

Abstract
Ataxia telangiectasia mutated (ATM), the deficiency of which causes a severe neurodegenerative disease, is a crucial mediator for the DNA double-strand break (DSB) response. We recently showed that transcription-blocking topoisomerase I cleavage complexes (TOP1cc) produce DSBs related to R-loop formation and activate ATM in post-mitotic neurons and lymphocytes. Here we discuss how TOP1cc can produce transcription arrest with R-loop formation and generate DSBs that activate ATM, as well as data suggesting that those transcription-dependent DSBs tend to form at the IgH locus and at specific genomic sites. We also address the potential roles of ATM in response to transcription-blocking TOP1cc.
Keywords: ATM, DNA double-strand breaks, R-loop, topoisomerase, transcription
Introduction
DNA double-strand breaks (DSBs) are among the most severe genomic lesions, and their repair requires the recruitment of DNA damage response (DDR) proteins. The loss of function of DDR proteins leads to genomic instability and human hereditary diseases such as ataxia telangiectasia (AT caused by ataxia telangiectasia mutated (ATM) deficiency).1 The gene product of ATM is a serine/threonine protein kinase activated by DSBs,2 which phosphorylates various DDR proteins in the vicinity of damaged sites including histone H2AX, checkpoint kinase 2 (Chk2), mediator of DNA damage checkpoint 1 (MDC1) and p53 binding protein 1 (53BP1).3 Phosphorylation of these proteins by ATM is critical for efficient DDR.3 DSBs can be produced directly by ionizing radiation and radiomimetic molecules or indirectly during replication of damaged DNA by DNA polymerase collisions or replication fork collapse.4 Until recently, it was not known whether transcription of damaged DNA templates could also lead to DSBs with ATM activation. This has been a longstanding concern as the main clinical feature of patients with AT is the acquisition of neuronal degeneration after full (post-replicative) development of the nervous system.1
Topoisomerase I (TOP1) removes DNA supercoiling generated by transcription by producing transient TOP1 cleavage complexes (TOP1cc), which are TOP1-linked DNA single-strand breaks.5,6 The rapid resealing of TOP1cc is inhibited by common DNA base alterations7 and by camptothecin (CPT) and its semi-synthetic derivatives topotecan and irinotecan, which are used to treat human cancers.8 Stabilized TOP1cc are potent transcription-blocking DNA lesions.9–11 Transcription complexes may be blocked physically a few base pair upstream of the TOP1cc12,13 by the accumulation of positive DNA supercoiling ahead of the transcribing RNA polymerase II (Pol II).14 Defective repair of transcription-blocking TOP1cc by inactivating mutation of tyrosyl-DNA-phosphodiesterase 1 (TDP1)15,16 can cause spinocerebellar ataxia with axonal neuropathy (SCAN), an autosomal recessive neurodegenerative syndrome.17 In our recent work, we used CPT to induce TOP1cc in non-replicating cells and determined whether transcription-blocking TOP1cc induce DSBs and activate the ATM-associated DDR.18
Transcription-Induced DNA Double-Strand Breaks and ATM Activation
We showed the induction of DSBs and DDR activation in post-mitotic primary neurons and lymphocytes treated with CPT, with the formation of large nuclear DDR foci containing activated ATM, phosphorylated H2AX at Serine 139 (γ-H2AX), activated Chk2, MDC1 and 53BP1.18 A single DDR focus reflects hundreds to thousands of DDR proteins concentrated around at least one DSB.4 The induction of γ-H2AX foci in neurons treated with CPT has simultaneously been reported by another group19 and can be also detected in the non-S phase population of replicating HeLa cells treated with CPT.18 Thus, stabilized TOP1cc produce replication-independent DSBs with activation of the ATM-associated DDR, asides from the well-established replication-associated DSBs in proliferating cells.20
Blocking Pol II by TOP1cc is likely the initial event for DSB production as selective inhibition of Pol II before the induction of TOP1cc suppresses DSB induction and ATM activation in post-mitotic cells as well as in the non-S phase population of replicating cells.18 Further support for the occurrence of transcription-dependent DSBs is provided by colocalization experiments showing the localization of the γ-H2AX foci in euchromatin regions at sites where transcribing Pol II is arrested by TOP1cc.18
How could blocking elongating Pol II by TOP1cc induce DSBs? R-loops (RNA: DNA hybrids) are known to induce DSBs and genomic instability.21,22 R-loops result from the extended pairing of nascent mRNA with the corresponding unwound DNA template behind the elongating Pol II.22 We have shown that γ-H2AX signals are reduced by RNase HI overexpression, which removes transcription-mediated R-loops, in both post-mitotic neurons treated with CPT as well as in HeLa cells in which replication was blocked before treatment with CPT.18 Thus, we proposed that R-loops generated by stalled TOP1cc induce the formation of DSBs (Fig. 1).18 R-loops may form as negative supercoiling accumulates behind the transcription complexes arrested by TOP1cc.23 Inhibition of the SR-kinase activity of TOPI by CPT24 may also promote R-loops by interfering with splicing.21 Although it is unclear how R-loops promote DSBs, CPT-induced R-loop-dependent DSBs may form in the 5’ region of actively transcribing genes as TOP1cc arrest Pol II early in transcription9,25 and DSBs seem to be produced within the region that forms R-loops.21
Figure 1.
