed cardioprotection against I/R injury, and we explored whether enhancement of AMPK activation reduces mitochondrial dysfunction by restoring impaired autophagic flux.
Material and Methods
Animals
All experiments were conducted in accordance with “the Guide for the Care and Use of Laboratory Animals (NIH publication vol. 25 no. 28, revised 1996) and policies of Soochow University. The study was approved by the Use of Experimental Animals Committee of the Medical College of Soochow University. We used only male rats because female rats have greater resistance against ischemia-reperfusion injury than males and volatile anesthetic preconditioning does not provide additional cardioprotection in female rats [15,16]. Adult male Sprague-Dawley rats (weight 280±50 g; age 10 to 12 weeks) were purchased from the Animal Center of Soochow University (Suzhou, China). All animals were treated as described elsewhere [4].
Sevoflurane preconditioning and in vivo myocardial I/R model
One day before surgery, animals spontaneously breathed 33% oxygen in an induction chamber with or without 1 MAC sevoflurane (2.4%; Abbott Laboratories, Shanghai, China) for 2 h. The myocardial I/R injury rat model was established as in our previous reports [4]. To detect the blood concentration of sevoflurane, blood collected from the left ventricle of 5 rats at the end of 2-h sevoflurane exposure in our preliminary study. The samples were measured by gas chromatography (GC-8AIF; Shimadzu Corp., Kyoto, Japan) as described previously [17]. The blood concentration of sevoflurane was 0.37±0.03 mM.
Experimental protocol
Figure 1 illustrates the experimental design. The sham group is not shown because there were no differences in hemodynamics between baseline and end time points of perfusion, the same as in our previous study [4]. We previously reported that inhaling 1 minimal alveolar concentration of sevoflurane for 2 h prior to 24 h before surgery conferred optimal cardioprotection [4]. All experimental rats received 33% oxygen (CON group) or plus 2.5% sevoflurane (SWOP group) for 2 h. We administered AMPK inhibitor compound C alone (CC, 250 μg/kg [18], Sigma-Aldrich, Inc, Saint Louis, USA, CC group) or CC with 2.5% sevoflurane for 2 h (CC+SWOP group). The AMPK inhibitor CC was diluted in dimethylsulfoxide (DMSO, 1%) injected via right jugular vein after occlusion (Figure 1A). We further blocked autophagic flux with chloroquine (CQ, 10 mg/kg [18], Sigma-Aldrich, Inc, St. Louis, USA) 1 h prior to surgery administered via intraperitoneal injection to observe if CQ abolished SWOP-induced cardioprotection in vivo (Figure 1B). All experimental rats underwent 30-min ischemia and 2-h reperfusion [4,19].
Figure 1.
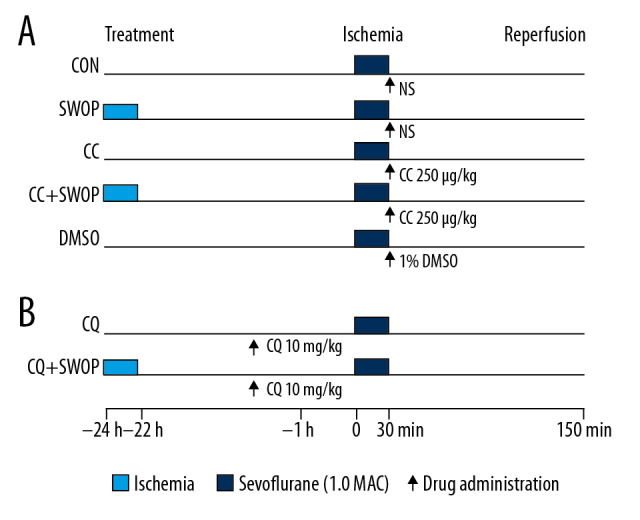
Schematic illustration of experimental protocols used to study the cardioprotection of second window of protection (SWOP) in vivo. Male rats inhaled 1 minimum alveolar concentration sevoflurane for 2 h. After 24 h, all rats underwent 30-min occlusion of left anterior descending coronary artery followed by 120 min of reperfusion (I/R). The putative AMP-activated protein kinase (AMPK) inhibitor compound C (CC, 250 μg/kg) and autophagic flux blocker chloroquine (CQ, 10 mg/kg) were administered in this protocol (A, B).
Myocardial infarct size measurement
After the end of the experiment, the left anterior descending (LAD) artery was re-occluded (n=8–10 per group). To stain the normal region of the left ventricle (LV), 5% Evans blue stain (Sinopharm Chemical Reagent Co., Beijing, China) was injected via the right jugular vein. The measurement of myocardial infarct size (IS) by 1% 2,3,5-triphenyltetrazolium chloride in 0.1 M phosphate buffer was performed as previously described [19], and the area at risk (AAR) was calculated [4].
