

Summary
ACOX1 (acyl-CoA oxidase 1) encodes the first and rate-limiting enzyme of the very-long-chain fatty acid (VLCFA) β-oxidation pathway in peroxisomes and leads to H2O2 production. Unexpectedly, dACOX1 is mostly expressed and required in glia, and loss of dACOX1 leads to developmental delay, pupal death, reduced lifespan, impaired synaptic transmission, and glial and axonal loss. Patients who carry a previously unidentified, de novo, dominant variant in ACOX1 (p.N237S) also exhibit glial loss. However, this mutation causes increased levels of ACOX1 protein and function resulting in elevated levels of reactive oxygen species in glia in flies and murine Schwann cells. ACOX1 (p.N237S) patients exhibit a severe loss of Schwann cells and neurons. However, treatment of flies and primary Schwann cells with an anti-oxidant suppressed the p.N237S induced neurodegeneration. In summary, both loss and gain of ACOX1 leads to glial and neuronal loss, but different mechanisms are at play and require different treatments.
Keywords: very long chain fatty acids, fatty acid peroxidation, ROS, axonal dystrophy, anti-oxidant NACA (NACA), Schwann cells, wrapping glia, Drosophila, ACOX1 deficiency
eTOC blurb
Chung et al. (2020) show that ACOX1, a peroxisomal protein required for degrading VLCFA, is mostly expressed in glial cells. Its loss causes an accumulation of VLCFA and glial loss. An ACOX1 gain-of-function mutation identified in three individuals leads to elevated ROS and glial loss, and is potently suppressed by anti-oxidants.
Introduction
Lipids are critical for neuronal development, synaptic plasticity, and function (Adibhatla and Hatcher, 2008; Tsui-Pierchala et al., 2002). Abnormal lipid metabolism contributes to the pathogenesis of several neurodegenerative disorders, including Alzheimer’s disease (Liu et al., 2017; Liu et al., 2015), Parkinson’s disease (Lin et al., 2018; Lin et al., 2019) and various diseases associated with glial dysfunction (Chrast et al., 2011; Toshniwal and Zarling, 1992). Mitochondria and peroxisomes have been implicated in some of these neurodegenerative diseases, but the molecular events that underlie the demise of neurons vary widely (Vishwanath, 2016). Some neurodegenerative diseases have been associated with defects in the degradation of fatty acids by β-oxidation, and these disorders share some common features (Eaton et al., 1996; Poirier et al., 2006; Wanders et al., 2010). In yeast and plants, peroxisomes are the only site of β-oxidation (Hu et al., 2012; Kao et al., 2018). However, in vertebrates and insects, peroxisomes have more restricted functions as they are the site of β-oxidation of VLCFA whereas long and medium chain fatty acids are processed in mitochondria (Wanders et al., 2010).
Peroxisomal ACOX1 (acyl‐CoA oxidase 1) is the first and rate‐limiting enzyme in fatty acid β‐oxidation of VLCFA and a major producer of hydrogen peroxide (H2O2) (Schrader and Fahimi, 2006). ACOX1 deficiency (OMIM #264470) in humans is an autosomal recessive disorder that causes a rapid and severe loss of nervous system function (Ferdinandusse et al., 2007). Newborns with ACOX1 deficiency exhibit hypotonia and seizures, then they experience a gradual loss of learning and speaking skills, usually beginning between the ages of 1 and 3 (Ferdinandusse et al., 2007). As the condition worsens, children develop exaggerated reflexes, have an increase in VLCFA, increased muscle tone, severe and recurrent seizures, and loss of vision and hearing (El Hajj et al., 2012). Most children with peroxisomal ACOX1 deficiency do not survive past early childhood (Ferdinandusse et al., 2007).
We and others have previously reported that loss of various peroxisomal proteins and enzymes causes severe neuronal defects in flies (Di Cara et al., 2019; Mast et al., 2011; Wangler et al., 2017). Exactly how loss of peroxisomes contributes to severe neurologic phenotypes in flies, mice, and humans is still poorly explored. Here, we report that dACOX1 is expressed in glia and that loss of dACOX1 severely affects lifespan, increases VLCFA, causes loss of vision, and leads to glial loss and reduced neuronal survival in flies. In contrast, a novel variant, p.N237S, identified in 3 patients with progressive ataxia and hearing loss, stabilizes ACOX1 as an active dimer, acts as a gain of function mutation, produces elevated levels of reactive oxygen species (ROS) in insulating glia, does not alter VLCFA levels, and leads to the demise of wrapping glia in flies and primary Schwann cells in mouse. A potent anti-oxidant, N-acetyl cysteine amide (NACA), strongly suppresses these effects. Our data show that proper activity of peroxisomal β-oxidation is essential for glial survival, and both loss and gain of ACOX1 severely affects glial function in flies and humans albeit via different pathways.
Results
Loss-of-function mutations in ACOX1 in flies cause semi-lethality
To assess the consequences of loss of dACOX1 in flies, we created two different alleles based on recently developed CRISPR methods (Lee et al., 2018; Li-Kroeger et al., 2018). To create a null allele, we replaced the entire open reading frame via homology-directed repair with the dominant yellow wing+ (ywg+) marker (Figure 1A (bottom) and Figure S1A, STAR Methods). Homozygous dACOX1ywg mutants die as pupae, and no escapers are observed (Figure 1B; 0/859 expected progeny). For the second loss of function allele we introduced attP-FRT-SA-T2A-Gal4-polyA-3xP3-EGFP-polyA-FRT-attP (T2A-GAL4) into the first coding intron of dACOX1, referred to as dACOX1T2A (Figure 1A (top) and Figure S1B). The SA-T2A-GAL4-polyA is the core of this artificial exon. It leads to a truncation of the transcript at the polyA and a truncated protein in conjunction with the production of GAL4 in the proper spatial and temporal pattern (Figure S1B) (Lee et al., 2018). GAL4 is a transcriptional activator that binds UAS enhancer sequences and can be used to drive UAS-nls-mCherry to assess which cells express dACOX1 (Figure S1B).
Figure 1.

Loss of function mutations in dACOX1 in flies cause lethality
(A) Schematic representation of the molecular lesion in dACOX1, the dACOX1 deletion (dACOX1ywg), and the insertional mutation (dACOX1T2A). (B) Summary of complementation tests. GR: Genomic Rescue construct. The numbers indicate the # of observed flies versus # of expected flies (C) Western blot of dACOX1 with protein lysates from 3rd instar larvae of dACOX1T2A;GR (control), dACOX1ywg and dACOX1T2A showing diminished dACOX1 level with both mutant alleles. Mean ± s.e.m. ***p< 0.001. (D) Larval growth curves of control, dACOX1ywg and dACOX1T2A animals. (E) dACOX1 is expressed in glia of larval CNS. The expression of dACOX1T2A>nls-mCherry (magenta) colocalized with anti-Repo (green), marking the glia nuclei, in larval CNS. Scale bar: 15μm. (F) dACOX1 is expressed in most glia of adult flies. The expression of dACOX1T2A>UAS-nls-mCherry (magenta) colocalized with anti-Repo (green), marking the glia nuclei, in the adult fly brain. Scale bar: 5μm
Both alleles correspond to severe loss of function mutations based on Western blots when probed with a polyclonal antibody against human ACOX1 (Figure 1C). Quantification of the faint doublet in dACOX1T2A homozygote animals reveals that this is not a null allele as about 510% of protein can be detected (Figure 1C). Approximately 90% of the homozygous dACOX1T2A animals die as pupae but about ~10% eclose as adults. This semi-lethality is also observed in dACOX1T2A/dACOX1ywg flies (Figure 1B). Both mutant alleles cause severe developmental delay of the larva-to-pupal transition after egg deposition (AED) (Figure 1D). Both CRISPR/Cas9 induced alleles do not carry any other recessive lethal or visible mutation as the lethality and other phenotypes discussed below are fully rescued by a 20 KB P[acman] BAC genomic rescue (GR) construct that carries the dACOX1 locus (Venken et al., 2009) (Figure 1B, STAR Methods).
dACOX1 is highly enriched in glia in the nervous system
To assess the expression patterns of endogenous dACOX1 in the central nervous system (CNS) and peripheral nervous system (PNS) of flies, we crossed dACOX1T2A with UAS-nls-mCherry to mark the nuclei of cells expressing dACOX1 (Figure 1A and Figure S1B). We observed a remarkable co-localization of nls-mCherry and Repo, a nuclear glial marker in the CNS (Figure 1E, white arrows) and PNS (Figure S2A) including perineural glia and wrapping glia. In the adult CNS, nls-mCherry is also expressed in most glial cells and very few neurons (Figure 1F and Figure S2B). These observations are surprising as we anticipated that dACOX1, a peroxisomal enzyme, would be expressed in most or all cells.
dACOX1 is required for the maintenance of glia
To examine the developmental phenotypes associated with homozygous dACOX1T2A, we counted the number of glial cells in larval brains and found no difference with dACOX1T2A;GR (GR, Genomic Rescue) larvae (Figure 2A). Hence, loss of dACOX1 does not affect the development of glial cells up to this stage. To assess the behavioral phenotypes of homozygous dACOX1T2A flies that escape pupal lethality, we first performed lifespan assays. Homozygote dACOX1T2A flies are short-lived, and die before day 25 (Figure 2B). In contrast, 70% of dACOX1T2A;GR flies live about 50 days. Climbing assays revealed that young flies (day 3) are not impaired, but by day 15 nearly all dACOX1T2A homozygote flies exhibit severe climbing defects (Figure 2C). Hence, homozygous dACOX1T2A flies progressively lose their motor skills and die prematurely.
Figure 2. dACOX1 is required for maintenance of glia.

(A) The number of glial cells in larval brains of homozygous dACOX1T2A are similar with control flies (dACOX1T2A;GR). (B) Homozygous dACOX1T2A flies are short lived compared to control flies (n = 100 for control and dACOX1T2A). (C) Homozygous dACOX1T2A flies exhibit progressive climbing defects. Time (seconds) required for flies of the indicated genotypes to climb past 7 cm (n > 25 per genotype). Statistical analyses are one-way ANOVA followed by a Tukey post-hoc test. Results are mean ± s.e.m. (***p< 0.001). (D) Electron microscopy of the wing nerves, which contain the axons of the peripheral neurons of the anterior wing margin. Quantification of total axon number and the number of wrapped axons per nerve (n= 3 (control), n= 2 (dACOX1T2A)), of TEM images from (C). Mean ± s.e.m. ***p< 0.001, Statistical analyses were determined by 2-sided Student’s t-test.