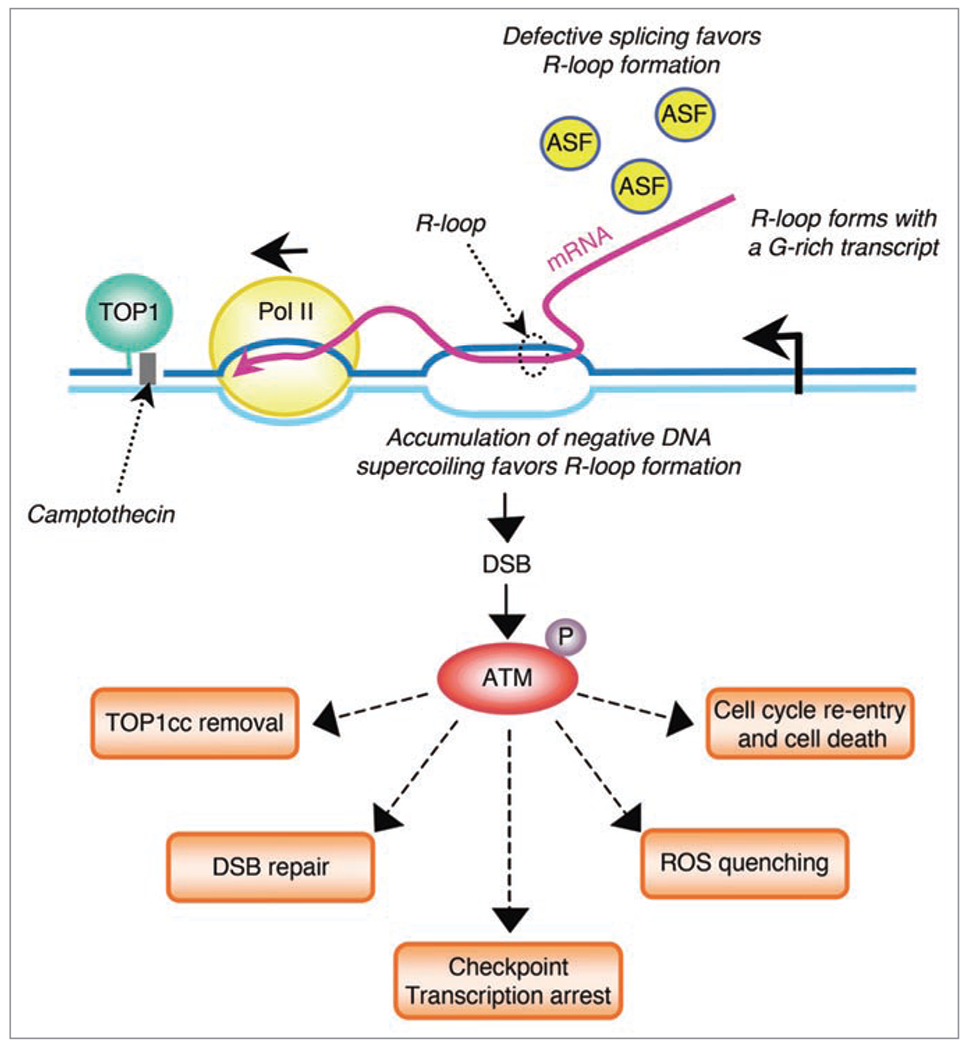
Potential roles of ATM activated by transcription- and TOP1cc-induced DSBs. Stabilization of TOP1cc by CPT interferes with transcription elongation and generates a hybrid RNA-DNA (R-loop). R-loops may form as negative supercoiling accumulates behind the transcription complexes arrested by TOP1cc. Inhibition of TOPI SR-kinase activity by CPT may also promote R-loops by interfering with splicing because of alternative splicing factor (ASF) hypophosphorylation. R-loops form with a G-rich transcript. R-loop promotes the induction of a DSB, which leads to autophosphorylation/activation of ATM in the vicinity of the DSB. ATM may promote TOP1cc removal, DSB repair, transcription arrest and ROS quenching. Besides its protective role, ATM may also promote cell cycle re-entry of post-mitotic neurons leading to neuronal death.
