

Abstract
Electron paramagnetic resonance (EPR) spectroscopy combined with site-directed spin labelling is applicable to biomolecules and their complexes irrespective of system size and in a broad range of environments. Neither short-range nor long-range order is required to obtain structural restraints on accessibility of sites to water or oxygen, on secondary structure, and on distances between sites. Many of the experiments characterize a static ensemble obtained by shock-freezing. Compared with characterizing the dynamic ensemble at ambient temperature, analysis is simplified and information loss due to overlapping timescales of measurement and system dynamics is avoided. The necessity for labelling leads to sparse restraint sets that require integration with data from other methodologies for building models. The double electron–electron resonance experiment provides distance distributions in the nanometre range that carry information not only on the mean conformation but also on the width of the native ensemble. The distribution widths are often inconsistent with Anfinsen's concept that a sequence encodes a single native conformation defined at atomic resolution under physiological conditions.
Keywords: crystallography, EPR spectroscopy, intrinsically disordered proteins, molecular modelling, protein structure, transmembrane proteins
Introduction
During the past two decades, electron paramagnetic resonance (EPR) spectroscopy combined with site-directed spin labelling [1,2] has become an important tool in structural biology that complements higher-resolution techniques, such as X-ray crystallography (XRD), cryo-electron microscopy (cryo-EM), and NMR (nuclear magnetic resonance) [3,4]. The advent of integrative structural biology [5] has increased demand for EPR restraints on structure and requires better understanding of their accuracy and precision. Two types of EPR restraints are suitable for model building: accessibility restraints [6] and distance distribution restraints in the nanometre range [7,8]. Accessibility of a site to water or oxygen and spin label mobility are periodic in a site scan along a secondary structure element [9]. Label mobility influences the EPR line shape, allowing to study conformational equilibria and the kinetics of conformational change [10,11].
This review focuses on distance distribution restraints obtained with standard methanethiosulfonate spin labels (MTSSLs). Such distance distribution restraints are a unique contribution of EPR to structural biology. They encode information on the width of a conformational ensemble and thus on order–disorder phenomena. Evidence is growing that such phenomena underlie mechanisms of function and regulation of many proteins. To study them, methodologies for determining a single structure at atomic resolution must be complemented by methodologies that yield information on the conformational ensemble in an approximately native environment. Measurements of distance distributions by EPR techniques have already provided such information for many systems. Ongoing method development is extending the scope for such studies and will offer access to better approximations of the native environment.
EPR distance distribution restraints
Application of pulsed dipolar spectroscopy to biomolecules
Spin labels carry magnetic dipoles. The distribution of dipole–dipole couplings can be measured by pulsed dipolar spectroscopy (PDS) [12] if the exchange coupling between the two spins is negligible, as is the case at distances longer than 15 Å. Reorientation of the spin–spin vector must be slow on the timescale of the dipolar interaction, which extends from 100 ns at a distance of 20 Å to 100 µs at a distance of 160 Å. Limited excitation bandwidth sets a lower limit of the reliable distance range at 18–20 Å. The upper limit depends on the maximum observation time of the spins and thus on their decoherence time. For typical nitroxide labels, the decoherence time increases with decrease in temperature down to ∼60–40 K. At such temperatures, distances up to at least 50 Å can be measured for membrane proteins and the range extends up to 160 Å for soluble proteins in favourable cases [13] if both the solvent and the protein are deuterated [14]. For ambient temperature measurements on immobilized biomolecules labelled with slowly relaxing trityl radicals [15,16], the upper distance limit reduces to 30–50 Å. Whether biomolecule conformation is more strongly perturbed by immobilization at ambient temperature or by shock-freezing in the presence of a cryoprotectant is still a matter of debate and may depend on the system under investigation. It is advisable to check consistency between EPR data obtained at cryogenic temperatures and data from other techniques obtained at ambient temperature, whenever this is possible.
While the necessity for cryogenic temperatures is a matter of concern, among all techniques in structural biology EPR spectroscopy probably poses the least restrictions on the environment of the biomolecules. Most biomolecules and small ligands are EPR-silent and, since distance distributions are measured, neither long-range nor short-range order is required. PDS can be applied to membrane proteins reconstituted into liposomes and even to cells [17–19]. The only requirement posed on the biomolecules is that they can be spin-labelled or bind a spin-labelled ligand.
Usually, the distribution of dipolar couplings is measured by the double electron–electron resonance (DEER) experiment that is also called pulsed electron–electron double resonance (PELDOR) [7]. For this experiment, the functional form of the background contribution from remote spin-labelled molecules is known and can thus be separated from the intramolecular contribution. If local concentration of spin-labelled biomolecules is known to be sufficiently low, single-frequency PDS techniques, such as the double-quantum coherence (DQC) experiment, the single-frequency technique for refocusing (SIFTER), or relaxation-induced dipolar modulation enhancement (RIDME), can also be applied and may provide higher sensitivity under some conditions.
From primary data to distance distributions
Except for orientation selection, which is often negligible or can be averaged [4], the normalized dipolar evolution function D(t) can be computed from the distance distribution P(r) by relying only on fundamental constants and the known g value of the spin label. The normalized primary signal V(t) is given by B(t) [1 − λ + λ D(t)]. Modulation depth λ depends on experimental parameters and the background is usually a stretched exponential function B(t) = exp[−(kt)ξ], with ξ ≍ 1 for soluble proteins and 2/3 < ξ ≤ 1 for membrane proteins. The inverse problem of computing P(r) from V(t) is fraught with two complications. First, reliable fitting of the background parameters k and λ and possibly ξ requires that V(t) be observed up to 2 µs for distances up to 4 nm. The mean distance can be estimated up to 5 µs for such data, but between 4 and 5 nm the width of the distribution is unreliable. Background correction usually suppresses distances longer than 5 nm for a data trace length of 2 μs, but can also generate artefact peaks at long distances. The limits scale with the cubic root of the trace length. The most widely used software package DeerAnalysis [20] colour-codes ranges where information on the distribution shape (green), width (yellow), and mean distance (orange) is still reliable.
As a second complication, conversion of D(t) into P(r) is a mathematically ill-posed problem. Tikhonov regularization stabilizes the solution without imposing restraints on the distribution shape and at the expense of only weak or moderate broadening. However, if both narrow and broad peaks occur, as is the case for equilibria between ordered and disordered conformational sub-ensembles, regularization either strongly broadens the narrow peaks or cannot suppress artificial splitting of broad peaks. Fitting by a few Gaussian peaks, also implemented in DeerAnalysis and supplemented by statistical analysis in GLADD [21], may then be advantageous. An overview of further software packages is provided in ref. [21].
