

Abstract
Aortic insufficiency (AI) or regurgitation is caused by the malcoaptation of the aortic valve (AV) cusps due to intrinsic abnormalities of the valve itself, a dilatation or geometric distortion of the aortic root, or by some combination thereof. In recent years, there has been an increase in the number of studies suggesting that AI is an active disease process caused by a combination of factors including but not limited to alteration of specific molecular pathways, genetic predisposition, and changes in the mechanotransductive properties of the AV apparatus. As the surgical management of AV disease continues to evolve, increasingly sophisticated surgical and percutaneous techniques for AV repair and replacement, including transcatheter aortic valve replacement (TAVR), have become more commonplace and will likely continue to expand as new devices are introduced. However, these techniques necessitate frequent reappraisal of the biological and mechanobiological mechanisms underlying AV regurgitation to better understand the risk factors for AI development and recurrence following surgical intervention as well as expand our limited knowledge on patient selection for such procedures. The aim of this review is to describe some of the putative mechanisms implicated in the development of AI, dissect some of the cross-talk among known and possible signaling pathways leading to valve remodeling, identify association between these pathways and pharmacological approaches, and discuss the implications for surgical and percutaneous approaches to AV repair in replacement in the TAVR era.
Keywords: Cardiac and Cardiothoracic Surgery, Non-invasive and minimally invasive Cardiology, Clinical Cardiology
Introduction
Aortic insufficiency (AI) is characterized by a pathologic derangement of the aortic valve (AV) or valvular apparatus resulting in retrograde blood flow into the left ventricle during diastole. AI is caused by the malcoaptation of the AV cusps due to intrinsic abnormalities with the valve or valve apparatus, the aortic root, or a combination of the two1,2. AI is a relatively prevalent source of cardiovascular morbidity and mortality and may be found in up to 13.0% and 8.5% of American men and women, respectively3. Traditional causes of AI include diseases such as rheumatic fever and endocarditis, congenital defects, cusp perforation, and degenerative processes2. However, diseases of the AV cusps have traditionally been poorly characterized and incompletely understood. Recent close examination of such pathological processes has demonstrated a wide array of complex, active remodeling mechanisms affecting the valve and surrounding structures. These pathological processes are examples of cardiac remodeling, which is defined as a group of molecular, cellular, and interstitial changes that clinically manifest as changes in size, shape, and function of the heart resulting from cardiac insult4.
New and ongoing research into the pathophysiology underlying these remodeling mechanisms has facilitated important discoveries pertaining to preventative, diagnostic, and mitigating strategies for patients with AI. This review will aim to describe various mechanisms involved in AI development, dissect some of the cross-talk among known and possible signaling pathways leading to valve remodeling, identify an association between these pathways and possible pharmacological approaches, and discuss surgical implications for management of these pathological processes. Considering the increasing popularity of TAVR, we will also discuss the burgeoning roles of percutaneous AV repair and replacement for patients with AI.
AI Classification
In the diagnosis of AI, the surgical El Khoury classification is employed to differentiate between different types of AI, each with their own clinical manifestations5. Accurate classification of the type of AI is essential for surgical treatment, as certain contraindications, such as severe calcification, active infection in the valve, extremely restricted cusp motion, severely dilated sinotubular junction (STJ), and decreased cusp height, preclude a durable repair6,7. According to this classification paradigm, AI is characterized by 3 discrete types: Type I, Type II, and Type III. Type I AI indicates normal cusp motion with a dilatation of the ascending aorta (Type-Ia), aortic sinus (Type-Ib), aortic annulus (Type-Ic), and cusp perforation (Type-Id)5,7. These patients normally have relatively healthy valve leaflets and valve sparing procedures represent the gold standard for treatment. Type II AI is characterized by prolapse or excessive motion of the cusp. Type II AI can also be treated with a valve repair, as opposed to complete valve replacement, allowing for preservation of the patient’s native valve6,7. Type III indicates restricted cusp mobility due to fibrosis and/or calcification5,7. Type III AI constitutes a particularly significant patient cohort, as calcification can restrict cusp mobility and may necessitate complete replacement.
Diagnostic Criteria
According to the American Heart Association (AHA) Valvular Heart Disease Guidelines, the diagnostic criteria for chronic AI are characterized by anatomic and hemodynamic changes to the AV in conjunction with clinical symptomatology. The most common causes of AI have been reported be congenital defects - especially bicuspid AV - and calcific AV disease. The AHA guidelines define four stages of chronic AI, ranging from “at risk,” to “progressive,” to “asymptomatic severe,” “symptomatic severe”8. As the disease is largely asymptomatic in most patients, the hemodynamic properties of the valve - obtained primarily via ultrasound imaging - are utilized to diagnose the severity of AI in patients. Notably, left ventricle ejection fraction (LVEF), regurgitation fraction (RF), and doppler jet width are frequently utilized as radiographic markers of AI. Intervention for severe AI in asymptomatic patients is recommended, as per the AHA guidelines, when LV systolic disfunction is present (LVEF <50%) or if severe LV dilation (left ventricular end-systolic dimension >50mm and left ventricular end-diastolic dimension >65mm) is present under normal LV systolic function8. The diagnostic criteria for patients at risk for AI development is currently aimed at patients with prior anatomical valve abnormalities, such as congenital diseases, or prior heart diseases including AV sclerosis, rheumatic heart disease, and infective endocarditis8. Given the asymptomatic nature of the disease, management is largely reactive to certain criteria rather than preventative.
As aforementioned, AI is a complex cardiovascular disease encompassing a diversity or even absence of clinical signs or symptoms, and functional or radiologic evaluation may illustrate a spectrum of hemodynamic derangements. At first, chronic AI is largely asymptomatic, including during exercise, in the “at risk” and “progressive” stages even if cardiac changes are occurring8,9. Once the disease progresses, patients previously diagnosed with moderate or mild AI may develop new symptoms, suggesting that AI is progressing towards or has reached the “severe” stage (see Table 1). In the severe AI, multiple symptoms present themselves including heart failure symptoms, such as exertional dyspnea, palpitations, or angina, at which point valve replacement is likely the only viable treatment option8,9.
Table 1:
Condensed Criteria for Intervention in Various AI Patient Subgroups8
| AI Stage | Symptoms | Hemodynamic Findings | Potential Pathological Markers |
|---|---|---|---|
| At Risk | None | <Trace AI | Markers for ECM disorganization13,15,50:
|
| Progressive | None | RF <50% Normal LV systolic function ≤Mild LV dilation |
|
| Severe | Symptomatic: Heart failure symptoms Asymptomatic: None |
Symptomatic: LV Dilation RF ≥ 50% Asymptomatic: LV Dilation RF ≥ 50% LV systolic disfunction -LVEF <50% Severe LV dilation present under normal LV systolic function -LVEF ≥ 50%, LVESD >50mm, LVEDD >65mm |
AI-Aortic Insufficiency
ECM-Extracellular Matrix
HA-Hyaluronic Acid
LV-Left Ventricle
LVEF-Left Ventricle Ejection Fraction
LVEDD-Left Ventricle End Diastolic Diameter
LVESD-Left Ventricle End Systolic Diameter
NAR-Nuclear Aspect Ratio
RF-Regurgitant Fraction
TGF-β-Transforming Growth Factor Beta
Discussion
Pathophysiology
On a cellular level, the AV is comprised of valvular interstitial cells (VICs) and valvular endothelial cells (VECs), both of which are seeded within intricate, interactive networks of extracellular matrices. These matrices provide a scaffolding upon which local and systemic cellular and extracellular signaling cascades contribute to the active maintenance of the three layers of the AV: the fibrosa, the spongiosa, and the ventricularis10. Considering the dynamic microenvironment present within the AV, the hemodynamic changes demonstrated in patients with AI can contribute strongly to the progression of the disease, even in the setting of an asymptomatic patient. Ultimately, these hemodynamic changes to the local environment can then lead to further remodeling in the AV through an interplay between molecular, cellular, and structural signaling pathways.
Molecular Components
Before discussing cardiac remodeling further, it is important to define and identify the key proteins and signaling molecules involved in AV remodeling and regulation. At the molecular level, the AV contains various protein families that regulate and maintain valve structure and function, primarily through interactions with the extracellular matrix (ECM). Hyaluronan, a glycosaminoglycan (GAG), constitutes 60% of the total GAG content in the heart and carries out various functions including cell interactions and linking proteins with aggrecan, making it an important component of the valvular ECM11,12. Hyaluronic Acid (HA), an acidic glycosaminoglycan (AGAG), makes up roughly half the total AGAG composition in the heart. HA disruptions have been associated with both ECM remodeling and VIC activation11,13–15.