We have also detected transcription-induced DSBs in response to the transcription-coupled nucleotide excision repair (TCR) poison, ecteinascidin (Et-743) although those DSBs are dependent of the TCR and the MRE11-RAD50-NBS1 (MRN) complex.26 Likewise, the histone deacetylase inhibitor, SAHA also induces transcription-dependent DSBs (Conti and Pommier, submitted). Hence, the formation of DSBs appears as a general response to DNA lesions and chromatin modifications that interfere with the translocation of Pol II through expressed genes.
Enrichment of Transcription-Dependent DSBs at Specific Genomic Regions
Transcription-dependent DDR foci score low as compared to replication-dependent DDR foci. An average of 2 and 5 γ-H2AX foci form per nucleus in response to CPT in post-mitotic lymphocytes and neurons, respectively.18 Under similar conditions, multiple γ-H2AX foci (at least 50) form in the nuclei of replicating cells.4,18,20 The low frequency of transcription-dependent γ-H2AX foci may reflect the induction of DSBs only at few specific genomic sites such as the transcribed regions prone to form R-loops.
R-loops tend to form at sequences that generate a G-rich transcript,27–29 and such sequences are found at the mammalian immunoglobulin heavy (IgH) class switch regions.29 Class switch recombination (CSR) is a recombination process that enables mammalian B-cells to generate antibody isotypes. CSR requires activation-induced cytidine deaminase (AID), an enzyme that requires DNA to be single-stranded and involves transcription and the generation of and R-loops at switch regions (reviewed in ref. 30). The induction of DSBs at switch regions has been visualized by the formation of γ-H2AX foci at the IgH locus in B cells activated for CSR.31
Owing to the susceptibility of IgH locus to form R-loops, we tested whether the DSBs induced by TOP1cc were enriched at that locus. We used immunocytochemistry staining followed by fluorescence in situ hybridization (ICC-FISH) to simultaneously visualize DSBs (detected by γ-H2AX foci) and specific DNA regions (IgH) in post-mitotic cells treated with CPT. Figure 2 shows an enrichment of γ-H2AX foci associated with sites of IgH in unstimulated splenocytes treated by CPT indicating that transcription-associated DSBs induced by TOP1cc form at specific genomic sites.
Figure 2.
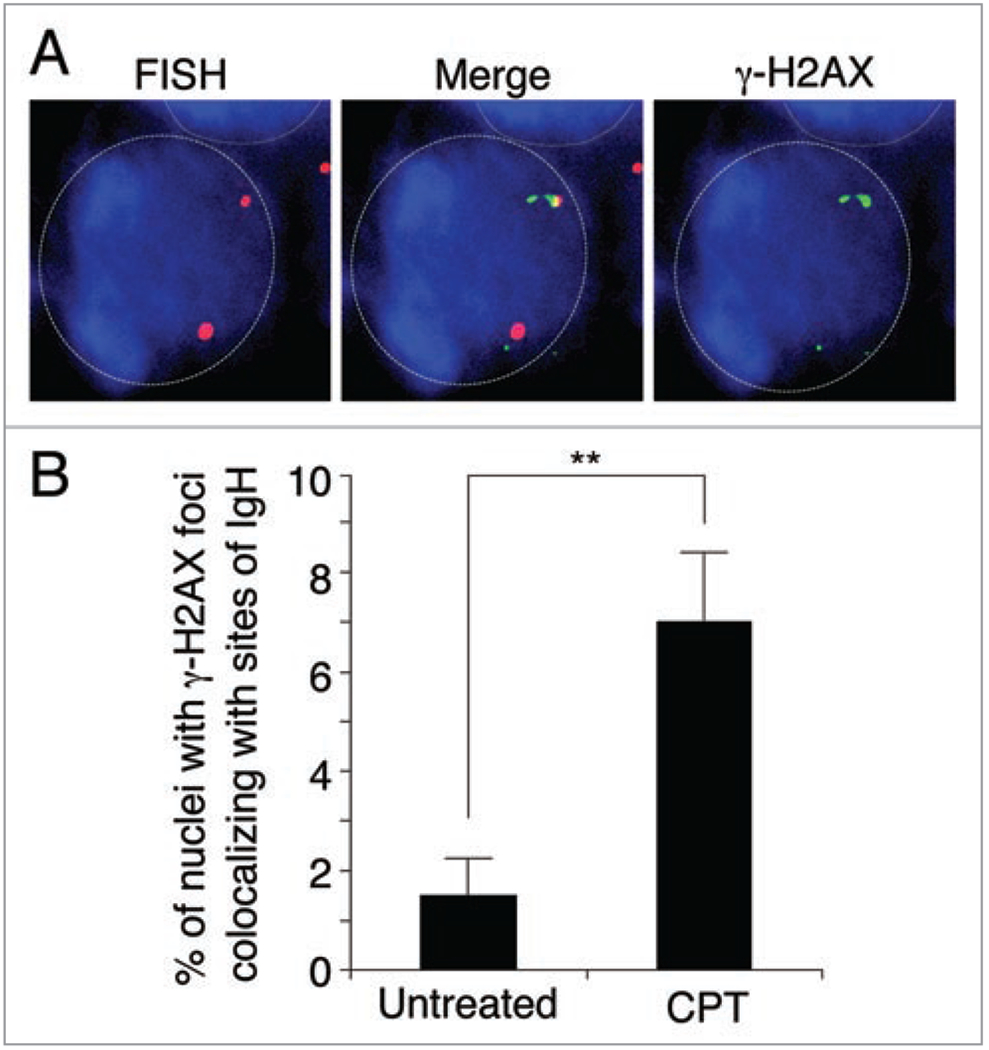
