

Abstract
Mitochondrial DNA (mtDNA) — which is well known for its role in oxidative phosphorylation and maternally inherited mitochondrial diseases — is increasingly recognized as an agonist of the innate immune system that influences antimicrobial responses and inflammatory pathology. On entering the cytoplasm, extracellular space or circulation, mtDNA can engage multiple pattern-recognition receptors in cell-type- and context-dependent manners to trigger pro-inflammatory and type I interferon responses. Here, we review the expanding research field of mtDNA in innate immune responses to highlight new mechanistic insights and discuss the physiological and pathological relevance of this exciting area of mitochondrial biology.
Mitochondria are ubiquitous eukaryotic organelles that originated from an ancient α-proteobacterium more than 2 billion years ago. They have a unique, double-membrane structure and are central sites of metabolism with cell- and tissue-specific morphology, dynamics and function1. Mitochondria have maintained DNA (mitochondrial DNA (mtDNA)) (BOX 1; FIG. 1), which encodes essential protein subunits of the oxidative phosphorylation system. This consists of the electron transport chain (ETC; complexes I–IV) and ATP synthase (complex V)2, which drive mitochondrial respiration and ATP production. Mitochondria have many other functions in cells, which include myriad anabolic and catabolic pathways, regulation of apoptosis and calcium homeostasis, and reactive oxygen species (ROS) signalling3. More recently, mitochondria have been demonstrated to have various roles in host immune responses. For example, they orchestrate signalling and effector functions to boost immune cell activation and antimicrobial defence, and trigger inflammation in response to cell and tissue damage4,5.
Box 1 |. Expression and replication of mitochondrial DNA.
The nucleotide sequence of human mitochondrial DNA (mtDNA) was determined in 1981; it is a circular molecule of 16,569 bp, which encodes 37 genes2. The 13 mRNAs direct synthesis of an essential subset of oxidative phosphorylation complex subunits by dedicated ribosomes in the mitochondrial matrix. The two ribosomal RNA components (12S and 16S) and the requisite 22 tRNAs are encoded by mtDNA, whereas all additional proteins needed for mitochondrial transcription, translation and mtDNA replication are the products of nuclear genes and are co- or post-translationally imported into the organelle. A major regulatory site of mtDNA (called the D-loop region) harbours the promoters for transcription (the heavy-strand promoter (HSP) and the light-strand promoter (LSP)) and is the origin of heavy-strand replication (OH) and other conserved cis-acting elements96 (see the figure).
Expression begins with transcription of almost full-length primary transcripts (both mtDNA strands), which are then processed into the mature mRNA, ribosomal RNA and tRNA species by various RNase enzymes. During the asymmetric mode of mtDNA replication, transcripts from the LSP are used as primers for leading-strand replication, the 3’ ends of which are generated by transcription termination and/or specific RNA processing events downstream of the LSP and extended by mtDNA polymerase γ (Pol γ). After DNA synthesis begins, Pol γ is efficiently stalled or terminated ~1 kb downstream and the nascent DNA remains stably bound to the template, forming a stable three-stranded D-loop structure — a hallmark of mammalian mtDNA, the precise biological relevance of which still remains a mystery2,96. A productive replication event requires synthesis past the 3’ end of the D-loop and then priming of the lagging strand, which occurs at multiple sites. One major site is called OL (origin of light-strand replication), which is located ~12 kb away from OH, and hence leading and lagging strand replication can occur asynchronously, with large stretches of single-stranded DNA (ssDNA) and RNA–DNA hybrids (shown in red) persisting as intermediates in the process. Other models of mtDNA replication (for example, the ‘bootlace’ model) have also been proposed97.
In addition to the immunostimulatory aspects of the mtDNA molecule itself, transcription and replication of mtDNA involves the formation of unique nucleic acid species that may also engage host nucleic acid sensors. We propose that ssDNA, RNA–DNA hybrids and perhaps other higher-order nucleic acid structures derived from mtDNA, such as triplexes, R-loops and four-way junctions (not shown), may be detected by cyclic GMP–AMP synthase (cGAS) or other pattern-recognition receptors (PRRs) of the innate immune system.

Figure 1 |. Immunostimulatory features of mitochondrial DNA and related species.