Another family of proteins implicated in AV remodeling and regulation are matricellular proteins. Matricellular proteins are a family of heterogeneous, structurally-unrelated extracellular molecules that interact with cell surface receptors, growth factors, proteases, and other bioactive effectors and serve as a link between cells and the ECM16. Matricellular proteins do not serve a role in tissue structure or architecture and expression in healthy adult cardiac tissue is normally low. However, matricellular proteins modulate cellular behavior in response to external stimuli and act as integrators of signaling cascades16. Following cardiac insult, matricellular protein expression is significantly upregulated, facilitating modulation of cell migration, adhesion, and proliferation during the wound repair process16.
In the regulation of the AV structure, the transforming growth factor beta (TGF-β) superfamily is one of the largest molecular components. TGF-β is a key cytokine involved in various biological processes including cellular proliferation and differentiation and is known to strongly affect the composition of the ECM through potent stimulation of the production and deposition of ECM components such as collagen, fibronectin, and integrins17. Additionally, TGF-β is known to signal through various pathways, depending on the isoform of TGF-β, and has been shown to be upregulated in AV development17,18. In fibrotic diseases, increased TGF-β production follows tissue injury before ECM production increases17. As a result of strong effects on ECM composition, TGF-β likely takes an active role in the development and remodeling of AI tissue.
The final major regulators of AV structure are small leucine-rich proteoglycans (SLRPs), which help trigger multiple cellular processes, and its complex signaling network provides added regulation during tissue morphogenesis, native immunity, and other functions19. Additionally, many SLRP gene family members bind to and modulate TGF-β and bone morphogenic proteins (BMPs), a member of the TGF-β superfamily and an inducer for osteoblastic differentiation19,20. The interactions with ECM components like TGF-β suggests that a wide network of different proteins interact together to maintain ECM structure and organization and show that many of these proteins could also lead to dysregulation, which ultimately may result in AI development.
Although many of these proteins, among others, are important in normal valvular organization and function, tissue injury is often a catalyst for protein overexpression, which subsequently leads to different biological processes including ECM disorganization and sustained VIC activation. Among these molecular changes, certain key proteins involved in AV regulation have been implicated in the dysregulation and remodeling of AV structure. As discussed, certain matricellular proteins, are known to have significantly upregulated expression following cardiac injury16. As matricellular proteins provide the link between the ECM and cells, potential changes in the activity of these proteins highlight the wide network of changes that could potentially affect the biomechanics of the native valve. Thrombospondin (TSP-1), SPARC, and Tenascin C (TenC) are known to be actively involved in a process of de-adhesion, by which cells undergo a change from strong adhesion to the ECM to weak adhesion16,21. Other proteins such as osteopontin (OPN) and periostin, have also been implicated in other valvular remodeling processes indicating that the family of matricellular proteins affects different mechanisms, not just a single one, despite serving similar functions16,19,22–24. As numerous matricellular proteins have been implicated in either a specific type of valvular disease or ECM remodeling, these proteins likely take an active role in the remodeling process. However, further research is required to clarify how and why these proteins alter the biomechanical function of the native valve.
As aforementioned, a major protein with significant impact on the ECM is TGF-β. Given TGF-β’s important role in ECM organization, any valvular remodeling will likely be associated with altered TGF-β expression, thereby directly impacting the molecular structure of the AV. Overexpression of TGF-β is evident in many cardiac diseases, and various proteins implicated in other pathways and mechanisms are known to activate TGF-β, including matricellular proteins TSP-1 and SPARC, as well as serotonin (5-HT)16–18,25–27. Additionally, TGF-β has been suggested to regulate VIC activation following cardiac injury, which marks the first step in the wound repair process28. Once remodeling has begun following tissue injury, protease activities are triggered, which lead to a rapid release of signaling molecules which then allows for a local, growth-mediated activation of cellular functions29. TGF-β overexpression has the ability to directly affect ECM components, making it an important protein in the initial process of cardiac remodeling, which can lead to AV structural changes.
In one specific example of valvulopathy – calcific disease – SLRPs, members of the TGF-β family, and matricellular proteins have been shown to interact together. In calcified valves, activated VICs, osteogenic growth factors, TGF-β1, BMPs, and tumor necrosis factor-alpha (TNF-α) are abundantly present20. In a recent study, TNF-α, a key inflammatory cytokine in stenotic AVs, has been associated with an increase the oxidative stress, a previously known contributor to the calcification of the AV, in the valve endothelium30. The increased oxidative stress was associated with a decrease in VEC secretion of the protective agent nitric oxide, leading to ECM disorganization30. Increased TNF-α has been shown to interact with the NF-kB pathway, which in turn elevates expression of BMP-2, an osteogenic growth factor and inducer of osteoblast differentiation31,32. BMP-2 is of clinical importance as it has been found to be overexpressed in stenotic AVs and upregulates expression of Runx2 (an osteogenic transcription factor) and OPN levels in VICs32–34. BMP-2 was also found to activate the Smad1 pathway via CD44, a transmembrane glycoprotein with a critical role in various cellular functions35. As demonstrated by Yang and colleagues, silencing this pathway reduced the expression of early osteogenic factors after BMP-2 stimulation, indicating its role in modulating the expression of these factors induced by BMP-2 stimulation32. Finally, OPN is known to also signal through integrin or CD44-mediated pathways as well as interact with decorin in order to modulate cell adhesion, gene expression, and survival as well as modulating bone turnover by inhibiting mineralization and by promoting osteoclast differentiation and activity16,36.
Another calcific mechanism caused by molecular changes revolves around lipoproteins, specifically low-density lipoprotein (LDL) and lipoprotein(a) (Lp(a)). Lrp1, an LDL receptor protein, has been found to interact with decorin, a collagen binding SLRP, and directly modulate TGF-β pathway in order to regulate ECM organization24,37. In another study decorin and bigylcan, another SLRP, were found to bind to LDL in excess, causing LDL retention in sites including the aortic wall and valve cusps24. The matricellular protein OPN has also been shown to interact with decorin, indicating a potential link between SLRPs, LDL, and matricellular proteins16,24. Finally, LDL has been found to bind to biglycan, which possesses a tissue-specific function in BMP signaling, in excess causing LDL retention in aortic wall and valve cusps19,24. Lp(a) has a structure similar to LDL and contains apolipoprotein B-100 (apoB), which when elevated, has been shown to be causally associated with an increased risk in cardiovascular disease, including in the development of aortic stenosis38,39. Lp(a) is known to increase the cholesterol deposition in the arterial wall, promote smooth muscle cell proliferation, and is a carrier of various oxidized phospholipids38.
In addition to the basic science studies discussed, various translational studies have highlighted other significant protein mediators and have also reinforced the respective roles of other known proteins that may contribute to AV dysregulation. One molecule involved prominently in AI development is angiotensin II (AngII). Activation of AngII promotes a host of responses, including cardiac hypertrophy, collagen synthesis, and significant valve thickening in different models40–43. In a study conducted by Driesbaugh and colleagues, AngII infusion showed remodeling of the mitral valve tissue via a TGF-β pathway44. Another study, conducted on apolipoprotein-E deficient mice, observed significant AV thickening upon AngII administration41. These findings indicate that AngII may have a major role in the remodeling of cardiac valves and VIC differentiation.
The rise of proteomics and knockout studies on individual proteins in mice over recent years has demonstrated that numerous different proteins can contribute to AV remodeling and AI when inactivated. In murine models, knockout of Lrp1 has been associated with age-dependent aortic root dilatation, with no cardiac abnormalities detected prior to the onset of the disease. Subsequently, secondary AI developed in these murine models which led to further cardiac remodeling and ultimately resulted in secondary cardiomyopathy45. Given Lrp1’s interactions with other proteins, Lrp1 likely represents an active and key protein involved in the calcification process of the AV. In multiple studies conducted by Théron and colleagues, the Krox20 protein was inactivated leading to both VIC alteration and ECM disorganization46,47. As a result, it was concluded that complete Krox20 expression is likely implicated in AV development. Additionally, the AVs in these Krox20 knockout models were found to contain significant AV thickening46,47. In another murine model, inactivation of the retinoblastoma protein tumor suppressor (pRb) in the AV led to increased cusp thickness and AI development, likely due to ECM disorganization48. Although these knockout models provide significant findings of possible proteins involved in AI mechanisms, it is important to remember the limitations that murine models carry, including anatomical differences to humans, and further research is required to verify the role of these proteins in human AI development as well.
Given the wide array of genomic and proteomic pathways involved in AV dysregulation (See Figure 1), primarily through ECM remodeling, it becomes clear that many more genes and proteins likely contribute to this process as well. Additionally, other proteins may contribute to other different pathological mechanisms implicated in AI development, such as myxomatous valve degeneration or calcification.