Ill-posedness of converting D(t) into P(r) and uncertainty in background separation require validation of distance distributions, as implemented in DeerAnalysis, or statistical error analysis, as implemented in GLADD. Validation of the conversion of D(t) into P(r) by a Bayesian approach has been demonstrated [22]. The ill-posed problem can be avoided and background correction stabilized by fitting structure models to primary data V(t) [23]. Often, restraints must be supplied in terms of mean distances and standard deviations of the distance. It is then advisable to refine models against primary data or to check their consistency with primary data.
From distance distributions to models
For dilute biomolecules labelled at two sites, PDS provides the distribution P(ree) of the distance ree between the unpaired electrons of the two spin labels. If the unpaired electrons are delocalized on length scales comparable to ree, P(ree) corresponds to an 1/r3-weighted average over their spatial distribution. For the commonly used nitroxide labels, this complication can be safely neglected at distances r > 15 Å, but for trityl spin labels it needs to be taken into account up to 30 Å [24].
For structure modelling, the distance rαα between the Cα atoms of the labelled residues or an analogous backbone–backbone distance for nucleotides are of interest. Since even for the smallest nitroxide labels, ree may differ from rαα by up to 10 Å, it is mandatory to include the spin label into modelling. The possible conformations of the label side chain can be approximated by a moderate number of rotameric states [23,25]. In the statistical mechanics-based approach in the software package MMM [25,26], the rotamer distribution of the free label is estimated once for all from molecular dynamics (MD) trajectories or by Monte Carlo sampling and stored in a library. The influence of the biomolecule on this distribution is computed by adding non-bonding interaction energies with protein, nucleic acid, and cofactor atoms to the free energy. A variant estimates the rotamer distribution from a set of crystal structures of spin-labelled proteins [27], and a simpler but also less physical approach samples the accessible volume by taking into account only repulsive interactions and ignoring the torsion potentials [28]. All these approaches end up with similar accuracy of ∼3 Å for predicting the mean distance 〈ree〉 from a known high-resolution structure [29]. Computationally more elaborate procedures sample the energy hypersurface of the labelled protein in a Monte Carlo approach [30] or by enhanced sampling MD [31], but do not improve strongly on that accuracy. Hence, at present, an accuracy of ∼2–3 Å must be accepted, which precludes building atomic-resolution models from only EPR restraints.
Forward computation of ree from a structural model requires information on only local structure around the labelling sites. This simplifies the treatment of large-scale conformational transitions, where rigid domains move with respect to each other [4], and of rigid-body docking [23,26,32]. In integrative modelling, the approximation of the local environment can be iteratively improved [33]. Rotamer library-based predictions are a computationally inexpensive approach for testing many models in ensemble refinement [34] and for simulating PDS data from MD trajectories of unlabelled biomolecules with the Python package RotamerConvolveMD [35].
Model building with EPR restraints
Stand-alone modelling
Typically, only one or two EPR restraints are obtained per expressed and purified mutated protein sample. Therefore, the number of EPR restraints is much smaller than that of residues and building of a model from only these restraints is precluded even on a coarse-grained level. In that sense, structure modelling with EPR restraints is always hybrid modelling. Stand-alone modelling denotes approaches based on experimental structures from XRD, NMR, or cryo-EM or on structures predicted from homology or de novo modelling without fitting other experimental data simultaneously. It is assumed that processing of experimental data from different methodologies is separable, in contrast with integrative modelling, where this is not the case. Stand-alone modelling presumes that the EPR data only weakly or only locally perturb the initial structure, which is often a good approximation for biomolecules containing rigid domains.
A case in point is binding of small-molecule ligands or metal ions. If the ligand can be spin-labelled or the metal ion is paramagnetic, they can be localized by a distance matrix approach [36] or by an approach akin to the global positioning system (GPS), where the position of an observer can be determined by at least three distance measurements to reference sites [37]. The distance matrix approach tests and relaxes the assumption of a rigid reference domain at the expense of additional measurements between reference points. If rigidity of the reference domain has been established by NMR or small-angle X-ray scattering (SAXS) measurements, the GPS-like approach may be advantageous. Both approaches have been implemented in MMM [26]; the GPS-like approach is available in mtsslSuite [38]. The same approach can localize labelled sites in a flexible domain by measurements to reference sites in a rigid domain [39].
For many transporters and quite a few enzymes, ligand binding is coupled to relative movement of domains. In modelling such conformational transitions, the rigid-domain assumption can be somewhat relaxed and subjective identification of the domains can be avoided by using an elastic network model [40]. An implementation into MMM that accounts for the spin labels [41] has revealed conformational changes in the nucleotide-gated HCN ion channel upon binding of 3′,5′-cyclic adenosine monophosphate [42].
The elastic network model approach performs well only for simple hinge motion or combination of a hinge motion with rotation of one domain [41]. For more complex conformational transitions, one may resort to comparative modelling by satisfaction of spatial restraints as implemented in MODELLER [43]. Conformational changes in the multidrug transporter EmrE with respect to the crystal structure and upon protonation were assessed using the MMM interface to MODELLER that allows for imposing distance restraints between spin-labelled sites [44]. By the same approach, additional restraints on secondary structure can be included, as shown for the external loop eL4 in proline/sodium symporter PutP [45]. A model study on T4 lysozyme demonstrated structure refinement by restraining an MD ensemble simulation by label-to-label distances [31].
If several structure models are available, the problem may boil down to discriminating between them at a sample composition not amenable to other techniques and to testing whether the model is consistent. For this approach, optimal labelling sites can be selected by analysing the distance matrix [46].
Rigid-body docking for homo-oligomers, protein complexes, or rigid domains connected by flexible linkers requires only few restraints. The two-body problem has three translational and three rotational degrees of freedom, and each additional body adds six more degrees of freedom. For homo-oligomers with known symmetry, only two translations and two rotations are free irrespective of the number of protomers. Such problems can be overdetermined by distance distribution restraints. The two-body problem can be solved by a genetic algorithm implemented in mtsslSuite [32], by a complete grid search [23,26], or by an Xplor–NIH [47]-based protocol [48]. For more than two bodies, the RigiFlex approach has been proposed [26].