Colocalization of γ-H2AX and IgH increases after CPT treatment in post-mitotic splenocytes. (A) Representative image of immunostaining for γ-H2AX (green) and fluorescent in situ hybridization (FISH) for IgH (red). DNA was counterstained with DAPI (blue). Nuclear outlines are highlighted. Mouse splenocytes were treated with 25 μM CPT for 1 h and slides were prepared as described previously.31 (B) Percentage of cells in which γ-H2AX and IgH colocalized. The data shows the average of two independent experiments in which 100 cells were counted for each experiment. Error bars signify standard deviation. Statistical analysis was carried out using Fishers exact test. A double asterisk indicates a p-value <0.005.
ATM Response to Transcription-Induced DNA Double-Strand Breaks
In our recent study,18 we concluded that ATM is the main transducer of transcription-dependent DSBs in post-mitotic cells treated with CPT. Although some ATM substrates can also be phosphorylated by ataxia telangiectasia and Rad3 related (ATR)32 and DNA-dependent protein kinase (DNA-PK),33 ATR is not or only weakly expressed in post-mitotic lymphocytes18,34 and neurons, respectively.18,35 Also, CPT-induced γ-H2AX foci formation was unaffected by DNA-PK kinase inhibition in post-mitotic cells.18 This is in marked contrast with replication-associated DSBs produced in CPT-treated proliferating cells where γ-H2AX is primarily induced by ATR and DNA-PK and in a lesser extend by ATM.20
It is well established that ATM signals DSB repair.3 However, ATM seems primarily involved in the repair of DSBs in heterochromatin regions36 whereas transcription-associated DSBs form in euchromatin.18 We have shown that ATM activates DNA-PK in post-mitotic cells treated by CPT.18 Although the DNA-PK-dependent nonhomologous end-joining (NHEJ) pathway is considered to be the prevalent pathway for DSB repair in G0 and G1leading to cell death phases of the cell cycle,37 DNA-PK does not seem to be involved in the repair of transcription-associated DSBs in post-mitotic cells exposed to CPT.18 Thus, other pathways are likely involved in repairing the transcription-associated DSBs in post-mitotic cells. Other potential roles of ATM activated by transcription-dependent DSBs include ROS quenching38 and cell cycle re-entry of post-mitotic neurons leading to cell death19,39 (Fig. 1). As detailed below, recent data suggest that ATM may also serve to repair or remove transcription-blocking TOP1cc as well as to prevent cumulative DNA lesions by inhibiting transcription (Fig. 1).