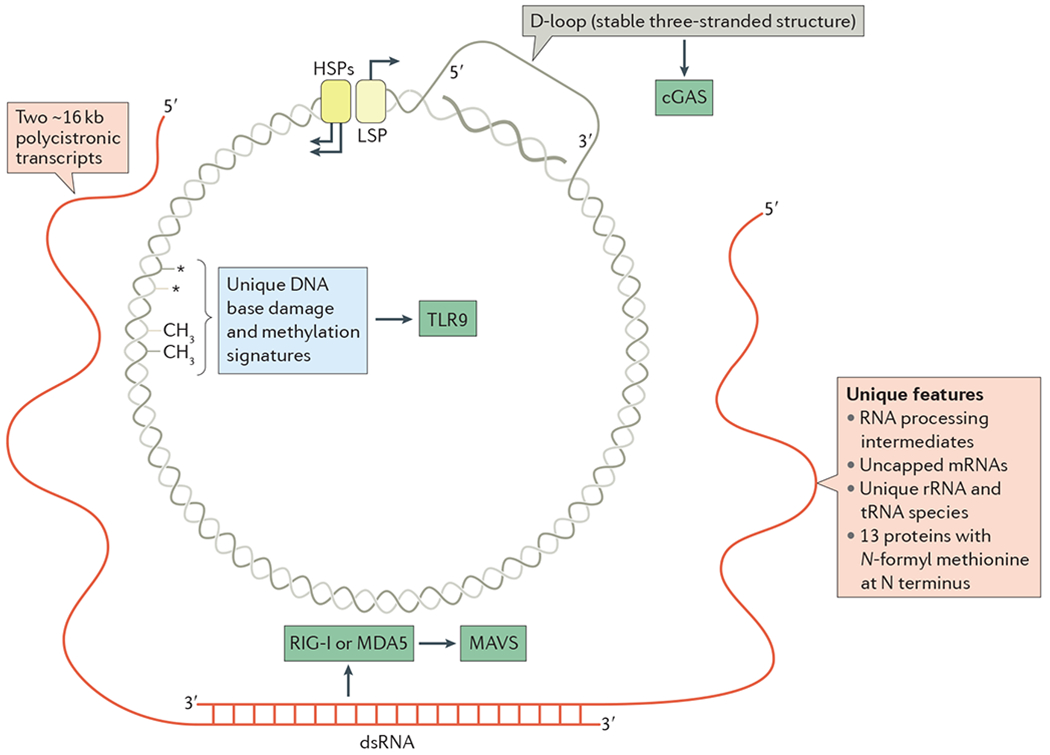
The circular mitochondrial DNA (mtDNA) of mammals is depicted with nucleic acid species generated during transcription and replication (BOX 1). Transcription of mtDNA involves the divergent light-strand promoter (LSP) and heavy-strand promoters (HSPs), which are shown at the top with the direction of transcription indicated by arrows. The two polycistronic primary transcripts from each strand are shown in red. Because almost the entire genome is transcribed in both directions, there is ample opportunity for double-stranded RNA (dsRNA) to be formed, as depicted at the bottom, which could engage retinoic acid-inducible gene I protein (RIG-I; also known as DDX58) or melanoma differentiation-associated protein 5 (MDA5; also known as IFIH1) and trigger mitochondrial antiviral signalling protein (MAVS). Replication of mtDNA initiates downstream of the LSP, but is paused or terminated frequently, forming a stable ~1 kb nascent DNA strand that remains associated with the template and displaces the non-template strand. This forms the hallmark three-stranded D-loop structure of mammalian mtDNA, and may be the source of cytosolic DNA that activates cyclic GMP–AMP synthase (cGAS). Other potentially unique features of mtDNA and mtDNA-encoded proteins are indicated, as are the innate immune sensors that we postulate might sense these unique features. rRNA, ribosomal RNA; TLR9, Toll-like receptor 9.
Since our efforts in 2011 to document the many roles for mitochondria in the innate immune system, there has been rapid growth in understanding how mitochondrial constituents, which are normally obscured from host pattern-recognition receptors (PRRs), trigger innate immune responses when exposed during cellular stress, infection or injury4. These so-called mitochondrial alarmins or damage-associated molecular patterns (DAMPs) stimulate the innate immune system by multiple routes and are implicated in a growing list of inflammatory diseases. Although a summation of these advances is not the aim of this article, we direct readers to several comprehensive reviews that broadly characterize the roles of mitochondria and mitochondrial DAMPs in mammalian immune responses and human diseases5,6. Because much of the growth in this field has centred on mtDNA, this Review characterizes the expanding roles for mtDNA as an endogenous trigger of both pro-inflammatory and type I interferon (IFN) responses. We discuss the detection of mtDNA by host PRRs, focusing largely on mechanisms that depend on Toll-like receptors (TLRs), NOD-like receptors (NLRs) and IFN stimulatory DNA receptors, and we detail recent efforts to reveal how mtDNA is exposed in the cytoplasm or extracellular space during stress and discuss the relevance of these processes to human disease.