In the absence of any related experimental structure, EPR restraints can be used to steer de novo modelling by the Rosetta approach [49]. Favourable labelling sites are selected by maximizing sequence separation under the constraint that predicted secondary structure elements are pairwise connected [50]. With a moderately sized set of 25 EPR distance distribution restraints, such de novo modelling provided useful results for the soluble form of Bax with 192 residues [51].
Distance distributions are particularly valuable for recognizing disorder, as illustrated in Figure 1, and for restraining the ensemble of internal or terminal intrinsically disordered domains [52]. They can be complemented by restraints on bilayer immersion depth from accessibility measurements and on secondary structure from spin labelling site scans [26]. An ensemble model for N-terminal residues 3–13 of the light-harvesting complex LHCII of green plants was generated by this approach [53].
Figure 1. Label-to-label distance distributions reveal backbone disorder.
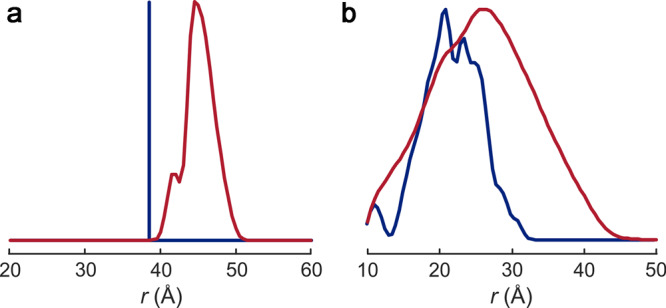
(a) Distribution of the Cα–Cα backbone distance rαα (blue) and the label-to-label distance ree (red) for residue pair 96/143 in the rigid core of plant light-harvesting complex LHCII. The width of P(ree) is well below 10 Å. (b) Distributions for residue pair 3/34 in the flexible N-terminal domain. Label-induced broadening is enhanced by the variation of side chain orientations of disordered residue 3. The width of P(ree) is well above 10 Å. The distributions were simulated in MMM [26] based on PDB structure 2BHW and the restraints reported in [53].
Integrative modelling
The Xplor–NIH-based protocol for determining relative domain orientation and translation in proteins with two domains [48], developed in a pioneering study on a tandem of polypeptide transport-associated domains in Omp85 [54], integrates EPR distances with NMR nuclear Overhauser Effect (NOE) restraints. The structure was still ill-defined with 2907 short-range NOE restraints and could be fixed by only three additional DEER restraints. Distance distributions showed that the linker is not flexible as had been assumed before. A similar protocol supplements medium-range paramagnetic relaxation enhancement (PRE) restraints from NMR by DEER restraints [55]. Since PRE restraints are more informative on domain–domain placement than NOE restraints, the improvement is less impressive, but still the backbone root mean square deviation of an ensemble of 10 models could be improved from 4.2 to 3 Å by adding only three DEER restraints. Integration of PRE and DEER restraints had been demonstrated before [56] using the CYANA software [57].
More complex models can also be built by integrating solution-state NMR and DEER data. The structure of the 70 kDa protein/RNA complex RmsE/RmsZ was modelled with a CYANA-based protocol [58] from 4664 intramolecular NOE restraints for the protein, ∼850 intramolecular NOE restraints for the RNA, and 21 distance distribution restraints between RNA stem loops [59]. Underlying assumptions and technical issues have been discussed in detail [60]. The structure consists of three rigid protein domains and a 72 nucleotide long non-coding RNA that binds to the protein via four stem loops. Bimodal distance distributions showed that the complex adopts two major conformations.
For discoidal high-density lipoprotein particles, six DEER distance distribution restraints fixed the shape of the apolipoprotein ring enclosing the lipid bilayer sheet, which was ill-defined when using only PRE and NOE restraints [33]. Integration of EPR distance restraints with sparse pseudo-contact shift data from solution-state NMR reduces the number of free parameters in fitting the susceptibility tensor of the lanthanide probe from 8 to 5 [61].
Integration of solid-state NMR and DEER data may require diamagnetic dilution of the spin-labelled protein. This was demonstrated for the oligomeric structure of sensory rhodopsin reconstituted into lipid bilayers [62]. A combination of solid-state PRE and DEER restraints ruled out dimer formation, whereas analysis of DEER modulation depth ruled out higher oligomers than trimers. By a CNS-based [63] protocol, the trimer structure could be modelled from short-range distance restraints, torsion angle restraints, and PRE restraints. The addition of the DEER restraints led to significant refinement, especially on the cytoplasmic side.
Distance distribution restraints are a nice complement to small-angle scattering curves, which provide information on the global shape of a macromolecule on the same length scales where PDS provides a few sequence-assigned distances. The tetramer structure of histone chaperones in solution was modelled from an XRD-based dimer structure and DEER restraints, whereas SAXS curve fitting was used for testing for the correct global shape [64]. A similar approach was applied to a chromatin-remodelling enzyme [65]. Integration of SAXS and DEER data in modelling a minimal ensemble of conformations, combined with tests against single-molecule fluorescence resonance energy transfer data, was performed by unbiased replica exchange Monte Carlo simulations [66]. Structural integrity of spin-labelled mutants of the FnIII-3,4 domains of integrin α6β4 was checked by monitoring their SAXS curves in solution [67]. A combination of DEER and SAXS data to determine relative orientation and translation of the two domains led to lower uncertainty than achieved with only the 13 DEER distance distributions.
PDS-based restraints have proved valuable in validating and even refining XRD and cryo-EM structures. Even in the age of high-resolution cryo-EM, not all systems yield high-resolution images and DEER data may be useful for resolving ambiguities in data interpretation [68]. Ambiguity was also encountered in solving the structure of arrestin bound to rhodopsin by XRD on micrometre-sized crystals using a free electron laser [69]. The crystals were twinned and DEER could validate the structure. Except for small single-domain globular proteins, one may need to test whether a crystal structure is representative of the solution structure. PDS-based distance distributions can provide this information [70], which is particularly useful when different techniques result in different predictions of solution structure [71] or when a shortened construct had to be used for crystallization [46,72].
Based on DEER distance distributions and coarse models derived from them [39,73], the first all-atom model was proposed for the mitochondrial pore formed by the pro-apoptotic protein Bax [74]. Together with MD simulations of pathogenic mutants, the model could be used for assessing the influence of the mutations on pore formation.
