

Abstract
Knowledge of the possible ionization states of a pharmaceutical substance, embodied in the pKa values (logarithm of the acid dissociation constant), is vital for understanding many properties essential to drug development. We compare nine commercially available or free programs for predicting ionization constants. Eight of these programs are based on empirical methods: ACD/pKa DB 12.0, ADME Boxes 4.9, ADMET Predictor 3.0, Epik 1.6, Marvin 5.1.4, Pallas pKalc Net 2.0, Pipeline Pilot 5.0, and SPARC 4.2; one program is based on a quantum chemical method: Jaguar 7.5. We compared their performances by applying them to 197 pharmaceutical substances with 261 carefully determined and highly reliable experimental pKa values from a literature source. The programs ADME Boxes 4.9, ACD/pKa DB 12.0, and SPARC 4.2 ranked as the top three with mean absolute deviations of 0.389, 0.478, and 0.651 and r2 values of 0.944, 0.908, and 0.894, respectively. ACD/pKa DB 12.0 predicted all sites, whereas ADME Boxes 4.9 and SPARC 4.2 failed to predict 5 and 18 sites, respectively. The performance of the quantum chemical-based program Jaguar 7.5 was not as expected, with a mean absolute deviation of 1.283 and an r2 value of 0.579, indicating the potential for further development of this type of approach to pKa prediction.
INTRODUCTION
Most pharmaceutical substances will be protonated or deprotonated in aqueous solution; for example, data from the 1999 World Drug Index suggest that 63% of the 51600 listed drugs are ionizable, of which 15% are acids, 67% bases, and 18% ampholytes.1 The ionization ability is quantified by a parameter, the (logarithm of the) acid ionization constant (pKa), which is also called the protonation constant, equilibrium constant, or (acid) dissociation constant in the literature. Along with the partition coefficient, solubility, and reaction rate, pKa is the most important physicochemical property of a substance to be formulated into a useful medicine. As a function of its intrinsic pKa value(s) and the pH value of the solution, the extent of ionization of a drug controls its solubility, dissolution rate, and consequently has great impact on gastrointestinal uptake into the bloodstream, distribution, cell permeability, drug–receptor binding, reaction kinetics, metabolism, elimination, etc.2,3 In the preformulation stage, knowledge of ionization constants is useful when trying to form a salt in order to obtain biopharmaceutical properties and solid-state characteristics that may be lacking in the free form of the compound.
pKa values can be either measured or calculated. There are a number of methods to use for the experimental determination of pKa values and closely related quantities such as pH values.4–6 These methods have been used extensively in drug discovery and various development stages in the pharmaceutical industry. The highly accurate measurement methods include conductance methods (reliable to ±0.0001 pK units or better) and electrochemical cells without liquid junction potentials (reliable to ±0.001 pK units or better), both of which, unfortunately, have been rarely used to measure ionization data for pharmaceutically relevant organic acids and bases. For this class of substances, most pKa measurements are based on relationships between the measured solution pH and a measured physicochemical quantity such as added titrant concentration, solubility, etc., which limits the expected accuracy. Measurement of pKa values has become easier and more convenient over recent years. Nevertheless, in early drug discovery, measuring millions of compounds in large screening libraries is costly and simply not practical, and outright impossible for virtual libraries, which makes in silico prediction of pKa values vital in modern drug discovery.
At present, standard methods for pKa prediction for pharmaceutical substances can be classified into two major groups: empirical methods and quantum chemical methods. On the basis of the detailed approach used, the empirical methods can be further divided into three groups: (1) linear free-energy relationships (LFER), methods utilizing the empirical relations of Hammett and Taft, (2) quantitative structure–property relationships (QSPR), methods correlating calculated structural descriptors with pKa values, and (3) database lookup, i.e., methods searching of similar structures in a predetermined database of molecules with known measured pKa values.7 One of the strengths of the empirical methods is their high speed, useful when processing large databases of drug-like molecules. The alternative to empirical methods, quantum chemical methods, are supposed to have higher accuracy because they are based on, or closer to, first principles when calculating quantum mechanical descriptors. However, these methods are much more time-consuming than empirical methods.
While a significant number of recent publications can be found reporting on new and better programs and methods for pKa prediction (see refs 8–16 and 17–21 for additional approaches using empirical or quantum chemical methods, respectively), albeit mostly for specific classes of compounds, much less prior work exists comparing such approaches. Melun et al. used the REGDIA regression diagnostics algorithm in S-Plus to examine the accuracy of pKa predictions of four programs: ACD/pKa, Marvin, PALLAS, and SPARC.22 Three different validation data sets were taken from literature, including 64 pKa values for different drugs. We therefore felt there was the need to not only compare a larger number of available programs but also use more pKa values for pharmaceutical substances in order to cover as much as possible of the drug-like chemical space. In this article, we use eight empirical programs and one quantum chemical program (Table 1) to predict 261 carefully determined pKa values of 197 pharmaceutical substances in order to compare the predictive power of the nine programs.
Table 1.
Programs Used For This Study
| program | version | company | method |
|---|---|---|---|
| ACD/pKa DB23 | 12.00 | Advanced Chemistry Development, Inc. | LFER |
| ADME Boxes24 | 4.9 | Pharma Algorithms, Inc. | QSPR |
| ADMET Predictor25 | 3.0 | Simulations Plus, Inc. | QSPR |
| Epik26,27 | 1.6 | Schrödinger, LLC. | LFER |
| Jaguar28,29 | 7.5 | Schrödinger, LLC. | quantum–chemical method (DFT) with empirical correction |
| Marvin30 | 5.1.4 | ChemAxon Ltd. | QSPR |
| Pallas pKalc31 | Net 2.0 | CompuDrug International, Inc. | LFER |
| Pipeline Pilot32 | 5.0 | SciTegic, Inc. | QSPR |
| SPARC33,34 | 4.2 | University of Georgia/U.S. Environmental Protection Agency | blend of LFER and perturbed molecular orbital (PMO) method |
While we have tried to bring together as many of the existing programs that have some usage, we do not claim that we have tested each and every code that predicts pKa values. One program, for example, that we are aware of but which was not included is MoKa,13 marketed by Molecular Discovery, Ltd.35 Unfortunately, a mutually satisfactory agreement to gain access to the program for the purpose of this study could not be reached with the company.
DATA SET
Significant numbers of experimental pKa data on aqueous ionization chemicals have been collected over the decades. Some of them have found their way into literature compilations36–40 which, however, are not focused on pharmaceutical substances, although they do include some such compounds. In 2007, a book titled Profiles of Drug Substances, Excipients, and Related Methodology: Critical Compilation of pKa Values for Pharmaceutical Substances was published.41 The author of this book systematically collected nearly 3500 reported pKa values for drugs and related compounds from the pertinent primary and secondary literature and then, using the IUPAC classification and guidelines given below, assessed the reliability of these reported pKa values. On the basis of the aspects of a pKa measurement, such as the experimental method, mathematical definition selected to calculate the value from the raw data, and degree to which technical refinements have been applied, the IUPAC established in the 1960s the criteria for its compilations of dissociation constants for weak organic acids and bases: very reliable (VR; pKa error < ±0.005), reliable (R; pKa error ±0.005 to ±0.02), approximate (A; pKa error ±0.02 to ±0.04), and uncertain (U; pKa, error > ±0.04).36,37 The author of this book set the cutoff for uncertain to > ±0.06 pKa unit. On the basis of this modified IUPAC criteria, about 74% of the collected pKa, values in this book were found to be of uncertain quality, whereas only 0.1% qualified as very reliable and 0.33% qualified as reliable, which left ~25% being classified as approximate. The compilation has two sections: Appendix A comprises pKa values for which the measurements were sufficiently well described for the data to be assessed for reliability, and Appendix B comprises those pKa values for which little reliability data could be assessed, which were mostly from the secondary literature.
