

Abstract
Objective:
This study investigates the effectiveness and tolerability of switching to a darunavir/cobicistat (DRV/c)-based antiretroviral regimen from a ritonavir-boosted protease inhibitor (PI/r)-based regimen in virologically suppressed HIV-positive patients. DRV trough values were also investigated.
Setting:
Prospective, multicenter, single-country, noninterventional cohort study.
Methods:
This study included patients on a PI/r-based ART for at least 12 months having plasma HIV-1 RNA <50 copies/mL since at least 6 months. The primary endpoint, defined as HIV-1 RNA <50 copies/mL, was measured at 48 ± 6 weeks from baseline. A secondary analysis was performed using the time to loss of virological response algorithm. Biochemical parameters, including DRV trough samples, were collected as per clinical practice and measured using high-performance liquid chromatography.
Results:
Of 336 patients enrolled, 282 completed the study: 70.8% had plasma HIV-1 RNA <50 copies/mL at 48 weeks; using the time to loss of virological response algorithm, 82.7% maintained virological suppression. Virological failure was observed in 6 patients (1.8%). Adverse event–related discontinuations were 4.5%. After 48 weeks, we found a significant improvement in both triglycerides (median, 130 to 113.5 mg/dL, P = 0.0254) and high-density lipoprotein cholesterol (48 to 49 mg/dL, P < 0.0001) but no change in other biomarkers. DRV trough concentrations in 56 subjects showed a median value of 2862.5 (1469.5–4439) ng/mL, higher in women than in men (4221 vs. 2634 ng/mL, P = 0.046).
Conclusions:
In stable HIV-1 positive virologically suppressed patients, the switch to DRV/c-based ART was beneficial in terms of low rates of virological failure and adverse events due to its high tolerability and improvement in triglycerides.
Key Words: darunavir/cobicistat, darunavir/ritonavir, effectiveness, HIV, STORE, virologically suppressed
INTRODUCTION
The current treatment of HIV-positive patients requires lifelong administration of combination antiretroviral (ARV) drugs, which is extremely effective and well tolerated, but several factors may decrease its long-term efficacy. As patient adherence is crucial to maintain the efficacy and immunological benefits of ARV treatment,1 it is essential to adopt effective and well-tolerated ARV strategies, including a reduced number of pills. Regarding this latter point, the adoption of fixed-dose combinations and single-tablet regimens has a decisive role in improving patient compliance and adherence to antiretroviral therapy (ART).2
For the above reason, a coformulation of the protease inhibitor (PI) darunavir (DRV) with the pharmacoenhancer cobicistat has been developed.2 Cobicistat, at the dose of 150 mg once a day, while not having antiviral activity, is more selective in terms of enzyme inhibition, thus reducing the number of clinically relevant drug-drug interactions compared with the previous pharmacoenhancer ritonavir (RTV).3 This fixed-dose combination, reducing the pill burden, allows to improve adherence to ART.4 To date, few clinical trials have studied DRV/c in a real-world clinical setting. Most data on DRV/c were obtained from the phase 3 Study GS-US-216-0130,5 but real-life data were available only in 2017.6
The primary aim of this study was to describe the effectiveness of DRV/c-based regimens in terms of maintenance of virological suppression after 48 ± 6 weeks from study enrollment. Furthermore, we described steady-state DRV trough values in a subgroup of patients, if collected per clinical practice.
METHODS
This study, named STart Of REzolsta (STORE), was a prospective, multicenter, noninterventional cohort study conducted between 2016 and 2018 with HIV-1–infected adult outpatients referred to the Italian Infectious Disease Hospital departments in the centers participating in this study. All patients were on stable RTV-boosted ART with PIs for at least 12 months and virologically suppressed (HIV-1 RNA < 50 copies/mL) for at least 6 months. Patients were offered to enter this study once their treating physician had considered them eligible for DRV/c-based treatment as per the DRV/c Summary of Product Characteristics.7
The main exclusion criteria were (1) estimated glomerular filtration rate <70 mL/min if any coadministered agent (eg, emtricitabine, lamivudine, tenofovir disoproxil fumarate, or adefovir dipivoxil) required dose adjustment based on creatinine clearance, (2) pregnancy or breastfeeding at the time of enrollment, (3) a history of allergy or intolerance to sulfonamides, (4) receiving DRV/r 600/100 bis in die or PI/r monotherapy, (5) being treated with directly active agents against hepatitis C virus within 1 year of enrollment, and (6) scheduled chemotherapy.