Figure 1:
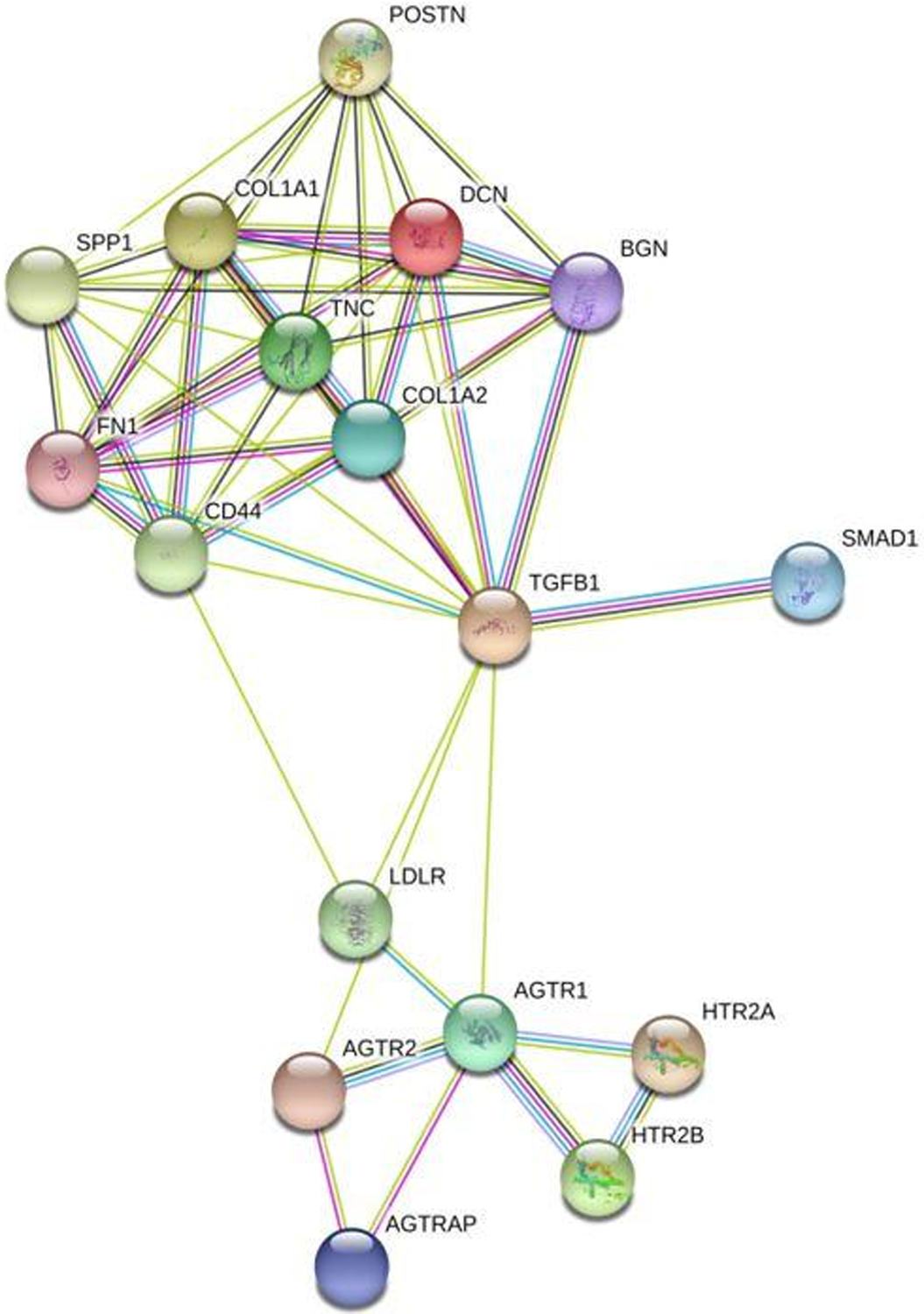
Network of AI-associated proteins created on https://string-db.org/. The lines highlight interactions between different proteins: Green lines are proteins interactions that have been associated with AI in various studies. Blue lines are known interactions found in curated databases. Pink lines represent proteins interactions that have been experimentally determined. Black lines represent gene co-expression. Purple lines indicate homologous proteins. Abbreviations are as follows: TGFB1, TGF- β; LDLR, LDL Receptor; HTR2B and HTR2A, 5-HT Receptors 2A and 2B; AGTRAP, AngII Receptor Associated Protein; AGTR1 and AGTR2, AngII Receptors Type 1 and Type 2; SPP1, Osteopontin; DCN, Decorin; BGN, Biglycan; POSTN, Periostin; TNC, Tenascin C; FN1, Fibronectin; COL1A1, Collagen Type 1 Alpha 1 101.
Cellular Components
As has been briefly mentioned, cellular regulation in the AV focuses around VICs. VICs are a specific population of cell types found in cardiac valves49. VICs have been found to possess the ability to sense and respond to their micromechanical environment in order to achieve valve homeostasis and during disease development10. VICs are responsible for valvular ECM maintenance through the synthesis and secretion of ECM components as well as through the secretion of matrix metalloproteinases (MMPs) and their inhibitors TIMPs49. As VICs are sensitive to their micromechanical environment, VIC deformation plays a crucial role in the regulation of the biosynthetic activity of the cells in addition to heart valve repair and remodeling mechanisms50. Studies have also shown that changes in the nuclear aspect ratio (NAR), a metric used for overall cell shape, were closely associated with geometric changes that occur in early pregnancy50,51 showing a concrete link between structural remodeling and VIC changes.
VICs become active once they are exposed to altered biomechanical stimuli or valvular insult28,44. Once activated, VICs exhibit the phenotype of myofibroblasts (through the expression of smooth muscle α-actin) or resemble osteoblast-like cells, through BMP-mediated processes already discussed10,32,49. This alteration in phenotype is typically associated with ECM remodeling, and the functional properties of activated VICs depend on which phenotype they exhibit44. For instance, collagen-secreting myofibroblast-like cells (activated VICs) could synthesize and secrete excessive amounts of ECM components, especially collagen, which could lead to increased tissue stiffness29. As VICs are sensitive to their local environments, changes in the strains they are exposed to directly affects their function. Due to the anatomical location of both aortic and mitral VICs, they are exposed to greater strains and transvalvular pressure than other valves, causing these VICs to have increased stiffness and higher collagen production50,52. Additionally, increasing the level of strain on mitral VICs, which are similar in structure to aortic VICs, have been found to cause VIC activation50,53. In diseased valves, VICs may also lose their homeostatic equilibrium and maintain an activated state54.
In addition to VICs, VECs populate the AV cusps. VECs are a highly proliferative endothelial population with specific functions in both AV disease and development, including valve homeostasis55. VECs cover valve cusps in a single layer on the ventricularis and fibrosa sides to serve as a physical barrier between the hemodynamic environment and VICs56,57. Given this location, VECs are exposed to shear stress as well as circulating signaling molecules, including cholesterol56. Since VICs are not directly exposed to these molecules, yet still have pathological responses to them, the role of VECs may be to mediate signaling between the hemodynamic environment and VICs as well as protect VICs from these molecular risk factors56. Furthermore, nitric oxide secreted by VECs has been shown to be a natural antagonist of calcific aortic valve disease (CAVD) pathogenesis by decreasing VIC activation (both osteoblastic and myofibroblastic) and matrix calcification, further highlighting the signaling mechanism between VICs and VECs58,59. VECs are also known to undergo an endothelial to mesenchymal transformation, resembling myofibroblastic VICs, after which they have been shown to affect collagen arraignment and possibly affect ECM organization as well through expression of MMP-9, TGF-β1, Notch1 and BMP-460,61. Additionally, the VEC transformation has been shown to be stimulated by TNF-α exposure60. Therefore, signaling between VICs and VECs has been found to be essential in VIC function during valve development and homeostasis, further corroborated by a VEC-specific TGF-β1 inactivation study56, in which endothelial cells were found to protect VICs against calcification through Sox9 nuclear localization maintenance55,56,61. In the same study, the loss of VEC-specific TGF- β1 signaling led to reduced Sox9 expression and increased calcification56. VEC function in protecting VICs as well as inhibiting VIC activation highlights a key mechanism that could lead to the onset of AI and other AV diseases. The signaling mechanism between the two suggests that there is a wide network of associated pathways and proteins that affect valvular remodeling through cellular interactions.
Extracellular Matrix Components
Following the cellular changes in the valve, the next level of structure involved in the remodeling process is the ECM. The ECM is an important regulator of cell and tissue function and may be defined as the diverse collection of proteins and sugars that surrounds cells in all solid tissues29,62. The ECM also plays a crucial role in wound healing and organ homeostasis, and any disorganization or dysregulation can result in potentially fatal pathological conditions29,62. The organized valvular ECM makes up the cusps of the valve and contains three overlapping layers, each with distinct properties: the ventricularis, the spongiosa, and the fibrosa63,64. Valve ECM is composed of a dense network of collagen, elastin, and glycosaminoglycans (GAGs), making it functionally and mechanically different from other cardiovascular structures65.