A fuzzy relation of structure to function
Atomic-resolution models not only are aesthetically pleasing, but also provide a wealth of information for generating hypotheses on function. To obtain them, it has become quite usual to truncate or otherwise mutate constructs or rely on constructs augmented with an easy-crystallizing domain. Based on the Anfinsen dogma, it became customary to explain function in terms of a single conformation or of well-defined transitions between a few conformations defined at atomic resolution. While this is certainly a reasonable approximation in some cases [75–77], availability of distance distributions demonstrates that rather often conformation transitions are coupled to order–disorder transitions or are shifts in disorder equilibria [39,78–94]. Among the systems addressed by PDS to date, the fraction where at least one state is genuinely disordered is surprisingly large. Although systems not fully amenable to structure determination at atomic resolution may preferably end up in EPR laboratories, this body of data suggests that the hitherto explored part of biomolecular structure space is biased towards the most strongly ordered systems. Even in these cases, conformational uncertainty may be underestimated compared with the one in a native environment. The available distance distributions hint at a structure–function relation that transcends not only the Anfinsen dogma of a single native conformation, but also the dichotomy between Anfinsen-type ordered and intrinsically disordered proteins. In the following, I discuss a few cases where this is apparent.
It is often assumed that intrinsically disordered proteins order upon binding to other proteins or to small-molecule ligands. The multidrug transporter EmrE is ordered in its substrate-bound form, but flexible when protonated at a pH of 5 [87]. Likewise, the homodimeric multidrug ABC transporter LmrA undergoes a disorder–order transition from a very broad conformational ensemble to a rather well-defined conformation upon nucleotide binding [80]. However, in the Toc34 GTPase homodimer, the GDP-bound state is tight and ordered and the GTB-bound state disordered [85]. In helicase PcrA from superfamily 1, the two motor domains are flexible with respect to each other in the apo- and ADP-bound states, while ATP-analogue binding brings them closer together and tightens the conformational bundle [94]. DNA binding further rigidifies the motor domains. In contrast, only slight additional narrowing of the already narrow ensemble and no significant mean distance changes are observed in helicase XPD from superfamily 2. The secondary sodium/aspartate transporter GltPh samples inward- and outward-facing conformations with almost equal probability and remains conformationally heterogeneous even in the presence of substrate and sodium ions [81,82]. Synaptotagmin 1 remains heterogeneous after binding to SNAREs [78]. In cardiac myosin-binding protein C, phosphorylation causes compaction and reduces disorder of the Pro/Ala-rich linker between two immunoglobin domains [88]. The human and teleost fish secretory components, which could be crystallized, exhibit well-defined conformations in the absence of their immunoglobulin ligands [95], whereas the unliganded avian secretory component is flexible, pointing to a distinct mechanism of ligand binding of secretory components among vertebrates [91]. Pro-apoptotic Bax exhibits disorder in the piercing domain that anchors Bax in the mitochondrial membrane, which may be related to its ability to form pores of different sizes by varying its degree of oligomerization [39]. The diversity of these examples indicates that disorder is a broadly used evolutionary strategy for optimizing protein function. Figure 2 depicts a possible strategy for tuning binding affinity by coupling an order–disorder transition to ligand binding.
Figure 2. Entropic tuning of affinity by coupling an order–disorder transition to binding.

(a) For a too low-affinity combination of ligand and binding site, coupling to an order–disorder transition increases the free energy gain upon binding. (b) For a too high-affinity combination, coupling to a disorder–order transition reduces the free energy gain.
Conclusion
EPR spectroscopy in conjunction with site-directed spin labelling provides structure restraints irrespective of system size and in a broad range of environments. Distance distributions from PDS match dimensions of protein domains or whole proteins and are a unique source of information on the width of conformation ensembles. Although modelling approaches that integrate EPR restraints are still in their infancy, they have already revealed several cases of conformational heterogeneity related to function. This relation is probably much more common than is suggested by the current body of structural data on biomolecules. The Anfinsen era of structural biology may be nearing its end.
Summary
EPR-based accessibility and distance distribution restraints complement information from X-ray crystallography, cryo-EM, and NMR.
Structures with atomic resolution can be built by integrating EPR with NMR restraints, and informative models can be built by integrating them with independently determined domain structures and small-angle scattering data.
The broad distance distributions observed in diverse systems cast doubt on the Anfinsen dogma that a sequence usually encodes a unique native conformation.
Acknowledgement
I thank Enrica Bordignon, Olivier Duss, Ines García Rubio, Christoph Gmeiner, Daniel Hilger, Benesh Joseph, Daniel Klose, Yevhen Polyhach, and Maxim Yulikov for helpful discussions during the past few years.
Abbreviations
- CNS
crystallography and NMR system
- Cryo-EM
cryo-electron microscopy
- DEER
double electron–electron resonance
- EPR
electron paramagnetic resonance
- GPS
global positioning system
- MD
molecular dynamics
- MTSSLs
methanethiosulfonate spin labels
- NMR
nuclear magnetic resonance
- NOE
Nuclear Overhauser Effect
- PDS
pulsed dipolar spectroscopy
- PRE
paramagnetic relaxation enhancement
- SAXS
small-angle X-ray scattering
- XRD
X-ray diffraction
Funding
This work was supported by SNSF grant [200020_169057].
Competing Interests
The Author declares that there are no competing interests associated with this manuscript.