For this project, the pKa values used to compare the abilities of the nine programs to predict pKa values for the nine programs were chosen according to the following criteria: (1) Only pKa values that came from Appendix A of the above-mentioned reference were chosen. (2) The data qualities had to be VR, R, or A. (3) The solvent contained only water except some possible inorganic ions. (4) The temperature at which the measurement was taken was in the range of 25 ± 2 °C (except for compound 134, measured at 20 °C, which was included because otherwise we would have lost the only thiol in the final set). These quite rigorous criteria finally left us with 261 pKa values measured for 197 compounds whose structures are shown in Figure 1. The molecular weight and pKa value distributions and numbers of acidic and basic sites of these compounds are shown in Figure 2 and Table 2, respectively, both of which demonstrate that, as far as chemical species, number, and value distribution are concerned, this data set can be considered high-quality for the purpose of this comparison. A cautionary note is warranted for compound 99, which may be capable of forming aggregates42,43 at high concentration because this compound likely has low solubility in water as a monomer. This might then affect the measured pKa values. Because firm evidence exists neither for nor against aggregation, however, we decided to keep it in our compound set (hoping that the authors of the original paper were aware of this issue and performed the necessary procedures to rule it out or prevent it).
Figure 1.
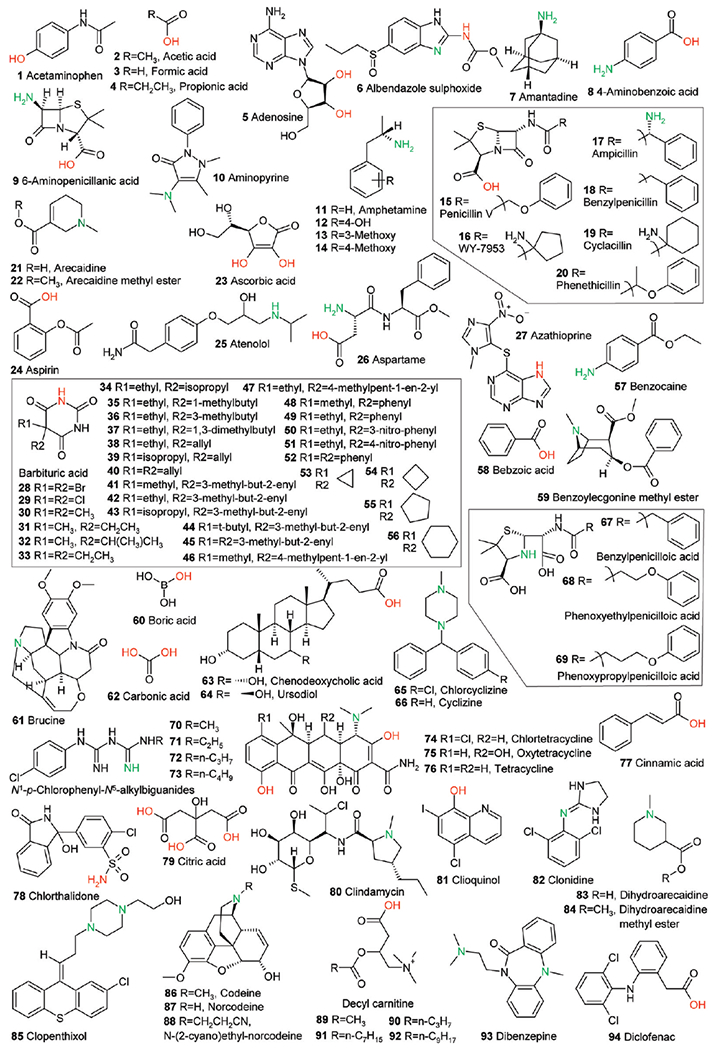
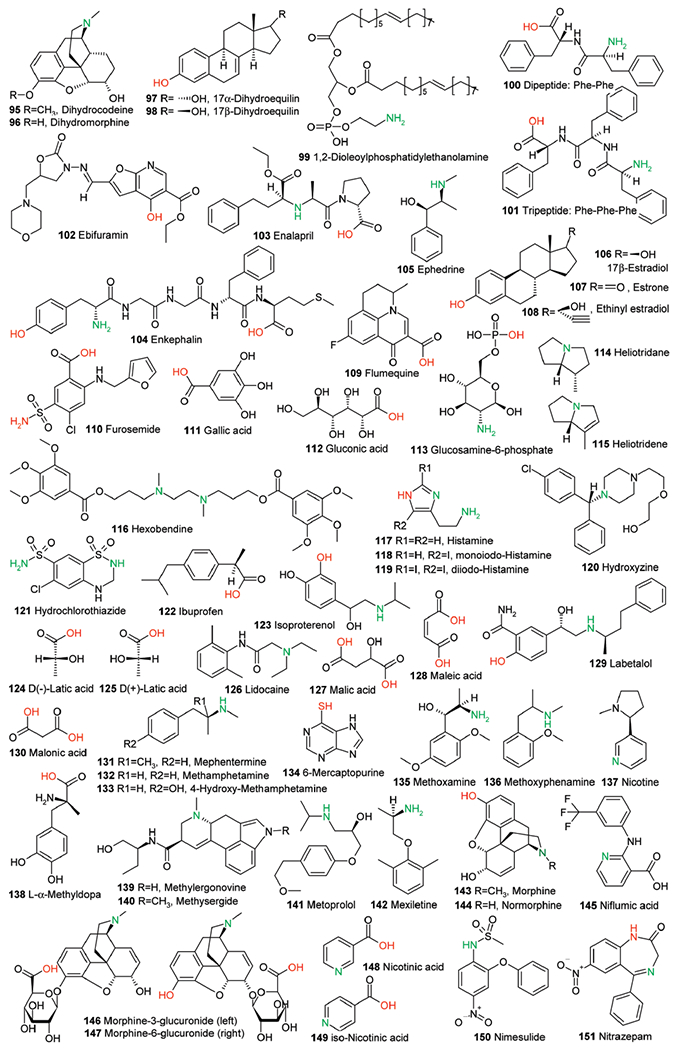
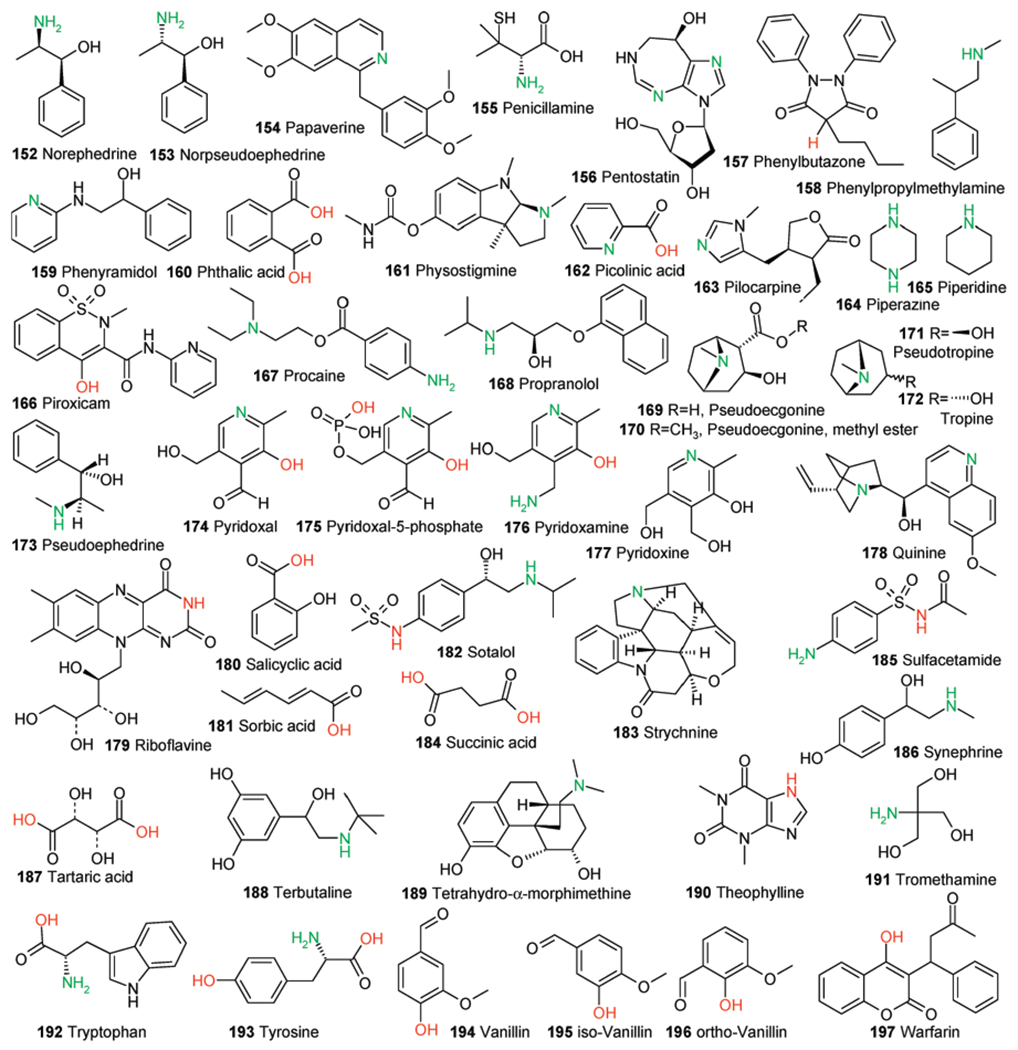
Part 3 of 3. Structure set used for this study. All structures were extracted from ref 41. Calculated acidic and basic atoms are indicated in red and green, respectively. Structures of compounds 23, 74, 140, and 154 in the original reference are wrong and are shown here in their corrected form.