Unique features of mtDNA
There are several unique features of mtDNA that are relevant to our discussion of its role in innate immune responses and inflammation. First, mtDNA is a small, double-stranded circular molecule that encodes 13 oxidative phosphorylation mRNAs as well as tRNAs and ribosomal RNAs that are needed for their translation in the mitochondrial matrix (BOX 1; FIG. 1). The remaining ~1,200 mitochondrial proteins are nuclear gene products that are imported into the organelle, including those needed for expression and maintenance of mtDNA7,8. Second, hundreds to thousands of mtDNA copies are present in each cell, and mtDNA copy number is regulated basally by cell-specific mechanisms and in response to various intrinsic and environmental stresses8. In many cells and tissues, mtDNA occurs in quantities that seem to be in excess of what is needed to sustain oxidative phosphorylation, which suggests that there are other evolutionary pressures for maintaining a high cellular mtDNA copy number, perhaps related to mitochondrial signalling and/or immune functions. Third, in addition to harbouring remnants of bacterial nucleic acid sequences, mtDNA is methylated in a different way from nuclear DNA, making it appear more like ‘foreign’ than ‘self’DNA (FIG. 1). There remains some uncertainty about the precise degree of CpG methylation in mammalian mtDNA: some reports have recorded none at all, whereas others have found a small, possibly regulated amount9–17. Recent studies indicate that isoforms of known nuclear DNA methyltransferases (DNMT1 and DNMT3b) are localized to mitochondria, which supports the notion that there is indeed some CpG mtDNA methylation in mammals12,13. There is also some evidence for non-CpG cytosine methylation and cytosine hydroxymethylation, the latter perhaps supported by the presence of Tet family demethylases in mitochondria12,13,18,19. It remains an open question whether other forms of DNA methylation — such as N6-adenine methylation, a common modification in bacteria and archaea that was recently discovered in mammalian nuclear DNA — occur in mtDNA20. However, although the molecular details of mtDNA methylation are clearly important to resolve, recognition of mtDNA by PRRs would probably occur regardless of whether methylation is zero, low or present in unique patterns. Fourth, owing to its oxidative environment and unique repertoire of DNA repair mechanisms, mtDNA might exhibit persistent, stereotypical oxidative damage modifications or mutagenic signatures that are immunostimulatory. Thus, mtDNA represents a source of endogenous ligands for DNA-sensing PRRs, and mtDNA is increasingly regarded as a mitochondrial DAMP and trigger of ‘hidden-self’ recognition. Last, during the process of mtDNA transcription and replication, many unique nucleic acid species with immunostimulatory potential are generated, such as long, double-stranded RNA, uncapped mRNAs and RNA-DNA hybrids (BOX 1; FIG. 1). This supports the idea that mitochondrial membrane integrity is an important barrier against self-derived innate immune activation in healthy cells and tissues.
Although it is often stated that mtDNA is prone to damage owing to its lack of packaging by histones and inefficient DNA repair mechanisms, such statements are somewhat misleading. In fact, mtDNA is not ‘naked’, but rather packaged into protein–DNA complexes called nucleoids8. The mtDNA-binding protein transcription factor A, mitochondrial (TFAM) — which was originally identified as a transcriptional activator for mtDNA promoters in humans and mice — is a major component that initiates and drives mtDNA packaging and overall nucleoid structure8,21. The TFAM concentration in cells and tissues, as well as its mtDNA-binding density, is likely to be regulated to enable different modes of packaging and precise regulation of mtDNA transcription22. Packaging by TFAM probably insulates mtDNA from oxidative damage to a certain degree — a mode of protection that is augmented by robust mitochondrial base-excision repair pathways to cope with oxidative and other non-bulky base damage. Thus, mammalian mtDNA is not devoid of protective or repair mechanisms. However, mtDNA repair pathways are not as extensive as those available for nuclear DNA, as mitochondria lack nucleotide excision repair and some other pathways that are active in the nucleus23–25. As such, the steady-state amount of cellular mtDNA damage is a balance between the number of insults endured in the oxidative environment of the mitochondrial matrix and/or imparted by environmental stress and the efficiency of mtDNA repair. These properties of mtDNA are relevant to this Review because the degree of packaging and oxidation of mtDNA have both been implicated in mtDNA-dependent innate immune signalling. Last, in a similar way to other high-mobility-group box proteins, TFAM possesses immunomodulatory potential, reinforcing the notion that mtDNA and its associated molecules serve as agonists of the innate immune system26,27.
mtDNA in pro-inflammatory responses
Collins et al.28 were the first to report the immunostimulatory potential of mtDNA in 2004; they found that mtDNA elicited secretion of tumour necrosis factor (TNF) when added to mouse splenocytes and induced arthritis when injected into the joints of mice. Since then, a number of other studies have substantiated these early observations and shown that mtDNA can directly engage PRRs of the innate immune system to enhance pro-inflammatory responses4 (TABLE 1). In this section, we discuss the detection of mtDNA by TLR9 and cytosolic inflammasomes, highlighting mechanistic aspects, open questions and disease relevance.
Table 1 |.