Histopathology and transmission electron microscopy
The hearts were harvested after the experiment, rinsed, and fixed in 10% formaldehyde solution (pH 7.4) for 24 h. After dehydration and embedding in paraffin wax, the heart was cut into 4-μm-thick slices, then stained with hematoxylin-eosin (HE) [20]. Transmission electron microscopy (TEM) was been used to detect myocardial ultrastructural alterations, as described our previous reports [4].
Western blot analysis and Immunohistochemistry
At the end of reperfusion, left ventricular tissues with infarcted area were separated from hearts and homogenized in RIPA lysis buffer (Millipore, Billerica, MA, USA) containing phenylmethanesulfonyl fluoride (Roche Diagnostics GmbH, Mannheim, Germany). Immunoblots were performed using standard techniques, as described previously [19,21]. Primary antibodies were probed with total AMPK, phosphorylated AMPK (p-AMPK, 1: 2000; Cell Signaling Technology, Danvers, MA, USA), microtubule-associated protein 1 light chain 3 (LC3, 1: 500; Abcam, Cambridgeshire, UK), Beclin 1 (1: 1000; Cell Signaling Technology, Danvers, MA, USA), cytochrome c and LAMP 2 (1: 500; Santa Cruz Biotechnology, California, USA), and P62 (1: 1000; Sigma-Aldrich, St. Louis, MO, USA). Secondary antibodies (1: 5000; Cell Signaling Technology, Danvers, MA, USA) were added at room temperature for 2 h. GAPDH (1: 2000; Beyotime, Shanghai, China) was assayed as a loading control. Immunoreactive bands were detected by an enhanced chemiluminescence system (ECL; Merck, Darmstadt, Germany) and images were measured using densitometric analysis software [4,22,23].
The heart tissues were sectioned and stained with LC3, as reported previously [24], and the antibodies were diluted at anti-LC3 1: 100. All sections were photographed under a microscope.
Determination of NAD+ and CK-MB levels
Nicotinamide adenine dinucleotide (NAD+) was measured 5 min after the heart was removed [25]. NAD+ activity was determined by fluorometric measurement at a wave length of 340 nm using a multi-frequency phase spectrofluorometer (DU-640, Beckman Coulter, Inc.). Plasma was collected at the end of reperfusion, and the levels of creatine kinase MB (CK-MB) were detected using a rat ELISA kit (Roche, Sweden).
Statistical analysis
All data are presented as mean±SD. We performed one-way ANOVA followed by Bonferroni post hoc test for multiple comparisons of multiple group means (GraphPad Software, Inc, San Diego, USA). Repeated-measures ANOVA was used to compare the differences in heart rate and MAP at different time points. A P value of < 0.05 was considered to be statistically significant.
Results
Systemic hemodynamics changes
Table 1 shows there were no differences in HR, MAP, and RPP observed at baseline, occlusion, and reperfusion time points (n=8–10) among all groups. Myocardial I/R significantly decreased MAP and RPP after occlusion (P<0.05 vs. baseline).
Table 1.
Systemic hemodynamic changes during experiments in intact ischemic reperfused rat hearts.
| Baseline | Occlusion | Reperfusion | ||
|---|---|---|---|---|
| 1 h | 2 h | |||
| HR (min−1) | ||||
| CON | 357±53 | 332±35 | 347±35 | 351±47 |
| SWOP | 367±44 | 341±53 | 341±46 | 353±42 |
| CC | 363±49 | 338±45 | 357±37 | 352±49 |
| CC+SWOP | 371±55 | 335±51 | 347±43 | 355±45 |
| DMSO | 361±47 | 344±42 | 352±45 | 352±30 |
| MAP (mmHg) | ||||
| CON | 113±12 | 112±14 | 81±12* | 78±12* |
| SWOP | 112±10 | 115±13 | 77±13* | 65±12* |
| CC | 105±13 | 121±11 | 76±13* | 68±14* |
| CC+SWOP | 103±9 | 108±7 | 78±9* | 69±10* |
| DMSO | 115±12 | 118±15 | 76±13* | 64±18* |
| RPP (min−1·mmHg·103) | ||||
| CON | 43±5 | 44±12 | 36±4* | 37±5* |
| SWOP | 44±7 | 45±7 | 38±4* | 38±8* |
| CC | 44±6 | 44±5 | 36±7* | 35±4* |
| CC+SWOP | 47±3 | 48±5 | 37±3* | 36±7* |
| DMSO | 46±10 | 48±7 | 36±8* | 33±4* |
Values are presented as mean±SD. The hemodynamics in the 5 experimental groups at baseline, during occlusion, and at reperfusion showed no between-group differences. HR – heart rate; MAP – mean arterial blood pressure; RPP – rate-pressure product; CON – ischemia and reperfusion; SWOP – the second window of protection with sevoflurane; CC – compound C; DMSO – dimethylsulfoxide;
P<0.05 versus baseline.