References
- 1.Hubbell W.L., Cafiso D.S. and Altenbach C. (2000) Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 7, 735–739 10.1038/78956 [DOI] [PubMed] [Google Scholar]
- 2.Klare J.P. and Steinhoff H.-J. (2015) Spin labeling studies of transmembrane signaling and transport: applications to phototaxis, ABC transporters and symporters. Methods Enzymol. 564, 315–347 10.1016/bs.mie.2015.05.025 [DOI] [PubMed] [Google Scholar]
- 3.Claxton D.P., Kazmier K., Mishra S. and Mchaourab H.S. (2015) Navigating membrane protein structure, dynamics, and energy landscapes using spin labeling and EPR spectroscopy. Methods Enzymol. 564, 349–387 10.1016/bs.mie.2015.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeschke G. (2012) DEER distance measurements on proteins. Annu. Rev. Phys. Chem. 63, 419–446 10.1146/annurev-physchem-032511-143716 [DOI] [PubMed] [Google Scholar]
- 5.Steven A.C. and Baumeister W. (2008) The future is hybrid. J. Struct. Biol. 163, 186–195 10.1016/j.jsb.2008.06.002 [DOI] [PubMed] [Google Scholar]
- 6.Oh K.J., Altenbach C., Collier R.J. and Hubbell W.L. (2000) Site-directed spin labeling of proteins. Applications to diphtheria toxin. Methods Mol. Biol. 145, 147–169 10.1385/1-59259-052-7:147 [DOI] [PubMed] [Google Scholar]
- 7.Jeschke G. and Polyhach Y. (2007) Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys. 9, 1895–1910 10.1039/b614920k [DOI] [PubMed] [Google Scholar]
- 8.Jeschke G. (2016) Dipolar spectroscopy — double-resonance methods. eMagRes 5, 1459–1476 10.1002/9780470034590.emrstm1518 [DOI] [Google Scholar]
- 9.Hubbell W.L., Gross A., Langen R. and Lietzow M.A. (1998) Recent advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 8, 649–656 10.1016/S0959-440X(98)80158-9 [DOI] [PubMed] [Google Scholar]
- 10.Klare J.P. and Steinhoff H.-J. (2009) Spin labeling EPR. Photosynth. Res. 102, 377–390 10.1007/s11120-009-9490-7 [DOI] [PubMed] [Google Scholar]
- 11.Klose D., Voskoboynikova N., Orban-Glass I., Rickert C., Engelhard M., Klare J.P. et al. (2014) Light-induced switching of HAMP domain conformation and dynamics revealed by time-resolved EPR spectroscopy. FEBS Lett. 588, 3970–3976 10.1016/j.febslet.2014.09.012 [DOI] [PubMed] [Google Scholar]
- 12.Jeschke G., Koch A., Jonas U. and Godt A. (2002) Direct conversion of EPR dipolar time evolution data to distance distributions. J. Magn. Reson. 155, 72–82 10.1006/jmre.2001.2498 [DOI] [PubMed] [Google Scholar]
- 13.Schmidt T., Wälti M.A., Baber J.L., Hustedt E.J. and Clore G.M. (2016) Long distance measurements up to 160 Å in the GroEL tetradecamer using Q-band DEER EPR spectroscopy. Angew. Chem., Int. Ed. Engl. 55, 15905–15909 10.1002/anie.201609617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El Mkami H. and Norman D.G. (2015) EPR distance measurements in deuterated proteins. Methods Enzymol. 564, 125–152 10.1016/bs.mie.2015.05.027 [DOI] [PubMed] [Google Scholar]
- 15.Yang Z., Liu Y., Borbat P., Zweier J.L., Freed J.H. and Hubbell W.L. (2012) Pulsed ESR dipolar spectroscopy for distance measurements in immobilized spin labeled proteins in liquid solution. J. Am. Chem. Soc. 134, 9950–9952 10.1021/ja303791p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shevelev G.Y., Krumkacheva O.A., Lomzov A.A., Kuzhelev A.A., Rogozhnikova O.Y., Trukhin D.V. et al. (2014) Physiological-temperature distance measurement in nucleic acid using triarylmethyl-based spin labels and pulsed dipolar EPR spectroscopy. J. Am. Chem. Soc. 136, 9874–9877 10.1021/ja505122n [DOI] [PubMed] [Google Scholar]
- 17.Igarashi R., Sakai T., Hara H., Tenno T., Tanaka T., Tochio H. et al. (2010) Distance determination in proteins inside Xenopus laevis oocytes by double electron–electron resonance experiments. J. Am. Chem. Soc. 132, 8228–8229 10.1021/ja906104e [DOI] [PubMed] [Google Scholar]
- 18.Joseph B., Sikora A., Bordignon E., Jeschke G., Cafiso D.S. and Prisner T.F. (2015) Distance measurement on an endogenous membrane transporter in E. coli cells and native membranes using EPR spectroscopy. Angew. Chem., Int. Ed. Engl. 54, 6196–6199 10.1002/anie.201501086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Theillet F.-X., Binolfi A., Bekei B., Martorana A., Rose H.M., Stuiver M. et al. (2016) Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530, 45–50 10.1038/nature16531 [DOI] [PubMed] [Google Scholar]
- 20.Jeschke G., Chechik V., Ionita P., Godt A., Zimmermann H., Banham J. et al. (2006) DeerAnalysis2006 — a comprehensive software package for analyzing pulsed ELDOR data. Appl. Magn. Reson. 30, 473–498 10.1007/BF03166213 [DOI] [Google Scholar]
- 21.Stein R.A., Beth A.H. and Hustedt E.J. (2015) A straightforward approach to the analysis of double electron-electron resonance data. Methods Enzymol. 563, 531–567 10.1016/bs.mie.2015.07.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edwards T.H. and Stoll S. (2016) A Bayesian approach to quantifying uncertainty from experimental noise in DEER spectroscopy. J. Magn. Reson. 270, 87–97 10.1016/j.jmr.2016.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hilger D., Polyhach Y., Padan E., Jung H. and Jeschke G. (2007) High-resolution structure of a Na+/H+ antiporter dimer obtained by pulsed electron paramagnetic resonance distance measurements. Biophys. J. 93, 3675–3683 10.1529/biophysj.107.109769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kunjir N.C., Reginsson G.W., Schiemann O. and Sigurdsson S.T. (2013) Measurements of short distances between trityl spin labels with CW EPR, DQC and PELDOR. Phys. Chem. Chem. Phys. 15, 19673–19685 10.1039/c3cp52789a [DOI] [PubMed] [Google Scholar]
- 25.Polyhach Y., Bordignon E. and Jeschke G. (2011) Rotamer libraries of spin labelled cysteines for protein studies. Phys. Chem. Chem. Phys. 13, 2356–2366 10.1039/C0CP01865A [DOI] [PubMed] [Google Scholar]
- 26.Jeschke G. (2018) MMM: a toolbox for integrative structure modeling. Protein Sci. 27, 76–85 10.1002/pro.3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beasley K.N., Sutch B.T., Hatmal M.M., Langen R., Qin P.Z. and Haworth I.S. (2015) Computer modeling of spin labels: NASNOX, PRONOX, and ALLNOX. Methods Enzymol. 563, 569–593 10.1016/bs.mie.2015.07.021 [DOI] [PubMed] [Google Scholar]