Ethics approval was obtained from each institutional review board, and patients signed written informed consent before being enrolled. The trial was registered on ClinicalTrials.gov (NCT02926456).
This study collected only data available per clinical practice. Patients were observed prospectively for 48 ± 6 weeks after starting the DRV/c-based regimen. The end of the study was defined as the last visit within the study for the last patient enrolled.
The primary endpoint, defined as HIV-1 RNA <50 copies/mL measured as per the FDA Snapshot algorithm, was measured at 48 ± 6 weeks from baseline. A secondary analysis was performed using the time to loss of virological response (TLOVR) algorithm.8 Virological failure (VF) was defined as the last plasma HIV-1 RNA value in the 48-week window ≥50 copies/mL as well as discontinuation before the end of the study for any reason with the last available viral load (VL) ≥50 copies/mL. Secondary endpoints included changes in CD4 and CD8 cell counts, in serum lipid levels, and in the values of routinely collected laboratory examinations.
Four visits were scheduled during the study following the clinical practice: visit 1 (enrollment), visit 2 (4–8 weeks from the start of DRV/c), visit 3 (24 ± 6 weeks from the start of DRV/c), and visit 4 (48 ± 6 weeks). The results described here are related to visit 4.
Where the plasma concentrations of ARVs were routinely measured, the values of cobicistat-boosted DRV trough concentrations (24 ± 3 hours after the last drug intake) were registered throughout the study and measured using high-performance liquid chromatography.
No data were available regarding the Ctrough of RTV-boosted PIs taken before the switch. To assess the adequacy of DRV exposure, a half-maximal effective concentration (EC50), adjusted for protein binding for both wild-type (55 ng/mL) and drug-resistant (550 ng/mL) HIV-1, was used.
A sample size of 300 patients was set with a prespecified target of 30% female patients. The original sample size calculation was based on feasibility criteria and assumed a 10% dropout rate.
Patient characteristics were described using standard descriptive statistics. Continuous variables are presented as mean values ± SD or median values and interquartile ranges and categorical variables as numbers and percentages. Data are presented as median and 25th–75th (Q1–Q3) percentiles. Nonparametric Mann–Whitney U, Wilcoxon signed-rank, and Kruskal–Wallis tests were used for the analysis.
RESULTS
Baseline
In this study, 348 patients were enrolled, and of these, 336 were included in the final analysis. Among the 336 patients, 282 completed the study, whereas 54 (16%) discontinued before week 48. The baseline characteristics are shown in Table 1. Female patients had longer duration of infection (16.9 vs. 13.2 years, P = 0.0006), longer ART use (13.9 vs. 10.7 years, P = 0.0002), and longer duration of viral suppression (5.8 vs. 4.7 years, P = 0.011); they acquired HIV through heterosexual contacts more frequently than male patients (72.9% vs. 26.6%, P < 0.0001). DRV/r was the most common PI used before enrollment (N = 274, 81.5%), followed by atazanavir/r (N = 33, 9.8%) and lopinavir/r (N = 21, 6.3%). Most patients (59.6%) were receiving 2 additional drugs besides boosted PIs: 16.7% received abacavir/lamivudine and 42.9% received tenofovir disoproxil fumarate /emtricitabine.
TABLE 1.
Main Demographic and HIV-Associated Patient Characteristics at Baseline

Efficacy
Of the 282 patients who completed the study, 242 had a VL measurement within the visit 4 (V4) window (48 ± 6 weeks). Of the 242 patients, 238 (98.3%) had a VL of <50 copies/mL. During the study period, 6 patients experienced VF; 5 of them were deemed incompletely adherent by the treating clinicians.