SWOP reduced myocardium infarct size and CK-MB levels
AAR in each group showed no differences following I/R (P>0.05, Figure 2A, 2C). SWOP attenuated the post-ischemic IS/AAR from 54±6 in the control group to 35±6, and SWOP attenuated the level of CK-MB. This protective effect was inhibited by the AMPK inhibitor CC and autophagic flux blocker CQ (Figures 2, 3, P<0.05, vs. CON). Sole administration of CC, CQ, and vehicle DMSO had no significant impact on IS or level of CK-MB (P>0.05, vs. CON).
Figure 2.

Sevoflurane preconditioning-induced decreases in infarct size were blocked by CC or CQ. (A, C) Representative area at risk (AAR), presented as the ratio of AAR/left ventricle (LV); (B, D) Representative slices of the LV stained by Evans blue and TTC, myocardial infarct size expressed as a percentage of AAR. Pentobarbital-anesthetized rats underwent 30 min of ischemia followed by 2 h of reperfusion with or without sevoflurane treatment. * P<0.05 versus CON group; & P<0.05 versus SWOP group (n=8 to 10 hearts per group).
Figure 3.

Sevoflurane preconditioning-induced attenuation of CK-MB level was blocked by CC (A) or CQ (B). The data are presented as means±SD. * P<0.05 vs. CON group; & P<0.05 vs. SWOP group (n=5 hearts/group).
SWOP alleviated post-ischemic myocardial histopathological changes and preserved ultrastructural integrity
To test whether SWOP improved myocardial histopathological and ultrastructural damage, we conducted HE staining and TEM in I/R rat hearts. Myocardial fibers were irregular following I/R injury, as shown in Figure 4A, and myocardium presented edema with large necrotic areas and neutrophil infiltration. These pathologic changes were improved in the SWOP group, as evidenced by the protection effect in myocardium. In addition, SWOP-alleviated histological outcomes were largely inhibited by the AMPK inhibitor CC and autophagic flux blocker CQ. Figure 4B shows ultrastructural sections of the CON group with marked ultrastructural damages, and autophagosomes were common. Electron photomicrographs of the SWOP group revealed relatively normal myofilaments ultrastructure, well-arranged sarcomeres, and mild rarefaction of myofilaments. The protective effects of SWOP on ultrastructural integrity of myocytes were blocked by CC and CQ.
Figure 4.

Sevoflurane attenuated histopathologic organ damage and ultrastructure following I/R. LV tissues were retrieved at the end of reperfusion, and paraffin sections were prepared and subjected to the hematoxylin-eosin (HE) staining. (A) Representative HE staining images are shown (magnification, 100×). (B) LV tissues were harvested for examination of myocardial ultrastructure by transmission electron microscopy. Scale bar, 0.5 μm. The arrowhead indicates that myofilaments were absent or fractured; the nuclear chromatin edge set, and an asterisk indicates mitochondrial edema near the autophagosomes.
SWOP enhanced AMPK activation following myocardial I/R
Sevoflurane preconditioning, administered 24 h before surgery, the ratio of phosphorylated AMPK to total AMPK (p-AMPK/AMPK) significantly increased in the ischemic hearts at 2 h after reperfusion compared to the rats receiving vehicle (Figure 5, P<0.05, SWOP vs. CON). CC blunted the effect of SWOP on AMPK activation (Figure 5A, P<0.05, CC+SWOP vs. SWOP) but not CQ (Figure 5B, P>0.05, CQ+SWOP vs. SWOP).
Figure 5.

Sevoflurane-induced increase in the phosphorylation of AMPK (p-AMPK) was abolished by inhibitor CC (A) but not autophagic flux blocker CQ (B) in intact hearts following myocardial I/R at 2-h reperfusion. Representative Western blot bands of p-AMPK, AMPK, and GAPDH as shown in the picture above. Bar graph represents the densitometric analysis of p-AMPK/AMPK in the ischemic heart at 2-h reperfusion. Data are presented as means±SD. * P<0.05 vs. CON group; & P<0.05 vs. SWOP group (n=5 hearts per group).