Figure 2.
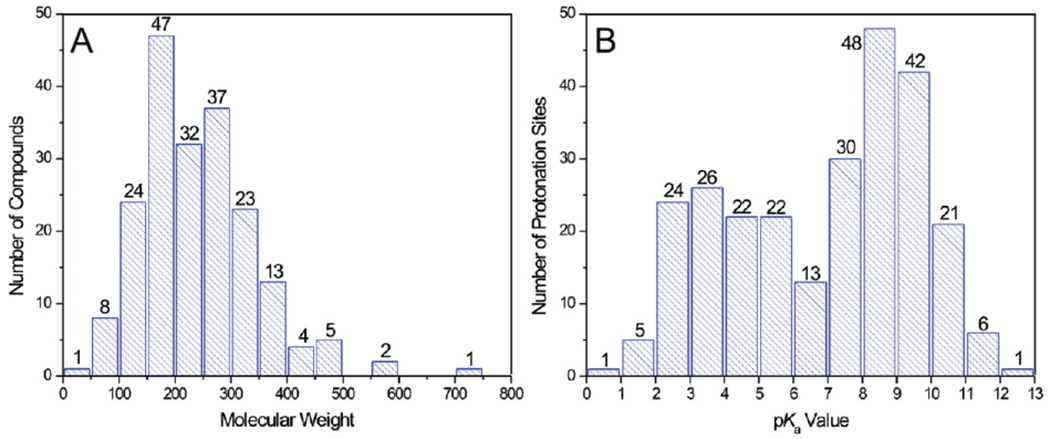
Distribution of (A) molecular weights of the calculated 197 compounds and (B) experimental pKa values of the 261 protonation sites used in the study.
Table 2.
Number of Different Acidic and Basic Sites Used for This Comparison
| acids | bases | ||
|---|---|---|---|
| alcohol | 1 | primary amines | 25 |
| enols | 7 | secondary amines | 24 |
| phenols | 24 | tertiary amines | 42 |
| carboxylic acids (not conjugated) | 42 | anilines | 5 |
| carboxylic acids (conjugated) | 17 | heterocycles | 17 |
| thiol | 1 | amidine | 1 |
| sulfonamides | 7 | benzodiazepine | 1 |
| imide | 1 | guanidines | 5 |
| barbituric acids | 29 | ||
| heterocycles | 4 | ||
| NH acids | 2 | ||
| CH acid | 1 | ||
| phosphoric acids | 2 | ||
| others | 3 | ||
| ∑ | 141 | ∑ | 120 |
It is worthwhile to point out that although this data set has dozens of multiprotic molecules, not all of the acidic and basic sites of every multiprotic molecule were predicted in this comparison. The main reason is that the book does not note every pKa value of every multiprotic molecule or that the data quality of some of these sites is below A.
It should also be pointed out and kept in mind by the reader, that these programs tend to be trained with as much of the data available in the literature as possible. Testing these programs using exclusively information from the literature, as we unavoidably had to do for this study, therefore entails the risk that the programs may effectively “look up” known pKa values rather than predict them. It would therefore not be a wrong strategy for the serious user to test any of these programs themselves using unpublished or private pKa data if possible.
COMPUTATIONAL METHODS
All eight programs based on empirical methods were executed one compound at a time or in batch mode. Some programs such as ACD/pKa, DB, ADME Boxes, Epik, Marvin, and SPARC can calculate different kinds of pKa values. For this study, only the pKa types that were designated as predicting experimental pKa values were calculated. ACD/pKa DB 12.00, ADME Boxes 4.9, ADMET Predictor 3.0, Marvin 5.1.4, Pallas pKalc Net 2.0, and Pipeline Pilot 5.0 were run using the default options in their respective graphical user interfaces on a Windows XP computer. SPARC 4.2 was run via a web-based interface (IE 7.0) on a Windows XP machine. Epik 1.6 was run via command line on a Linux system.
pKa values are in direct proportion to ΔG°, the free energy change for transition from the protonated state to the deprotonated state. A small calculation error of ΔG° (on the order of a few kcal/mol) can therefore lead to a significant prediction error for pKa for programs that are based on quantum–chemical methods. To correct for this deficiency that affects the quantum chemistry-based program Jaguar, this program employs two additional empirical parameters, scaling and additive factors.
pKa predictions of Jaguar 7.5 consist of a series of calculations on the protonated and deprotonated forms of the target molecule, followed by the aforementioned empirical correction. Because the calculated results partly depend on the conformation of the target molecule, first a conformational search was performed with MacroModel.44 As recoimnended by Schrödinger, the lowest-energy conformers found were used for further pKa calculations. Jaguar calculates microscopic (atomic) pKa values but not macroscopic (experimental) pKa values. If two or more microscopic pKa values lie within one pKa unit of each other, the macroscopic pKa values can markedly differ from the corresponding microscopic values. Therefore, for such a multiprotic molecule, in order to obtain the macroscopic pKa values, Schrödinger suggests in the Jaguar manual to run 2n states (n being the number of close pKa values in a multiprotic molecule) and then to assemble the titration curve. Given that each QM computation takes already orders of magnitude longer than the corresponding empirical calculation, this would have heavily increased the amount of calculation necessary for the project while creating hard-to-assess additional sources of potential error. We therefore decided to drop the calculation of such protonation sites. We set the cutoff here for two experimental pKa values in a multiprotic molecule being too close to each other if their difference was less than or equal to 2.5 pKa units. This led to a reduction of the number of calculated sites from 261 to 204. For some kinds of acidic or basic sites, Jaguar 7.5 does not have parameters for an aqueous solution, which led to another 11 sites being dropped to yield the final number of 193 sites included in the calculations. For those multiprotic molecules whose experimental pKa values are well separated (by >2.5 pKa units), when calculating the lowest pKa value, the sites with higher pKa values were in the protonated states; when calculating the middle pKa value, the sites with higher and lower pKa values were in the protonated and deprotonated states, respectively; and when calculating the highest pKa value, the sites with lower pKa values were in the deprotonated states.