According to the FDA snapshot analysis, 238 patients (70.8%) had a virological response with plasma HIV-1 RNA <50 copies/mL at V4. Considering the TLOVR algorithm, 278 patients (82.7%) maintained virological suppression. Both rates were higher in male patients than in female patients (FDA snapshot: 72.5% vs. 67.3%, P = 0.329; TLOVR: 85.6% vs. 76.6%, P = 0.043). The Kaplan–Meier curve, depicting the risk of withdrawal from the study for different reasons, is represented in Figure 1. Considering the patients who completed the study, including the ones having a VL measurement outside the V4 window (N = 282), 98% of them were virologically suppressed. At baseline, female patients showed a higher median CD4+ T-lymphocyte count (704 vs. 617 cells/mm3, P = 0.0149, Kruskal–Wallis test), a higher median CD4/CD8 ratio (0.9 vs. 0.7, P = 0.0003), and a lower median CD8+ T-cell count (758 vs. 893 cells/mm3, P = 0.0235). After 48 weeks, the difference between men and women in either CD4+ or CD8+ cell counts was lost (P = 0.36 and P = 0.06, respectively).
FIGURE 1.
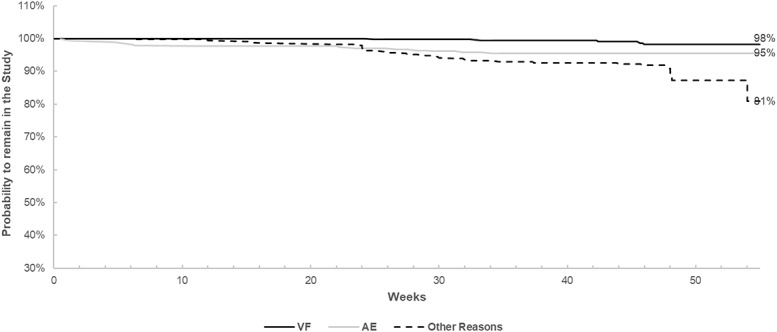
Kaplan–Meier curves depicting the probability of remaining in the study according to different causes of discontinuation.
Safety
Adverse events (AEs) and serious AEs (SAEs) were experienced by 69 of 336 (20.5%) and 17 of 336 (5.1%) patients, respectively. During the study, 120 AEs were reported; the most common were infections (6.8%), gastrointestinal events (6.8%, mostly nausea), and events involving the central nervous system (4.2%, mostly headache). No SAEs were considered ART-related. Fifteen patients were withdrawn from the study due to safety reasons, including 1 pregnancy and 2 patients with non–drug-related SAEs (hepatitis A and anaplastic lymphoma). The remaining 12 patients were withdrawn primarily due to gastrointestinal AEs, which were all grade 1 or 2 and deemed as either possibly or very likely to be related to DRV/c.
Serum creatinine showed a small expected median (Q1–Q3) increase during the first 4–8 weeks [from 0.8 (0.7–1) to 0.9 (0.8–1) mg/dL] and subsequently remained stable. This was paralleled by a slight decrease in the estimated glomerular filtration rate [from 98.5 (85.6–106.1) mL/min per 1.73 m2 to 94 (80.1–103.4) mL/min per 1.73 m2]. We observed a significant decrease in triglycerides [130 (97–180) mg/dL at baseline and 113.5 (83–165.5) mg/dL at week 48, P = 0.0254] and a slight increase in high-density lipoprotein cholesterol [48 (39–57) mg/dL at baseline and 49 (41–60) mg/dL at week 48, P = <0.0001].
We did not observe any significant changes in other biochemical markers (lipid, glucidic, and hepatic profile).
Pharmacokinetics
DRV trough concentrations were collected from 56 patients. These patients were mostly of European ancestry (n = 53, 94.6%) and men (n = 39, 69.6%), with a median age of 49 (38–54) years and a body mass index (BMI) of 24.1 (21.9–26.8) kg/m2. No concomitant drugs had any predicted significant effect on DRV exposure according to the Liverpool HIV Interactions Website.9
Median DRV Ctrough values were 2862.5 (1469.5–4439) ng/mL; values were higher in women than in men [4221 (2741–5622) ng/mL vs. 2634 (1430–3752) ng/mL, P = 0.046, Mann–Whitney U test]. BMI did not differ between genders. No correlation of Ctrough values with BMI, age, or patient-reported side effects was observed.
At the time of sampling, all patients were virologically suppressed. None showed DRV Ctrough values below the threshold of 55 ng/mL, whereas 6 patients had DRV Ctrough values below 550 ng/mL, despite maintaining virological suppression.