To determine if loss of dACOX1 affects glia we performed transmission electron microscopy (TEM) of the wing nerves of 10-day-old dACOX1T2A flies to examine the axons of the peripheral neurons of the anterior wing. The wing nerves contain three types of glia (Freeman, 2015; Freeman and Doherty, 2006; Keller et al., 2011): wrapping glia, perineural glia, and subperineural glia. As shown in Figure 2D, the number of axons in dACOX1T2A flies is severely decreased (top) compared to control, and many remaining axons are irregular in shape when compared to the control dACOX1T2A;GR (bottom). Note that most of the wrapping glia are absent or highly aberrant in size and shape (red arrows). However, the perineural and subperineural glial cells of the nerves are not or minimally affected when compared to the control (Figure S2C, black arrows). These data show that dACOX1 is required for the maintenance of wrapping glia and axons.
Loss of dACOX1 in glia induces progressive vision loss
Patients with ACOX1 deficiency gradually lose vision. Hence, we performed electroretinogram (ERG) recordings of homozygous dACOX1T2A flies to assess the vision in young and older flies. As shown in Figure 3A, between day 2 and day 15 dACOX1T2A mutant flies exhibit a strong progressive loss of ERG on- and off-transients and a severe decrease in amplitude when compared to controls. The on- and off-transients are a measure of synaptic transmission between the photoreceptor cells and the postsynaptic neurons in the lamina (Montell, 1999; Sanes and Zipursky, 2010), whereas the loss of amplitude of the ERG reveals a defect in phototransduction. Interestingly, the ERG defects are rescued by a UAS-human ACOX1 driven by the homozygous dACOX1T2A alleles. Hence, the human homologue can replace the loss of function of dACOX1. To assess the morphological demise of the retina we raised the flies in the dark and performed Toluidine blue staining on eye sections of 2 and 15 day old flies. As shown in Figure 3B, we observe a severe neurodegeneration of the retina between day 2 and day 15.
Figure 3. Homozygous dACOX1 mutants exhibit progressive neurodegenerative phenotypes via accumulation of VLCFA.

(A) ERG traces of homozygous dACOX1T2A flies and control flies (dACOX1T2A;GR) at day 2 and day 15 with quantification of ERG amplitudes. Quantification of depolarization amplitude and off transient (n = 12 per each genotype), of ERG traces from (A). Mean ± s.e.m. ***p < 0.001; n.s., not significant. Statistical analyses are one-way ANOVA followed by a Tukey post-hoc test. (B) Toluidine blue staining on eye section of day 2 and day 15 homozygous dACOX1T2A flies and control flies (dACOX1T2A;GR). (C) Gas chromatography mass spectrometry for total VLCFA, and the relative level of VLCFA (C28:0/C22:0 and C26:0/C22:0) Mean ± s.e.m. (***p< 0.001, **p< 0.01). (D) Bezafibrate is an inhibitor of ELOVL1, a VLCFA synthase. (E) Bezafibrate and ELOVL RNAi (STAR Methods) increased the viability of homozygous dACOX1T2A flies. Quantification of the percentage of expected homozygous dACOX1T2A flies per crosses (n = 10 (Vehicle), n = 10 (Bezafibrate 0.4μM), n = 11 (dACOX1T2A/dACOX1T2A; UAS-dELOVL RNAi), n = 5 (dACOX1T2A/dACOX1T2A;UAS-hELOVL1)). Statistical analyses are one-way ANOVA followed by a Tukey post-hoc test. Results are mean ± s.e.m., ***p< 0.001. (F) Bezafibrate and ELOVL RNAi restored climbing ability of dACOX1T2A flies (n > 15 per genotype). Statistical analyses are one-way ANOVA followed by a Tukey post-hoc test. Results are mean ± s.e.m. (***p< 0.001, **p< 0.01). (G) Bezafibrate and ELOVL RNAi rescued the amplitude defects of homozygous dACOX1T2A flies (n = 16 per genotype). Statistical analyses are one-way ANOVA followed by a Tukey post-hoc test. Results are mean ± s.e.m. (***p< 0.001).
Bezafibrate suppresses the loss of function phenotypes associated with loss of dACOX1
As dACOX1 is a rate-limiting enzyme for β-oxidation of VLCFA, dACOX1T2A should lead to an increase in VLCFA. To assess if peroxisomal β-oxidation was altered in dACOX1T2A mutants, we examined canonical peroxisomal lipids using the same methods as employed for clinical diagnosis (Hubbard et al., 2009; Moser et al., 1999). We used gas chromatograph mass spectrometry (GC-MS) (Moser et al., 1999) to measure VLCFA in adult flies and observed increased levels of total VLCFA, C28/C22, and C26/C22 in dACOX1T2A mutants (Figure 3C).
Bezafibrate, an agonist of PPARα used to treat hyperlipidemia, reduces VLCFA synthesis by inhibiting ELOVL1, a VLCFA synthase (Engelen et al., 2012a; Engelen et al., 2012b) (Figure 3D). To examine if the increase in VLCFA directly promotes the demise of glia and axons in dACOX1T2A animals, we supplemented the fly food with bezafibrate. We assessed a dose response using 0.2, 0.4, 0.8 and 1.6 μM bezafibrate. As shown in Figure S3A, 0.4 μM provided the best response to suppress the lethality of dACOX1T2A animals (Figure 3E). This bezafibrate treatment (0.4 μM) also improved climbing ability (Figure 3F) and loss of amplitude dACOX1T2A (Figure 3G) and OFF transients (Figure S3B) when compared to flies raised on normal food supplemented with vehicle.
To assess if lowering VLCFA using another strategy improved the phenotypes associated with loss of dACOX1, we reduced the level of dELOVL1 using an established RNAi (Gordon et al., 2018). Reducing dELOVL1 with this RNAi suppresses lethality (Figure 3E), improves climbing ability (Figure 3F), and reduces the amplitude reduction (Figure 3G) and OFF transient defects (Figure S3B). In contrast, overexpression of human ELOVL1 using dACOX1T2A is toxic and causes pupal lethality (Figure 3E). These data argue strongly that an increase in VLCFA promotes neurodegenerative defects and that reducing the elevated levels of VLCFA is beneficial.
Three patients with ACOX1 p.N237S display progressive axonal loss
Through the efforts of the Undiagnosed Diseases Network (Splinter et al., 2018) and by posting ACOX1 on Genematcher (Sobreira et al., 2015), we identified 3 patients with the same de novo missense variant in ACOX1 (chr17:73951722T>C [hg19]; NM_004035:c.710A>G, p.N237S). This variant is not present in the parents of any of the 3 families and has not been observed in over 140,000 control genomes and exomes of gnomAD (STAR methods) (Karczewski et al., 2019; Lek et al., 2016; Wang et al., 2017). Hence, they are a recurrent de novo event in three unrelated families. In each case the probands display remarkably similar phenotypes, including a progressive myeloneuropathy with sensorineural hearing loss (Table 1, STAR Methods), onset from age 3 to 12 years (STAR Methods). Of note, the ACOX1 variant was not initially flagged by diagnostic clinical sequencing as prior to our study, ACOX1 was only known to relate to an autosomal recessive Peroxisomal Acyl-CoA Oxidase Deficiency (STAR methods) (OMIM #264470), and the 3 patients described here are all heterozygous for the ACOX1 variant. Indeed, the phenotypes of the three cases contrasted sharply to patients with ACOX1 deficiency, and the de novo variant present in these probands does not cause an increase in VLCFA suggesting a new disease mechanism for ACOX1 (table S1). Finally, the de novo p.N237S variant is predicted ‘Damaging’ and ‘Deleterious’ in Poly-Phen (Adzhubei et al., 2010) and SIFT (Ng and Henikoff, 2003), and has a CADD score of 32 (Rentzsch et al., 2018). These in silico predictions are indicative that the p.N237S is deleterious.
Table 1.
Comparison of clinical features for each subject with ACOX1 deficiency
| Patient 1 | Patient 2 | Patient 3 | Peroxisomal ACOX1 deficiency (MIM: #264470) | |
|---|---|---|---|---|
| ACOX1 variant |
de novo
heterozygous Missense variant (p.N237S) |
de novo
heterozygous Missense variant (p.N237S) |
de novo
heterozygous Missense variant (p.N237S) |
Autosomal Recessive |
| Disease onset | 12y | 9y | 3y | 0–3y |
| Survival | 19y (Death) | 15y (Coma) | 9y (still alive) | Death by ~5y |
| Disease course | Progressive | Progressive | Progressive | Early onset and severe |
| Inflammatory response | No | No | No | Yes |
| VLCFA | Normal | Normal | Normal | Accumulation |
| White matter demyelination | Normal => Abnormal | Normal => Abnormal | Normal => Abnormal | Abnormal |
| Sensorimotor polyneuropathy | Decreased | Decreased | Decreased | N/A |
| Cognition | Normal => Decreased | Normal => Decreased | Decreased | Significantly Decreased |
| Ataxia | Yes | Yes | Yes | Yes |
| Notes | Chronic axon loss | Ongoing and chronic axon loss | Ongoing and chronic axon loss | First reported in 1988. Deletions, nonsense and missense mutations reported. |
All three patients with de novo ACOX1 p.N237S variant present similar clinical features but distinct from peroxisomal ACOX1 deficiency (MIM: #264470) (STAR Methods).
ACOX1 p.N237S is a gain of function mutation
The crystal structure of ACOX1 revealed that this enzyme acts as a homodimer bound to Flavin adenine dinucleotide (FAD) (Nakajima et al., 2002; Pedersen and Henriksen, 2005; Zhang et al., 2016) (Figure 4A). The amino acids that line the FAD binding pocket of ACOX1 include amino acid N237 (Figure 4B). To assess the functional consequences of the ACOX1 p.N237S (ACOX1N237S) variant, we ubiquitously overexpressed the human ACOX1N237S in flies using the daughterless-Gal4 (da-Gal4) driver and compared them to flies expressing the reference human protein. To determine the organelle where wild type and mutant ACOX1 proteins localize, we ubiquitously co-expressed an enhanced yellow fluorescent protein (eYFP) carrying a peroxisomal targeting signal (PTS1) (da>UAS-eYFP PTS1). As shown in Figure 4C, both hACOX1WT and hACOX1N237S are localized to peroxisomes. However, the signal for hACOX1N237S is strongly increased when compared to hACOX1WT despite a similar number of peroxisomes being labeled (Figure 4C). Expression of hACOX1N237S is also significantly increased in lysates of 3rd instar larvae (Figure 4D) or adult flies when probed on Western blots. Most of the ACOX1N237S protein is detected at ~140 kD, instead of the predicted 70 kD, consistent with the reported dimerization of the protein (Chen et al., 2018). In contrast, the levels of monomeric ACOX1N237S are comparable to those in control animals, both in larvae (Figure 4D) and adults. These data show that ACOX1N237S promotes the dimerization of ACOX1. In addition, this dimer appears to be resistant to protein turnover, as it is much more abundant.
Figure 4. hACOX1N237S promotes dimer formation and elevated ACOX1 protein levels.