As already mentioned, changes in ECM are critical in AI development and changes in the local environment of the valve are known to lead to disorganization and remodeling of the valvular ECM in order to maintain hemostasis and/or to recover from any insult20,29,47,49. As discussed, certain proteins and pathways are known to trigger or greatly affect these changes in the ECM, including TGF-β. ECM disorganization has also been implicated in valve disease through ECM-related gene mutations, however, their mechanisms are not known66. Importantly, VICs are responsible for the synthesis and secretion of ECM components, highlighting an important connection between how individual cells can greatly affect their surrounding structure, which in turn can affect the entire valve structure through cardiac remodeling. Given the highly interconnected network of proteins and pathways involved in ECM maintenance that have been implicated in AI development, it is safe to suggest that the ECM changes are at the center of the network from which valve remodeling occurs.
Cardiac Remodeling
Due to the interplay between these molecular, cellular, and structural changes, remodeling can dramatically alter the mechanical and functional properties of the valve. For instance, physiologic changes to heart valves occur during pregnancy to maintain valve function in a setting of increased hydrodynamic stress. In a bovine model, the mitral valve in pregnant cows were shown to undergo substantial remodeling resulting in structural and biochemical changes that dramatically alter valve leaflet tissue morphology51. It was shown that changes in collagen phase of leaflet tissue change during pregnancy, however, following the end of pregnancy, these processes reversed themselves and the valve regained relatively normal structure and function, despite the valve’s ECM not returning to a pre-pregnant state51. Given these results, it becomes clear that valve remodeling is an extremely active process which facilitates the ability to return to normal function following reversal in changes to its mechanical environment. It is important to note that this study conducted by Rego and colleagues was done on the mitral valve, and additional research is necessary to demonstrate these findings in the AV.
Cardiac remodeling resulting in AI can manifest itself in various ways, such as prolapse, degeneration, and calcification, which can result in changes in the hemodynamic properties of the valve. Important clinical pathways have been found to remodel the valve, including through calcification and LDL cholesterol, which has been well documented to be associated with atherosclerosis, which in turn has been shown to be strongly associated with the development and progression of CAVD67–71. Given the previously discussed molecular pathways and interactions with LDL and associated proteins, LDL may represent an important integrative clinical pathway in remodeling. Furthermore, the structurally similar protein Lp(a) has been shown, when elevated, to be strongly associated with aortic stenosis development as well as aortic valve calcium deposition38,72,73. Other structural results of AV remodeling include myxomatous degeneration which can result in valve prolapse, as previously been documented in the mitral valve44.
One particular pathway of interest that has been implicated in multiple AI studies is the 5-HT pathway. Given 5-HT’s role in treatment for other diseases, including but not limited to neurodegenerative diseases like Parkinson disease and Huntington disease as well as several highly-prevalent mental health disorders such as depression and anxiety, any cardiac remodeling involving 5-HT represents clinically significant processes with important ramifications. The 5-HT pathway has been implicated in both AI and mitral valve mechanisms, including VIC activation and ECM remodeling15,26,40,44. 5-HT signaling in VICs has been shown to interact with AngII, suggesting that 5-HT has an active impact on the AngII pathway already described which has been shown to affect valve structure40. Additionally, various translational studies have shown similar results. In a rat model, long-term 5-HT administration was found to induce carcinoid syndrome74 and there is strong evidence of association between 5-HT and gradual thickening of the leaflets followed by fibrosis and retraction of the valve40. In another study done in a sheep model, 5-HT was shown to upregulate TGF-β1 in aortic VICs, which resulted in increased amounts of both active and latent TGF-β115, which has been discussed to be involved in various valve diseases. Additionally, 5-HT2 receptor signaling has been associated with increased VIC mitogenesis and increased production of ECM proteins44. In particular, 5-HT2A and 5-HT2B have also been heavily linked to valve function and degeneration. 5-HT2B is thought to have an active role in cardiac development and the regulation of cardiovascular functions, and has been found to be expressed throughout cardiac tissue, as seen in various studies40,75,76. As shown by Driesbaugh and colleagues, these receptors have active roles in myxomatous mitral valve prolapse44. In particular, 5-HT2 receptors A and B have been implicated to have active roles in myxomatous mitral valve prolapse44. Additionally, another study found AI to be induced by a myxomatous degeneration of the cusps due to irregular accumulations of myxoid materials in the spongiosa and fibrosa layer of the valve77.
Given the various interconnected pathways, changes in VICs, including activation and alteration, have direct effects on ECM composition and the proteins in the ECM. The synthesis and secretion of these components, such as TGF-β, can lead to ECM disorganization, which has been implicated in multiple studies described previously as a key component to AI development across a wide range of models. Given the clinical significance of certain common pathways in AI progression, these pathological mechanisms that manifest themselves through cardiac remodeling processes across all types of AI (see Figure 2) have large implications on patient treatment.
Figure 2:
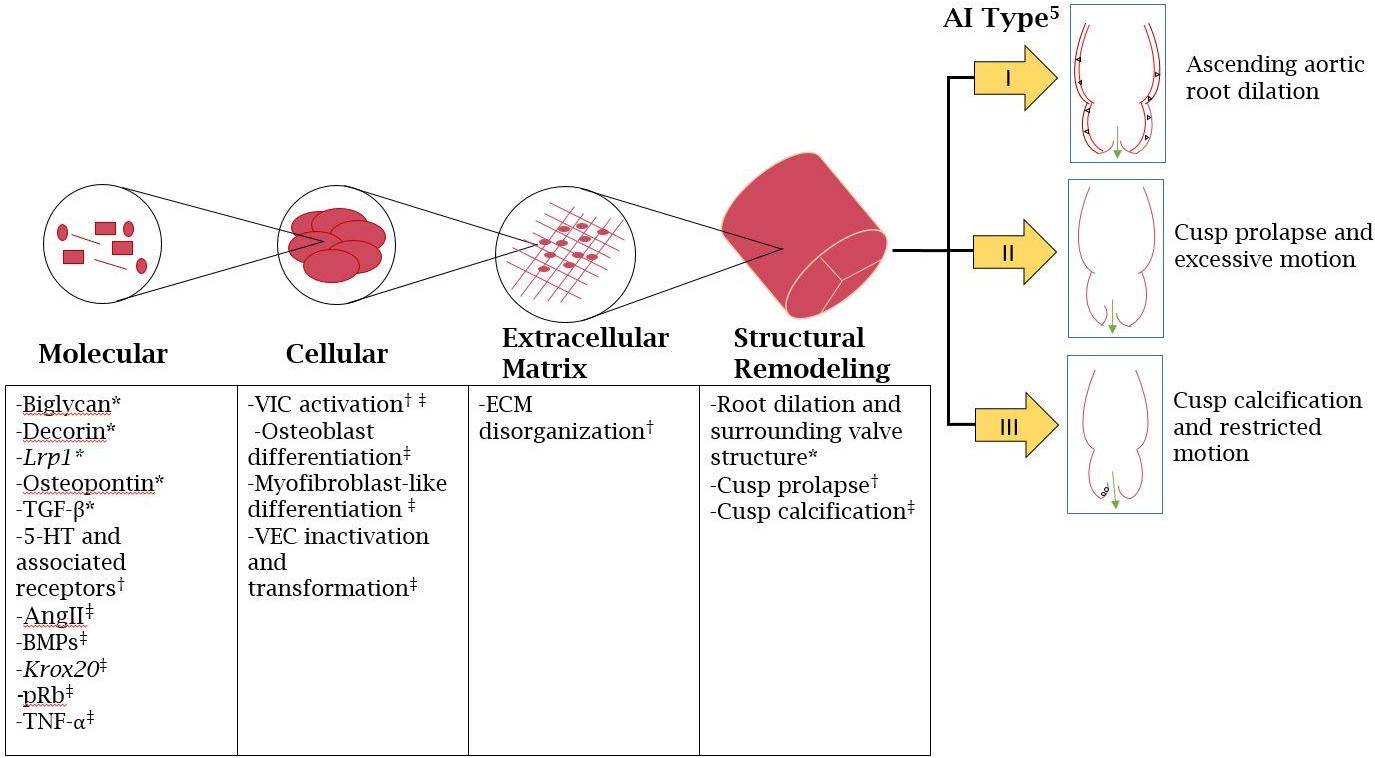
Schematic depiction of molecular, cellular, and extracellular signaling pathways and the subsequent structural remodeling, as per the El Khoury classification 5. *-Associated with Type I, †-Associated with Type II, ‡-Associated with Type III.
Management
Due to the largely asymptomatic natural history of the disease, management of AI is largely surgical in nature and frequently necessitates AV replacement or repair. However, given the wide network of proteins, pathways, and structural changes that occur in AI development, innovative ways to manage the disease should be researched (See Table 2). For example, certain proteins and structural changes could be used as markers of ECM disorganization, including HA, TGF-β, matricellular proteins, and NAR ratio13,15,50, which could guide clinical management.