Agonist activities of mitochondrial DNA in the innate immune system
| Pattern-recognition receptor | Stimulatory features | Cell of origin and/or responding cell type | Signalling pathway | Innate immune outcome |
|---|---|---|---|---|
| cGAS | • mtDNA • Oxidized mtDNA90 • mtDNA–mtRNA hybrid102 |
• MEF65,66,78 • Splenocytes65 • PBMC90 • cDC69 |
• STING–IRF3 and/or IRF7 • STING–NF-κB |
• Type I IFN responses • ISGs • Pro-inflammatory cytokines90 |
| TLR9 | • Hypomethylated CpG mtDNA • Oxidized mtDNA91,94 |
• Macrophages103 • Neutrophils30,104 • cDC105 • pDC91,106,107 • Hepatocytes103 • Cardiomyocytes32 • Kidney epithelial cells35 |
• MYD88–NF-κB108 • MYD88–MAPK • MYD88–IRF7 |
• Pro-inflammatory cytokines • Neutrophil chemoattraction and matrix metalloproteinase secretion30,31 • Type I IFN responses91 |
| NLRP3 inflammasome | • mtDNA • Oxidized mtDNA |
Macrophages38,39,42,43 | • ASC and caspase 1 • ASC, caspase 2 and BID41 |
IL-1β and IL-18 |
| NLRC4 inflammasome | • mtDNA • Oxidized mtDNA |
Macrophages | Caspase 1 | IL-1β44 |
| AIM2 inflammasome | mtDNA | Macrophages38 | ASC and caspase 1 | IL-1β and IL-18 |
AIM2, absent in melanoma 2; BID, BH3 interacting-domain death agonist; cDC, conventional dendritic cell; cGAS, cyclic GMP–AMP synthase; IFN, interferon; IL, interleukin; IRF, interferon regulatory factor; ISG, interferon-stimulated genes; MAPK, mitogen-activated protein kinase; MEF, murine embryonic fibroblast; mtDNA, mitochondrial DNA; mtRNA, mitochondrial RNA; MYD88, myeloid differentiation primary response protein 88; NF-κB, nuclear factor-κB; NLRC4, NLR family CARD domain-containing protein 4; NLRP3, NOD, LRR and Pyrin domain-containing protein 3; PBMC, peripheral blood mononuclear cell; pDC, plasmacytoid dendritic cell; STING, stimulator of interferon genes; TLR9, Toll-like receptor 9.
mtDNA as a pro-inflammatory TLR9 agonist.
TLR9 was the first TLR that was shown to sense nucleic acids, and it recognizes hypomethylated CpG motifs in DNA in the endolysosomal compartment29. TLR9 signalling proceeds through the adaptor myeloid differentiation primary response protein 88 (MYD88), which activates mitogen-activated protein kinases (MAPKs) and nuclear factor-κB (NF-κB) to trigger inflammatory responses, or through interferon regulatory factor 7 (IRF7) to enhance type I IFN responses in dendritic cells (DCs) or other immune cells. In 2010, two studies from the Hauser laboratory reported that CpG motifs from mtDNA could trigger TLR9 signalling to activate p38 and p42–44 MAPK activity, CXC-chemokine ligand 8 secretion and neutrophil chemotaxis30,31. Furthermore, they reported the presence of circulating mtDNA in the plasma of trauma patients and other individuals with non-infectious injury, thus implicating mtDNA as a DAMP. A large body of literature supports mtDNA as an endogenous TLR9 agonist (FIG. 2; TABLE 1), and circulating mtDNA has been implicated in the TLR9-dependent inflammatory pathology of diverse diseases such as rheumatoid arthritis, atherosclerosis, hypertension, acute liver injury and non-alcoholic steatohepatitis (TABLE 2). Circulating mtDNA seems to correlate with increased inflammatory phenotypes, which is also true for nuclear DNA, and there are potential limitations to the assays used to quantify mtDNA31 (BOX 2). It is therefore difficult to determine the relative contribution of circulating nuclear DNA versus mtDNA to inflammatory pathology, and thus further investigation is warranted in many cases.
Figure 2 |. Mitochondrial DNA in inflammasome activation and pro-inflammatory responses.