Transcription-blocking TOP1cc seem primarily removed by the TDP1 excision pathway. We refer the reader to recent reviews for a full description of TOP1 repair pathways.40,41 Briefly, TOP1 excision by TDP1 requires prior proteolysis of TOP1. Our group and others have reported that TOP1 ubiquitination and subsequent degradation by the 26 S proteasome is selectively transcription dependent following CPT exposure,25,42,43 as well as that TDP1 primarily repairs transcription-blocking TOP1cc as compared to replication-blocking TOP1cc.15,16 Consistent with the involvement of the TDP1 pathway in the removal of transcription-blocking TOP1cc, TOP1 degradation has been suggested to promote resumption of RNA synthesis and cell survival.43 Our recent observations suggest that ATM could activate the TDP1 excision pathway. First, TOP1 degradation is dependent of the DDR protein, BRCA1.25 BRCA1 is a known ATM substrate,44 although it is still unclear whether the ATM-dependent phosphorylation of BRCA1 is involved in TOP1 degradation. Second, TDP1 is phosphorylated/activated by ATM in response to CPT.45 Thus, a working model is that TOP1cc produce transcription arrest and generate DSBs that activate ATM, which in turn removes transcription-blocking TOP1cc by promoting TOP1 degradation and TDP1 action. Based on this model, ATM might serve to promote resumption of RNA synthesis and cell survival following transcription arrest by TOP1cc.
The ATM pathway has been shown to inhibit Pol I transcription in response to DSBs.46 These findings and our work showing the activation of ATM by transcription-induced DSBs,18 suggest the existence of an ATM-dependent transcription checkpoint pathway, in which few localized transcription-associated DSBs activate ATM, which in turn inhibits transcription more globally. In line with this hypothesis, approximately 80 to 90% of transcription is inhibited within minutes following CPT exposure,43 and some ATM substrates are involved in transcription regulation such as the corepressor of gene transcription Kap-1,47 and the transcription factor Spl.48 Thus, similar to replication checkpoint that arrest DNA replication and cell cycle progression, global transcription arrest could provide time for cells to repair the transcription-associated DNA lesions and thereby to prevent additional genomic alterations resulting from transcription of a damaged DNA template.
Conclusions and Perspectives
The transcription- and R-loop-dependent DSBs, which are potentially lethal if not repaired, might contribute to the cytotoxicity of TOP1cc in post-mitotic cells.19,39,49 Thus, defective response to TOP1-induced transcription-dependent DSBs could be involved in the death of neurons in AT and SCAN patients. As should be the case for novel insights, our findings raise a set of new inquiries. First, it will be important to know whether stabilization of TOP1cc by common DNA lesions (i.e., DNA oxidative lesions)7,40 can also promote the induction of transcription-dependent DSBs with ATM activation. Other issues concern the IgH locus being targeted in lymphoid cells treated with CPT in the absence of immune stimulation and the involvement of AID in the formation of the transcription-induced DSBs by TOP1cc. Finally, it will be relevant to know the distribution of the transcription-dependent DSBs across the genome and whether genome-wide analyses will identify hot-spot regions where transcription-associated DSBs are produced in response to CPT.