Table 2:
Molecular, Cellular, and Structural Changes per Class5 of Aortic Insufficiency (AI), and Implications For Future Therapy
| AI Class | I | II | III |
|---|---|---|---|
| Current Surgical Approach(es) | Valve sparing root remodeling/replacement | Prolapse repair Surgical valve replacement |
Leaflet repair Surgical valve replacement |
| Molecular Involvement | Biglycan24 Decorin16,24,37 Lrp124,37,45 Osteopontin16 TGF-β24,37 |
5-HT2 Receptors44 | 5-HT40 AngII41 Biglycan19,24 BMP and BMP-232–35 Krox2046,47 Lp(a)38,72,73 Osteopontin16 pRb48 TNF-α20,30–32 |
| Cellular Involvement | VIC activation44 | VIC activation Osteoblast differentiation16,19,24,32–35 Myofibroblast-like differentiation29 VEC inactivation55,56,61 VEC transformation60,61 |
|
| Valve Extracellular Matrix Involvement | ECM disorganization due to VIC activation44 | ECM disorganization30,46–48 | |
| Larger Pathways Implicated | LDL | 5-HT44 | CAVD67–71 LDL67,69–71 5-HT40 |
| Remodeling Location(s) and Mechanisms | Root dilation and surrounding valve structure | Cusp prolapse/coaptation | Cusp calcification and thickening |
| Proposed Future Treatment* | Valve sparing root remodeling/replacement | 5-HT antagonists80,81 TAVR |
5-HT antagonists80,81 ACE inhibitors45, 78,79 TAVR |
5-HT-Serotonin
AngII-Angiotensin II
BMP-Bone Morphogenic Protein
CAVD -Calcific Aortic Valve Disease
ECM-Extracellular Matrix
LDL-Low Density Lipoprotein
Lp(a)- Lipoprotein (a)
Lrp1-Low Density Lipoprotein Receptor-Related Protein 1
pRb-Retinoblastoma Protein Tumor Suppressor
TGF-β-Transforming Growth Factor Beta
TNF-α-Tumor Necrosis Factor Alpha
VEC-Valve Endothelial Cell
VIC-Valve Interstitial Cell
Further Research Necessary
Medical Regimens
Certain medications have been shown to actively affect pathways and proteins implicated in AI development and cardiac remodeling. For example, angiotensin-converting enzyme (ACE) generates AngII, which has been shown to lead to cusp thickening40–43. ACE inhibitors would block synthesis of AngII, potentially limiting the thickening of valve cusps78. However, no clinical trials have shown ACE inhibitors to be effective in targeting this valvular mechanism. ACE inhibitors have been suggested to affect disease progression and treatment through cardiac remodeling mechanisms. One study utilizing an LRP1 murine knockout model of secondary AI demonstrated phenotypic improvement with ACE inhibitor administration45. Another study found that administering captopril to patients mitigated the deleterious effects of AI by reducing left ventricle volume overload, lowering regurgitation fraction, and reduction of AngII levels79. Given the unclear role of ACE inhibitors in the mitigation of AI, additional prospective research is warranted.
Additionally, for the 5-HT pathway, certain 5-HT2 receptor antagonists have been suggested to mitigate valvular changes caused by 5-HT. In a study by Droogmans and colleagues, administration of the 5-HT2B antagonist cyproheptadine was found to prevent the development of the disease in a rat model, following Pergolide administration80. Additionally, another study found that the 5-HT2B atagonist Terguride was suggested to prevent 5-HT induced heart damage in a rat model81. Although certain studies have been shown to reduce disease and target these pathological mechanisms, medical treatment for AI patients requires more extensive research to understand if it represents a viable treatment option.
Despite these promising studies, other drugs have been shown to not be as effective in treating valvular remodeling pathways. For example, in a clinical trial examining patients with mild-to-moderate asymptomatic aortic stenosis, a combination of simvastatin and ezetimibe were shown to lower LDL cholesterol by an average of 50%, however, over a longer period of time no overall reduction of disease progression was observed, questioning the targeting of the LDL pathway82. As a result, further study is needed to understand how the LDL can be targeted medically to treat valvular diseases. In the absence of effective medical management of AI, treatment is largely dependent upon surgical or percutaneous intervention.
Surgical Treatment
For decades, the gold standard of AI treatment has traditionally been complete valve replacement, with either a biological or mechanical aortic prosthesis. Currently, the AHA guidelines recommend complete AV replacement in the majority of patients. Valve repair is also a viable option in select patients, and periodic monitoring is recommended for two at-risk patient cohorts: those presenting with progressive AI with no prior cardiac surgery, and those with asymptomatic severe AI (LVEF ≥ 50%, left ventricle end-systolic dimension (LVESD) ≤ 50 mm, and left ventricle end-diastolic dimension ≤ 65 mm)8. Innovative procedures such as valve-sparing aortic root replacement and AV repair are becoming more commonplace, allowing for the patient’s native valve to remain in place and avoid the need for lifelong anticoagulation in the case of mechanical valve implantation or further reoperations due to prosthetic valve degeneration7.
Several studies have shown that AV repair procedures have the potential to improve patient outcomes following AV disease with high survival rates6,83,84. As the mechanotransductive properties of VICs are changed during valve-sparing and valve-repair procedures, the fibrosa NAR levels could be of clinical significance in future AI studies as well to help identify patients at risk for further remodeling following surgical intervention. As shown by Ayoub and colleagues, valve repair was found to reduce circumferential strain and is estimated to reduce the fibrosa NAR53. However, certain advanced manifestations of AI cannot be repaired. For example, the mechanisms causing calcification can restrict the movement of the cusps and may require complete replacement if there is a lack of pliable tissue that can preclude a satisfactory repair6,7.
Despite surgical repair and replacement still being the major treatment for AI, the rise of percutaneous treatment has allowed for faster recovery times for patients as well as successful treatment options for high risk surgical candidates. Percutaneous mitral valve repair has been well-documented, however the experience with such techniques and technologies in the aortic position have been limited. In the EVEREST II study, MitraClip placement in high-risk patients with severe mitral regurgitation was associated with a significant improvement in NYHA functional class at both 30 days and 12 months relative to baseline in most patients, while mitral regurgitation grade was also improved at 12 months following placement85. Prior success of surgical AV repair, in conjunction with the continued sophistication of percutaneous valve techniques and technologies, may bolster the feasibility of percutaneous AV repair in select patients86–88.
As transcatheter aortic valve replacement (TAVR) has gained more traction as a successful alternative to surgical AV replacement following publication of PARTNER 3 trial89, its role in treating AI patients has been debated. Using older generation devices, including CoreValve and Sapien XT devices, AI was associated with increased complications for patients undergoing TAVR due to increased risk for embolization, valve migration, and post-procedural regurgitation87,90. This risk is accentuated by the lack of calcification present in patients with AI, as opposed to aortic stenosis TAVR patients, making device placement difficult as TAVR valves are anchored into place by native valve calcification86,90. Given the difficult management of associated aortic root diseases and necessity of large valve sizes, treating native AI with TAVR has been fairly limited91. However, newer generation transcatheter heart valves, including Evolut R, Sapien 3, JenaValve, Lotus, Direct Flow, Acurate, Portico, and J-Valve, have shown to be more effective and improve outcomes in treating native AI when compared to older generation devices due to improved anchoring mechanisms and repositioning capability86,87,90. The JenaValve, for example, has demonstrated efficacy in the management of native AI by utilizing a calcification-independent anchoring mechanism92. High-risk patients with native AI, including those with significant comorbidities and advanced age that preclude a safe open surgical intervention constitute a population that may benefit from a less invasive procedure such as TAVR93. In fact, a systematic review conducted by Wernly et al. demonstrated that TAVR in high-risk candidates with native AI obtained a 90% procedural success rate94. For patients with failing prosthetic AVs that result in AI, TAVR has been shown to be a viable option as well, with the original, surgically implanted valve providing better anchoring and visualization for TAVR as well as positive 30-day clinical outcomes90. Additionally, several “valve-in-valve” cases have been reported where percutaneous valve implantation has successfully replaced degenerated bioprosthesis, for example as a result of prolapse95 and suspected endocarditis96 which had caused severe AI, with strong hemodynamic outcomes reported97–99. Additionally, direct access surgical valve-in-valve procedures have also been shown to be feasible in cases where percutaneous intervention is contraindicated100. Despite these positive results, TAVR treatment in young patients would still likely necessitate a further reoperation due to bioprosthetic degeneration, unlike a surgical repair. Therefore, the effectiveness of TAVR as a treatment option for native AI in certain patient populations should still be questioned. With continued improvement in percutaneous techniques and technologies as well as increased outcomes data from such procedures, the utility TAVR for patients with native AI demands frequent reappraisal.