Among the 197 compounds, some molecules, for example, compounds 79, 128, and 164, have two equivalent sites for protonation or deprotonation. In this situation, the need for a statistical correction factor arises from the increased entropy of the appropriate species. A correction of +0.60 (log1022) or −0.60 was added by hand to the result obtained from running the pKa prediction module on the basis of whether the calculated molecule has two equivalent acidic sites or basic sites because Jaguar 7.5 does not automatically recognize equivalent sites.
RESULTS AND DISCUSSION
Overview.
Execution Speeds.
Program execution was very fast for all programs based on empirical methods except SPARC 4.2. For example, ADMET Predictor 3.0 and Pipeline Pilot 5.0 finished the calculation of predicting the pKa values of these 197 pharmaceutical substances in less than 1 s on a Windows computer. Epik 1.6 took 119 s on one CPU (AMD 64, dual core FX-61) of our Linux cluster. It is therefore possible to predict pKa values of millions of compounds in a tractable time by using these seven programs. The calculation speed of SPARC 4.2, because of its use of the PMO theory, was much slower than that of the other seven programs. For example, submission of compound 74, which has five protonation sites, resulted in the following message: “This will result in ~5120 calculations and may take as long as 51.87 minutes”. In fact, the server did not produce any result for this compound but an error message: “The request has exceeded the allowable time limit Tag”. Yet for simpler compounds with one or two protonation sites, the calculation time was acceptable. One-by-one submission and longer execution times make SPARC 4.2 not suitable for predicting pKa values of large number of compounds.
The pKa prediction of Jaguar 7.5 was very time-consuming, and it was strongly dependent on the size and flexibility of the molecule. For examples, on the above-mentioned AMD 64, dual core FX-61 CPU, it took about 4, 23, 96, 202, 1568, and 3831 min, respectively, to predict pKa values of the carboxylic acid sites in compounds 3 and 148, the barbituric acid site in compound 36, the pyridine site in compound 145, the carboxylic acid site in compound 104, and the tertiary amine site in compound 116. The molecular weights of these six compounds in the same order are 46.03, 123.11, 226.28, 282.22, 573.67, and 592.69, respectively. After more than three days of computation, Jaguar 7.5 failed for an unknown reason (but presumably due to resource exhaustion) in the prediction of the primary amine site of compound 99, which is the largest and most flexible molecule in the test set with a molecular weight of 744.05. With such slow calculation speeds, we can conclude that Jaguar 7.5 is not a practical solution to handle many compounds, especially when they are large, flexible, and/or multiprotic with close pKa values.
Prediction Results.
The prediction results for the 261 pKa protonation sites are shown in Table S1 of the Supporting Information and Figure 3 and summarized in Table 3 and Figure 4. Only ACD/pKa DB 12.0, ADMET Predictor 3.0, and Marvin 5.1.4 predicted all 261 protonation sites; ADME Boxes 4.9, Epik 1.6, Pallas pKalc Net 2.0, Pipeline Pilot 5.0 and SPARC 4.2 failed for 5, 4, 2, 9, and 18 sites, respectively. Jaguar 7.5 failed for 11 sites with the error message “No pKa functional group with parameters for water could be identified”.
Figure 3.
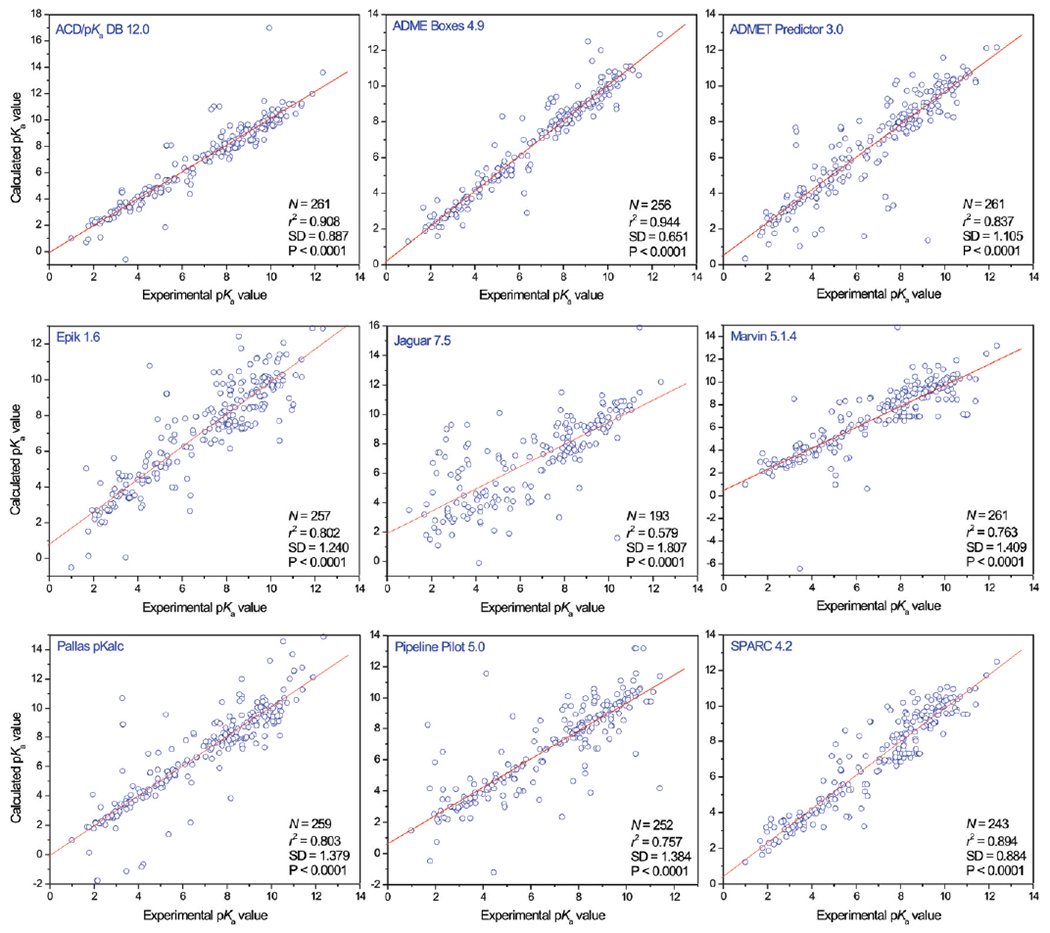
Predicted pKa values calculated by nine different programs versus experimental pKa values for 261 protonation sites.
Table 3.