(A) The current model for hACOX1 dimerization (left) and the 3D protein structure of a hACOX1 dimer bound to 2 flavine adenine dinucleotides (FAD) (right). (B) Enlarged view of a portion of the FAD contact region of hACOX1. The black arrow points to asparagine 237, and N237 is part of the FAD binding pocket of hACOX1. (C) hACOX1N237S protein accumulates in vivo when compared to hACOX1WT. Scale bar: 10μm. (D) hACOX1N237S accumulates in the form of dimers. hACOX1 protein levels are elevated in larval extracts in which N237S is expressed. Quantification of relative dimer/monomer ACOX1 level from the blot of (D). n = 4 independent blots, Mean ± s.e.m. ***p< 0.001, Statistical analyses were determined by 2-sided Student’s t-test. (E) Purified hACOX1N237S protein show higher activity than hACOX1WT when normalized for protein level. (n = 3 for each), Mean ± s.e.m. **p< 0.01, Statistical analyses were determined by 2-sided Student’s t-test. (F) Gas chromatography mass spectrometry for total VLCFA, and the relative level of VLCFA (C28:0/C22:0). n = 7 (da>dACOX1WT, 10 adult flies extracts). n = 9 (da>dACOX1N250S), Statistical analyses were determined by 2-sided Student’s t-test. n.s., not significant. (G) Quantification of relative 4HNE (STAR Methods) level normalized by actin, n = 4 independent blots (50 larvae per each blot), Statistical analyses are one-way ANOVA followed by a Tukey post-hoc test. Mean ± s.e.m. ***p < 0.001, n.s., not significant.
It has been documented that the dimer is the active form of the protein (Chen et al., 2018; Pedersen and Henriksen, 2005; Zhang et al., 2016). To compare the enzymatic activity of ACOX1WT and ACOX1N237S, we performed enzymatic activity assays in vitro (Chen et al., 2018; Small et al., 1985). The data show that ACOX1N237S is more active by ~40% than ACOX1WT protein when normalized for protein levels (Figure 4E). Given that we also observe a strong elevation of ACOX1N237S (da>ACOX1N237S) protein levels when compared to animals that express ACOX1WT (da>ACOX1WT), we argue that increased activity combined with elevated protein levels lead to strongly enhanced activity. The p.N237 residue is conserved in the fly homolog and corresponds to p.N250 (Figure S4). We therefore generated transgenic flies that carry wild type or mutant dACOX1 (da>dACOX1N250S). Expression of dACOX1N250S also leads to accumulation of a dimeric dACOX1 when compared to dACOX1WT overexpression in larvae (Figure S5A), suggesting that a role of N237 in dimer formation is evolutionarily conserved.
In contrast to flies that lose dACOX1 (dACOX1T2A), we observe no difference in VLCFA levels between these controls (da>dACOX1WT) and mutant flies (da>dACOX1N250S) (Figure 4F). Given the increase in active dimers we measured the levels of ROS (Chen et al., 2018; Schrader and Fahimi, 2006) using protein electrophoresis followed by detection of HNE (hydroxynonenal)-modified proteins using an anti-4HNE polyclonal antibody (STAR Methods). 4HNE is a highly toxic aldehyde product of lipid peroxidation and is a sensitive marker of oxidative damage and lipid peroxidation (Esterbauer et al., 1985). We found a mild increase of 4HNE signal when we overexpressed dACOX1WT. However, dACOX1N250S overexpression caused a dramatic increase in the levels of the 4-HNE-modified proteins arguing that overexpression of dACOX1N250S induces much more ROS than overexpression of dACOX1WT (Figure 4G, Figure S5B). The above data indicate that ACOX1 p.N237S is a gain of function variant that leads to the formation of a constitutively active dimer that generates excess ROS.
Expression of dACOX1N250S causes lethality that is potently suppressed by an anti-oxidant, N-acetyl cysteine amide (NACA)
To assess whether the expression of dACOX1N250S causes defects in flies we ubiquitously expressed the dACOX1WT and dACOX1N250S variants. Overexpression of dACOX1WT causes a reduction in eclosion by ~25%. The remaining 75% of flies eclose and do not exhibit any obvious behavioral defects (Figure S5C). In contrast, only 20% of the dACOX1N250S flies eclose (da>UAS-dACOX1N250S) but these flies are unable to fly, walk, or feed, and die in less than 2 days (Figure S5C, movie S1). To determine if these flies exhibit glial and/or axonal loss, we performed TEM of the wing nerves on day 1 in dACOX1N250S flies (da>UAS-dACOX1N250S). As shown in Figure S5D, the number of axons are severely decreased and we observed abnormal multi-lamella bodies (Figure S5D, red arrows) and aberrant membrane structures (Figure S5D, yellow arrow). Multilamellar bodies have been associated with neurodegenerative models (Nixon et al., 2005; Phillips et al., 2008; Tagliaferro and Burke, 2016). In addition, many axons are irregular in shape, and most of the wrapping glia are aberrant in size and shape. These data show that dACOX1N250S is toxic and leads to axonal loss as well as glial loss (Figure S5D).
The dimer of ACOX1 is the active form implicated in VLCFA β-oxidation and generates H2O2 during its enzymatic reaction. H2O2 is reduced by peroxisomal catalase, an abundant peroxisomal enzyme responsible for the conversion of H2O2 into O2 (Schrader and Fahimi, 2006). Hence, we determined whether anti-oxidants are able to suppress the lethality of dACOX1N250S. We tested NACA, N-acetyl cysteine amide. This low-molecular-weight molecule has been shown to be a potent anti-oxidant in flies and mice that efficiently crosses the bloodbrain-barrier, scavenges ROS, and increases intracellular glutathione (Amer et al., 2008; Grinberg et al., 2005; Liu et al., 2017; Liu et al., 2015). In the absence of NACA, no da>dACOX1N250S flies survive beyond day 2 as shown in Figure 5A (flat green line). Supplementing food with NACA (160 μg/ml) rescues the lethal phenotype of da>dACOX1N250S, and each vial typically produced 60% of the expected flies (3 repeats). Importantly, these flies were not only viable, but were able to walk, climb, fly, mate, and reproduce. Additional improvements in viability were not noted when using higher concentrations of NACA (320 μg/ml) (Figure S5E). Importantly, the lethality associated with the dACOX1 loss of function mutation was not suppressed by NACA at any concentration, showing that the specific nature of the lesion in the gain of function mutation (p.N250S) is elevated ROS (Figure 5A).
Figure 5. Decreased ROS level by N-acetyl cysteine amide (NACA) or catalase expression suppresses the lethality induced by expression of dACOX1N250S.

(A) The anti-oxidant NACA suppresses the lethality of da>dACOX1N250S, but cannot suppress the lethality of dACOX1T2A (n = 3 crosses, >50 flies were counted for one cross). (B) Lifespan of flies that co-express UAS-catalase (green) or maintained on NACA (red) or switched to normal food after eclosion (black) (n > 100 per each). (C) % of expected adult flies of the indicated genotypes. The dACOX1WT and dACOX1N250S were expressed using various drivers, including ubiquitous drivers (whole body) or drivers for neuronal, glial, ring gland (endocrine organ), hemocyte (blood cell), or fat body (adipose and metabolic organ) expression (n > 200 progenies were counted across 3 trials, STAR Methods). Mean ± s.e.m. ***p< 0.001, **p < 0.01, n.s., not significant. Statistical analyses were determined by 2-sided Student’s t-test. (D) Time to climb past 7 cm for the indicated fly genotypes (n > 50 per genotype). Statistical analyses are one-way ANOVA followed by a Tukey post-hoc test. Means ± s.e.m. ***p < 0.001; n.s., not significant. Western blot below indicated that dACOX1N250S expression is not affected by the expression of catalase. (E) Lifespan of flies of the indicated genotypes (n > 50 per each genotype).
To determine if reducing ROS can be beneficial for da>dACOX1N250S expressing flies post-development, we compared the life span of flies that are exposed to NACA during development and after eclosion with flies that are raised on NACA but switched to standard diet after eclosion (Figure 5B). Lack of NACA following eclosion leads to death by day 23. Conversely, animals that remained on NACA-containing food performed significantly better, and 50% lived beyond day 40. Hence, NACA provides a robust protection against insults induced by expression of dACOX1N250 in adult flies (Figures. 5A and B). Consistent with this, overexpression of peroxisomal catalase also rescued both the lethality and life span defects associated with dACOX1N250S (Figure 5B).
We next wished to determine which tissue/cells are affected by the gain of function mutation. Ubiquitous expression (da-Gal4; Act-Gal4 and Tub-Gal4) of dACOX1N250S, but not dACOX1WT (Figure 5C, STAR Methods) causes 100% lethality. We therefore carried out a Gal4screen with tissue specific drivers to determine which tissues or cells are susceptible to dACOX1N250S overexpression. We selected 20 different Gal4 drivers. Expression of dACOX1N250S by neuronal-specific Gal4 drivers (Elav-Gal4 and Nsyb-Gal4) cause a slight reduction (~5–15%) in viability but do not cause any obvious neurodegenerative phenotypes. However, overexpression of the glial specific driver Repo-Gal4 as well as the wrapping glia driver egr-Gal4 decreases viability by more than 50%. Yet, there are about 10 X more neurons than glia in flies (Freeman, 2015; Freeman and Doherty, 2006). In contrast, astrocyte (Alrm-Gal4) or ensheathing glia (Mz-Gal4) specific Gal4s lead to only mild effects on viability (10% and 15%, respectively). Expression of dACOX1N250S using other Gal4s including ring gland (Feb36-Gal4 and Mai60-Gal4), hemocyte (Hml-Gal4, He-Gal4 and Pxn-Gal4), and fat body (Cg-Gal4) drivers did not affect fly viability (Figure 5C). These data clearly indicate that fly wrapping glia are sensitive to the high ROS induced by dACOX1N250S.
Expression of dACOX1N250S in wrapping glia causes severe motor deficits that are restored by catalase overexpression
dACOX1N250S is toxic when expressed in glia (Figure 5C), and wild type dACOX1 is abundantly expressed in most glia cells in the adult brain (Figure 1F, Figure S2B). Therefore, we next assayed climbing in flies expressing dACOX1N250S driven by three specific glial drivers: Mz, ensheathing glia; alrm, astrocytes; and egr, wrapping glia. Overexpression of dACOX1N250S by Mz-Gal4 or Alrm-Gal4 did not cause obvious lethality or motor deficits, however, wrapping glia-specific expression by egr-Gal4 significantly affected survival and climbing (Figures 5D and E). To determine if these motor defects are caused by high ROS, we co-expressed UAS-catalase (Missirlis et al., 2001) together with UAS-dACOX1N250S. Consistent with the rescue of lethality shown previously with NACA administration (Figures 5A and B), overexpression of catalase rescued both the climbing defects and lethality associated with dACOX1N250S (Figures 5D and E).