Conclusions
With the discovery of various pathways and mechanisms for different types of AI, the next step must be to reevaluate the current classifications and guidelines. Although this review attempts to cover many of the known mechanisms already implicated, many more exist and more are not yet known. Additionally, ECM remodeling and VIC activation also require further research in order to solidify our understanding of valvular remodeling. New questions must be asked in order to determine if all patients necessarily require surgical treatment or if other medical treatments can be found that could limit, stop, or even reverse the effects of remodeling in the progression of AI, which could delay any surgical intervention. If surgical treatment is necessary, researching these pathological mechanisms is essential to understand why AI may preclude durable replacement as well as to expand accurate patient selection classifications for different procedures. Consequently, it is essential to continue pursuing further characterization of the pathophysiologic mechanisms underlying AI, and to further elucidate both modifiable and non-modifiable risk factors as well as novel strategies for disease mitigation. In the interim, surgical management remains the gold standard for AI management, and further evolution of percutaneous and techniques for AV repair and replacement will inevitably allow for decreased recovery times and hospital stays, and will furthermore offer surgical intervention for patients who are traditionally considered too high-risk for more invasive procedures.
Funding Sources
This work was supported by the following research grants and funds: National Institutes of Health [R01-HL131872, R01-HL143008, and R01-HL122805 to GF and 5T32HL007854-24 to APK], The Valley Hospital Foundation “Marjorie C Bunnel” charitable fund [to GF and JG] and The Kibel Fund for Aortic Valve Research.
Abbreviations
- AGAG
Acidic Glycosaminogylcan
- AngII
Angiotensin II
- AI
Aortic Insufficiency
- AV
Aortic Valve
- BMP
Bone Morphogenic Protein
- CAVD
Calcific Aortic Valve Disease
- ECM
Extracellular Matrix
- GAG
Glycosaminoglycan
- HA
Hyaluronic Acid
- LVEF
Left Ventricle Ejection Fraction
- LV
Left Ventricle
- Lpa(a)
Lipoprotein (a)
- LDL
Low Density Lipoprotein
- NAR
Nuclear Aspect Ratio
- OPN
Osteopontin
- 5-HT
Serotonin
- SLRP
Small Leucine-Rich Proteins
- TenC
Tenascin C
- TSP-1
Thrombospondin
- TAVR
Transcatheter Aortic Valve Replacement
- TGF-β
Transforming Growth Factor Beta
- TNF- α
Tumor Necrosis factor Alpha
- VIC
Valve Interstitial Cells
- VEC
Valve Endothelial Cells
Footnotes
Disclosures statement
The authors report no conflicts of interest.
References
- 1.Bekeredjian R, Grayburn PA. Valvular Heart Disease. Circulation. 2005;112(1):125 LP–134. [DOI] [PubMed] [Google Scholar]
- 2.Maurer G Aortic regurgitation. Heart. 2006;92(7):994–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh JP, Evans JC, Levy D, et al. Prevalence and clinical determinants of mitral, tricuspid, and aortic regurgitation (the Framingham Heart Study). The American Journal of Cardiology. 1999;83(6):897–902. [DOI] [PubMed] [Google Scholar]
- 4.Azevedo PS, Polegato BF, Minicucci MF, Paiva SAR, Zornoff LAM. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arquivos Brasileiros de Cardiologia. 2016;106(1):62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boodhwani M, de Kerchove L, Glineur D, et al. Repair-oriented classification of aortic insufficiency: Impact on surgical techniques and clinical outcomes. The Journal of Thoracic and Cardiovascular Surgery. 2009;137(2):286–294. [DOI] [PubMed] [Google Scholar]
- 6.Antoniou A, Harky A, Bashir M, El Khoury G. Why I choose to repair and not to replace the aortic valve? General Thoracic and Cardiovascular Surgery. 2018. [DOI] [PubMed] [Google Scholar]
- 7.Al-Atassi T, Boodhwani M. Aortic valve insufficiency in aortic root aneurysms: consider every valve for repair. Journal of Visualized Surgery. 2018;4:60–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Journal of the American College of Cardiology. 2014;63(22):2438–2488. [DOI] [PubMed] [Google Scholar]
- 9.Rosengart T, Anand J. Acquired heart disease: valvular. Sabiston Textbook of Surgery 20th ed Philadelphia, PA: Elsevier; 2017. [Google Scholar]
- 10.Wyss K, Yip CYY, Mirzaei Z, Jin X, Chen J-H, Simmons CA. The elastic properties of valve interstitial cells undergoing pathological differentiation. Journal of Biomechanics. 2012;45(5):882–887. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez KJ, Piechura LM, Masters KS. Regulation of Valvular Interstitial Cell Phenotype and Function by Hyaluronic Acid in 2-D and 3-D Culture Environments. Matrix biology : journal of the International Society for Matrix Biology. 2011;30(1):70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andhare RA, Takahashi N, Knudson W, Knudson CB. Hyaluronan Promotes the Chondrocyte Response to BMP-7. Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society. 2009;17(7):906–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riessen R, Wight TN, Pastore C, Henley C, Isner JM. Distribution of Hyaluronan During Extracellular Matrix Remodeling in Human Restenotic Arteries and Balloon-Injured Rat Carotid Arteries. Circulation. 1996;93(6):1141 LP–1147. [DOI] [PubMed] [Google Scholar]
- 14.Murata K Acidic glycosaminoglycans in human heart valves. Journal of Molecular and Cellular Cardiology. 1981;13(3):281–292. [DOI] [PubMed] [Google Scholar]
- 15.Jian B, Xu J, Connolly J, et al. Serotonin Mechanisms in Heart Valve Disease I: Serotonin-Induced Up-Regulation of Transforming Growth Factor-β1 via G-Protein Signal Transduction in Aortic Valve Interstitial Cells. The American Journal of Pathology. 2002;161(6):2111–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frangogiannis NG. Matricellular Proteins in Cardiac Adaptation and Disease. Physiological Reviews. 2012;92(2):635–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blobe GC, Schiemann WP, Lodish HF. Role of Transforming Growth Factor β in Human Disease. New England Journal of Medicine. 2000;342(18):1350–1358. [DOI] [PubMed] [Google Scholar]
- 18.Azhar M, Schultz JEJ, Grupp I, et al. Transforming growth factor beta in cardiovascular development and function. Cytokine & Growth Factor Reviews. 2003;14(5):391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaefer L, Iozzo RV. Biological Functions of the Small Leucine-rich Proteoglycans: From Genetics to Signal Transduction. The Journal of Biological Chemistry. 2008;283(31):21305–21309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez KJ, Masters KS. Regulation of Valvular Interstitial Cell Mineralization by Components of the Extracellular Matrix. Journal of biomedical materials research Part A. 2009;90(4):1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy-Ullrich JE. The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? The Journal of Clinical Investigation. 2001;107(7):785–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stansfield WE, Andersen NM, Tang R-H, Selzman CH. Periostin Is a Novel Factor in Cardiac Remodeling After Experimental and Clinical Unloading of the Failing Heart. The Annals of thoracic surgery. 2009;88(6):1916–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norris RA, Moreno-Rodriguez RA, Sugi Y, et al. PERIOSTIN REGULATES ATRIOVENTRICULAR VALVE MATURATION. Developmental biology. 2008;316(2):200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neufeld EB, Zadrozny LM, Phillips D, Aponte A, Yu Z-X, Balaban RS. Decorin and biglycan retain LDL in disease-prone valvular and aortic subendothelial intimal matrix. Atherosclerosis. 2014;233(1):113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sweetwyne MT, Murphy-Ullrich JE. Thrombospondin1 in tissue repair and fibrosis: TGF-β-dependent and independent mechanisms. Matrix Biology. 2012;31(3):178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Balachandran K, Hussain S, Yap C-H, Padala M, Chester AH, Yoganathan AP. Elevated cyclic stretch and serotonin result in altered aortic valve remodeling via a mechanosensitive 5-HT2A receptor-dependent pathway. Cardiovascular Pathology. 2012;21(3):206–213. [DOI] [PubMed] [Google Scholar]