Performance of the Nine pKa Prediction Programs Using a Set of 261 Protonation Sites, Sorted Alphabetically by Program Name
| program | number handled | predicted excellently (ADa ≤ 0.1) | predicted well (0.1 < AD ≤ 0.5) | predicted poorly (1.0 < AD ≤ 2.0) | predicted awfully (AD > 2.0) | r2 | MADb |
|---|---|---|---|---|---|---|---|
| ACD/pKa DB | 261 | 64 | 121 | 17 | 10 | 0.908 | 0.478 |
| ADME Boxes | 256 | 95 | 102 | 16 | 7 | 0.944 | 0.389 |
| ADMET Predictor | 261 | 40 | 119 | 28 | 18 | 0.837 | 0.659 |
| Epik | 257 | 30 | 87 | 64 | 23 | 0.802 | 0.893 |
| Jaguar | 193 | 24 | 38 | 36 | 40 | 0.579 | 1.283 |
| Marvin | 261 | 39 | 91 | 57 | 22 | 0.763 | 0.872 |
| Pallas pKalc | 259 | 48 | 105 | 32 | 26 | 0.803 | 0.787 |
| Pipeline Pilot | 252 | 77 | 74 | 46 | 24 | 0.757 | 0.769 |
| SPARC | 243 | 30 | 96 | 45 | 12 | 0.894 | 0.651 |
AD: absolute deviation.
MAD: mean absolute deviation.
Figure 4.
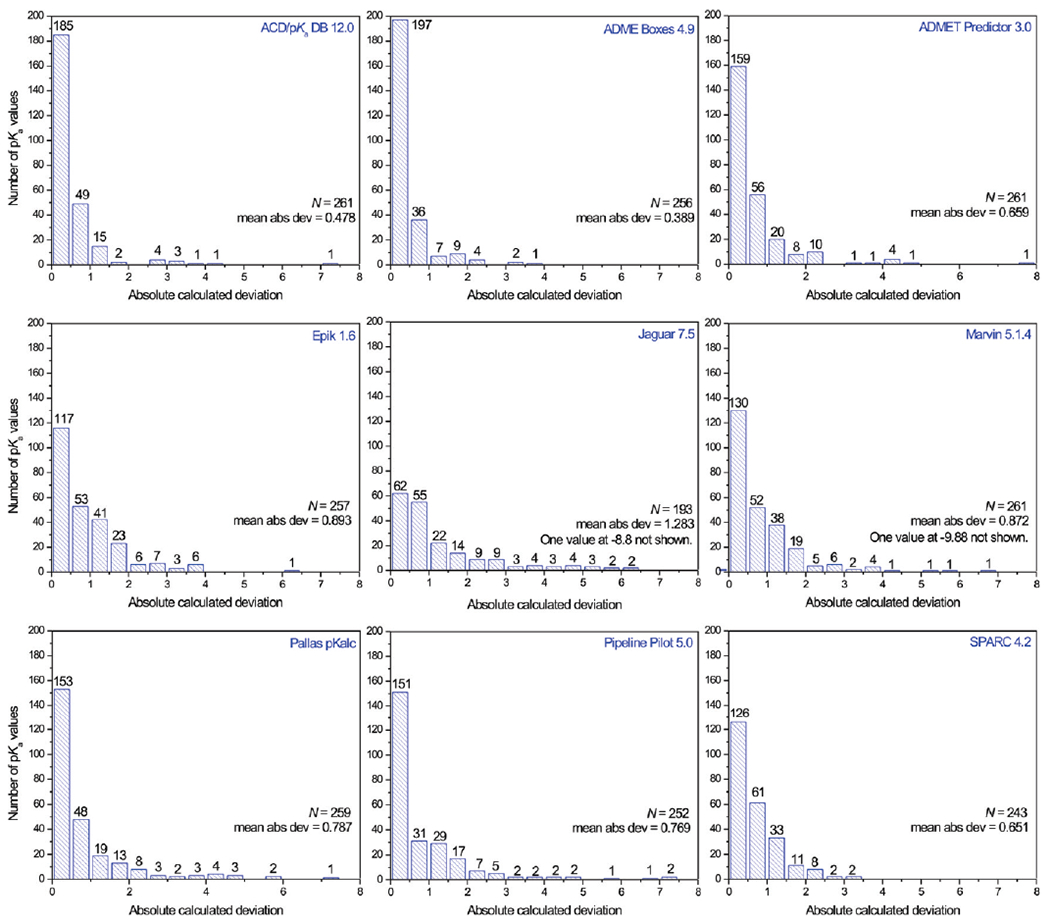
Summary of the pKa value predictions of 261 protonation sites. The predictions are binned by prediction accuracy with a resolution of 0.5 log unit.
When based on mean absolute deviation (MAD), the rank order of this comparison is ADME Boxes 4.9, ACD/pKa DB 12.0, SPARC 4.2, ADMET Predictor 3.0, Pipeline Pilot 5.0, Pallas pKalc Net 2.0, Marvin 5.1.4, Epik 1.6, and Jaguar 7.5. In terms of r2, the situation changes a little, but the top three are still ADME Boxes 4.9, ACD/pKa DB 12.0, and SPARC 4.2. It might be argued that this criteria is actually unfair for the three programs that predicted all protonation sites.
When predicting pKa values as part of the drug discovery process, researchers may typically be interested mostly in the protonation state at physiological pH, i.e., 7.4. Table 4 shows the performance of these nine programs, specifically for those 116 sites whose measured pKa values are in the range of 5.4–9.4. In general, on the basis of the mean absolute deviation, most programs performed worse in this range than for the full set of sites, except Jaguar 7.5 and Pallas pKalc Net 2.0. The top three performers in terms of MAD were ADME Boxes 4.9, ACD/pKa DB 12.00, and Pallas pKalc Net 2.0; and ADME Boxes 4.9, ACD/pKa DB 12.00, and SPARC 4.2 in terms of r2.
Table 4.
Performance for pKa Range 5.4–9.4; Programs Sorted Alphabetically
| program | number handled | predicted excellently (ADa ≤ 0.1) | predicted well (0.1 < AD ≤ 0.5) | predicted poorly (1.0 < AD ≤ 2.0) | predicted awfully (AD > 2.0) | r25.4–9.4 | MADb |
|---|---|---|---|---|---|---|---|
| ACD/pKa DB | 116 | 25 | 51 | 9 | 4 | 0.574 | 0.504 |
| ADME Boxes | 115 | 33 | 60 | 8 | 5 | 0.682 | 0.452 |
| ADMET Predictor | 116 | 14 | 53 | 15 | 8 | 0.350 | 0.744 |
| Epik | 114 | 9 | 29 | 39 | 9 | 0.403 | 0.979 |
| Jaguar | 84 | 10 | 16 | 20 | 9 | 0.422 | 1.044 |
| Marvin | 116 | 10 | 35 | 35 | 8 | 0.374 | 0.961 |
| Pallas pKalc | 115 | 21 | 45 | 19 | 6 | 0.469 | 0.680 |
| Pipeline Pilot | 114 | 35 | 30 | 28 | 10 | 0.422 | 0.762 |
| SPARC | 108 | 7 | 30 | 27 | 10 | 0.479 | 0.876 |
AD: absolute deviation.
MAD: mean absolute deviation.
About thirty of the compounds were consistently predicted poorly, i.e., more than six of the programs predicted them with an deviation of at least 0.5 log units or even failed to predict one or more sites of them altogether: 6, 9, 52, 53, 60, 62, 67–69, 74–76, 79, 85, 99, 102, 110, 119, 121, 128, 129, 134, 144, 155, 156, 174, 175, 178, and 188–190. It is not clear whether this points to a general weakness in the understanding and/or algorithms in the field for these molecules or if this may indicate potential problems with the experimental results.
ACD/pKa DB 12.0.
AC D/pKa DB 12.0 ranks second in this comparison with an MAD of 0.478 and an r2 of 0.908. A strong point of ACD/pKa DB was that it calculated all 261 protonation sites. It produced 64 (24.52%) and 185 (70.88%) predicted values with accuracies of ±0.1 and ±0.5 log unit, respectively. This makes it a well-built and robust program for the prediction of ionization states of pharmaceutical substances. Nevertheless, there are still 10 sites (3.83%) whose predicted accuracies were more than 2.0. The two largest deviations are 7.07 and −4.05 log units, which occurred on the general C substituted amide site of compound 6 and the substituted aniline site of compound 93, respectively. For the tertiary amine sites of the three tetracycline antibiotics (compounds 74–76), the prediction errors are significant: all of them are around +3.50 log units. The other very poorly predicted sites include the primary amine sites of three penicilloic acids (compounds 67–69), the phenol site of ebifuramin (compound 102), and the benzodiazepine site of compound 156.