Event 1: tissue pathology and cellular damage trigger necrosis and/or mitochondrial stress, resulting in the release of mitochondrial DNA (mtDNA) or mtDNA-containing microparticles into the extracellular milieu. Event 2: mtDNA in the plasma engages intracellular Toll-like receptor 9 (TLR9)–myeloid differentiation primary response protein 88 (MYD88)–nuclear factor-κB (NF-κB) signalling on circulating leukocytes, resulting in increased production of pro-inflammatory mediators, such as tumour necrosis factor (TNF), interleukin-6 (IL-6) and adhesion molecules. This enhances leukocyte differentiation and extravasation into tissues and causes inflammasome priming (signal 1) in tissue-resident cells. The NOD, LRR and Pyrin domain-containing protein 3 (NLRP3) inflammasome (signal 2) in tissue-resident cells may also be activated by mtDNA, resulting in caspase 1-mediated cleavage of IL-1β and IL-18 to further amplify inflammatory responses. Event 3: mtDNA that enters the endocytic pathway by endocytosis or through mitochondria-derived vesicles (MDVs) can engage TLR9 on tissue-resident macrophages, resulting in increased NF-κB signalling for pro-inflammatory gene expression (signal 1). Event 4: exposure to cellular stress, inflammasome agonists or intracellular bacteria can trigger mitochondrial damage and enhance production of mitochondrial reactive oxygen species (mROS), resulting in the release of oxidized mtDNA (OX-mtDNA) into the cytosol to trigger NLRP3-, NLR family CARD domain-containing protein 4 (NLRC4)- or absent in melanoma 2 (AIM2)-dependent activation of caspase 1 (signal 2), which increases the processing and secretion of mature IL-1β and IL-18, further enhancing tissue inflammation and pathology (event 5). Event 6: increased expression of sequestosome 1 (SQSTM1; also known as p62) through NF-κB signalling increases mitophagy to clear damaged mitochondria and dampen inflammasome activation.
Table 2 |.
Mitochondrial DNA-dependent inflammatory pathology in human disease and animal models
| Condition and/or disease | mtDNA stress signalling pathway | Pathology and/or inflammatory phenotypes reported | |
|---|---|---|---|
| Autoimmunity | SLE | • Neutrophil extrusion of oxidized mtDNA–TFAM complexes • RAGE–TLR9 signalling |
• Increased oxidized mtDNA is present in SLE neutrophils • pDC activation and type I IFN secretion91 |
| • Oxidized mtDNA released from neutrophils • cGAS–STING signalling (PBMC and/or macrophages)90 • TLR9 signalling (pDCs)89 |
• SLE ribonucleoprotein protein complexes induce mROS and NET formation • SLE NETs contain more mtDNA, which drives anti-mtDNA autoantibody formation to enhance lupus nephritis89 • NET mtDNA triggers increased pro-inflammatory cytokine, type I IFN and ISG expression/secretion • mROS scavengers decrease NET formation and lupus-like disease90 |
||
| Rheumatoid and inflammatory arthritis | Oxidized mtDNA in synovial fluid and plasma | • mtDNA levels in synovial fluid correlate with disease severity109 • Monocyte and/or macrophage infiltration into joints • NF-κB-dependent pro-inflammatory cytokine production28 |
|
| Bacterial infection | Caecal ligation and puncture-induced sepsis | • Increased mtDNA in circulation • TLR9 signalling |
• Systemic pro-inflammatory cytokine secretion • Splenic apoptosis • Acute kidney injury110 |
| Streptococcus pneumoniae | • Increased mtDNA in circulation • TLR9 signalling |
• Pro-inflammatory cytokine expression • Myocardial immune cell infiltration and injury111 |
|
| Pseudomonas aeruginosa | • Oxidized mtDNA in the lung • TLR9 signalling |
• Increases in the lung vascular filtration coefficient • Acute lung injury112 |
|
| Cardiovascular diseases | Hypertension | • Increased mtDNA in plasma • TLR9 signalling |
• Impaired DNase activity • Increased arterial pressure and vascular dysfunction, which is inhibited by intraperitoneal injection of TLR9 antagonist113 |
| Atherosclerosis | • Intracellular and extracellular mtDNA • TLR9 signalling |
• Incomplete degradation of mtDNA by autophagy causes aortic inflammation and atherosclerosis in low-density lipoprotein receptor knockout mice114 • mtDNA–LL37 complexes accumulate in atherosclerotic plaques and trigger inflammatory cytokines and immune cell recruitment115 |
|
| • Oxidized mtDNA • NLRP3 activation |
Increased mtDNA oxidation due to 8-oxoguanine glycosylase deficiency increases inflammasome activation, plaque formation and lipid content in atherosclerosis-prone mice43 | ||
| Myocarditis and dilated cardiomyopathy | • Intracellular mtDNA • TLR9 signalling |
• Haemodynamic stress triggers intracellular mtDNA accumulation in DNase II-deficient murine cardiomyocytes • Increased pro-inflammatory cytokine secretion and immune cell infiltration in the heart, which is inhibited by intravenous injection of TLR9 antagonist32 |
|
| Diseases of ageing | Age-related macular degeneration | • Intracellular mtDNA • STING-dependent NF-κB activation • NLRP3 activation |
• mtDNA damage in retinal pigment epithelium increases with age116,117 • Caspase 1 activation45 • Pro-inflammatory cytokine production |
| Age-related systemic inflammation | Increased mtDNA in circulation | • Increased levels of pro-inflammatory cytokines and chemokines in plasma118 • Increased frequency and phenotypic alterations of blood neutrophils119 |
|