SWOP enhanced autophagic flux following myocardial I/R
The LC3II/I ratio is a marker for autophagosome formation. LAMP 2 is essential for autophagosome clearance [26], Ma et al. [10] showed that autophagic flux was impaired by upregulating Beclin 1 and downregulating LAMP 2 in a myocardial I/R model. P62 is a well-established adapter protein that indicates autophagic flux that connects aggregated proteins that are sequestered in autophagosomes and degraded in autolysosomes [26]. Compared to the CON group, we found that sevoflurane preconditioning, administered 24 h before surgery, blunted I/R-induced increases in LC3II/I ratios, Beclin 1, and P62 levels and decrease in LAMP 2 protein levels (Figures 6, 7, P<0.05, SWOP vs. CON). We found that SWOP restored I/R-impaired autophagy flux. The AMPK inhibitor CC and autophagic flux blocker CQ inhibited the effects of SWOP on LC3II/I ratios, Beclin 1, P62, and LAMP 2. At baseline conditions, CC and DMSO had no significant effects on LC3II/I ratios, Beclin 1, P62, and LAMP 2 protein levels (P>0.05 vs. CON, respectively). Sevoflurane attenuated LC3 immunohistochemistry staining, and the protective effects of SWOP were inhibited by CC and CQ (Figure 8).
Figure 6.

Sevoflurane-enhanced autophagic flux was cancelled by AMPK inhibitor CC in intact hearts following myocardial I/R at 2-h reperfusion. (A) Representative immunoblots and densitometric analysis of light chain 3 (LC3) I and LC3 II in the ischemic/reperfused heart. (B) Representative immunoblots and densitometric analysis of P62 in the ischemic/reperfused heart. (C) Representative immunoblots and densitometric analysis of Beclin 1 in the ischemic/reperfused heart. (D) Representative immunoblots and densitometric analysis of lysosome-associated membrane protein-2 (LAMP 2) in the ischemic/reperfused heart. Data are presented as means±SD. * P<0.05 vs. CON group; & P<0.05 vs. SWOP group (n=5 hearts per group).
Figure 7.

Sevoflurane-enhanced autophagic flux was reversed by autophagic flux blocker CQ in intact hearts following myocardial I/R at 2-h reperfusion. (A) Representative immunoblots and densitometric analysis of LC3 I and LC3 II in the ischemic/reperfused heart. (B) Representative immunoblots and densitometric analysis of P62 in the ischemic/reperfused heart. (C) Representative immunoblots and densitometric analysis of Beclin 1 in the ischemic/reperfused heart. (D) Representative immunoblots and densitometric analysis of LAMP 2 in the ischemic/reperfused heart. Data are presented as the means±SD. * P<0.05 vs. CON group; & P<0.05 vs. SWOP group (n=5 hearts per group).
Figure 8.

Sevoflurane attenuated LC3 immunohistochemistry staining and the protective effects of SWOP were cancelled by CC and CQ (magnification, 400×).
SWOP mediated attenuation of myocardial NAD+ content loss and cytochrome c release
As shown in Figure 9, SWOP induced by sevoflurane significantly increased the mitochondrial NAD+ level (P<0.05, SWOP group vs. CON group) and decreased cytochrome c expression. CC and CQ attenuated the sevoflurane-mediated increase in mitochondrial NAD+ and decrease in cytosolic cytochrome c release.
Figure 9.

(A–D) SWOP induced by sevoflurane reduced nicotinamide adenine dinucleotide (NAD+) content loss and cytochrome c release due to I/R in the in vivo model. (A) SWOP reduced mitochondrial NAD+ loss. (B) SWOP decreased cytoplasm cytochrome c release. SWOP-induced mitochondrial protection was inhibited by AMPK inhibitor CC and autophagic flux blocker CQ. The data are presented as means±SD. * P<0.05 vs. CON group; & P<0.05 vs. SWOP group (n=5 hearts/group).
Discussion
Our study demonstrated that the second window of protection with sevoflurane reduced myocardial infarct size, CK-MB level, NAD+ content loss, and cytochrome c release. These effects may due to the increased AMPK activation and subsequent restoration of autophagic flux, as evidenced by increased p-AMPK and LAMP 2 protein level and decreased LC3II/I ratios and Beclin 1 and P62 protein levels. AMPK inhibitor CC administration after occlusion not only blocked AMPK phosphorylation, but also impaired autophagic flux and abolished the delayed cardioprotection. In addition, pretreatment with the autophagic flux inhibitor CQ abolished SWOP-induced reduction in infarct size, CK-MB level, NAD+ content loss, and cytochrome c release, along with the accumulation of autophagosomes. This indicates that activation of AMPK and restoration autophagic flux are essential mechanisms for anesthetics-induced delayed cardioprotection.