- 27.Xu J, Jian B, Chu R, et al. Serotonin Mechanisms in Heart Valve Disease II: The 5-HT2 Receptor and Its Signaling Pathway in Aortic Valve Interstitial Cells. The American Journal of Pathology. 2002;161(6):2209–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu AC, Gotlieb AI. Transforming Growth Factor-β Regulates in Vitro Heart Valve Repair by Activated Valve Interstitial Cells. The American Journal of Pathology. 2008;173(5):1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Disease Models & Mechanisms. 2011;4(2):165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farrar EJ, Huntley GD, Butcher J. Endothelial-derived oxidative stress drives myofibroblastic activation and calcification of the aortic valve. PLoS One. 2015;10(4):e0123257–e0123257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu Z, Seya K, Daitoku K, Motomura S, Fukuda I, Furukawa K-I. Tumor Necrosis Factor-α Accelerates the Calcification of Human Aortic Valve Interstitial Cells Obtained from Patients with Calcific Aortic Valve Stenosis via the BMP2-Dlx5 Pathway. Journal of Pharmacology and Experimental Therapeutics. 2011;337(1):16 LP–23. [DOI] [PubMed] [Google Scholar]
- 32.Yang X, Meng X, Su X, et al. Bone morphogenic protein 2 induces Runx2 and osteopontin expression in human aortic valve interstitial cells: Role of Smad1 and extracellular signal-regulated kinase 1/2. The Journal of Thoracic and Cardiovascular Surgery. 2009;138(4):1008–1015.e1001. [DOI] [PubMed] [Google Scholar]
- 33.Song R, Zeng Q, Ao L, et al. Biglycan induces the expression of osteogenic factors in human aortic valve interstitial cells via Toll-like receptor 2. Arteriosclerosis, thrombosis, and vascular biology. 2012;32(11):2711–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang X, Fullerton DA, Su X, Ao L, Cleveland JC, Meng X. Pro-Osteogenic Phenotype of Human Aortic Valve Interstitial Cells Is Associated With Higher Levels of Toll-Like Receptors 2 and 4 and Enhanced Expression of Bone Morphogenetic Protein 2. Journal of the American College of Cardiology. 2009;53(6):491–500. [DOI] [PubMed] [Google Scholar]
- 35.Tanikawa R, Tanikawa T, Hirashima M, Yamauchi A, Tanaka Y. Galectin-9 induces osteoblast differentiation through the CD44/Smad signaling pathway. Biochemical and Biophysical Research Communications. 2010;394(2):317–322. [DOI] [PubMed] [Google Scholar]
- 36.Chellaiah MA, Hruska KA. The Integrin {alpha}v{beta}3 and CD44 Regulate the Actions of Osteopontin on Osteoclast Motility. Calcified Tissue International. 2003;72(3):197–205. [DOI] [PubMed] [Google Scholar]
- 37.Cabello-Verrugio C, Brandan E. A Novel Modulatory Mechanism of Transforming Growth Factor-β Signaling through Decorin and LRP-1. Journal of Biological Chemistry. 2007;282(26):18842–18850. [DOI] [PubMed] [Google Scholar]
- 38.Gencer B, Kronenberg F, Stroes ES, Mach F. Lipoprotein(a): the revenant. European Heart Journal. 2017;38(20):1553–1560. [DOI] [PubMed] [Google Scholar]
- 39.Tsimikas S A test in context: lipoprotein (a): diagnosis, prognosis, controversies, and emerging therapies. Journal of the American College of Cardiology. 2017;69(6):692–711. [DOI] [PubMed] [Google Scholar]
- 40.Goldberg E, Grau JB, Fortier JH, Salvati E, Levy RJ, Ferrari G. Serotonin and catecholamines in the development and progression of heart valve diseases. Cardiovascular Research. 2017;113(8):849–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujisaka T, Hoshiga M, Hotchi J, et al. Angiotensin II promotes aortic valve thickening independent of elevated blood pressure in apolipoprotein-E deficient mice. Atherosclerosis. 2013;226(1):82–87. [DOI] [PubMed] [Google Scholar]
- 42.Hingtgen SD, Tian X, Yang J, et al. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiological Genomics. 2006;26(3):180–191. [DOI] [PubMed] [Google Scholar]
- 43.Xie C, Shen Y, Hu W, Chen Z, Li Y. Angiotensin II promotes an osteoblast-like phenotype in porcine aortic valve myofibroblasts. Aging clinical and experimental research. 2016;28(2):181–187. [DOI] [PubMed] [Google Scholar]
- 44.Driesbaugh KH, Branchetti E, Grau JB, et al. Serotonin receptor 2B signaling with interstitial cell activation and leaflet remodeling in degenerative mitral regurgitation. Journal of Molecular and Cellular Cardiology. 2018;115:94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Basford JE, Koch S, Anjak A, et al. Smooth Muscle LDL Receptor-Related Protein-1 Deletion Induces Aortic Insufficiency and Promotes Vascular Cardiomyopathy in Mice. PLoS ONE. 2013;8(11):e82026–e82026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Odelin G, Faure E, Kober F, et al. Loss of Krox20 results in aortic valve regurgitation and impaired transcriptional activation of fibrillar collagen genes. Cardiovascular Research. 2014;104(3):443–455. [DOI] [PubMed] [Google Scholar]
- 47.Théron A, Odelin G, Faure E, Avierinos J-F, Zaffran S. Krox20 heterozygous mice: A model of aortic regurgitation associated with decreased expression of fibrillar collagen genes. Archives of Cardiovascular Diseases. 2016;109(3):188–198. [DOI] [PubMed] [Google Scholar]
- 48.Freytsis M, Baugh L, Liu Z, et al. Conditional deletion of RB1 in the Tie2 lineage leads to aortic valve regurgitation. PLoS ONE. 2018;13(1):e0190623–e0190623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor PM, Batten P, Brand NJ, Thomas PS, Yacoub MH. The cardiac valve interstitial cell. The International Journal of Biochemistry & Cell Biology. 2003;35(2):113–118. [DOI] [PubMed] [Google Scholar]
- 50.Khang A, Buchanan RM, Ayoub S, et al. Chapter 8 - Mechanobiology of the heart valve interstitial cell: Simulation, experiment, and discovery In: Verbruggen SW, ed. Mechanobiology in Health and Disease. Academic Press; 2018:249–283. [Google Scholar]
- 51.Rego BV, Wells SM, Lee C-H, Sacks MS. Mitral valve leaflet remodelling during pregnancy: insights into cell-mediated recovery of tissue homeostasis. Journal of The Royal Society Interface. 2016;13(125):20160709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sakamoto Y, Buchanan RM, Sacks MS. On intrinsic stress fiber contractile forces in semilunar heart valve interstitial cells using a continuum mixture model. Journal of the Mechanical Behavior of Biomedical Materials. 2016;54:244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ayoub S, Lee C-H, Driesbaugh KH, et al. Regulation of valve interstitial cell homeostasis by mechanical deformation: implications for heart valve disease and surgical repair. Journal of The Royal Society Interface. 2017;14(135):20170580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang H, Leinwand LA, Anseth KS. Cardiac valve cells and their microenvironment--insights from in vitro studies. Nat Rev Cardiol. 2014;11(12):715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu B, Wang Y, Xiao F, Butcher JT, Yutzey KE, Zhou B. Developmental Mechanisms of Aortic Valve Malformation and Disease. Annual Review of Physiology. 2017;79(1):21–41. [DOI] [PubMed] [Google Scholar]
- 56.Huk DJ, Austin BF, Horne TE, et al. Valve Endothelial Cell-Derived Tgfβ1 Signaling Promotes Nuclear Localization of Sox9 in Interstitial Cells Associated With Attenuated Calcification. Arteriosclerosis, thrombosis, and vascular biology. 2016;36(2):328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gomel MA, Lee R, Grande-Allen KJ. Comparing the Role of Mechanical Forces in Vascular and Valvular Calcification Progression. Front Cardiovasc Med. 2019;5:197–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Richards J, El-Hamamsy I, Chen S, et al. Side-Specific Endothelial-Dependent Regulation of Aortic Valve Calcification: Interplay of Hemodynamics and Nitric Oxide Signaling. The American Journal of Pathology. 2013;182(5):1922–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gould ST, Matherly EE, Smith JN, Heistad DD, Anseth KS. The role of valvular endothelial cell paracrine signaling and matrix elasticity on valvular interstitial cell activation. Biomaterials. 2014;35(11):3596–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farrar EJ, Butcher JT. Heterogeneous Susceptibility of Valve Endothelial Cells to Mesenchymal Transformation in Response to TNFα. Annals of Biomedical Engineering. 2014;42(1):149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rutkovskiy A, Malashicheva A, Sullivan G, et al. Valve Interstitial Cells: The Key to Understanding the Pathophysiology of Heart Valve Calcification. Journal of the American Heart Association. 2017;6(9):e006339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nature reviews Molecular cell biology. 2014;15(12):786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hinton RB, Adelman-Brown J, Witt S, et al. Elastin haploinsufficiency results in progressive aortic valve malformation and latent valve disease in a mouse model. Circulation research. 2010;107(4):549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schoen FJ. Evolving Concepts of Cardiac Valve Dynamics. Circulation. 2008;118(18):1864–1880. [DOI] [PubMed] [Google Scholar]