- 28.Hagelueken G., Ward R., Naismith J.H. and Schiemann O. (2012) Mtsslwizard: in silico spin-labeling and generation of distance distributions in PyMOL. Appl. Magn. Reson. 42, 377–391 10.1007/s00723-012-0314-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeschke G. (2013) Conformational dynamics and distribution of nitroxide spin labels. Prog. Nucl. Magn. Reson. Spectrosc. 72, 42–60 10.1016/j.pnmrs.2013.03.001 [DOI] [PubMed] [Google Scholar]
- 30.Fajer P., Fajer M., Zawrotny M. and Yang W. (2015) Full atom simulations of spin label conformations. Methods Enzymol. 563, 623–642 10.1016/bs.mie.2015.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Islam S.M., Stein R.A., Mchaourab H.S. and Roux B. (2013) Structural refinement from restrained-ensemble simulations based on EPR/DEER data: application to T4 lysozyme. J. Phys. Chem. B 117, 4740–4754 10.1021/jp311723a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hagelueken G., Abdullin D., Ward R. and Schiemann O. (2013) Mtsslsuite: in silico spin labelling, trilateration and distance-constrained rigid body docking in PyMOL. Mol. Phys. 111, 2757–2766 10.1080/00268976.2013.809804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bibow S., Polyhach Y., Eichmann C., Chi C.N., Kowal J., Albiez S. et al. (2017) Solution structure of discoidal high-density lipoprotein particles with a shortened apolipoprotein A-I. Nat. Struct. Mol. Biol. 24, 187–193 10.1038/nsmb.3345 [DOI] [PubMed] [Google Scholar]
- 34.Boura E., Rozycki B., Herrick D.Z., Chung H.S., Vecer J., Eaton W.A. et al. (2011) Solution structure of the ESCRT-I complex by small-angle X-ray scattering, EPR, and FRET spectroscopy. Proc. Natl Acad. Sci. U.S.A. 108, 9437–9442 10.1073/pnas.1101763108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stelzl L.S., Fowler P.W., Sansom M.S.P. and Beckstein O. (2014) Flexible gates generate occluded intermediates in the transport cycle of LacY. J. Mol. Biol. 426, 735–751 10.1016/j.jmb.2013.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gaffney B.J., Bradshaw M.D., Frausto S.D., Wu F., Freed J.H. and Borbat P. (2012) Locating a lipid at the portal to the lipoxygenase active site. Biophys. J. 103, 2134–2144 10.1016/j.bpj.2012.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdullin D., Florin N., Hagelueken G. and Schiemann O. (2015) EPR-based approach for the localization of paramagnetic metal ions in biomolecules. Angew. Chem., Int. Ed. Engl. 54, 1827–1831 10.1002/anie.201410396 [DOI] [PubMed] [Google Scholar]
- 38.Hagelueken G., Abdullin D. and Schiemann O. (2015) Mtsslsuite: probing biomolecular conformation by spin-labeling studies. Methods Enzymol. 563, 595–622 10.1016/bs.mie.2015.06.006 [DOI] [PubMed] [Google Scholar]
- 39.Bleicken S., Jeschke G., Stegmueller C., Salvador-Gallego R., García-Sáez A.J. and Bordignon E. (2014) Structural model of active Bax at the membrane. Mol. Cell 56, 496–505 10.1016/j.molcel.2014.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng W. and Brooks B.R. (2006) Modeling protein conformational changes by iterative fitting of distance constraints using reoriented normal modes. Biophys. J. 90, 4327–4336 10.1529/biophysj.105.076836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeschke G. (2012) Characterization of protein conformational changes with sparse spin-label distance constraints. J. Chem. Theory Comput. 8, 3854–3863 10.1021/ct300113z [DOI] [PubMed] [Google Scholar]
- 42.Puljung M.C., DeBerg H.A., Zagotta W.N. and Stoll S. (2014) Double electron–electron resonance reveals cAMP-induced conformational change in HCN channels. Proc. Natl Acad. Sci. U.S.A. 111, 9816–9821 10.1073/pnas.1405371111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fiser A. and Šali A. (2003) Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 374, 461–491 10.1016/S0076-6879(03)74020-8 [DOI] [PubMed] [Google Scholar]
- 44.Dastvan R., Brouwer E.-M., Schuetz D., Mirus O., Schleiff E. and Prisner T.F. (2016) Relative orientation of POTRA domains from cyanobacterial Omp85 studied by pulsed EPR spectroscopy. Biophys. J. 110, 2195–2206 10.1016/j.bpj.2016.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raba M., Dunkel S., Hilger D., Lipiszko K., Polyhach Y., Jeschke G. et al. (2014) Extracellular loop 4 of the proline transporter PutP controls the periplasmic entrance to ligand binding sites. Structure 22, 769–780 10.1016/j.str.2014.03.011 [DOI] [PubMed] [Google Scholar]
- 46.Hagelueken G., Hoffmann J., Schubert E., Duthie F.G., Florin N., Konrad L. et al. (2016) Studies on the X-ray and solution structure of FeoB from Escherichia coli BL21. Biophys. J. 110, 2642–2650 10.1016/j.bpj.2016.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwieters C.D., Kuszewski J.J., Tjandra N. and Clore G.M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 10.1016/S1090-7807(02)00014-9 [DOI] [PubMed] [Google Scholar]
- 48.Hammond C.M., Owen-Hughes T. and Norman D.G. (2014) Modelling multi-protein complexes using PELDOR distance measurements for rigid body minimisation experiments using XPLOR-NIH. Methods 70, 139–153 10.1016/j.ymeth.2014.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hirst S.J., Alexander N., Mchaourab H.S. and Meiler J. (2011) RosettaEPR: an integrated tool for protein structure determination from sparse EPR data. J. Struct. Biol. 173, 506–514 10.1016/j.jsb.2010.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kazmier K., Alexander N.S., Meiler J. and Mchaourab H.S. (2011) Algorithm for selection of optimized EPR distance restraints for de novo protein structure determination. J. Struct. Biol. 173, 549–557 10.1016/j.jsb.2010.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fischer A.W., Bordignon E., Bleicken S., García-Sáez A.J., Jeschke G. and Meiler J. (2016) Pushing the size limit of de novo structure ensemble prediction guided by sparse SDSL-EPR restraints to 200 residues: the monomeric and homodimeric forms of BAX. J. Struct. Biol. 195, 62–71 10.1016/j.jsb.2016.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jeschke G. (2016) Ensemble models of proteins and protein domains based on distance distribution restraints. Proteins: Struct., Funct., Bioinf. 84, 544–560 10.1002/prot.25000 [DOI] [PubMed] [Google Scholar]