Although many studies have shown the delayed cardioprotective effect of anesthetics following myocardial I/R, the underlying mechanisms are still poorly understood. Several mechanisms have been implicated in ATP-sensitive potassium channel, inducible nitric oxide synthase, and cyclooxygenase-2 [27,28]. AMPK has been shown to be involved in regulating cellular energy metabolism, and AMPK activation plays a key role in upregulating endogenous anti-oxidants and mitochondrial biogenesis [29,30]. Mitochondria supply energy to cells, and damaged mitochondria increase mitochondrial permeability transition pore (mPTP) opening, and release of reactive oxygen species (ROS) and cytochrome c, finally resulting in cell death. Maintenance of cell viability depends on functional mitochondria [31–33]. Some studies showed that delayed mPTP opening reduced mitochondrial membrane potential collapse, Ca2+ and cytochrome c efflux, and matrix swelling [32,34]. The NAD+ level is an important index to assess the opening of mPTP, and it is usually drastically decreased and subsequently leads to cytochrome c release [35]. We previously showed that NADH reached a peak value 5 min after ischemia, followed by a gradual decline [25]. We recently found that volatile anesthetic isoflurane has remarkable effects on different states of mitochondrial respiration under oxidation of complex I substrates [23]. In the present study, SWOP increased NAD+ content in the cytoplasm and reduced cytochrome c release. These beneficial effects may contribute to mitochondrial protection. The mitochondria protective effect by SWOP was inhibited by CC and CQ, indicating that mitochondria is a target of SWOP-derived cardioprotection.
Recent studies demonstrated that sevoflurane activation of AMPK during ischemia is beneficial. However, whether AMPK activation is responsible for sevoflurane-mediated cardioprotection during reperfusion remains actively debated [36]. AMPK is a trimeric protein complex that is formed by a functional subunit (α) and 2 regulatory subunits (β, γ) [37]. Some studies showed that AMPK activation by sevoflurane protects the heart against I/R injury and is dependent on upstream production of ROS [38]. In the present study, we observed that 1 day before surgery, 2-h sevoflurane exposure upregulated AMPK activation, and the AMPK inhibitor CC decreased AMPK phosphorylation and attenuated the cardioprotective effects of SWOP. Our results suggested that the AMPK activation may be an underlying mechanism involved in the cardioprotection conferred by SWOP.
The AMPK signaling pathway induces autophagy by inhibiting mTOR activation [39]. Autophagy is a self-degradative process, degrading damaged proteins and organelles into new synthetic substrates [12], including autophagosomes induction and degradation [40]. This complete process is termed autophagic flux and has been linked to intracellular homeostasis and cardiac myocyte survival [41]. The mechanism of autophagy underlying myocardial I/R injury remains unclear [42,43]. It was reported that ischemia induced by autophagy alone was upregulated and beneficial during the ischemic phase, which is dependent on the activation of AMPK [9]. Autophagosome accumulation in myocardial I/R is mediated by upregulation of Beclin 1 and downregulation of LAMP 2, but AMPK activation is not enhanced during the reperfusion phase [18]. In contrast to ischemia-induced autophagy, autophagy enhanced during the reperfusion phase exacerbates infarct damage [9]. Moreover, impairment of autophagy flux and autophagosome clearance can exacerbate myocardial ischemia injury following reperfusion because myocardial I/R stimulates autophagosome formation [12,44].
Our hypothesis in the present study was that the activation of AMPK can restore myocardial I/R-impaired autophagic flux, which can delay cardioprotection by anesthetic preconditioning after myocardial I/R. In support of this hypothesis, we found that SWOP significantly enhanced AMPK phosphorylation and remarkably restored autophagic flux that was impaired by I/R. Furthermore, CC markedly inhibited the beneficial effects of SWOP on autophagosome clearance. More relevantly, both AMPK inhibitor CC and autophagic flux blocker CQ inhibited SWOP-induced cardioprotection after myocardial I/R. Taken together, our study demonstrates that the restoration of I/R-impaired autophagic flux by SWOP may be dependent on AMPK activation and is critical to cardiac protection of SWOP against myocardial I/R.
Recent evidence shows that the protection of sevoflurane postconditioning (SPC) can restore impaired autophagic flux in myocardial I/R [44]. Although it is well-established that AMPK activation is important for the initiation of autophagy by inhibiting mTOR to promote autophagosome formation, AMPK activation may also be critical to autophagosome clearance in the ischemic heart [10]. Indeed, it has been reported that reduction in LAMP 2, a key factor in autophagosome-lysosome fusion [18], is responsible for impaired autophagosome clearance and autophagic flux following myocardial I/R. Here, we observed SWOP enhanced I/R-reduced LAMP 2 expression in the I/R heart in an AMPK-dependent manner. Our results suggest that SWOP can promote autophagosome clearance by enhancing LAMP 2 expression. A previous study demonstrated that CQ induced autophagosomes accumulation, which increases mPTP opening and ROS and ATP production [45]. In contrast, the favorable autophagic flux process can decrease mitochondrial membrane potential loss and reduce ROS production by removing damaged mitochondria, thereby reducing deleterious autophagosomes accumulation.