- 65.Sacks MS, Yoganathan AP. Heart valve function: a biomechanical perspective. Philos Trans R Soc Lond B Biol Sci. 2007;362(1484):1369–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hinton RB, Lincoln J, Deutsch GH, et al. Extracellular Matrix Remodeling and Organization in Developing and Diseased Aortic Valves. Circulation Research. 2006;98(11):1431 LP–1438. [DOI] [PubMed] [Google Scholar]
- 67.Pohle K, Mäffert R, Ropers D, et al. Progression of aortic valve calcification: association with coronary atherosclerosis and cardiovascular risk factors. Circulation. 2001;104(16):1927–1932. [DOI] [PubMed] [Google Scholar]
- 68.Sathyamurthy I, Alex S. Calcific aortic valve disease: Is it another face of atherosclerosis? Indian Heart Journal. 2015;67(5):503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stewart BF, Siscovick D, Lind BK, et al. Clinical Factors Associated With Calcific Aortic Valve Disease. Journal of the American College of Cardiology. 1997;29(3):630 LP–634. [DOI] [PubMed] [Google Scholar]
- 70.Bergheanu SC, Bodde MC, Jukema JW. Pathophysiology and treatment of atherosclerosis. Netherlands Heart Journal. 2017;25(4):231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ference BA, Ginsberg HN, Graham I, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. European Heart Journal. 2017;38(32):2459–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG. Elevated lipoprotein (a) and risk of aortic valve stenosis in the general population. Journal of the American College of Cardiology. 2014;63(5):470–477. [DOI] [PubMed] [Google Scholar]
- 73.Hojo Y, Kumakura H, Kanai H, Iwasaki T, Ichikawa S, Kurabayashi M. Lipoprotein(a) is a risk factor for aortic and mitral valvular stenosis in peripheral arterial disease. European Heart Journal - Cardiovascular Imaging. 2016;17(5):492–497. [DOI] [PubMed] [Google Scholar]
- 74.Gustafsson BI, Tømmerås K, Nordrum I, et al. Long-Term Serotonin Administration Induces Heart Valve Disease in Rats. Circulation. 2005;111(12):1517–1522. [DOI] [PubMed] [Google Scholar]
- 75.Nebigil CG, Choi D-S, Dierich A, et al. Serotonin 2B receptor is required for heart development. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(17):9508–9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nebigil CG, Hickel P, Messaddeq N, et al. Ablation of serotonin 5-HT2B receptors in mice leads to abnormal cardiac structure and function. Circulation. 2001;103(24):2973–2979. [DOI] [PubMed] [Google Scholar]
- 77.Tamura K, I-ida T, Fujii T, Tanaka S, Asano G. Floppy Aortic Valves Without Aortic Root Dilatation: Clinical, Histologic, and Ultrastructural Studies. Journal of Nippon Medical School. 2002;69(4):355–364. [DOI] [PubMed] [Google Scholar]
- 78.Borer JS, Sharma A. Drug Therapy for Heart Valve Diseases. Circulation. 2015;132(11):1038–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reske SN, Heck I, Kropp J, et al. Captopril mediated decrease of aortic regurgitation. British Heart Journal. 1985;54(4):415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Droogmans S, Roosens B, Cosyns B, et al. Cyproheptadine prevents pergolide-induced valvulopathy in rats: an echocardiographic and histopathological study. American Journal of Physiology-Heart and Circulatory Physiology. 2009;296(6):H1940–H1948. [DOI] [PubMed] [Google Scholar]
- 81.Hauso Ø, Gustafsson BI, Loennechen JP, Stunes AK, Nordrum I, Waldum HL. Long-term serotonin effects in the rat are prevented by terguride. Regulatory Peptides. 2007;143(1):39–46. [DOI] [PubMed] [Google Scholar]
- 82.Rossebø AB, Pedersen TR, Boman K, et al. Intensive Lipid Lowering with Simvastatin and Ezetimibe in Aortic Stenosis. New England Journal of Medicine. 2008;359(13):1343–1356. [DOI] [PubMed] [Google Scholar]
- 83.Komiya T Aortic valve repair update. General Thoracic and Cardiovascular Surgery. 2015;63(6):309–319. [DOI] [PubMed] [Google Scholar]
- 84.Kari FA, Siepe M, Sievers H-H, Beyersdorf F. Repair of the Regurgitant Bicuspid or Tricuspid Aortic Valve. Circulation. 2013;128(8):854–863. [DOI] [PubMed] [Google Scholar]
- 85.Glower DD, Kar S, Trento A, et al. Percutaneous Mitral Valve Repair for Mitral Regurgitation in High-Risk Patients. Results of the EVEREST II Study. 2014;64(2):172–181. [DOI] [PubMed] [Google Scholar]
- 86.Franzone A, Piccolo R, Siontis GCM, et al. Transcatheter Aortic Valve Replacement for the Treatment of Pure Native Aortic Valve Regurgitation: A Systematic Review. JACC: Cardiovascular Interventions. 2016;9(22):2308–2317. [DOI] [PubMed] [Google Scholar]
- 87.Yoon S-H, Schmidt T, Bleiziffer S, et al. Transcatheter Aortic Valve Replacement in Pure Native Aortic Valve Regurgitation. Journal of the American College of Cardiology. 2017;70(22):2752–2763. [DOI] [PubMed] [Google Scholar]
- 88.Pellikka PA, Dangas G. TAVR for Severe Aortic Regurgitation: Advancing the Frontier*. Journal of the American College of Cardiology. 2017;70(22):2764–2765. [DOI] [PubMed] [Google Scholar]
- 89.Mack MJ, Leon MB, Thourani VH, et al. Transcatheter Aortic-Valve Replacement with a Balloon-Expandable Valve in Low-Risk Patients. New England Journal of Medicine. 2019;380(18):1695–1705. [DOI] [PubMed] [Google Scholar]
- 90.Sawaya FJ, Deutsch M-A, Seiffert M, et al. Safety and Efficacy of Transcatheter Aortic Valve Replacement in the Treatment of Pure Aortic Regurgitation in Native Valves and Failing Surgical Bioprostheses: Results From an International Registry Study. JACC: Cardiovascular Interventions. 2017;10(10):1048–1056. [DOI] [PubMed] [Google Scholar]
- 91.Bonow RO, Leon MB, Doshi D, Moat N. Management strategies and future challenges for aortic valve disease. The Lancet. 2016;387(10025):1312–1323. [DOI] [PubMed] [Google Scholar]
- 92.Silaschi M, Conradi L, Wendler O, et al. The JUPITER registry: One-year outcomes of transapical aortic valve implantation using a second generation transcatheter heart valve for aortic regurgitation. Catheterization and Cardiovascular Interventions. 2018;91(7):1345–1351. [DOI] [PubMed] [Google Scholar]
- 93.Rawasia WF, Khan MS, Usman MS, et al. Safety and efficacy of transcatheter aortic valve replacement for native aortic valve regurgitation: A systematic review and meta-analysis. Catheterization and Cardiovascular Interventions. 2019;93(2):345–353. [DOI] [PubMed] [Google Scholar]
- 94.Wernly B, Eder S, Navarese EP, et al. Transcatheter aortic valve replacement for pure aortic valve regurgitation: “on-label” versus “off-label” use of TAVR devices. Clinical Research in Cardiology. 2019;108(8):921–930. [DOI] [PubMed] [Google Scholar]
- 95.Krumsdorf U, Bekeredjian R, Korosoglou G, et al. Percutaneous Aortic “Valve in Valve” Implantation for Severe Aortic Regurgitation in a Degenerated Bioprosthesis. Circulation: Cardiovascular Interventions. 2010;3(3):e6–e7. [DOI] [PubMed] [Google Scholar]
- 96.Wenaweser P, Buellesfeld L, Gerckens U, Grube E. Percutaneous aortic valve replacement for severe aortic regurgitation in degenerated bioprosthesis: The first valve in valve procedure using the corevalve revalving system. Catheterization and Cardiovascular Interventions. 2007;70(5):760–764. [DOI] [PubMed] [Google Scholar]
- 97.Latib A, Ielasi A, Montorfano M, et al. Transcatheter valve-in-valve implantation with the Edwards SAPIEN in patients with bioprosthetic heart valve failure: the Milan experience. EuroIntervention. 2012;7(11):1275–1284. [DOI] [PubMed] [Google Scholar]
- 98.Attias D, Himbert D, Hvass U, Vahanian A. “ Valve-in-valve” implantation in a patient with stentless bioprosthesis and severe intraprosthetic aortic regurgitation. The Journal of thoracic and cardiovascular surgery. 2009;138(4):1020–1022. [DOI] [PubMed] [Google Scholar]
- 99.Bapat V, Attia R, Redwood S, et al. Use of transcatheter heart valves for a valve-in-valve implantation in patients with degenerated aortic bioprosthesis: Technical considerations and results. The Journal of Thoracic and Cardiovascular Surgery. 2012;144(6):1372–1380. [DOI] [PubMed] [Google Scholar]
- 100.Kossar AP, Borger M, George I. Direct access valve-in-valve implantation for management of complex valvulopathy. Catheterization and Cardiovascular Interventions. 2019;93(7):1385–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Research. 2015;43(Database issue):D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]