- 53.Fehr N., Dietz C., Polyhach Y., von Hagens T., Jeschke G. and Paulsen H. (2015) Modeling of the N-terminal section and the lumenal loop of trimeric light harvesting complex II (LHCII) by using EPR. J. Biol. Chem. 290, 26007–26020 10.1074/jbc.M115.669804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ward R., Zoltner M., Beer L., El-Mkami, H., Henderson I.R., Palmer T. et al. (2009) The orientation of a tandem POTRA domain pair, of the β-barrel assembly protein BamA, determined by PELDOR spectroscopy. Structure 17, 1187–1194 10.1016/j.str.2009.07.011 [DOI] [PubMed] [Google Scholar]
- 55.Wu K., Shi C., Li J., Wang H., Shi P., Chen L. et al. (2013) Efficient long-distance NMR-PRE and EPR-DEER restraints for two-domain protein structure determination. Protein Cell 4, 893–896 10.1007/s13238-013-3109-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Y., Ramelot T.A., McCarrick R.M., Ni S., Feldmann E.A., Cort J.R. et al. (2010) Combining NMR and EPR methods for homodimer protein structure determination. J. Am. Chem. Soc. 132, 11910–11913 10.1021/ja105080h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 10.1385/1-59259-809-9:353 [DOI] [PubMed] [Google Scholar]
- 58.Duss O., Yulikov M., Jeschke G. and Allain F.H.-T. (2014) EPR-aided approach for solution structure determination of large RNAs or protein–RNA complexes. Nat. Commun. 5, 3669 10.1038/ncomms4669 [DOI] [PubMed] [Google Scholar]
- 59.Duss O., Michel E., Yulikov M., Schubert M., Jeschke G. and Allain F.H.-T. (2014) Structural basis of the non-coding RNA RsmZ acting as a protein sponge. Nature 509, 588–592 10.1038/nature13271 [DOI] [PubMed] [Google Scholar]
- 60.Duss O., Yulikov M., Allain F.H.T. and Jeschke G. (2015) Combining NMR and EPR to determine structures of large RNAs and protein-RNA complexes in solution. Methods Enzymol. 558, 279–331 10.1016/bs.mie.2015.02.005 [DOI] [PubMed] [Google Scholar]
- 61.Abdelkader E.H., Yao X., Feintuch A., Adams L.A., Aurelio L., Graham B. et al. (2016) Pulse EPR-enabled interpretation of scarce pseudocontact shifts induced by lanthanide binding tags. J. Biomol. NMR 64, 39–51 10.1007/s10858-015-0003-z [DOI] [PubMed] [Google Scholar]
- 62.Milikisiyants S., Wang S., Munro R.A., Donohue M., Ward M.E., Bolton D. et al. (2017) Oligomeric structure of anabaena sensory rhodopsin in a lipid bilayer environment by combining solid-state NMR and long-range DEER constraints. J. Mol. Biol. 429, 1903–1920 10.1016/j.jmb.2017.05.005 [DOI] [PubMed] [Google Scholar]
- 63.Brünger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 54, 905–921 10.1107/S0907444998003254 [DOI] [PubMed] [Google Scholar]
- 64.Bowman A., Hammond C.M., Stirling A., Ward R., Shang W., El-Mkami H. et al. (2014) The histone chaperones Vps75 and Nap1 form ring-like, tetrameric structures in solution. Nucleic Acids Res. 42, 6038–6051 10.1093/nar/gku232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sundaramoorthy R., Hughes A.L., Singh V., Wiechens N., Ryan D.P., El-Mkami H. et al. (2017) Structural reorganization of the chromatin remodeling enzyme Chd1 upon engagement with nucleosomes. eLife 6, e22510 10.7554/eLife.22510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boura E., Różycki B., Chung H.S., Herrick D.Z., Canagarajah B., Cafiso D.S. et al. (2012) Solution structure of the ESCRT-I and -II supercomplex: implications for membrane budding and scission. Structure 20, 874–886 10.1016/j.str.2012.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alonso-García N., García-Rubio I., Manso J.A., Buey R.M., Urien H., Sonnenberg A. et al. (2015) Combination of X-ray crystallography, SAXS and DEER to obtain the structure of the FnIII-3,4 domains of integrin α6β4. Acta Crystallogr., Sect. D: Biol. Crystallogr. 71, 969–985 10.1107/S1399004715002485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Celia H., Noinaj N., Zakharov S.D., Bordignon E., Botos I., Santamaria M. et al. (2016) Structural insight into the role of the Ton complex in energy transduction. Nature 538, 60–65 10.1038/nature19757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kang Y., Zhou X.E., Gao X., He Y., Liu W., Ishchenko A. et al. (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 10.1038/nature14656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yin D.M., Hannam J.S., Schmitz A., Schiemann O., Hagelueken G. and Famulok M. (2017) Studying the conformation of a receptor tyrosine kinase in solution by inhibitor-based spin labeling. Angew. Chem., Int. Ed. Engl. 56, 8417–8421 10.1002/anie.201703154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pliotas C., Ward R., Branigan E., Rasmussen A., Hagelueken G., Huang H. et al. (2012) Conformational state of the MscS mechanosensitive channel in solution revealed by pulsed electron-electron double resonance (PELDOR) spectroscopy. Proc. Natl Acad. Sci. U.S.A. 109, E2675–E2682 10.1073/pnas.1202286109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vercellino I., Rezabkova L., Oliéric V., Polyhach Y., Weinert T., Kammerer R.A. et al. (2017) Role of the nucleotidyl cyclase helical domain in catalytically active dimer formation. Proc. Natl Acad. Sci. U.S.A. 114, E9821–E9828 10.1073/pnas.1712621114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aluvila S., Mandal T., Hustedt E., Fajer P., Choe J.Y. and Oh K.J. (2014) Organization of the mitochondrial apoptotic BAK pore: oligomerization of the BAK homodimers. J. Biol. Chem. 289, 2537–2551 10.1074/jbc.M113.526806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang M., Zheng J., Nussinov R. and Ma B. (2016) Oncogenic mutations differentially affect Bax monomer, dimer, and oligomeric pore formation in the membrane. Sci. Rep. 6, 33340 10.1038/srep33340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang X., de Vera I.M.S., Veloro A.M., Blackburn M.E., Kear J.L., Carter J.D. et al. (2012) Inhibitor-induced conformational shifts and ligand-exchange dynamics for HIV-1 protease measured by pulsed EPR and NMR spectroscopy. J. Phys. Chem. B 116, 14235–14244 10.1021/jp308207h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kazmier K., Sharma S., Islam S.M., Roux B. and Mchaourab H.S. (2014) Conformational cycle and ion-coupling mechanism of the Na+/hydantoin transporter Mhp1. Proc. Natl Acad. Sci. U.S.A. 111, 14752–14757 10.1073/pnas.1410431111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Glaenzer J., Peter M.F., Thomas G.H. and Hagelueken G. (2017) PELDOR spectroscopy reveals two defined states of a sialic acid TRAP transporter SBP in solution. Biophys. J. 112, 109–120 10.1016/j.bpj.2016.