Our study has several limitations. First, we did not perform two-dimensional echocardiography to evaluate the parameters of cardiac function in vivo, which could further support the beneficial effects of the sevoflurane in I/R animals because it has been reported that SPC improved the hemodynamic parameters, cardiac dysfunction [44]. Second, some studies have shown that sevoflurane preconditioning plays a regulatory role in multiple types of cardiomyocyte death such as apoptosis, necrosis, and autophagic death [38,46]. Therefore, sevoflurane preconditioning can also protect the heart against ischemia reperfusion injury by regulating other categories of cardiomyocyte death beyond autophagy. Further research is warranted to explore these issues.
Conclusions
Our study demonstrates that sevoflurane preconditioning conferred delayed cardioprotection against I/R injury by AMPK activation-dependent autophagic flux restoration, which offers a potential therapeutic approach in the treatment of perioperative myocardial I/R injury.
Footnotes
Conflict of interest
None.
Source of support: This work was supported by grants from the Suzhou Science and Technology Development Plan (no. SS201613 to Chen Wang and no. SS201756 to Jian-Zhong An), the Jiangsu Natural Science Foundation (no. BK20141187 to Chen Wang), the Suzhou Gaoxin District Science and Technology Plan (no. 2017Q003 to Lei Hong, 2017Z004 to Jian-Zhong An), the Suzhou Science and Technology Development Plan (no. SYS2019017 to Lei Hong), the National Natural Science Foundation of China (no. 81703501 to Shi-Gang Qiao), the Jiangsu Key Talent Youth Awards in Medicine (no. QNRC2016219 to Shi-Gang Qiao), and the Gusu Key Talent Youth Awards in Medicine (no. GSWS2019092 to Shi-Gang Qiao)
Data availability
The raw data used to support the findings of this study are available from the corresponding author upon request
References
- 1.Pollak A, Merin G, Horowitz M, et al. Heat acclimatization protects the left ventricle from increased diastolic chamber stiffness immediately after coronary artery bypass surgery: A lesson from 30 years of studies on heat acclimation mediated cross tolerance. Front Physiol. 2017;8:1022. doi: 10.3389/fphys.2017.01022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi Y, Hutchins WC, Su J, et al. Delayed cardioprotection with isoflurane: Role of reactive oxygen and nitrogen. Am J Physiol Heart Circ Physiol. 2005;288(1):H175–84. doi: 10.1152/ajpheart.00494.2004. [DOI] [PubMed] [Google Scholar]
- 3.Lutz M, Liu H. Inhaled sevoflurane produces better delayed myocardial protection at 48 versus 24 hours after exposure. Anesth Analg. 2006;102(4):984–90. doi: 10.1213/01.ane.0000198568.79079.4c. [DOI] [PubMed] [Google Scholar]
- 4.Qiao S, Xie H, Wang C, et al. Delayed anesthetic preconditioning protects against myocardial infarction via activation of nuclear factor-κB and upregulation of autophagy. J Anesth. 2013;27(2):251–60. doi: 10.1007/s00540-012-1494-3. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh R, Pattison JS. Macroautophagy and chaperone-mediated autophagy in heart failure: The known and the unknown. Oxid Med Cell Longev. 2018;2018 doi: 10.1155/2018/8602041. 8602041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang S, Ping Z, Ge J. Coenzyme Q10 regulates antioxidative stress and autophagy in acute myocardial ischemia-reperfusion injury. Oxid Med Cell Longev. 2017;2017 doi: 10.1155/2017/9863181. 9863181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rohailla S, Clarizia N, Sourour M, et al. Acute, delayed and chronic remote ischemic conditioning is associated with downregulation of mTOR and enhanced autophagy signaling. PLoS One. 2014;9(10):e111291. doi: 10.1371/journal.pone.0111291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao Z, Pan X, Zhang H, et al. Isoflurane preconditioning alleviated murine liver ischemia and reperfusion injury by restoring AMPK/mTOR-mediated autophagy. Anesth Analg. 2017;125(4):1355–63. doi: 10.1213/ANE.0000000000002385. [DOI] [PubMed] [Google Scholar]
- 9.Matsui Y, Takagi H, Qu X, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914–22. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 10.Ma X, Liu H, Foyil SR, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125(25):3170–81. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu M, Yang L, Rong JG, et al. Inhibition of cysteine cathepsin B and L activation in astrocytes contributes to neuroprotection against cerebral ischemia via blocking the tBid-mitochondrial apoptotic signaling pathway. Glia. 2014;62(6):855–80. doi: 10.1002/glia.22645. [DOI] [PubMed] [Google Scholar]