Acknowledgements
We thank Dr. A. Nussenzweig for providing the IgH DNA FISH probe. This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Abbreviations:
- AID
activation-induced cytidine deaminase
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia and Rad3 related
- Chk2
checkpoint kinase 2
- CPT
camptothecin
- DDR
DNA damage response
- DNA-PK
DNA-dependent protein kinase
- CSR
class switch recombination
- DSB
DNA double-strand break
- γ-H2AX
phosphorylated histone H2AX on Ser139
- MDC1
mediator of DNA damage checkpoint 1
- 53BP1
p53 binding protein 1
- Pol II
RNA polymerase II
- SCAN
spinocerebellar ataxia with axonal neuropathy
- TCR
transcription-coupled nucleotide excision repair
- TDP1
tyrosyl-DNA-phosphodiesterase 1
- TOP1
topoisomerase I
- TOP1cc
topoisomerase I cleavage complexes
References
- 1.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995; 268:1749–53. [DOI] [PubMed] [Google Scholar]
- 2.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003; 421:499–506. [DOI] [PubMed] [Google Scholar]
- 3.Shiloh Y The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci 2006; 31:402–10. [DOI] [PubMed] [Google Scholar]
- 4.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. gammaH2AX and cancer. Nat Rev Cancer 2008; 8:957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Champoux JJ. DNA topoisomerase I-mediated nicking of circular duplex DNA. Methods Mol Biol 2001; 95:81–7. [DOI] [PubMed] [Google Scholar]
- 6.Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nature reviews 2002; 3:430–40. [DOI] [PubMed] [Google Scholar]
- 7.Pourquier P, Pommier Y. Topoisomerase I-mediated DNA damage. Adv Cancer Res 2001; 80:189–216. [DOI] [PubMed] [Google Scholar]
- 8.Pommier Y Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 2006; 6:789–802. [DOI] [PubMed] [Google Scholar]
- 9.Capranico G, Ferri F, Fogli MV, Russo A, Lotito L, Baranello L. The effects of camptothecin on RNA polymerase II transcription: roles of DNA topoisomerase I. Biochimie 2007; 89:482–9- [DOI] [PubMed] [Google Scholar]
- 10.Ljungman M, Lane DP. Transcription—guarding the genome by sensing DNA damage. Nat Rev Cancer 2004; 4:727–37. [DOI] [PubMed] [Google Scholar]
- 11.Baranello L, Bertozzi D, Fogli MV, Pommier Y, Capranico G. DNA topoisomerase I inhibition by camptothecin induces escape of RNA polymerase II from promoter-proximal pause site, antisense transcription and histone acetylation at the 5 human HIF-1 {alpha} gene locus. Nucleic Acids Res 2009; In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bendixen C, Thomsen B, Alsner J, Westergaard O. Camptothecin-stabilized topoisomerase I-DNA adducts cause premature termination of transcription. Biochemistry 1990; 29:5613–9- [DOI] [PubMed] [Google Scholar]
- 13.Wu J, Liu LF. Processing of topoisomerase I cleavable complexes into DNA damage by transcription. Nucleic Acids Res 1997; 25:4181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mondal N, Zhang Y,Jonsson Z, Dhar SK, Kannapiran M, Parvin JD. Elongation by RNA polymerase II on chromatin templates requires topoisomerase activity. Nucleic Acids Res 2003; 31-5016–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miao ZH, Agama K, Sordet O, Povirk L, Kohn KW, Pommier Y. Hereditary ataxia SCAN1 cells are defective for the repair of transcription-dependent topoisomerase I cleavage complexes. DNA repair 2006. [DOI] [PubMed] [Google Scholar]
- 16.El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, et al. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 2005; 434:108–13 [DOI] [PubMed] [Google Scholar]
- 17.Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet 2002; 32:267–72. [DOI] [PubMed] [Google Scholar]
- 18.Sordet O, Redon CE, Guirouilh-Barbat J, Smith S, Solier S, Douarre C, et al. Ataxia telangiectasia mutated activation by transcription-and topoisomerase I-induced DNA double-strand breaks. EMBO Rep 2009; 10:887–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian B, Yang Q, Mao Z. Phosphorylation of ATM by Cdk5 mediates DNA damage signalling and regulates neuronal death. Nat Cell Biol 2009; 11:211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, et al. Phosphorylation of histone H2AX and activation of Mre11, Rad50 and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J Biol Chem 2003; 278:20303–12. [DOI] [PubMed] [Google Scholar]
- 21.Li X, Manley JL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 2005; 122:365–78. [DOI] [PubMed] [Google Scholar]
- 22.Huertas P, Aguilera A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Molecular cell 2003; 12:711–21. [DOI] [PubMed] [Google Scholar]
- 23.Drolet M, Broccoli S, Rallu F, Hraiky C, Fortin C, Masse E, et al. The problem of hypernegative supercoiling and R-loop formation in transcription. Front Biosci 2003; 8:210–21. [DOI] [PubMed] [Google Scholar]