ADME Boxes 4.9.
This program ranks first if one only focuses on the MAD (0.389) or r2 (0.944) values. However, it failed to predict five protonation sites: one enol site of compound 23 (another enol site was successfully predicted), one acid site of compound 60 (which actually is an inorganic acid), the substituted aniline site of compound 93, and the two heterocycle acid sites of compounds 118 and 119. Apart from these cases, ADME Box 4.9 predicted normal organic compounds very well: it produced 95 (37.11%) and 197 (76.95%) predicted values with accuracies of ±0.1 and ±0.5 log unit, respectively. Each of these two values is at the top of its list for the nine programs. There are seven sites (2.73%) whose prediction accuracies were worse than 2.0 log units, which also represents the top spot among the nine compared programs. The largest deviation was −3.5, which occurred for one of the two acid sites of carbonic acid (compound 62). The other very poorly predicted six sites include the three phenol sites of the three tetracycline antibiotic compounds 74 – 76, which all were predicted higher than experimentally measured, one tertiary amine site of the antipsychotic compound 85, the phenol site of compound 102, and one carboxylic acid site of compound 128.
ADMET Predictor 3.0.
This program ranks fourth with a MAD of 0.659 and an r2 of 0.837. It also is one of the three programs that did not fail to predict even one site. It gave 40 (15.33%) and 159 (60.92%) predicted values with accuracies of ±0.1 and ±0.5 log unit, respectively. There were 18 sites (6.90%) whose predicted accuracies were more than 2.0 log units. The calculated largest deviation is −7.86, which occurred for compound 60. Besides this site, there are 17 additional rather poorly predicted sites: both of the two acid sites of carbonic acid, compound 62; the three primary amine sites of the three penicilloic acids (compounds 67–69); the three enol sites, and the three tertiary amine sites of compounds 74–76 (whereas the three phenol sites of these three compounds were predicted quite well, with absolute deviations between 0.24 and 0.79); the substituted aniline site of compound 93; the thiol site of compound 134; the pyridine site of compound 145; the primary amine sites of compounds 155 and the 175; and the heterocycle acid site of compound 190.
Epik 1.6.
The r2 and MAD values of this program were 0.802 and 0.893, respectively, which represents the sixth and last rank, respectively, among the eight programs utilizing empirical methods. The prediction of two protonation sites, the acid site of the inorganic acid compound 60, and the thiol site of compound 134 were not completed by this program. The enol sites of compounds 75 and 102 were tautomerized into carbonyl groups, which are not protonation sites (it is interesting to note that the enol sites of compounds 74 and 76 were not tautomerized in the pKa prediction process by the program, although the structures of the compounds 74–76 are almost identical to each other). All in all, Epik predicted 257 sites and failed for four sites. It produced 30 (11.67%) and 117 (45.52%) predicted values with accuracies of ±0.1 and ±0.5 log unit, respectively. There were 23 sites (8.95%) whose predicted accuracies were off by more than 2.0 log units. The largest deviation was 6.26, which occurred for the CH acid site of compound 157. The other very poorly predicted protonation sites comprised both of the two acid sites of carbonic acid, compound 62; the three primary amine sites of the three penicilloic acids (compounds 67–69); the three guanidine sites of compounds 70–72; the two tertiary amine sites of compounds 74 and 76; the phenol sites of compounds 81, 144, and 176; the tertiary amine site of compound 88; the substituted aniline site of compound 93; the primary amine sites of compounds 9 and 99; the secondary amine site of compound 103; the carboxylic acid site of compound 109; the imine site of compound 151; the heterocycle site of compound 156; and the heterocycle acid site of compound 190.
Jaguar 7.5.
Even though based on a more sophisticated fundamental approach than the other eight programs, which are based on empirical methods, Jaguar 7.5 did not pull ahead of any one of them on the basis of our analysis. Only 193 protonation sites were predicted. One reason is the lack of parameters for some less frequently occurring sites, the other is the closeness of the pKa values for some sites in a multiprotic molecule. The r2 value for Jaguar 7.5 was 0.579, 0.178 lower than the worst-performing one of the other eight programs. Likewise, its MAD was 1.283, 0.390 higher than the next-ranked program. Jaguar 7.5 produced 24 (12.44%) and 62 (32.12%) predicted values with accuracies of ±0.1 and ±0.5 log units, respectively. The 24 sites whose predicted values were very close to the best literature values include the barbituric acid site of compound 37, the carboxylic acid site of compound 77, the phenol site of compound 81, the secondary amine sites of compounds 87, 105, and 131, the tertiary amine site of compound 114, the heterocycle site and the primary amine site of compound 118, one carboxylic acid site of compound 130, and several others. One strong point of Jaguar 7.5 over the other eight programs is that it can distinguish diastereomers. For example, compounds 152 and 153 are diastereomers of each other. The experimental and predicted pKa values for the two primary amine sites of compound 152 are 9.05 and 9.0, respectively, and for compound 153, they are 9.19 and 9.3, respectively. Nevertheless, this version of Jaguar would not appear to be an ideal tool for predicting pKa values of pharmaceutical substances. There were 40 sites (20.73%) whose predicted accuracies were off by more than 2.0 log units. Frequently, Jaguar 7.5 worked incorrectly when there were one or more charged groups in the molecule, even though this molecule is not multiprotic. For example, for the three antibiotic compounds 15, 18, and 20, which are monoprotic, the predictions for the three carboxylic acid sites were acceptable: The calculated deviations were 0.4, 0.7, and 0.9 respectively. However, the deviations for the other three antibiotic compounds 16, 17, 19 were 4.5, 5.7, and 6.3, which are clearly not acceptable. Each of these three compounds has an −NH2 group, which we set as protonated and then maintained positively charged for the prediction of the pKa value of the carboxylic acid site. When calculating the pKa value of the second acid group of carbonic acid (compound 62), the first carboxylic acid was deprotonated. This led to the pKa value being predicted as 1.6, which is even lower than the first calculated one and then brings the deviation to −8.8. The compounds 89–92 are acids with quaternary ammonium groups; however, they are monoprotic, not diprotic. The positive quaternary ammonium groups induced big deviations for every carboxylic acid site: 3.3, 4.2, 5.7, and 4.4, respectively. In fact, among the 40 very poorly predicted sites (>2.0 log units), 29 were calculated when a charge existed in the molecule. Schrödinger’s explanation in the Jaguar manual acknowledges this shortcoming: “When the ionziable groups are close together in the molecule, the calculated pKa may not be as accurate because the two groups could interact in ways that the existing parameterization cannot handle.”
Marvin 5.1.4.
With no sites with failed prediction attempts, the r2 and MAD values for the program were 0.763 and 0.872, respectively, which makes this program rank seventh. It produced 39 (14.94%) and 130 (49.81%) predicted values with accuracies of ±0.1 and ±0.5 log units, respectively. Marvin calculated barbituric acid sites relatively poorly. The average deviation for the 29 barbituric acid sites was 1.254; two deviations were more than 2.0 log units. There were 22 sites (8.43%) whose predicted accuracies were more than 2.0 log units. The worst five predicted deviations were −9.88, 6.93, −5.88, 5.34, and −4.08, which happened on the substituted aniline site of compound 93, the primary amine site of compound 26, the pyridine site of compound 159, the carboxylic acid site of compound 26, and the tertiary amine site of compound 10, respectively. The other very poorly estimated sites include the four guanidine sites of the four biguanidine compounds 70–73, the primary amine site of compound 9, one enol site of compound 23, the phenol sites of compounds 75 and 102, one tertiary amine site of compound 85, the tertiary amine site of compound 115, the pyridine site of compound 149, the benzodiazepine site of compound 156, the enol site of compound 166, the imide site of compound 179, and the heterocycle site of compound 190.