- 12.Godar RJ, Ma X, Liu H, et al. Repetitive stimulation of autophagy-lysosome machinery by intermittent fasting preconditions the myocardium to ischemia-reperfusion injury. Autophagy. 2015;11(9):1537–60. doi: 10.1080/15548627.2015.1063768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W, Siraj S, Zhang R, Chen Q, et al. Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy. 2017;13(6):1080–81. doi: 10.1080/15548627.2017.1300224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie H, Liu Q, Qiao S, et al. Delayed cardioprotection by sevoflurane preconditioning: a novel mechanism via inhibiting Beclin 1-mediated autophagic cell death in cardiac myocytes exposed to hypoxia/reoxygenation injury. Int J Clin Exp Pathol. 2015;8(1):217–26. [PMC free article] [PubMed] [Google Scholar]
- 15.Humphreys RA, Kane KA, Parratt JR. The influence of maturation and gender on the anti-arrhythmic effect of ischaemic preconditioning in rats. Basic Res Cardiol. 1999;94(1):1–8. doi: 10.1007/s003950050120. [DOI] [PubMed] [Google Scholar]
- 16.Wang C, Chiari PC, Weihrauch D, et al. Gender-specificity of delayed preconditioning by isoflurane in rabbits: Potential role of endothelial nitric oxide synthase. Anesth Analg. 2006;103(2):274–80. doi: 10.1213/01.ANE.0000230389.76351.0C. table of contents. [DOI] [PubMed] [Google Scholar]
- 17.Sedlic F, Pravdic D, Ljubkovic M, et al. Differences in production of reactive oxygen species and mitochondrial uncoupling as events in the preconditioning signaling cascade between desflurane and sevoflurane. Anesth Analg. 2009;109(2):405–11. doi: 10.1213/ane.0b013e3181a93ad9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie H, Xu Q, Jia J, et al. Hydrogen sulfide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem Biophys Res Commun. 2015;458(3):632–38. doi: 10.1016/j.bbrc.2015.02.017. [DOI] [PubMed] [Google Scholar]
- 19.Qiao S, Mao X, Wang Y, et al. Remifentanil preconditioning reduces postischemic myocardial infarction and improves left ventricular performance via activation of the Janus activated kinase-2/signal transducers and activators of transcription-3 signal pathway and subsequent inhibition of glycogen synthase kinase-3β in rats. Crit Care Med. 2016;44(3):e131–45. doi: 10.1097/CCM.0000000000001350. [DOI] [PubMed] [Google Scholar]
- 20.Huang Z, Zhong X, Irwin MG, et al. Synergy of isoflurane preconditioning and propofol postconditioning reduces myocardial reperfusion injury in patients. Clin Sci. 2011;121(2):57–69. doi: 10.1042/CS20100435. [DOI] [PubMed] [Google Scholar]
- 21.Qiao SG, Sun Y, Sun B, et al. Sevoflurane postconditioning protects against myocardial ischemia/reperfusion injury by restoring autophagic flux via an NO-dependent mechanism. Acta Pharmacol Sin. 2019;40(1):35–45. doi: 10.1038/s41401-018-0066-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiao S, Olson JM, Paterson M, et al. MicroRNA-21 mediates isoflurane-induced cardioprotection against ischemia-reperfusion injury via Akt/nitric oxide synthase/mitochondrial permeability transition pore pathway. Anesthesiology. 2015;123(4):786–98. doi: 10.1097/ALN.0000000000000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu F, Qiao S, Li H, et al. The effect of mitochondrial complex I-linked respiration by isoflurane is independent of mitochondrial nitric oxide production. Cardiorenal Med. 2018;8(2):113–22. doi: 10.1159/000485936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sung HK, Chan YK, Han M, et al. Lipocalin-2 (NGAL) attenuates autophagy to exacerbate cardiac apoptosis induced by myocardial ischemia. J Cell Physiol. 2017;232(8):2125–34. doi: 10.1002/jcp.25672. [DOI] [PubMed] [Google Scholar]
- 25.An J, Camara AK, Riess ML, et al. Improved mitochondrial bioenergetics by anesthetic preconditioning during and after 2 hours of 27 degrees C ischemia in isolated hearts. J Cardiovasc Pharmacol. 2005;46(3):280–87. doi: 10.1097/01.fjc.0000175238.18702.40. [DOI] [PubMed] [Google Scholar]
- 26.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 27.Jiang X, Shi E, Nakajima Y, Sato S, et al. Inducible nitric oxide synthase mediates delayed cardioprotection induced by morphine in vivo: Evidence from pharmacologic inhibition and gene-knockout mice. Anesthesiology. 2004;101(1):82–88. doi: 10.1097/00000542-200407000-00014. [DOI] [PubMed] [Google Scholar]