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lai A.L., Huang H., Herrick D.Z., Epp N. and Cafiso D.S. (2011) Synaptotagmin 1 and SNAREs form a complex that is structurally heterogeneous. J. Mol. Biol. 405, 696–706 10.1016/j.jmb.2010.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dockter C., Müller A.H., Dietz C., Volkov A., Polyhach Y., Jeschke G. et al. (2012) Rigid core and flexible terminus: structure of solubilized light-harvesting chlorophyll a/b complex (LHCII) measured by EPR. J. Biol. Chem. 287, 2915–2925 10.1074/jbc.M111.307728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hellmich U.A., Lyubenova S., Kaltenborn E., Doshi R., van Veen H.W., Prisner T.F. et al. (2012) Probing the ATP hydrolysis cycle of the ABC multidrug transporter LmrA by pulsed EPR spectroscopy. J. Am. Chem. Soc. 134, 5857–5862 10.1021/ja211007t [DOI] [PubMed] [Google Scholar]
- 81.Georgieva E.R., Borbat P.P., Ginter C., Freed J.H. and Boudker O. (2013) Conformational ensemble of the sodium-coupled aspartate transporter. Nat. Struct. Mol. Biol. 20, 215–221 10.1038/nsmb.2494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hänelt I., Wunnicke D., Bordignon E., Steinhoff H.-J. and Slotboom D.J. (2013) Conformational heterogeneity of the aspartate transporter GltPh. Nat. Struct. Mol. Biol. 20, 210–214 10.1038/nsmb.2471 [DOI] [PubMed] [Google Scholar]
- 83.Georgieva E.R., Xiao S., Borbat P.P., Freed J.H. and Eliezer D. (2014) Tau binds to lipid membrane surfaces via short amphipathic helices located in its microtubule-binding repeats. Biophys. J. 107, 1441–1452 10.1016/j.bpj.2014.07.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cafiso D.S. (2014) Identifying and quantitating conformational exchange in membrane proteins using site-directed spin labeling. Acc. Chem. Res. 47, 3102–3109 10.1021/ar500228s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lumme C., Altan-Martin H., Dastvan, R., Sommer M.S., Oreb M., Schuetz D. et al. (2014) Nucleotides and substrates trigger the dynamics of the Toc34 GTPase homodimer involved in chloroplast preprotein translocation. Structure 22, 526–538 10.1016/j.str.2014.02.004 [DOI] [PubMed] [Google Scholar]
- 86.Sun Y., Borbat P.P., Grigoryants V.M., Myers W.K., Freed J.H. and Scholes C.P. (2015) Pulse dipolar ESR of doubly labeled mini TAR DNA and its annealing to mini TAR RNA. Biophys. J. 108, 893–902 10.1016/j.bpj.2014.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dastvan R., Fischer A.W., Mishra S., Meiler J. and Mchaourab H.S. (2016) Protonation-dependent conformational dynamics of the multidrug transporter EmrE. Proc. Natl Acad. Sci. U.S.A. 113, 1220–1225 10.1073/pnas.1520431113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Colson B.A., Thompson A.R., Espinoza-Fonseca L.M. and Thomas D.D. (2016) Site-directed spectroscopy of cardiac myosin-binding protein C reveals effects of phosphorylation on protein structural dynamics. Proc. Natl Acad. Sci. U.S.A. 113, 3233–3238 10.1073/pnas.1521281113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Georgieva E.R., Borbat P.P., Grushin K., Stoilova-McPhie S., Kulkarni N.J., Liang Z. et al. (2016) Conformational response of influenza A M2 transmembrane domain to amantadine drug binding at Low pH (pH 5.5). Front. Physiol. 7, 317 10.3389/fphys.2016.00317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Krug U., Alexander N.S., Stein R.A., Keim A., Mchaourab H.S., Sträter N. et al. (2016) Characterization of the domain orientations of E. coli 5′-nucleotidase by fitting an ensemble of conformers to DEER distance distributions. Structure 24, 43–56 10.1016/j.str.2015.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stadtmueller B.M., Yang Z., Huey-Tubman K.E., Roberts-Mataric H., Hubbell W.L. and Bjorkman P.J. (2016) Biophysical and biochemical characterization of avian secretory component provides structural insights into the evolution of the polymeric Ig receptor. J. Immunol. 197, 1408–1414 10.4049/jimmunol.1600463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eschmann N.A., Georgieva E.R., Ganguly P., Borbat P.P., Rappaport M.D., Akdogan Y. et al. (2017) Signature of an aggregation-prone conformation of tau. Sci. Rep. 7, 44739 10.1038/srep44739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Verhalen B., Dastvan, R., Thangapandian S., Peskova Y., Koteiche H.A., Nakamoto R.K. et al. (2017) Energy transduction and alternating access of the mammalian ABC transporter P-glycoprotein. Nature 543, 738–741 10.1038/nature21414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Constantinescu-Aruxandei D., Petrovic-Stojanovska B., Schiemann O., Naismith J.H. and White M.F. (2016) Taking a molecular motor for a spin: helicase mechanism studied by spin labeling and PELDOR. Nucleic Acids Res. 44, 954–968 10.1093/nar/gkv1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stadtmueller B.M., Huey-Tubman K.E., López C.J., Yang Z., Hubbell W.L. and Bjorkman P.J. (2016) The structure and dynamics of secretory component and its interactions with polymeric immunoglobulins. eLife 5, e10640 10.7554/eLife.10640 [DOI] [PMC free article] [PubMed] [Google Scholar]