Overexpression of ACOX1WT and ACOX1N237S causes Schwann cell death which is suppressed by an anti-oxidant
To investigate the role of ACOX1 in glia in vertebrates, we characterized its expression pattern in the rat sciatic nerve. Immunohistochemistry of Acox1 in transverse sections of the sciatic nerve revealed that it co-localizes with S100+ myelinating Schwann cells (Figure 6A). However, we did not observe Acox1 in cells co-stained with Neurofilament-H, an axonal marker (Figure 6A, STAR Methods).
Figure 6. Overexpression of ACOX1WT and ACOX1N237S causes Schwann cell death but is suppressed by an anti-oxidant.

(A) Endogenous Acox1 (green) is expressed in myelinating Schwann cells (S100, red) but not in axons (NF, red). Scale bar: 10μm. (B) Analysis of Schwann cell survival upon overexpression of ACOX1WT and ACOX1N237S. The cell death caused by either expression of hACOX1WT or hACOX1N237S in Schwann cells is significantly reduced upon treatment with the anti-oxidant NACA (1.5 mM). The quantification results are merged data from 2 independently derived sets of cultures and are representative of 3 experiments, each performed in triplicate. Results are mean ± s.e.m., n.s., not significant, **p < 0.001, ***p < 0.0001). Scale bar: 50μm. (C) The dimers of hACOX1N237S are elevated in primary Schwann cells when compared to hACOX1WT, Quantification of relative dimer/monomer ACOX1 level (n = 3 independent blots, Mean ± s.e.m. ***p< 0.001, Statistical analyses were determined by 2-sided Student’s t-test. Western blot below indicated that hACOX1N237S expression does not affect the expression of peroxisomal marker, PMP70. (D) H2O2 increase when hACOX1WT and hACOX1N250S (red) are overexpressed in Schwann cells is suppressed by 1.5 mM NACA (green). Statistical analyses are one-way ANOVA followed by a Tukey post-hoc test. Means ± s.e.m. ***p < 0.001, **p < 0.01, n.s., not significant. (E) Co-immunoprecipitation experiments in primary Schwann cells. 4 different combinations of ACOX1 constructs with different tags were transfected into primary Schwann cells as described in table. (F) The effect of MG132 on the expression of ACOX1WT and ACOX1N237S. Quantification of relative level of monomeric (top) and dimeric ACOX1 (bottom) from the blot (F) (n = 3 independent blots), Mean ± s.e.m. ***p< 0.001, n.s., not significant. Statistical analyses were determined by 2-sided Student’s t-test.
Given that dACOX1 plays an important role in wrapping glia in flies, and murine Acox1 is expressed in Schwann cells, we next overexpressed hACOX1WT in rat primary Schwann cells using a lentivirus and observed a reduction in the number of S100-expressing cells (Figure 6B). This suggests that overexpression of hACOX1WT can affect Schwann cell viability, consistent with the elevated ROS observed when dACOX1WT is expressed in flies. Expression of hACOX1N237S in rat Schwann cells caused an even more severe increase in apoptotic cell death than the wt isoform (Figure 6B). Interestingly, the toxic effects of hACOX1WT and hACOX1N237S in rat Schwann cells are strongly suppressed by NACA (Figure 6B) when compared to vehicle control. The ACOX1N237S protein is more abundant as a dimer similar as what was observed in flies (Figure 4D, Figure 6C), and we also did not observe a change in expression of the peroxisomal marker, PMP70 (Figure 6C). We also observe an increase in H2O2 when we overexpress hACOX1WT and hACOX1N250S in Schwann cells and this increase is again suppressed by NACA (1.5 mM) (Figure 6D).
To assess if the ACOX1N237S protein creates a homodimer or heterodimer with the wild type isoform we performed co-immunoprecipitation experiments in primary Schwann cells. We transfected 4 different combinations of ACOX1 constructs with different tags into primary Schwann cells 1) Flag-ACOX1WT + V5-ACOX1WT; 2) Flag-ACOX1WT + V5-ACOX1N237S; 3) Flag-ACOX1N237S + V5-ACOX1WT; and 4) Flag-ACOX1N237S + V5-ACOX1N237S. As shown in Figure 6E, the mutant homodimer is clearly much more abundant than the wild type homodimer or the heterodimers. We further examined the effects of protease inhibitor, MG132 (STAR Methods) on the expression of ACOX1. As shown in Figure 6F, the levels of monomeric ACOX1WT were increased by MG132 but the levels of dimeric ACOX1WT were not changed when compared to control without MG132. However, the levels of monomeric ACOX1N237S were not changed but the levels of dimeric ACOX1N237S were slightly increased by MG132. This suggests that monomeric ACOX1N237S appears to be resistant to protein turnover, and preferentially exists as a mutant homodimer that is a toxic (Figure 6E).
ACOX1N237S causes a disorder associated with severe Schwann cell loss and myelination defects
Patient 1 displayed a progressive neuropathy and received serial nerve conduction studies and a sural nerve biopsy as part of his clinical care, allowing examination of the human Schwann cells. We observed a prominent loss of large and small myelinated and small non-myelinated axons in the sural nerve of Patient 1 when compared to a normal nerve section (Figures 7A, Figure S6A and B). As shown in Figure 7A, Toluidine blue showed actively degenerating axons and abnormal myelin folding. Transmission electron microscopy (Figure 7B) showed degenerating Schwann cells with lipid droplets and myelin debris (Figure S7). We labeled a transverse section of the nerve for neurofilament (NF), an axon marker (Nixon and Shea, 1992), as well as for neural cell adhesion molecule (NCAM), a marker of non-myelinating Schwann cells (Figure S6A) (Rutishauser et al., 1988). Staining for myelin basic protein (MBP) showed an obvious loss of large myelinated axons (fig S6A). Büngner Schwann cells, usually observed with chronic axon loss, are not present in normal sural nerves (Arthur-Farraj et al., 2012; Shy, 2006). However, we observed an increased number of Büngner Schwann cells in the sural nerve of Patient 1, consistent with his chronic axon loss (Figure S6A-S6C and table S2).
Figure 7. Patient 1 exhibits a progressive myelin loss.

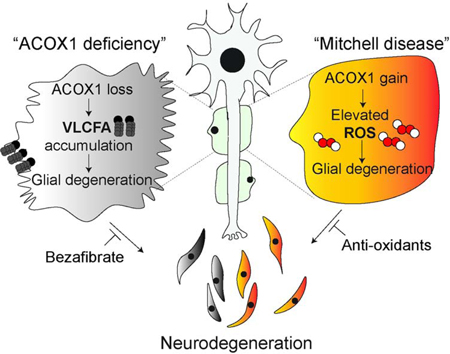
(A) Toluidine blue stained plastic sections of sural nerve. Healthy axons have normal myelin folding (Control, left), but there is a moderate loss of large and small myelinated axons and 2 actively degenerating axons (red arrows) with associated cells containing lipid and myelin debris in nerve from Patient 1 (right). (B) TEM ultrastructural findings. Normal/healthy large and small myelinated axons are demonstrated in the Control (left), but the nerve of Patient 1 (right) exhibits active axonal degeneration at various stages (red arrows). (C) Proposed model of ACOX1 lossand gain-of function diseases.
Our data demonstrate that NACA is an effective drug to reverse the effects of ACOX1 gain of function in fly and primary Schwann cells (Figures 5 and 6), and this drug can penetrate the blood-brain-barrier (Amer et al., 2008; Grinberg et al., 2005). Given that NACA is not yet approved for human use, during a severe disease flare Patient 1 was started on very high doses of the NAC as an alternative. His disease course initially stabilized (Figure S8, table S3), and he had an 11 month phase of improvement. However, use of NAC, which does not penetrate the blood-brain-barrier, was unable to prevent another severe disease flare of the central nervous system. Therefore, after 7 years of a waxing and waning course of peripheral disease, Patient 1 succumbed over the course of weeks to the acute and rapid onset of central nervous system disease. With high regard to his immeasurable contributions to help us understand the pathogenesis of ACOX1 gain of function, we propose to name this disease Mitchell disease in honor of Patient 1.
Discussion
Role of VLCFA in myelin and axonal insulation: vertebrates versus flies
Flies have no myelin and this has often been raised as a concern for modeling glial disease in flies. However, our data indicate striking parallels between wrapping glia and Schwann cells. Myelin is rich in VLCFA, and ELOVL1, an enzyme that synthesizes VLCFA from LCFA, and VLCFA are required for myelin maintenance (Imgrund et al., 2009; Ohno et al., 2010; Sassa and Kihara, 2014). Indeed, VLCFA are incorporated in glycerophospholipids and most VLCFAs in mammals are present in sphingolipids (Kihara, 2012), lipids that are abundant in the outer leaflet of the plasma membrane (Kihara, 2012), especially in lipid rafts and myelin where they reduce the fluidity of membranes. The ratio of C24 sphingolipids/total sphingolipids varies in different tissues. In mice, C24 sphingomyelin accounts for ~25% of the sphingomyelin in the brain (Laviad et al., 2008) and VLCFA are converted to C24 sphingolipids by ceramide synthase 2 (CerS2) (Ohno et al., 2010), which is required for myelin maintenance (Imgrund et al., 2009). Loss of CerS2 leads to an 80% reduction of myelin basic protein. Hence, VLCFA are critical for myelin maintenance. Interestingly, a form of the progressive neuropathy Charcot-Marie-Tooth disease (CMT1A) (Hong et al., 2016) is caused by a mutation in PMP2 which encodes Fatty Acid Binding Protein 8 that is abundantly expressed in Schwann cells and binds LCFA (Smathers and Petersen, 2011). Glia in flies do not contain myelin, but the wrapping glia that insulate axons are rich in the ceramide phosphoethanolamine (CPE) a lipid that is very similar to sphingomyelin (Faust et al., 2014; Freeman and Doherty, 2006; Kretzschmar et al., 1997). Depletion of CPE results in defasciculation and failure of wrapping glia to enwrap peripheral axons similar to CMT (Ghosh et al., 2013). We have previously shown that VLCFA are present in CPE in flies (Lin et al., 2018); based on these data we estimate that 67% of all CPEs contain VLCFA above C22 in length and 19% contain FA longer than C24. Our data here indicate that an increase in VLCFA upon loss of ACOX1 also causes a defasciculation and failure of wrapping glia to ensheath axons. Similarly, patients with loss of ACOX1 also exhibit demyelinating peripheral and central neuropathies (El Hajj et al., 2012; Ferdinandusse et al., 2007; Raas et al., 2018). Hence, not being able to break down VLCFA is toxic (Khan et al., 2005; Schonfeld and Reiser, 2016; Wanders et al., 2010) and most likely severely affects the membrane composition of wrapping glia and myelin (Lingwood and Simons, 2010; Sassa et al., 2012; Simons and Ikonen, 1997), drawing striking parallels between myelin in vertebrate glia and CPE in wrapping glia in insects. Our findings that bezafibrate (Figure 3), a drug that inhibits the synthesis of VLCFA, strongly suppresses the phenotypes in ACOX1 deficient flies clearly supports the evidence that elevated levels of VLCFA in glia may be a key trigger for glial and axonal loss.