- 28.Tonkovic-Capin M, Gross GJ, Bosnjak ZJ, et al. Delayed cardioprotection by isoflurane: Role of K(ATP) channels. Am J Physiol Heart Circ Physiol. 2002;283(1):H61–68. doi: 10.1152/ajpheart.01040.2001. [DOI] [PubMed] [Google Scholar]
- 29.Dugan LL, You YH, Ali SS, et al. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest. 2013;123(11):4888–99. doi: 10.1172/JCI66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li S, Wu H, Han D, et al. ZP2495 protects against myocardial ischemia/reperfusion injury in diabetic mice through improvement of cardiac metabolism and mitochondrial function: The possible involvement of AMPK-FoxO3a signal pathway. Oxid Med Cell Longev. 2018;2018 doi: 10.1155/2018/6451902. 6451902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pagliaro P, Femminò S, Popara J, Penna C. Mitochondria in cardiac postconditioning. Front Physiol. 2018;9:287. doi: 10.3389/fphys.2018.00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cadenas S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic Biol Med. 2018;117:76–89. doi: 10.1016/j.freeradbiomed.2018.01.024. [DOI] [PubMed] [Google Scholar]
- 33.Xie H, Zhang J, Zhu J, et al. Sevoflurane post-conditioning protects isolated rat hearts against ischemia-reperfusion injury via activation of the ERK1/2 pathway. Acta Pharmacol Sin. 2014;35(12):1504–13. doi: 10.1038/aps.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ong SB, Samangouei P, Kalkhoran SB, Hausenloy DJ, et al. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol. 2015;78:23–34. doi: 10.1016/j.yjmcc.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 35.Petrosillo G, Di Venosa N, Moro N, et al. In vivo hyperoxic preconditioning protects against rat-heart ischemia/reperfusion injury by inhibiting mitochondrial permeability transition pore opening and cytochrome c release. Free Radic Biol Med. 2011;50(3):477–83. doi: 10.1016/j.freeradbiomed.2010.11.030. [DOI] [PubMed] [Google Scholar]
- 36.Lotz C, Ping P, Kehl F. Letter by Lotz et al. regarding article, “Reactive oxygen species-induced stimulation of 5’ AMP-activated protein kinase mediates sevoflurane-induced cardioprotection”. Circulation. 2010;121(18):e399. doi: 10.1161/CIR.0b013e3181df9134. author reply e400. [DOI] [PubMed] [Google Scholar]
- 37.Towler M, Hardie D. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100(3):328–41. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 38.Shiomi M, Miyamae M, Takemura G, et al. Sevoflurane induces cardioprotection through reactive oxygen species-mediated upregulation of autophagy in isolated guinea pig hearts. J Anesth. 2014;28(4):593–600. doi: 10.1007/s00540-013-1755-9. [DOI] [PubMed] [Google Scholar]
- 39.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dutta D, Xu J, Kim JS, et al. Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity. Autophagy. 2013;9(3):328–44. doi: 10.4161/auto.22971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loos B, Engelbrecht AM, Lockshin RA, et al. The variability of autophagy and cell death susceptibility: Unanswered questions. Autophagy. 2013;9(9):1270–85. doi: 10.4161/auto.25560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Nadtochiy SM, Urciuoli WR, Brookes PS. The cardioprotective compound cloxyquin uncouples mitochondria and induces autophagy. Am J Physiol Heart Circ Physiol. 2016;310(1):H29–38. doi: 10.1152/ajpheart.00926.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang S, et al. N-acetylcysteine attenuates diabetic myocardial ischemia reperfusion injury through inhibiting excessive autophagy. Mediators Inflamm. 2017;2017 doi: 10.1155/2017/9257291. 9257291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu P, Zhang J, Yu S, et al. Protective effect of sevoflurane postconditioning against cardiac ischemia/reperfusion injury via ameliorating mitochondrial impairment, oxidative stress and rescuing autophagic clearance. PLoS One. 2015;10(8):e0134666. doi: 10.1371/journal.pone.0134666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu Q, Gao C, Wang H, et al. Mdivi-1 alleviates blood-brain barrier disruption and cell death in experimental traumatic brain injury by mitigating autophagy dysfunction and mitophagy activation. Int J Biochem Cell Biol. 2018;94:44–55. doi: 10.1016/j.biocel.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 46.Lu X, Moore PG, Liu H, Schaefer S. Phosphorylation of ARC is a critical element in the antiapoptotic effect of anesthetic preconditioning. Anesth Analg. 2011;112(3):525–31. doi: 10.1213/ANE.0b013e318205689b. [DOI] [PubMed] [Google Scholar]