DISCUSSION
In this study, we describe the effectiveness of switching to DRV/c in people living with HIV treated with boosted PI/r-based regimens. We observed a low rate of VFs and few discontinuations due to side effects. Antiviral efficacy was maintained in most patients, with rates comparable with those in other PI-based regimens. Furthermore, in our observational setting, the FDA snapshot analysis may have underestimated the virological results: 40 patients had results outside the 48-week window, leading to an apparent lower rate of virological suppression. However, the TLOVR algorithm showed a satisfactory rate of HIV-1 RNA suppression at week 48 (82.7%). In addition, confirmed VFs were uncommon (1.8%), and no treatment-emergent resistance-associated mutations were reported.
In this study, the DRV/c-based ART was well tolerated; 4.5% (15/336) of study patients stopped the study drug because of AEs. After excluding SAEs or special situations unrelated to DRV (such as lymphoma, hepatitis A, and pregnancy), most side effects were related to the gastrointestinal system (specifically, nausea), as previously reported.5 The observed discontinuation rate is not quite different from what has been observed in recent studies, where stable patients were switched to other ARV drugs.10–12 Laboratory abnormalities were not observed, except an expected small serum creatinine increase, attributed to the well-known MATE1 inhibition by cobicistat.7 Serum lipid levels were stable with a small but significant improvement in high-density lipoprotein cholesterol and a decrease in triglyceride levels, as already reported elsewhere.6 The data regarding a neutral-positive impact on the lipid profile in our observational trial support DRV/c as a therapeutic option for both dyslipidemic and normolipidemic HIV patients.
The real-life setting of this study is relevant and explains some of the observed results. Notably, we enrolled a high proportion of women (31.8%), often underrepresented in clinical trials. This allowed us to notice some differences in virological efficacy, lower in female patients, whereas the immunological profile was quite similar between genders.
In this study, the CD4/CD8 ratio increased in both men and women, and this may show a positive impact on the immune outcomes associated with the use of DRV/c.
Few real-life data on cobicistat-boosted DRV Ctrough values have been published. In this study, the measurements of DRV Ctrough have shown DRV exposure to be similar to that observed in patients receiving a low-dose RTV but higher than that reported in other studies assessing DRV exposure when administered with cobicistat.13–15
Six of the 56 samples showed DRV Ctrough values below the target concentration for drug-resistant viruses (550 ng/mL), and none showed DRV Ctrough values below the target concentration for wild-type viruses (55 ng/mL), confirming the high inhibitory quotient obtained by DRV. The data showing higher DRV concentrations in female patients are partially unexplained, but these were consistent with earlier studies.16,17 The significant difference between men and women in our study could be ascribed to the small sample size.
The main limitation of this study is that it is a single-arm study with no comparison.
In conclusion, stable HIV-positive patients receiving boosted PIs may benefit from switching to DRV/c in terms of low rates of VF and AEs due to its high tolerability and in terms of reduced levels of certain lipid biomarkers, particularly triglycerides.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank all the patients and all the clinicians who participated in this study.
Footnotes
Supported by Janssen-Cilag SpA.
A.G. received a grant from Janssen-Cilag, ViiV Healthcare, MSD, BMS, Abbvie, Gilead Sciences, Novartis, Pfizer, Astellas, AstraZeneca and Angelini; he received consulting fees or honorarium from Janssen-Cilag, ViiV Healthcare, MSD, BMS, Abbvie, Gilead Sciences, Novartis, Pfizer, Astellas, AstraZeneca and Angelini. A.A. received a grant from Gilead Sciences, ViiV Healthcare and Janssen-Cilag; he received consulting fees or honorarium from ViiV Healthcare, Janssen-Cilag and Merck. S.R. received grants research support, compensation for CME activities or advisory boards form ViiV Healthcare, MSD, Gilead Sciences and Janssen. S.B. has received grants, travel grants and consultancy fees from Abbvie, BMS, MSD, Gilead, Janssen-Cilag, and ViiV. A.U., R.T., and D.M. are Janssen employees. The remaining authors have no conflicts of interest to disclose.