| Liver diseases | Hepatocellular carcinoma | • Intracellular mtDNA release • Endosomal HMGB1–TLR9 signalling |
MAPK and NF-κB-dependent pro-tumorigenic and pro-inflammatory signalling33 |
| Acute liver injury | • Increased mtDNA in circulation • TLR9 signalling |
• Neutrophil influx • Local and systemic pro-inflammatory chemokine and/or cytokine secretion • Acute lung injury120 |
|
| NASH | • Increased mtDNA in plasma • Circulating microparticles containing oxidized mtDNA • TLR9 signalling |
• Liver steatosis, ballooning and inflammation • Pro-inflammatory cytokine expression • Weekly administration of TLR9 antagonist protects against high-fat diet-induced NASH103 |
|
| Trauma and sterile injury | SIRS | • Increased mtDNA in plasma • TLR9 activation |
• Pro-inflammatory cytokine secretion • Neutrophil activation and infiltration, hepatic inflammation, lung injury30,31 • NET formation104 |
cGAS, cyclic GMP–AMP synthase; HMGB1; high-mobility-group protein B1; IFN; interferon; ISG, interferon-stimulated gene; MAPK, mitogen-activated protein kinase; mROS, mitochondrial reactive oxygen species; mtDNA, mitochondrial DNA; NASH, non-alcoholic steatohepatitis; NET, neutrophil extracellular trap; NFκB, nuclear factor-κB; NLRP3, NOD, LRR and Pyrin domain-containing protein 3; PBMC, peripheral blood mononuclear cell; pDC, plasmacytoid dendritic cell; RAGE, receptor for advanced glycosylation end products; SIRS, systemic inflammatory response syndrome; SLE, systemic lupus erythematosus; STING, stimulator of interferon genes; TFAM, transcription factor A, mitochondrial; TLR9, Toll-like receptor 9.
Box 2 |. Experimental and interpretive issues when analysing mitochondrial DNA.
There has been an explosion of interest in mitochondrial DNA (mtDNA) as an immunostimulatory and pro-inflammatory agent, and here we summarize some important experimental considerations when studying mtDNA. First, PCR is commonly used to detect and quantify cytosolic or circulating, cell-free mtDNA. It should be noted that mtDNA has been transferred to the nucleus many times during evolution, and hence there are hundreds of mtDNA sequences resident in mammalian genomes, some of which are close to full length98. Without proper controls for nuclear contamination, these so-called nuclear mitochondrial sequences (NUMTs) will be amplified by PCR using mtDNA-specific primers and can lead to false-positive results seemingly demonstrating that mtDNA has been released into the cytoplasm or into circulation. NUMTs are often mutated, and therefore, in the absence of controls to eliminate nuclear contamination, can also lead to similar false conclusions that mtDNA mutations are involved in a given process. Second, it is well-accepted that the release of mtDNA into the cytoplasm can activate nucleic acid sensors such as Toll-like receptor 9 (TLR9) and cyclic GMP–AMP synthase (cGAS). However, it should be noted that release per se has never been demonstrated in a real-time assay. Release can only be surmised if highly purified cytoplasmic fractions are generated that are devoid of nuclear (that is, NUMT) contamination. The use of multiplexed mtDNA probes to ensure that the amplification of all probes scales equally can help to circumvent these issues, as can the use of specific primers or methods designed to avoid NUMT contamination as described99–101. Even this approach has the caveat that mitochondria can be preferentially broken during preparation, and various experimental manipulations could lead to fragile mitochondria that rupture more readily during fractionation when compared with controls. Either of these scenarios can lead to the false conclusion that mtDNA is released into the cytoplasm from the organelle itself in vivo.
To circumvent these issues, many investigators have turned to using compounds such as dideoxycytosine or ethidium bromide, which inhibit mtDNA replication and deplete mtDNA, or have used cells that are completely devoid of mtDNA (rho0 cells) to more conclusively implicate mtDNA in the responses of interest. Although these are valid approaches, they too are not without caveats. For example, ethidium bromide is an intercalating agent that also binds to RNA and DNA in the cytoplasm, which, in principle, could directly influence the stability or detection of any nucleic acid, mitochondrial or nuclear, by host pattern-recognition receptors. In addition, most rho0 lines are derived from immortalized cancer cells and are products of strong selection for cells that can survive this harsh metabolic challenge, possibly resulting in populations with adaptive nuclear mutations and/or dramatically altered innate immune responses. Furthermore, rho0 cells, and those treated with mtDNA-depleting compounds for long periods, are devoid of not only mtDNA but also all mtDNA-derived RNA and protein species. These cells are deficient in oxidative phosphorylation and hence have completely altered cellular metabolism, ATP production and mitochondrial reactive oxygen species profiles. Thus, using rho0 conditions alone to conclude that a given phenotype is due specifically to the absence of mtDNA is tenuous. Despite these caveats, mtDNA can be confidently implicated in innate immune responses by pursuing the proper controls and gathering multiple lines of evidence. However, better techniques to detect mtDNA in situ and visualize mtDNA release in real time are needed.