Pallas pKalc Net 2.0.
This program failed to predict two sites: the acid site of compound 60 and the enol site of compound 166. With this, it garnered an r2 value of 0.803 and an MAD value 0.787, which helped it rank fifth and sixth, respectively, among the nine programs. It produced 48 (18.53%) and 153 (59.07%) predicted values with accuracies of ±0.1 and ±0.5 log units, respectively. There were 26 sites (10.04%) whose calculated deviations were more than 2.0 log units, among which the worst three predicted deviations occurred on the enol sites of the antibiotic compounds 74–76. The other 23 very poorly predicted protonation sites included the alcohol sites of compound 5, the general C substituted amide site and the heterocycle site of compound 6, the primary amine site of compound 9, one acid site of compound 62, the tertiary amine sites of compounds 65 and 66, the guanidine site of compound 70, the phenol sites of compound 81, 102, and 176, the substituted aniline site of compound 93, the secondary amine sites of compounds 103, 123, and 188, one carboxylic acid site of compound 128, the thiol site of compound 134, the pyridine site of compound 145, the quinolin-2-one site of compound 151, the pyridine sites of compounds 174 and 175, the imide site of compound 179, and the heterocycle site of compound 190.
Pipeline Pilot 5.0.
With nine failed sites, the r2 and MAD values of this program were 0.757 and 0.769, respectively, which made it rank eighth and fifth in these two categories. The failed nine sites were the carboxylic acid site of compound 3, the alcohol sites of compound 5, the two sites of compound 6, the heterocycle sites of compounds 27, 118, 119, and 190, and the quinolin-2-one site of compound 151. Pipeline Pilot 5.0 produced 77 (29.62%) and 151 (57.85%) predicted values with accuracies of ±0.1 and ±0.5 log units, respectively. The quality of the predictions of Pipeline Pilot for the pKa values of the barbituric acids varied strongly. Some compounds, such as 36 and 56, were predicted well, with deviations close or equal to zero, whereas other barbituric acids were predicted very poorly. For example, the deviations for compounds 30 and 52 were −4.62 and −4.96, respectively. The predicted values of the two enol sites of compound 23 were very close to the experimental values; unfortunately, Pipeline Pilot assigned them to the wrong sites, which led to the two largest deviations, 7.43 and −7.22. Besides these four sites, there were 20 sites whose predicted accuracies were off by more than 2.0 log units. These were the carboxylic acid sites of compounds 9, 79 and 149, the barbituric acid sites of compounds 53 and 54, the tertiary amine sites of compounds 61, 74, and 183, the two acid sites of compound 62, the primary amine site of compound 99, the phenol sites of compound 102, 106–108, one tertiary amine site of compound 120, the thiol site of compound 134, the pyridine site of compound 145, the heterocycle site of compound 156, and the sulfonamide site of compound 185.
SPARC 4.2.
This program gave up on the prediction of 18 protonation sites, mostly because it exceeded its time limit. It ranks third with an r2 value of 0.894 and an MAD value of 0.651 in this comparison. This program produced 30 (12.35%) and 126 (51.85%) predicted values with accuracies of ±0.1 and ±0.5 log units, respectively. These two values are actually the second worst ones in each category among the eight empirical methods; nevertheless, this program did not produce too exorbitantly bad deviations. Only 12 deviations (4.94%) were larger than 2.0 log units. The worst two were −3.11 and 3.10, which happened for one acid site of compound 62 and the benzodiazepine site of compound 156. This would seem to confirm that the PMO method is effective in preventing big deviations when it is used to predict pKa values of pharmaceutical substances, albeit at the cost of prolonging calculation times. The other 10 poorly predicted sites involve the primary amine sites of compounds 9 and 99, the guanidine site of compound 82, one tertiary amine site of compound 85, one carboxylic acid site of compound 128, the phenol site of compound 129, the tertiary amine sites of compounds 139 and 140, the enol site of compound 166, and the heterocycle site of compound 190.
CONCLUSION
Predicting pKa values of pharmaceutical substances is both challenging and important in drug development and thus is an intriguing task in computational chemistry. We have compared nine currently available programs, including eight based on empirical methods and one based on a quantum chemical approach as to their ability to accurately predict 261 carefully experimentally measured pKa values of 197 pharmaceutical compounds. It had been suggested that an approach based on a quantum chemical method would have led to better predictions, but our study did not bear this out. On the contrary, the only program in this comparison based on a higher level of theory than empirical methods ranked last in essentially all respects, indicating that the more recently introduced quantum chemical approach for predicting pKa values has not yet reached the maturity level of the empirical methods.
Among the eight programs that are based on empirical methods, we found several that performed very well with this test set, with two predicting all or nearly all protonation sites, and doing so with an r2 value of better than 0.9 and a mean absolute deviation of less than half a log unit. While extrapolation to any possible compound of interest in drug development is obviously risky, we believe that the best pKa predicting programs currently available are useful tools in the arsenal of the drug developer.
Supplementary Material
ACKNOWLEDGMENT
We would like to acknowledge Advanced Chemistry Development, Inc., Pharma Algorithms, Inc., ChemAxon, Ltd., and CompuDrug International, Inc. for giving us access to their programs.
Footnotes
Note Added after ASAP Publication. This paper was published ASAP on December 4, 2009 with an incorrect version of Figure 1. The corrected version was published ASAP on December 9, 2009.