REFERENCES
- 1.Altice F, Evuarherhe O, Shina S, et al. Adherence to HIV treatment regimens: systematic literature review and meta-analysis. Patient Prefer Adherence. 2019;13:475–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curran A, Pérez-Valero I, Moltó J. Rezolsta® (darunavir/cobicistat): first boosted protease inhibitor co-formulated with cobicistat. AIDS Rev. 2015;17:114–120. [PubMed] [Google Scholar]
- 3.Tseng A, Hughes CA, Wu J, et al. Cobicistat versus ritonavir: similar pharmacokinetic enhancers but some important differences. Ann Pharmacother. 2017;51:1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capetti A, Cossu MV, Rizzardini G. Darunavir/cobicistat for the treatment of HIV-1: a new era for compact drugs with high genetic barrier to resistance. Expert Opin Pharmacother. 2015;16:2689–2702. [DOI] [PubMed] [Google Scholar]
- 5.Tashima K, Crofoot G, Tomaka FL, et al. Cobicistat-boosted darunavir in HIV-1-infected adults: week 48 results of a phase IIIb, open-label single-arm trial. AIDS Res Ther. 2014;11:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Echeverría P, Bonjoch A, Puig J, et al. Significant improvement in triglyceride levels after switching from ritonavir to cobicistat in suppressed HIV-1-infected subjects with dyslipidaemia. HIV Med. 2017;18:782–786. [DOI] [PubMed] [Google Scholar]
- 7.Available at: https://www.ema.europa.eu/en/documents/product-information/rezolsta-epar-product-information_en.pdf. Accessed July 2019.
- 8.US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry. Antiretroviral Drugs Using Plasma HIV RNA Measurements—Clinical Considerations for Accelerated and Traditional Approval; 2002. Available at: www.fda.gov/cder/guidance/index.htm. Accessed July 2019. [Google Scholar]
- 9.Available at: www.hiv-druginteractions.org. Accessed July 2019.
- 10.Orkin C, Molina JM, Negredo E, et al. Efficacy and safety of switching from boosted protease inhibitors plus emtricitabine and tenofovir disoproxil fumarate regimens to single-tablet darunavir, cobicistat, emtricitabine, and tenofovir alafenamide at 48 weeks in adults with virologically suppressed HIV-1 (EMERALD): a phase 3, randomised, non-inferiority trial. Lancet HIV. 2018;5:e23–e34. [DOI] [PubMed] [Google Scholar]
- 11.Daar ES, DeJesus E, Ruane P, et al. Efficacy and safety of switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide from boosted protease inhibitor-based regimens in virologically suppressed adults with HIV-1: 48-week results of a randomised, open-label, multicentre, phase 3, non-inferiority trial. Lancet HIV. 2018;5:e347–e356. [DOI] [PubMed] [Google Scholar]
- 12.Trottier B, Lake JE, Logue K, et al. Dolutegravir/abacavir/lamivudine versus current ART in virally suppressed patients (STRIIVING): a 48-week, randomized, non-inferiority, open-label, Phase IIIb study. Antivir Ther. 2017;22:295–305. [DOI] [PubMed] [Google Scholar]
- 13.Kakuda TN, Opsomer M, Timmers M, et al. Pharmacokinetics of darunavir in fixed-dose combination with cobicistat compared with coadministration of darunavir and ritonavir as single agents in healthy volunteers. J Clin Pharmacol. 2014;54:949–957. [DOI] [PubMed] [Google Scholar]
- 14.Kakuda T, Sekar V, Vis P, et al. Pharmacokinetics and pharmacodynamics of darunavir and etravirine in HIV-1-infected, treatment-experienced patients in the gender, race, and clinical experience (GRACE) trial. AIDS Res Treat. 2012;2012:186987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moltó J, Curran A, Miranda C, et al. Pharmacokinetics of darunavir/cobicistat and etravirine alone and co-administered in HIV-infected patients. J Antimicrob Chemother. 2018;73:732–737. [DOI] [PubMed] [Google Scholar]
- 16.Madruga JV, Berger D, McMurchie M, et al. Efficacy and safety of darunavir-ritonavir compared with that of lopinavir-ritonavir at 48 weeks in treatment-experienced, HIV-infected patients in TITAN: a randomised controlled phase III trial. Lancet. 2007;370:49–58. [DOI] [PubMed] [Google Scholar]
- 17.Cahn P, Fourie J, Grinsztejn B, et al. Week 48 analysis of once-daily vs. twice-daily darunavir/ritonavir in treatment-experienced HIV-1-infected patients. AIDS. 2011;25:929–939. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.