Several reports have shown that inflammation and disease can be promoted not only by stimulation of TLR9 through an extracellular release mechanism but also by cell-autonomous ligation of TLR9 by mtDNA. Oka et al.32 demonstrated that in DNase II-deficient hearts, mtDNA is inefficiently degraded by autophagy and engages TLR9-mediated inflammatory responses in cardiomyocytes to induce myocarditis and dilated cardiomyopathy. Moreover, under hypoxic conditions, high-mobility-group protein B1 (HMGB1) and mtDNA can form a complex in hepatocellular carcinoma cells and bind to TLR9 to enhance pro-tumorigenic signalling and inflammation33. This study did not delineate how mtDNA, which presumably enters the cytosol first, is trafficked into the endolysosomal compartment to engage TLR9. Autophagy is the most obvious route; however, other pathways may have a role. For example, mitochondria-derived vesicles (MDVs) — which have been recently shown to mediate the trafficking of mitochondrial proteins to endosomes to facilitate antigen presentation — could in principle introduce mtDNA into the endocytic machinery, where it could engage TLR9 (REFS 34,35). MDVs might also provide a direct route for the release of mtDNA into the cytoplasm (FIG. 2).
mtDNA as an inflammasome agonist.
Inflammasomes are multi-subunit, cytoplasmic protein complexes that consist of receptor and sensor molecules, the adaptor protein ASC and the inflammatory cysteine protease caspase 1. Four receptors have been shown to form inflammasomes — including NOD, LRR and Pyrin domain-containing protein 1 (NLRP1), NLRP3, NLR family CARD domain-containing protein 4 (NLRC4) and absent in melanoma 2 (AIM2) — all of which are activated by exogenous pathogen-associated molecular patterns (PAMPs) and/or endogenous DAMPs released during necrosis or cellular stress36. Receptor clustering promotes inflammasome complex assembly and caspase 1 activation, leading to processing of the cytokines pro-interleukin-1β (pro-IL-1β) and pro-IL-18 into mature, secreted forms. Production of mitochondrial ROS (mROS), release of mitochondrial DAMPs and altered mitochondrial dynamics have been linked to inflammasome activation, although the exact mechanisms by which mitochondria engage NLRP3 and other inflammasomes remain under investigation37.
Substantial evidence supports mtDNA as an endogenous agonist of inflammasomes (FIG. 2; TABLE 1). A 2010 report by Nakahira et al. was one of the first to link mtDNA to NLRP3 inflammasome activation, showing that depletion of the autophagy proteins beclin 1 and LC3B in mouse bone marrow-derived macrophages (BMDMs) results in enhanced caspase 1 activation and IL-1β and IL-18 secretion38. This response requires mROS as an upstream activator of NLRP3, which they proposed leads to increased mtDNA release into the cytoplasm to enhance IL-1β and/or IL-18 secretion. Interestingly, they also observed mtDNA release in wild-type BMDMs in response to ATP and lipopolysaccharide (LPS), but not to urate crystals. Moreover, cytosolic mtDNA was detected in beclin 1- and LC3B-deficient cells, even in the absence of ATP and LPS stimulation, which suggests that mtDNA release selectively induces a subset of inflammasome agonists and is basally inhibited by autophagy. Last, Nakahira et al.38 demonstrated a role for AIM2 in caspase 1 activation downstream of mtDNA, showing that AIM2-deficient BMDMs secreted less IL-1β in response to mtDNA transfection than control BMDMs. Shortly thereafter, Shimada et al.39 demonstrated that NLRP3 binds mtDNA and suggested that the requirement for mROS in this response is likely to be due to the preference of NLRP3 for oxidized mtDNA species. Their work suggests that NLRP3 might not promote mtDNA release per se, but instead stabilize it in the cytoplasm after release, although other reports have strengthened the notion that mitochondrial damage and mtDNA release are amplified by NLRP3 inflammasome activation40,41. Several other research groups have implicated mROS and oxidized mtDNA in NLRP3 inflammasome activation42,43, and Jabir et al.44 have recently provided evidence of mtDNA binding to NLRC4 complexes. Furthermore, several studies have shown that mtDNA-release-associated inflammasome activation is probably involved in several pathogenic states, including atherosclerosis, age-related macular degeneration, mevalonate kinase deficiency and certain bacterial infections (TABLE 2), which supports mtDNA as an inflammasome agonist with implications for disease43–46
Despite clear evidence of mtDNA involvement in inflammasome activation, many mechanistic questions remain. First, it is unclear how mtDNA enters the cytosol, and the specific sequences and structural features of these immunostimulatory molecules are unknown. Some data indicate that mtDNA sequences several hundred base pairs in length are released to activate inflammasomes39. This suggests that membrane damage and mitochondrial rupture is probably responsible for the activation, because although the gated release of mtDNA of this length is possible, it seems unlikely (FIG. 2). Second, the precise role of mROS in inflammasome activation is controversial47. Oxidation of mtDNA by mROS seems to be required, but ROS also have context-specific signalling and damaging roles, so activation of other ROS-mediated inflammatory pathways might explain some of the conflicting results3. Third, although complexes of NLRP3 and NLRC4 seem to bind to mtDNA or oxidized mtDNA in co-immunoprecipitation experiments, it is not clear whether the NLRs themselves bind directly or whether other factors are needed. Furthermore, the role of autophagy and/or mitophagy in limiting inflammasome activation by removing damaged mtDNA needs to be investigated further, as do the consequences of mitochondrial damage and mtDNA release upstream and downstream of inflammasome-dependent caspase 1 activation40,42. Last, more effort is needed to unravel the dynamic physical association of NLRs with mitochondria and how this imparts signalling specificity47,48.