- 24.Soret J, Gabut M, Dupon C, Kohlhagen G, Stevenin J, Pommier Y, et al. Altered serine/arginine-rich protein phosphorylation and exonic enhancer-dependent splicing in Mammalian cells lacking topoisomerase I. Cancer research 2003; 63:8203–11. [PubMed] [Google Scholar]
- 25.Sordet O, Larochelle S, Nicolas E, Stevens EV, Zhang C, Shokat KM, et al. Hyperphosphorylation of RNA polymerase II in response to topoisomerase I cleavage complexes and its association with transcription-and BRCA1-dependent degradation of topoisomerase I. J Mol Biol 2008; 381:540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guirouilh-Barbat J, Redon C, Pommier Y. Transcription-coupled DNA double-strand breaks are mediated via the nucleotide excision repair and the Mre11-Rad50-Nbs1 complex. Mol Biol Cell 2008; 19:3969–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee DY, Clayton DA. Properties of a primer RNA-DNA hybrid at the mouse mitochondrial DNA leading-strand origin of replication. J Biol Chem 1996; 271:24262–9. [DOI] [PubMed] [Google Scholar]
- 28.Masukata H, Tomizawa J. A mechanism of formation of a persistent hybrid between elongating RNA and template DNA. Cell 1990; 62:331–8. [DOI] [PubMed] [Google Scholar]
- 29.Roy D, Yu K, Lieber MR. Mechanism of R-loop formation at immunoglobulin class switch sequences. Mol Cell Biol 2008; 28:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaudhuri J, Alt FW. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol 2004; 4:541–52. [DOI] [PubMed] [Google Scholar]
- 31.Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature 2001; 414:660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160–6. [DOI] [PubMed] [Google Scholar]
- 33.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer research 2004; 64:2390–6. [DOI] [PubMed] [Google Scholar]
- 34.Jones GG, Reaper PM, Pettitt AR, Sherrington PD. The ATR-p53 pathway is suppressed in noncycling normal and malignant lymphocytes. Oncogene 2004; 23:1911–21. [DOI] [PubMed] [Google Scholar]
- 35.Carlessi L, De Filippis L, Lecis D, Vescovi A, Delia D. DNA-damage response, survival and differentiation in vitro of a human neural stem cell line in relation to ATM expression. Cell Death Differ 2009; 16:795–806. [DOI] [PubMed] [Google Scholar]
- 36.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, et al. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Molecular cell 2008; 31:167–77. [DOI] [PubMed] [Google Scholar]
- 37.Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 2003; 4:712–20. [DOI] [PubMed] [Google Scholar]
- 38.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004; 431:997–1002. [DOI] [PubMed] [Google Scholar]
- 39.Morris EJ, Geller HM. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell cycle-independent toxicity. J Cell Biol 1996; 134:757–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pommier Y, Barcelo JM, Rao VA, Sordet O, Jobson AG, Thibaut L, et al. Repair of topoisomerase I-mediated DNA damage. Prog Nucleic Acid Res Mol Biol 2006; 81:179–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pommier Y, Redon C, Rao VA, Seiler JA, Sordet O, Takemura H, et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat Res 2003; 532:173–203. [DOI] [PubMed] [Google Scholar]
- 42.Desai SD, Li TK, Rodriguez-Bauman A, Rubin EH, Liu LF. Ubiquitin/26S proteasome-mediated degradation of topoisomerase I as a resistance mechanism to camptothecin in tumor cells. Cancer research 2001; 61:5926–32. [PubMed] [Google Scholar]
- 43.Desai SD, Zhang H, Rodriguez-Bauman A, Yang JM, Wu X, Gounder MK, et al. Transcription-dependent degradation of topoisomerase I-DNA covalent complexes. Mol Cell Biol 2003; 23:2341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999; 286:1162–6. [DOI] [PubMed] [Google Scholar]
- 45.Das BB, Antony S, Gupta S, Dexheimer TS, Redon CE, Garfield S, et al. Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. EMBO J 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kruhlak M, Crouch EE, Orlov M, Montano C, Gorski SA, Nussenzweig A, et al. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 2007; 447:730–4. [DOI] [PubMed] [Google Scholar]
- 47.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM-and KAP-1 dependent pathway. Nat Cell Biol 2006; 8:870–6. [DOI] [PubMed] [Google Scholar]
- 48.Olofsson BA, Kelly CM, Kim J, Hornsby SM, Azizkhan-Clifford J. Phosphorylation of sp1 in response to DNA damage by ataxia telangiectasia-mutated kinase. Mol Cancer Res 2007; 5:1319–30. [DOI] [PubMed] [Google Scholar]
- 49.Keramaris E, Hirao A, Slack RS, Mak TW, Park DS. Ataxia telangiectasia-mutated protein can regulate p53 and neuronal death independent of Chk2 in response to DNA damage. J Biol Chem 2003; 278:37782–9. [DOI] [PubMed] [Google Scholar]