Energy metabolism in neurons and glia
Our data show that ACOX1 is abundant in glia and poorly expressed in neurons in both flies and mice (Figures1 and 6). This finding is in agreement with previous observations in vertebrates that peroxisomes are abundant in Schwann cells, but absent in the axons they enwrap (Kassmann et al., 2011; Kleinecke et al., 2017). The breakdown of myelin leads to the production of VLCFA that, through β-oxidation in peroxisomes produce LCFA that can be used as an energy source in the mitochondria of glial cells. Glial cells are known to provide lactate to neurons as an energy source in vertebrates (Pellerin and Magistretti, 1994) and flies (Liu et al., 2017; Liu et al., 2015; Saab et al., 2016). (Kassmann, 2014) proposed that LCFA produced in glia may correspond to a second energy source for axons (Kohlwein et al., 2013; Reddy and Hashimoto, 2001). We have previously shown that lipids can be transferred from neurons to glia, requiring fatty acid transport protein and apolipoprotein D (Dourlen et al., 2012; Liu et al., 2017; Liu et al., 2015). A similar export pathway may transport fatty acids from glia to neurons (Kis et al., 2015). Hence, the reduction in LCFA in glia upon loss of ACOX1 may also be detrimental for axons and deprive them of an energy source.
In mammals, peroxisomes are observed in astrocytes, oligodendrocytes, microglia, and Schwann cells (Kassmann et al., 2007; Kassmann et al., 2011; Kleinecke et al., 2017). Although mice that lack the peroxisomal proteins Pex5 (Baes et al., 1997; Dodt et al., 1995) or Pex11 (Hanson et al., 2014) display severe neurological defects, removal of Pex5 specifically from neurons or from astrocytes does not cause obvious phenotypes (Bottelbergs et al., 2010). In contrast, removal of Pex5 from oligodendrocytes (CNP-Pex5 knock-out) induces demyelination and severe axonal loss (Kassmann et al., 2007) suggesting that axonal loss is caused by peroxisome-deficient oligodendrocytes. In addition, mice that lack Mfp2, a VLCFA metabolizing enzyme that acts downstream of Acox1, also exhibit severe loss of axons (Turk et al., 2017; Verheijden et al., 2015). Again, these defects are not observed when Mfp2 is only absent from neurons or astrocytes (Beckers et al., 2018). However, mice deficient for Mfp2 in oligodendrocytes do not show prominent pathology in the first year of life. This is inconsistent with observations in flies and humans (McMillan et al., 2012), suggesting some redundancy for Mfp2 in mice. Finally, loss of Acox1 in mice has not been associated with axonal loss in the first 6 months of life (Fan et al., 1996), nor has a neurodegenerative phenotype been documented yet (Moreno-Fernandez et al., 2018). Hence, humans and flies share similar phenotypes in the absence of ACOX1 that have not yet been observed in mice. We propose this may be due to redundancy with murine Acox2 and Acox3.
STAR Methods text
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hugo J Bellen (hbellen@bcm.edu). All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human patients
A detail information on how three patients were allocated to experimental groups are described in Table 1 and Supplementary material: extended case reports.
Primary Schwann cell culture
4 weeks old female SAS Sprague Dawley Rats were obtained from Charles River Laboratories, and Schwann cells from adult sciatic nerves were cultured as previously described (Lee et al., 2007; Lee et al., 2009). Briefly, the sciatic nerves of adult Sprague-Dawley rats were axotomized to enhance the Schwann cell population. Sciatic nerves were isolated and submitted to a chemical digestion in 0.2% collagenase in calcium/magnesium-free Hank’s buffered solution at 37°C for 2 hrs. The nerves were then dissociated by gentle shaking for 2 min followed by 2 or 3 triturations using a flame-polished Pasteur pipette. The cell pellets obtained after centrifugation were resuspended in DMEM containing 10% FBS and plated for 2 d. After 2 sub-cultures, the identity and purity of Schwann cells were assessed by immunostaining with an antibody against S100. For viral infection of Schwann cells, cells were dissociated and plated in culture media with forskolin (10 μM) and NRG-1 (200 ng/mL), then subsequently infected with either hACOX1WT or hACOX1N237S-lenti-pHAGE or control lentivirus for 14 hrs. For the chemical treatment experiments, Schwann cells were transfected using ImFectin (GenDepot, STAR Methods) transfection reagent for 6 hrs and harvested for analysis 3 days after transfection. NACA (1.5 mM) was treated on a daily basis for 3 days after transfection. MG132 (20 μM, Sigma) were treated 20 hrs before harvesting cells.
METHOD DETAILS
Fly strains and genetics
dACOX1ywg and dACOX1T2A alleles were generated as described by (Li-Kroeger et al., 2018) and (Lee et al., 2018), respectively. The following oligonucleotides were used to generate the dACOX1ywg allele:
Upstream sgRNA
FW: 5’ – CAACGAAGACGGACGCTTTG – 3’
Upstream HMA forward oligo:
FW: 5’ – atatatGGTCTCtGACCggaatctcacagtcactggtatc – 3’ Upstream HMA reverse oligo:
FW: 5’ – gagagagGGTCTCgATCCagcgtccgtcttcgttG – 3’
Downstream sgRNA
FW: 5’ – GCAATAGGATATGAATCTAGAGG – 3’
Downstream HMA forward oligo:
FW: 5’ – atatatGGTCTCtTTCCgattcatatcctattgctctggaag – 3’
Downstream HMA reverse HMA oligo
FW: 5’ – agagagGGTCTCgTATAcagaagcttttgctgaagg – 3’
The genetically modified regions of these alleles were confirmed by Sanger DNA sequencing. The dACOX1 genomic rescue line was generated by injection of the 20kb P[acman] clone CH322–174F04 (Venken et al., 2009) into the y,w,ΦC31; VK33 target site at 65B2 (Venken et al., 2006). Transgenic lines harboring the P[acman] construct were selected based on their eye color (white+). All the fly stocks were maintained at room temperature (22°C). Flies were raised at 25°C or 29°C.
Transmission electron microscopy
Adult fly retinas were processed for TEM imaging as described (Luo et al., 2017). Samples were processed using a Ted Pella Bio Wave microwave oven with vacuum attachment. Adult fly heads were dissected at 25 °C in 4 % paraformaldehyde, 2 % glutaraldehyde, and 0.1 M sodium cacodylate (pH 7.2). Samples were subsequently fixed at 4 °C for 48 hr. 1 % osmium tetroxide was used for secondary fixation and subsequently dehydrated in ethanol and propylene oxide, and then embedded in Embed-812 resin (Electron Microscopy Science, Hatfield, PA). 50 nm ultra-thin sections were obtained with a Leica UC7 microtome and collected on Formvar-coated copper grids (Electron Microscopy Science, Hatfield, PA). Specimens were stained with 1 % uranyl acetate and 2.5 % lead citrate and imaged using a JEOL JEM 1010 transmission electron microscope with an AMT XR-16 mid-mount 16 mega-pixel CCD camera. For quantification of ultrastructural features, electron micrographs were examined from 3 different animals per treatment.
Transmission electron microscopy for wing margin
Drosophila wing margins were imaged following standard electron microscopy procedures using a Ted Pella Bio Wave processing microwave with vacuum attachments. The fly was covered in 2% paraformaldehyde, 2.5% glutaraldehyde, in 0.1 M sodium cacodylate buffer at pH 7.2. Briefly, the thorax was dissected under the fixative away from the head and abdomen leaving the wings on the thorax. After dissection the thorax was incubated overnight up to 3 days in the fixative on a rotator. The pre-fixed thorax with wings was then fixed again, rinsed by 3x with Millipore water, post-fixed with 1% aqueous osmium tetroxide, and rinsed again 3x with Millipore water. Concentrations from 25–100% of ethanol were used for the initial dehydration series, followed with propylene oxide as the final dehydrant. Samples were gradually infiltrated with 3 ratios of propylene oxide and Embed 812, finally going into 3 changes of pure resin under vacuum. Samples were allowed to infiltrate in pure resin overnight on a rotator. The samples were embedded into flat silicone molds arranged so that the sample could be cross sectioned in the wing margin area. The samples were then cured in the oven at 62°C for three days. Thinsections of the polymerized samples were cut at 48–50 nm and stained with 1% uranyl acetate for 10 minutes followed by 2.5% lead citrate for 2 minutes before TEM examination. Grids were viewed in a JEOL 1400+ transmission electron microscope at 80kV. Images were captured using an AMT XR-16 mid-mount 16 mega-pixel digital camera.
Cloning and transgenesis
Site-directed mutagenesis was performed using iProof™ High-Fidelity DNA Polymerase (Bio-Rad) as previously described (Ansar et al., 2018). Briefly, using the full-length cDNA of dACOX1 in the pENTR223 Gateway donor vector (GH07485 from Drosophila Genomics Resource Center) and hACOX1 in the pENTRY221 Gateway donor vector (closed clone, IOH27813 from Invitrogen). The information for the primers to generate dACOX1 p.N250S and hACOX1 p.N237S, is described in STAR methods. The fully sequenced pENTR223 dACOX1WT, pENTR223 dACOX1N250S, pDON221 hACOX1WT and pDON221 hACOX1N237S clones were subcloned into the pGW-attB HA with NEB® 5-alpha Competent E. coli (STAR Methods) (Bischof et al., 2013) vector to generate pGW-attB-dACOX1WT, pGW-attB-dACOX1N250S, pGW-attB-hACOX1WT and pGWattB-hACOX1N237S. These constructs were injected into y,w,ΦC31;VK33 embryos (Venken et al., 2006) and transgenic flies were selected by the w+ marker on the pGW-attB-HA vector. Although the plasmid contains a C’−3xHA tag, this tag is not added to the ACOX1 proteins due to a stop codon present before the tag. pDON221 hACOX1WT and pDON221 hACOX1N237S clones were subcloned into the pDEST-CMV-3xFLAGgateway-EGFP (Addgene) and pCDNA-DEST40 (Invitrogen) vectors for the co-immunoprecipitation experiments in primary Schwann cells.
Drosophila behavioral assays
Climbing was performed as previously described (Marcogliese et al., 2018). Briefly, flies were anesthetized 24 hrs before testing using CO2, then housed individually. Flies were transferred to an empty vial and given 1 min to habituate before being tapped to the bottom of the vial and examined for a negative geotaxis (climbing) response. Flies were given a maximum of 30 s to reach 7 cm. For lifespan, freshly eclosed flies were separated by genotype and sex and incubated at 25°C. Flies were flipped into a fresh vial every 3 days and survival was determined once a day. At least 100 flies were tested in each assay.