Supporting Information Available: The data set used in this study and calculation results. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES AND NOTES
- (1).Comer J; Tam K In Pharmacokinetic Optimization in Drug Research: Biological, Physicochemical, and Computational Strategies; Testa B, Waterbeemd H, Folkers G, Guy R, Eds.; Wiley-VCH: Weinheim; New York, 2001; pp 275–304. [Google Scholar]
- (2).Comer JEA In Comprehensive Medicinal Chemistry II. Testa B, Waterbeemd H, Eds.; Elsevier: Oxford, UK, 2007; Vol. 5, pp 357–398. [Google Scholar]
- (3).Mitra R; Shyam R; Mitra I; Miteva MA; Alexov E Calculating the protonation states of proteins and small molecules: Implications to ligand-receptor interactions. Curr. Comput-Aided Drug Des. 2008, 4, 169–179. [Google Scholar]
- (4).Prue JE Ionic Equilibria. Pergamon Press: Oxford, New York, 1966. [Google Scholar]
- (5).Poole SK; Patel S; Dehring K; Workman H; Poole CF Determination of acid dissociation constants by capillary electrophoresis. J. Chromatogr., A 2004, 1037, 445–454. [DOI] [PubMed] [Google Scholar]
- (6).Meloun M; Bordovska S; Galla L The thermodynamic dissociation constants of four non-steroidal anti-inflammatory drugs by the least-squares nonlinear regression of multiwavelength spectrophotometric pH-titration data. J. Phann. Biomed. Anal 2007, 45, 552–564. [DOI] [PubMed] [Google Scholar]
- (7).Fraczkiewicz R In Comprehensive Medicinal Chemistry II. Testa B, Waterbeemd H, Eds.; Elsevier: Oxford, UK, 2007; Vol. 5, pp 603–626. [Google Scholar]
- (8).Sipila J; Nurmi H; Kaukonen AM; Hirvonen J; Taskinen J; Yli-Kauhaluoma J A modification of the Hammett equation for predicting ionisation constants of p-vinyl phenols. Eur. J. Pharm. Sci 2005, 25, 417–425. [DOI] [PubMed] [Google Scholar]
- (9).Kaminski GA Accurate prediction of absolute acidity constants in water with a polarizable force field: Substituted phenols, methanol, and imidazole. J. Phys. Chem. B 2005, 109, 5884–5890. [DOI] [PubMed] [Google Scholar]
- (10).Zhang J; Kleinoder T; Gasteiger J Prediction of pKa values for aliphatic carboxylic acids and alcohols with empirical atomic charge descriptors. J. Chem. Inf. Model. 2006, 46, 2256–2266. [DOI] [PubMed] [Google Scholar]
- (11).Brown TN; Mora-Diez N Computational determination of aqueous pKa values of protonated benzimidazoles (part 1). J. Phys. Chem. B 2006, 110, 9210–9219. [DOI] [PubMed] [Google Scholar]
- (12).Brown TN; Mora-Diez N Computational determination of aqueous pKa values of protonated benzimidazoles (Part 2). J. Phys. Chem. B 2006, 110, 20546–20554. [DOI] [PubMed] [Google Scholar]
- (13).Milletti F; Storchi L; Sforna G; Cruciani G New and original pKa prediction method using grid molecular interaction fields. J. Chem. Inf. Model. 2007, 47, 2172–2181. [DOI] [PubMed] [Google Scholar]
- (14).Meloun M; Bordovská S; Syrový T A novel computational strategy for the pKa estimation of drugs by non-linear regression of multiwavelength spectrophotometric pH-titration data exhibiting small spectral changes. J. Phys. Org. Chem 2007, 20, 690–701. [Google Scholar]
- (15).Jelfs S; Ertl P; Selzer P Estimation of pKa for druglike compounds using semiempirical and information-based descriptors. J. Chem. Inf. Model. 2007, 47, 450–459. [DOI] [PubMed] [Google Scholar]
- (16).Jover J; Bosque R; Sales J Neural network-based QSPR study for predicting pKa of phenols in different solvents. QSAR & Combin. Sci. 2007, 26, 385–397. [Google Scholar]
- (17).Caballero NA; Melendez FJ; Munoz-Caro C; Nino A Theoretical prediction of relative and absolute pKa values of aminopyridines. Biophys. Chem 2006, 124, 155–160. [DOI] [PubMed] [Google Scholar]
- (18).Lu H; Chen X; Zhan CG First-principles calculation of pKa for cocaine, nicotine, neurotransmitters, and anilines in aqueous solution. J. Phys. Chem. B 2007, 111, 10599–10605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bryantsev VS; Diallo MS; Goddard WA pKa calculations of aliphatic amines, diamines, and aminoamides via density functional theory with a Poisson–Boltzmann continuum solvent model. J. Phys. Chem. A 2007, 111, 4422–4430. [DOI] [PubMed] [Google Scholar]
- (20).Trummal A; Rummel A; Lippmaa E; Burk P; Koppel IA IEF-PCM calculations of absolute pKa for substituted phenols in dimethyl sulfoxide and acetonitrile solutions. J. Phys. Chem. A 2009,113, 6206–6212. [DOI] [PubMed] [Google Scholar]
- (21).Verdolino V; Cammi R; Munk BH; Schlegel HB Calculation of pKa values of nucleobases and the guanine oxidation products guanidinohydantoin and spiroiminodihydantoin using density functional theory and a polarizable continuum model. J. Phys. Chem. B 2008, 112, 16860–16873. [DOI] [PubMed] [Google Scholar]
- (22).Meloun M; Bordovska S Benchmarking and validating algorithms that estimate pKa values of drugs based on their molecular structures. Anal. Bioanal. Chem 2007, 389, 1267–1281. [DOI] [PubMed] [Google Scholar]
- (23).Predict Ionization Constant, Acid–Base Dissociation Constant, pKa, Experimental pKa. http://www.acdlabs.com/products/phys_chem_lab/pka/ (accessed September 17, 2009).
- (24).Physicochemical and ADMET Laboratory. http://pharma-algorithms.com/ionization.htm (accessed September 17, 2009).
- (25).Simulations Plus. http://www.simulations-plus.com (accessed September 17, 2009).
- (26).Epik, Schrödinger, LLC: New York, 2008. [Google Scholar]
- (27).Shelley JC; Cholleti A; Frye LL; Greenwood JR; Timlin MR; Uchimaya M Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des 2007, 21, 681–691. [DOI] [PubMed] [Google Scholar]
- (28).Jaguar, version 7.5; Schrödinger, LLC: New York, 2008. [Google Scholar]
- (29).Klicić JJ; Friesner RA; Liu S-Y; Guida WC Accurate prediction of acidity constants in aqueous solution via density functional theory and self-consistent reaction field methods. J. Phys. Chem. A 2002, 106, 1327–1335. [Google Scholar]
- (30).pKa Plugin ionization equilibrium partial charge distribution. http://www.chemaxon.com/product/pka.html (accessed September 17, 2009).
- (31).CompuDrug, Latest Upgrades. http://www.compudrug.com (accessed September 17, 2009).
- (32).Pipeline Pilot data analysis and reporting platform. http://accelrys.com/products/scitegic/ (accessed September 17, 2009).
- (33).Sparc On-Line Calculator. http://ibmlc2.chem.uga.edu/sparc/ (accessed September 17, 2009).
- (34).Lee PH; Ayyampalayam SN; Carreira LA; Shalaeva M; Bhattachar S; Coselmon R; Poole S; Gifford E; Lombardo F In silico prediction of ionization constants of drugs. Mol. Pharmacol 2007, 4, 498–512. [DOI] [PubMed] [Google Scholar]
- (35).MoKa–pKa Modeling. http://www.moldiscovery.com/soft_moka.php (accessed September 30, 2009).
- (36).Dissociation Constants of Organic Acids in Aqueous Solution Kortüm G, Ed.; International Union of Pure and Applied Chemistry, Commission on Electrochemical Data; Butterworths: London, 1961. [Google Scholar]
- (37).Perrin DD In Dissociation Constants of Organic Bases in Aqueous Solution. Butterworths: London, 1965. [Google Scholar]
- (38).Perrin DD; Dempsey B; Serjeant EP In pKa Prediction for Organic Acids and Bases. Chapman and Hall: London, New York, 1981. [Google Scholar]
- (39).Ionization Constants of Organic Acids in Aqueous Solution Serjeant EP; Dempsey B, Eds.; International Union of Pure and Applied Chemistry, Commission on Electrochemical Data; Pergamon Press: Oxford, New York, 1979. [Google Scholar]
- (40).Perrin DD Dissociation Constants of Weak Bases in Aqueous Solution. Butterworths: London, 1972. [Google Scholar]
- (41).Prankerd RJ In Profiles of Drug Substances, Excipients, and Related Methodology. Elsevier Academic Press: San Diego, CA, 2007; Vol. 33. [DOI] [PubMed] [Google Scholar]
- (42).Frenkel YV; Clark AD Jr.; Das K; Wang YH; Lewi PJ; Janssen PA; Arnold E Concentration and pH dependent aggregation of hydrophobic drug molecules and relevance to oral bioavailability. J. Med. Chem 2005, 48, 1974–1983. [DOI] [PubMed] [Google Scholar]
- (43).Coan KE; Maltby DA; Burlingame AL; Shoichet BK Promiscuous aggregate-based inhibitors promote enzyme unfolding. J. Med. Chem 2009, 52, 2067–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Macromodel, version 9.6; Schrödinger, LLC: New York, 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.