ERG Recording of Fly Eye
ERG recordings were performed as described in (Verstreken et al., 2003). In brief, flies were glued to a slide with Elmer’s Glue. A recording electrode filled with 100 mM NaCl was placed on the eye, and a reference electrode was placed on the fly head. During the recording, a 1 s pulse of light stimulation was given, and the ERG traces of ten flies for each genotype were recorded and analyzed with WinWCP v.5.3.3 software.
Glutaraldehyde cross-linking experiment
The cross-linking experiment was performed with minor modifications to a previously described protocol (Chen et al., 2018). In brief, 100μl of whole fly lysates extracted from modified RIPA buffer (50 mM Tris-Cl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 50 mM NaF, 1 mM Na3VO4, 10% glycerol and Roche protease inhibitor mix, STAR methods) were treated with 5 μl of 2% glutaraldehyde at 37°C for 3 min. The reaction was terminated by adding 15μl of 1 M Tris–HCl (pH 8.0) and then subjected to Western blot analysis.
ACOX1 enzymatic activity assay
ACOX1 enzyme activity was determined as described previously (Chen et al., 2018; Small et al., 1985). hACOX1WT and hACOX1N237S were incubated for 30 mins in enzymatic activity reaction solution (150 μl) (0.8 mM 4-aminoantipyrine (Sigma), 11 mM phenol solution, 0.04% Triton X100, 0.05% (w/v) palmitoyl-CoA (Sigma), 5 μM FAD (Sigma), 1 purpurogallin units’ peroxidase enzyme (Sigma) in 50 mM 2-(N-morpholino) ethanesulfonic acid (MES) buffer (Sigma)), and activity of ACOX1 was measured by the ratio of A520 change in 5 min. Both human ACOX1 WT and human ACOX1 N237S protein were extracted using TNT® Quick Coupled Transcription/Translation System (Promega, STAR Methods), and purified using Flag Immunoprecipitation Kit (Sigma, STAR Methods). Each sample was analyzed in triplicate.
Protein electrophoresis
Western blot was performed as previously described (Kanca et al., 2019). Briefly, the whole bodies of adult flies or larvae were homogenized in modified RIPA buffer (50 mM Tris-Cl, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 50 mM NaF, 1 mM Na3VO4, 10% glycerol and Roche protease inhibitor mix) on ice. Tissue or cell debris were removed by centrifugation (15,000rpm). For co-immunoprecipitation in primary Schwann cells, cells were lysed on ice using a Dounce homogenizer in 0.1% CHAPS buffer (STAR Methods) (50 mM HEPES (pH7.5), 50 mM KCl, 2 mM MgCl2, 1 mM EDTA (pH 8.0), 0.1% CHAPS (Sigma), 1 mM DTT, 1 mM PMSF, proteinase inhibitor and phosphatase inhibitor cocktail (Roche)), and the lysates were immunoprecipitated with 1μg of rabbit anti-Flag (M2, Sigma, STAR Methods) bound to Dynabeads (Dynabeads™ Protein G, Invitrogen, STAR Methods) at 4 °C for overnight. The immunocomplexes were washed five times with cold IP buffer using magnet (DynaMag, Invitrogen, STAR Methods) at 4 °C. The samples were boiled in protein loading buffer at 94 °C for 3 mins, and isolated lysates were subject to electrophoresis using a 4%–15% MiniPROTEAN TGX precast native gel (Biorad, STAR Methods) and 10x Tris/Glycine Buffer (nonSDS, Biorad, STAR Methods). The native gels were transferred to Immobilon-FL polyvinylidene difluoride membranes.
Drug administration in fly food
NACA (STAR Methods) was added freshly to regular fly food at the indicated concentrations: 40 μg/ml, 80 μg/ml, 160 μg/ml, and 320 μg/ml dissolved in distilled water (Liu et al., 2015). For life-span analysis, flies were transferred to freshly prepared food supplemented with NACA every 2 days. Bezafibrate (Sigma, STAR Methods) was added freshly to regular fly food at the indicated concentrations: 0.2 μM, 0.4 μM, 0.8 μM, 1.6 μM, dissolved in DMSO.
Immunocytochemistry
In brief, salivary gland staining was performed as previously described (Chung et al., 2016). Briefly, glands were fixed with 4% paraformaldehyde in phosphate buffered saline (PBS) for 30 min and then permeabilized in 0.1% Triton X-100 in PBS at room temperature for 1 hr. After permeabilization, the tissues were used for immunofluorescence staining with the following antibodies: Rabbit anti-DLG (from Kwang-Wook Choi (Lee et al., 2003), STAR Methods), Mouse anti-DLG (4F3, DSHB (Parnas et al., 2001), STAR Methods). For immunocytochemistry of fly brains, tissues were dissected in PBS and fixed in 4% paraformaldehyde in PBS at 4°C on a nutating platform, then transferred to 0.2% Triton X-100 in PBS (0.2% PBST) at 4°C on a nutating platform for overnight incubation. For immunostaining, the samples were blocked in 5% BSA/2% PBST and incubated with the primary antibodies: Rat anti-Elav (1:500, 7E8A10, DSHB (O’Neill et al., 1994), STAR Methods) and Mouse anti-Repo (1:50, 8D12, DSHB, (Alfonso and Jones, 2002), STAR Methods) diluted in 5% BSA/0.2% PBST at 4°C for 48 hrs with nutation, then washed 3x with 0.2% PBST for 5 min. The secondary antibodies Donkey anti-Mouse (Cy3) and Donkey anti-Rat (Alexa-647) (Jackson ImmunoResearch, STAR Methods) were diluted 1:250 in 5% BSA/0.2% PBST and incubated with the samples at 4°C for 48 hrs on a rotating platform. Samples were cleared and mounted in RapiClear (SunJin Lab Co.) and imaged with a Leica SP8 Confocal Microscope under a 20x objective lens and analyzed using Fiji (Schindelin et al., 2012).
H2O2 absorbance assay
To measure H2O2 absorbance in primary Schwann cells, cells were lysed on ice using a Dounce homogenizer in 0.1% CHAPS buffer (50 mM HEPES (pH7.5), 50 mM KCl, 2 mM MgCl2, 1 mM EDTA (pH 8.0), 0.1% CHAPS, 1 mM DTT, 1 mM PMSF, proteinase inhibitor and phosphatase inhibitor cocktail (Roche)), and the lysates were directly applied to H2O2 assay kit (ab102500; Abcam, STAR Methods) and were analyzed with a fluorescence spectrophotometer (FLUOstar OPTIMA, STAR Methods).
Sciatic nerve staining
Mouse sciatic nerves were fixed in 4% paraformaldehyde and cryo-protected in 20% sucrose. To detect the endogenous expression of ACOX1 in sciatic nerve, the following antibodies were used: Mouse anti-hACOX1 (1:500; HPA021195, Sigma-aldrich, STAR Methods), Rabbit antiS100 (1:1000; GA50461–2, DAKO, STAR Methods), Chick anti-Neurofilament-H (1:5000; 835601, BioLegend, STAR Methods).
QUANTIFICATION AND STATISTICAL ANALYSIS
Results are presented as dot or bar plots, in which the mean +/− standard error of the mean (SEM) are depicted. All statistical analysis was performed using Graphpad Prism (GraphPad Software, Inc., Ca, US). When the means of 2 groups were compared, a two-tailed unpaired t-test was used, and when the means of 2 variables of more than 2 groups were compared, a two way ANOVA with Tukey’s multiple comparisons post-test was used. Results were designated significant when the P-value (p)<0.05: *= p<0.05, **= p<0.01, ***= p<0.001, n.s. = non-significant
Supplementary Material
Highlights.
Loss of dACOX1 leads to glial degeneration via accumulation of VLCFA
A de novo ACOX1N237S gain-of-function mutation also leads to glial loss
Glial loss in ACOX1N237S is associated with elevated ROS, not an increase in VLCFA
N-acetyl cysteine amide, an antioxidant, suppresses glial defects in ACOX1N237S flies
Acknowledgments:
We thank Drs. Alex Sweeney and Kevin Baszis for clinical expertise. We thank Hongling Pan and Yuchun He for injections to create transgenic flies. H.K.L. is supported by NMSS via RG1508–08406. M.F.W., S.Y and H.J.B. are supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number(s) U01HG007709, U54NS093793 and by ORIP via R24OD022005. Confocal microscopy was performed in the Neurovisualization core of the BCM IDDRC (supported by NICHD U54HD083092). P.C.M. is funded by CIHR (MFE-164712). Drosophila stocks were obtained from the Bloomington Stock Center (NIH P40OD018537) at Indiana University. Monoclonal antibodies were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD and maintained at The University of Iowa. H.J.B. is an Investigator of the Howard Hughes Medical Institute. T.A.R. is supported by The Cullen Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing offered by Baylor Genetics Laboratories.
Footnotes
Declaration of Interests
The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted by Baylor Genetics Laboratories.
DATA AND CODE AVAILABILITY
The fly dACOX1 cDNA (GH07485) sequence is available in Drosophila Genomic Resource Center (DGRC, https://dgrc.bio.indiana.edu/), and the sequence of human ACOX1 cDNA (BC010425.1) is available in NCBI. All the reagents and stocks that were created will be deposited in the DGRC or BDSC (https://bdsc.indiana.edu/). Those that are not accepted by these centers will be distributed from the Bellen lab.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes:
- Adibhatla RM, and Hatcher JF. (2008). Altered lipid metabolism in brain injury and disorders. Subcell Biochem 49, 241–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR. (2010). A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso TB, and Jones BW. (2002). gcm2 promotes glial cell differentiation and is required with glial cells missing for macrophage development in Drosophila. Dev Biol 248, 369–383. [DOI] [PubMed] [Google Scholar]
- Amer J, Atlas D, and Fibach E. (2008). N-acetylcysteine amide (AD4) attenuates oxidative stress in beta-thalassemia blood cells. Biochim Biophys Acta 1780, 249–255. [DOI] [PubMed] [Google Scholar]
- Ansar M, Chung H, Waryah YM, Makrythanasis P, Falconnet E, Rao AR, Guipponi M, Narsani AK, Fingerhut R, Santoni FA, et al. (2018). Visual impairment and progressive phthisis bulbi caused by recessive pathogenic variant in MARK3. Hum Mol Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur-Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, Woodhoo A, Jenkins B, Rahman M, Turmaine M, et al. (2012). c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baes M, Gressens P, Baumgart E, Carmeliet P, Casteels M, Fransen M, Evrard P, Fahimi D, Declercq PE, Collen D, et al. (1997). A mouse model for Zellweger syndrome. Nat Genet 17, 49–57. [DOI] [PubMed] [Google Scholar]
- Beckers L, Stroobants S, D’Hooge R, and Baes M. (2018). Neuronal Dysfunction and Behavioral Abnormalities Are Evoked by Neural Cells and Aggravated by Inflammatory Microglia in Peroxisomal beta-Oxidation Deficiency. Front Cell Neurosci 12, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Bjorklund M, Furger E, Schertel C, Taipale J, and Basler K. (2013). A versatile platform for creating a comprehensive UAS-ORFeome library in Drosophila. Development 140, 2434–2442. [DOI] [PubMed] [Google Scholar]
- Bottelbergs A, Verheijden S, Hulshagen L, Gutmann DH, Goebbels S, Nave KA, Kassmann C, and Baes M. (2010). Axonal integrity in the absence of functional peroxisomes from projection neurons and astrocytes. Glia 58, 1532–1543. [DOI] [PubMed] [Google Scholar]
- Chen XF, Tian MX, Sun RQ, Zhang ML, Zhou LS, Jin L, Chen LL, Zhou WJ, Duan KL, Chen YJ, et al. (2018). SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrast R, Saher G, Nave KA, and Verheijen MH. (2011). Lipid metabolism in myelinating glial cells: lessons from human inherited disorders and mouse models. J Lipid Res 52, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HL, Augustine GJ, and Choi KW. (2016). Drosophila Schip1 Links Expanded and Tao-1 to Regulate Hippo Signaling. Dev Cell 36, 511–524. [DOI] [PubMed] [Google Scholar]
- Di Cara F, Rachubinski RA, and Simmonds AJ. (2019). Distinct Roles for Peroxisomal Targeting Signal Receptors Pex5 and Pex7 in Drosophila. Genetics 211, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt G, Braverman N, Wong C, Moser A, Moser HW, Watkins P, Valle D, and Gould SJ. (1995). Mutations in the PTS1 receptor gene, PXR1, define complementation group 2 of the peroxisome biogenesis disorders. Nat Genet 9, 115–125. [DOI] [PubMed] [Google Scholar]
- Dourlen P, Bertin B, Chatelain G, Robin M, Napoletano F, Roux MJ, and Mollereau B. (2012). Drosophila fatty acid transport protein regulates rhodopsin-1 metabolism and is required for photoreceptor neuron survival. PLoS Genet 8, e1002833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton S, Bartlett K, and Pourfarzam M. (1996). Mammalian mitochondrial beta-oxidation. Biochem J 320 ( Pt 2), 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hajj HI, Vluggens A, Andreoletti P, Ragot K, Mandard S, Kersten S, Waterham HR, Lizard G, Wanders RJ, Reddy JK, et al. (2012). The inflammatory response in acyl-CoA oxidase 1 deficiency (pseudoneonatal adrenoleukodystrophy). Endocrinology 153, 2568–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelen M, Schackmann MJ, Ofman R, Sanders RJ, Dijkstra IM, Houten SM, Fourcade S, Pujol A, Poll-The BT, Wanders RJ, et al. (2012a). Bezafibrate lowers very long-chain fatty acids in X-linked adrenoleukodystrophy fibroblasts by inhibiting fatty acid elongation. J Inherit Metab Dis 35, 1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelen M, Tran L, Ofman R, Brennecke J, Moser AB, Dijkstra IM, Wanders RJ, Poll-The BT, and Kemp S. (2012b). Bezafibrate for X-linked adrenoleukodystrophy. PLoS One 7, e41013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterbauer H, Zollner H, and Lang J. (1985). Metabolism of the lipid peroxidation product 4-hydroxynonenal by isolated hepatocytes and by liver cytosolic fractions. Biochem J 228, 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan CY, Pan J, Chu R, Lee D, Kluckman KD, Usuda N, Singh I, Yeldandi AV, Rao MS, Maeda N, et al. (1996). Hepatocellular and hepatic peroxisomal alterations in mice with a disrupted peroxisomal fatty acyl-coenzyme A oxidase gene. J Biol Chem 271, 24698–24710. [DOI] [PubMed] [Google Scholar]
- Faust JE, Manisundaram A, Ivanova PT, Milne SB, Summerville JB, Brown HA, Wangler M, Stern M, and McNew JA. (2014). Peroxisomes are required for lipid metabolism and muscle function in Drosophila melanogaster. PLoS One 9, e100213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandusse S, Denis S, Hogenhout EM, Koster J, van Roermund CW,L,IJ, Moser AB, Wanders RJ, and Waterham HR. (2007). Clinical, biochemical, and mutational spectrum of peroxisomal acyl-coenzyme A oxidase deficiency. Hum Mutat 28, 904–912. [DOI] [PubMed] [Google Scholar]
- Freeman MR. (2015). Drosophila Central Nervous System Glia. Cold Spring Harb Perspect Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR, and Doherty J. (2006). Glial cell biology in Drosophila and vertebrates. Trends Neurosci 29, 82–90. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Kling T, Snaidero N, Sampaio JL, Shevchenko A, Gras H, Geurten B, Gopfert MC, Schulz JB, Voigt A, et al. (2013). A global in vivo Drosophila RNAi screen identifies a key role of ceramide phosphoethanolamine for glial ensheathment of axons. PLoS Genet 9, e1003980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon HB, Valdez L, and Letsou A. (2018). Etiology and treatment of adrenoleukodystrophy: new insights from Drosophila. Dis Model Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg L, Fibach E, Amer J, and Atlas D. (2005). N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic Biol Med 38, 136–145. [DOI] [PubMed] [Google Scholar]
- Hanson MG, Fregoso VL, Vrana JD, Tucker CL, and Niswander LA. (2014). Peripheral nervous system defects in a mouse model for peroxisomal biogenesis disorders. Dev Biol 395, 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong YB, Joo J, Hyun YS, Kwak G, Choi YR, Yeo HK, Jwa DH, Kim EJ, Mo WM, Nam SH, et al. (2016). A Mutation in PMP2 Causes Dominant Demyelinating Charcot-Marie-Tooth Neuropathy. PLoS Genet 12, e1005829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Baker A, Bartel B, Linka N, Mullen RT, Reumann S, and Zolman BK. (2012). Plant peroxisomes: biogenesis and function. Plant Cell 24, 2279–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard WC, Moser AB, Liu AC, Jones RO, Steinberg SJ, Lorey F, Panny SR, Vogt RF Jr., Macaya D, Turgeon CT, et al. (2009). Newborn screening for X-linked adrenoleukodystrophy (X-ALD): validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method. Mol Genet Metab 97, 212–220. [DOI] [PubMed] [Google Scholar]
- Imgrund S, Hartmann D, Farwanah H, Eckhardt M, Sandhoff R, Degen J, Gieselmann V, Sandhoff K, and Willecke K. (2009). Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J Biol Chem 284, 33549–33560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanca O, Zirin J, Garcia-Marques J, Knight SM, Yang-Zhou D, Amador G, Chung H, Zuo Z, Ma L, He Y, et al. (2019). An efficient CRISPR-based strategy to insert small and large fragments of DNA using short homology arms. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao YT, Gonzalez KL, and Bartel B. (2018). Peroxisome Function, Biogenesis, and Dynamics in Plants. Plant Physiol 176, 162–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-offunction intolerance across human protein-coding genes bioRxiv. [Google Scholar]
- Kassmann CM. (2014). Myelin peroxisomes - essential organelles for the maintenance of white matter in the nervous system. Biochimie 98, 111–118. [DOI] [PubMed] [Google Scholar]
- Kassmann CM, Lappe-Siefke C, Baes M, Brugger B, Mildner A, Werner HB, Natt O, Michaelis T, Prinz M, Frahm J, et al. (2007). Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat Genet 39, 969–976. [DOI] [PubMed] [Google Scholar]
- Kassmann CM, Quintes S, Rietdorf J, Mobius W, Sereda MW, Nientiedt T, Saher G, Baes M, and Nave KA. (2011). A role for myelin-associated peroxisomes in maintaining paranodal loops and axonal integrity. FEBS Lett 585, 2205–2211. [DOI] [PubMed] [Google Scholar]
- Keller LC, Cheng L, Locke CJ, Muller M, Fetter RD, and Davis GW. (2011). Glial-derived prodegenerative signaling in the Drosophila neuromuscular system. Neuron 72, 760–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Haq E, Giri S, Singh I, and Singh AK. (2005). Peroxisomal participation in psychosinemediated toxicity: implications for Krabbe’s disease. J Neurosci Res 80, 845–854. [DOI] [PubMed] [Google Scholar]
- Kihara A. (2012). Very long-chain fatty acids: elongation, physiology and related disorders. J Biochem 152, 387–395. [DOI] [PubMed] [Google Scholar]
- Kis V, Barti B, Lippai M, and Sass M. (2015). Specialized Cortex Glial Cells Accumulate Lipid Droplets in Drosophila melanogaster. PLoS One 10, e0131250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinecke S, Richert S, de Hoz L, Brugger B, Kungl T, Asadollahi E, Quintes S, Blanz J, McGonigal R, Naseri K, et al. (2017). Peroxisomal dysfunctions cause lysosomal storage and axonal Kv1 channel redistribution in peripheral neuropathy. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlwein SD, Veenhuis M, and van der Klei IJ. (2013). Lipid droplets and peroxisomes: key players in cellular lipid homeostasis or a matter of fat--store ‘em up or burn ‘em down. Genetics 193, 1–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar D, Hasan G, Sharma S, Heisenberg M, and Benzer S. (1997). The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J Neurosci 17, 7425–7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviad EL, Albee L, Pankova-Kholmyansky I, Epstein S, Park H, Merrill AH Jr., and Futerman AH. (2008). Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J Biol Chem 283, 5677–5684. [DOI] [PubMed] [Google Scholar]
- Lee HK, Seo IA, Park HK, Park YM, Ahn KJ, Yoo YH, and Park HT. (2007). Nidogen is a prosurvival and promigratory factor for adult Schwann cells. J Neurochem 102, 686–698. [DOI] [PubMed] [Google Scholar]
- Lee HK, Seo IA, Suh DJ, Hong JI, Yoo YH, and Park HT. (2009). Interleukin-6 is required for the early induction of glial fibrillary acidic protein in Schwann cells during Wallerian degeneration. J Neurochem 108, 776–786. [DOI] [PubMed] [Google Scholar]
- Lee OK, Frese KK, James JS, Chadda D, Chen ZH, Javier RT, and Cho KO. (2003). Discs-Large and Strabismus are functionally linked to plasma membrane formation. Nat Cell Biol 5, 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PT, Zirin J, Kanca O, Lin WW, Schulze KL, Li-Kroeger D, Tao R, Devereaux C, Hu Y, Chung V, et al. (2018). A gene-specific T2A-GAL4 library for Drosophila. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Kroeger D, Kanca O, Lee PT, Cowan S, Lee MT, Jaiswal M, Salazar JL, He Y, Zuo Z, and Bellen HJ. (2018). An expanded toolkit for gene tagging based on MiMIC and scarless CRISPR tagging in Drosophila. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Lee PT, Chen K, Mao D, Tan KL, Zuo Z, Lin WW, Wang L, and Bellen HJ. (2018). Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to alpha-Synuclein Gain. Cell Metab 28, 605–618 e606. [DOI] [PubMed] [Google Scholar]
- Lin G, Wang L, Marcogliese PC, and Bellen HJ. (2019). Sphingolipids in the Pathogenesis of Parkinson’s Disease and Parkinsonism. Trends Endocrinol Metab 30, 106–117. [DOI] [PubMed] [Google Scholar]
- Lingwood D, and Simons K. (2010). Lipid rafts as a membrane-organizing principle. Science 327, 46–50. [DOI] [PubMed] [Google Scholar]
- Liu L, MacKenzie KR, Putluri N, Maletic-Savatic M, and Bellen HJ. (2017). The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab 26, 719–737 e716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, et al. (2015). Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Rosenfeld JA, Yamamoto S, Harel T, Zuo Z, Hall M, Wierenga KJ, Pastore MT, Bartholomew D, Delgado MR, et al. (2017). Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially. PLoS Genet 13, e1006905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcogliese PC, Shashi V, Spillmann RC, Stong N, Rosenfeld JA, Koenig MK, Martinez-Agosto JA, Herzog M, Chen AH, Dickson PI, et al. (2018). IRF2BPL Is Associated with Neurological Phenotypes. Am J Hum Genet 103, 245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast FD, Li J, Virk MK, Hughes SC, Simmonds AJ, and Rachubinski RA. (2011). A Drosophila model for the Zellweger spectrum of peroxisome biogenesis disorders. Dis Model Mech 4, 659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan HJ, Worthylake T, Schwartzentruber J, Gottlieb CC, Lawrence SE, Mackenzie A, Beaulieu CL, Mooyer PA, Consortium FC, Wanders RJ, et al. (2012). Specific combination of compound heterozygous mutations in 17beta-hydroxysteroid dehydrogenase type 4 (HSD17B4) defines a new subtype of D-bifunctional protein deficiency. Orphanet J Rare Dis 7, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missirlis F, Phillips JP, and Jackle H. (2001). Cooperative action of antioxidant defense systems in Drosophila. Curr Biol 11, 1272–1277. [DOI] [PubMed] [Google Scholar]
- Montell C. (1999). Visual transduction in Drosophila. Annu Rev Cell Dev Biol 15, 231–268. [DOI] [PubMed] [Google Scholar]
- Moreno-Fernandez ME, Giles DA, Stankiewicz TE, Sheridan R, Karns R, Cappelletti M, Lampe K, Mukherjee R, Sina C, Sallese A, et al. (2018). Peroxisomal beta-oxidation regulates whole body metabolism, inflammatory vigor, and pathogenesis of nonalcoholic fatty liver disease. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser AB, Kreiter N, Bezman L, Lu S, Raymond GV, Naidu S, and Moser HW. (1999). Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann Neurol 45, 100–110. [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Miyahara I, Hirotsu K, Nishina Y, Shiga K, Setoyama C, Tamaoki H, and Miura R. (2002). Three-dimensional structure of the flavoenzyme acyl-CoA oxidase-II from rat liver, the peroxisomal counterpart of mitochondrial acyl-CoA dehydrogenase. J Biochem 131, 365–374. [DOI] [PubMed] [Google Scholar]
- Ng PC, and Henikoff S. (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31, 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, and Shea TB. (1992). Dynamics of neuronal intermediate filaments: a developmental perspective. Cell Motil Cytoskeleton 22, 81–91. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, and Cuervo AM. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64, 113–122. [DOI] [PubMed] [Google Scholar]
- O’Neill EM, Rebay I, Tjian R, and Rubin GM. (1994). The activities of two Ets-related transcription factors required for Drosophila eye development are modulated by the Ras/MAPK pathway. Cell 78, 137–147. [DOI] [PubMed] [Google Scholar]
- Ohno Y, Suto S, Yamanaka M, Mizutani Y, Mitsutake S, Igarashi Y, Sassa T, and Kihara A. (2010). ELOVL1 production of C24 acyl-CoAs is linked to C24 sphingolipid synthesis. Proc Natl Acad Sci U S A 107, 18439–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas D, Haghighi AP, Fetter RD, Kim SW, and Goodman CS. (2001). Regulation of postsynaptic structure and protein localization by the Rho-type guanine nucleotide exchange factor dPix. Neuron 32, 415–424. [DOI] [PubMed] [Google Scholar]
- Pedersen L, and Henriksen A. (2005). Acyl-CoA oxidase 1 from Arabidopsis thaliana. Structure of a key enzyme in plant lipid metabolism. J Mol Biol 345, 487–500. [DOI] [PubMed] [Google Scholar]
- Pellerin L, and Magistretti PJ. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 91, 10625–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips SE, Woodruff EA 3rd, Liang P, Patten M, and Broadie K. (2008). Neuronal loss of Drosophila NPC1a causes cholesterol aggregation and age-progressive neurodegeneration. J Neurosci 28, 6569–6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier Y, Antonenkov VD, Glumoff T, and Hiltunen JK. (2006). Peroxisomal beta-oxidation--a metabolic pathway with multiple functions. Biochim Biophys Acta 1763, 1413–1426. [DOI] [PubMed] [Google Scholar]
- Raas Q, Saih FE, Gondcaille C, Trompier D, Hamon Y, Leoni V, Caccia C, Nasser B, Jadot M, Menetrier F, et al. (2018). A microglial cell model for acyl-CoA oxidase 1 deficiency. Biochim Biophys Acta Mol Cell Biol Lipids. [DOI] [PubMed] [Google Scholar]
- Reddy JK, and Hashimoto T. (2001). Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr 21, 193–230. [DOI] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, and Kircher M. (2018). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutishauser U, Acheson A, Hall AK, Mann DM, and Sunshine J. (1988). The neural cell adhesion molecule (NCAM) as a regulator of cell-cell interactions. Science 240, 53–57. [DOI] [PubMed] [Google Scholar]
- Saab AS, Tzvetavona ID, Trevisiol A, Baltan S, Dibaj P, Kusch K, Mobius W, Goetze B, Jahn HM, Huang W, et al. (2016). Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 91, 119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, and Zipursky SL. (2010). Design principles of insect and vertebrate visual systems. Neuron 66, 15–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassa T, and Kihara A. (2014). Metabolism of very long-chain Fatty acids: genes and pathophysiology. Biomol Ther (Seoul) 22, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassa T, Suto S, Okayasu Y, and Kihara A. (2012). A shift in sphingolipid composition from C24 to C16 increases susceptibility to apoptosis in HeLa cells. Biochim Biophys Acta 1821, 1031–1037. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonfeld P, and Reiser G. (2016). Brain Lipotoxicity of Phytanic Acid and Very Long-chain Fatty Acids. Harmful Cellular/Mitochondrial Activities in Refsum Disease and X-Linked Adrenoleukodystrophy. Aging Dis 7, 136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader M, and Fahimi HD. (2006). Peroxisomes and oxidative stress. Biochim Biophys Acta 1763, 1755–1766. [DOI] [PubMed] [Google Scholar]
- Shy ME. (2006). Peripheral neuropathies caused by mutations in the myelin protein zero. J Neurol Sci 242, 55–66. [DOI] [PubMed] [Google Scholar]
- Simons K, and Ikonen E. (1997). Functional rafts in cell membranes. Nature 387, 569–572. [DOI] [PubMed] [Google Scholar]
- Small GM, Burdett K, and Connock MJ. (1985). A sensitive spectrophotometric assay for peroxisomal acyl-CoA oxidase. Biochem J 227, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smathers RL, and Petersen DR. (2011). The human fatty acid-binding protein family: evolutionary divergences and functions. Hum Genomics 5, 170–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, and Hamosh A. (2015). GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter K, Adams DR, Bacino CA, Bellen HJ, Bernstein JA, Cheatle-Jarvela AM, Eng CM, Esteves C, Gahl WA, Hamid R, et al. (2018). Effect of Genetic Diagnosis on Patients with Previously Undiagnosed Disease. N Engl J Med 379, 2131–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliaferro P, and Burke RE. (2016). Retrograde Axonal Degeneration in Parkinson Disease. J Parkinsons Dis 6, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshniwal PK, and Zarling EJ. (1992). Evidence for increased lipid peroxidation in multiple sclerosis. Neurochem Res 17, 205–207. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Encinas M, Milbrandt J, and Johnson EM Jr. (2002). Lipid rafts in neuronal signaling and function. Trends Neurosci 25, 412–417. [DOI] [PubMed] [Google Scholar]
- Turk BR, Theisen BE, Nemeth CL, Marx JS, Shi X, Rosen M, Jones RO, Moser AB, Watkins PA, Raymond GV, et al. (2017). Antioxidant Capacity and Superoxide Dismutase Activity in Adrenoleukodystrophy. JAMA Neurol 74, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJ, Carlson JW, Schulze KL, Pan H, He Y, Spokony R, Wan KH, Koriabine M, de Jong PJ, White KP, et al. (2009). Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nat Methods 6, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJ, He Y, Hoskins RA, and Bellen HJ. (2006). P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 314, 1747–1751. [DOI] [PubMed] [Google Scholar]
- Verheijden S, Beckers L, Casazza A, Butovsky O, Mazzone M, and Baes M. (2015). Identification of a chronic non-neurodegenerative microglia activation state in a mouse model of peroxisomal beta-oxidation deficiency. Glia 63, 1606–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Koh TW, Schulze KL, Zhai RG, Hiesinger PR, Zhou Y, Mehta SQ, Cao Y, Roos J, and Bellen HJ. (2003). Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron 40, 733–748. [DOI] [PubMed] [Google Scholar]
- Vishwanath VA. (2016). Fatty Acid Beta-Oxidation Disorders: A Brief Review. Ann Neurosci 23, 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanders RJ, Ferdinandusse S, Brites P, and Kemp S. (2010). Peroxisomes, lipid metabolism and lipotoxicity. Biochim Biophys Acta 1801, 272–280. [DOI] [PubMed] [Google Scholar]
- Wang J, Al-Ouran R, Hu Y, Kim SY, Wan YW, Wangler MF, Yamamoto S, Chao HT, Comjean A, Mohr SE, et al. (2017). MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am J Hum Genet 100, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangler MF, Chao YH, Bayat V, Giagtzoglou N, Shinde AB, Putluri N, Coarfa C, Donti T, Graham BH, Faust JE, et al. (2017). Peroxisomal biogenesis is genetically and biochemically linked to carbohydrate metabolism in Drosophila and mouse. PLoS Genet 13, e1006825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li K, Jones RA, Bruner SD, and Butcher RA. (2016). Structural characterization of acyl-CoA oxidases reveals a direct link between pheromone biosynthesis and metabolic state in Caenorhabditis elegans. Proc Natl Acad Sci U S A 113, 10055–10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.