mtDNA haplogroups and inflammatory disease.
Different human populations have acquired sequence polymorphisms in mtDNA that characterize maternally inherited genetic haplotypes. Clusters of these single-nucleotide polymorphisms (SNPs) define related mtDNA haplotypes, called haplogroups. Haplogroups have enabled tracking of global migrations of human populations, and there is substantial evidence that they are drivers of evolutionary adaptation and disease susceptibility49. In addition to these inherited SNPs, somatic mtDNA mutations accumulate with age. The involvement of mtDNA in innate immune responses begs the question of whether mtDNA haplotypes or somatic mtDNA mutations contribute to inflammatory pathology and/or the increase in chronic inflammation with age. Although more investigation into this area is certainly warranted, the concept has been supported by identifying that a mutation in the mtDNA-encoded cytochrome B gene correlates with NLRP3 inflammasome activation in people with fibromyalgia and that mtDNA haplotypes are associated with sepsis susceptibility and other inflammatory pathways and disorders in humans49–52. Therefore, we envision that inherited mtDNA SNPs or acquired somatic mtDNA mutations could influence inflammatory pathways directly by differentially affecting how mtDNA engages cytoplasmic PRRs and/or by imparting functional changes in mitochondria that could influence downstream inflammatory signalling events. These could include alterations in oxidative phosphorylation and/or mROS generation, which might result in heightened immune cell function on PRR ligation or pathogen infection.
mtDNA in type I interferon responses
In addition to triggering pro-inflammatory responses, recent reports have demonstrated that both cytosolic and extracellular mtDNA engage PRRs and trigger type I IFNs and interferon-stimulated gene (ISG) expression. Although some of the mechanistic details warrant clarification, the identification of mtDNA as a type I IFN-inducing DAMP raises important implications for understanding the pathobiology of infectious and non-infectious diseases involving both mitochondrial stress and type I IFN signatures53. In this section, we discuss the emerging concept that mtDNA serves as an agonist of membrane-bound and cytosolic PRRs regulating type I IFN responses such as those mediated by TLR9, cyclic GMP–AMP synthase (cGAS) and stimulator of interferon genes (STING) (TABLE 1).
Intracellular mtDNA engages the cGAS–STING axis.
The cGAS–STING signalling axis has emerged as a crucial regulator of type I IFN responses to both exogenous and endogenous DNA53–55 (FIG. 3a). The enzyme cGAS detects cytoplasmic DNA and generates the cyclic dinucleotide cGAMP, which serves as a second messenger that binds to and activates STING56. Activated STING engages TANK-binding kinase 1 (TBK1), which phosphorylates interferon regulatory factor 3 (IRF3) to promote its homodimerization and translocation to the nucleus, where it induces expression of IFNβ and ISGs. Several research groups have demonstrated that cGAS functions as the predominant sensor of viral and bacterial DNA in the cytoplasm of infected cells56–60. In addition, cGAS–STING signalling plays a vital part in orchestrating type I IFN and inflammatory responses to self DNA, driving pathology in type I interferonopathies, such as Aicardi–Goutières syndrome, as well as regulating inflammatory responses in the tumour microenvironment53,55,61–64. Although most endogenous ligands for cGAS are presumed to originate from nuclear DNA, several lines of evidence now suggest that mtDNA also serves as a cell-intrinsic cGAS ligand in certain contexts (FIG. 3a).
Figure 3 |. Mitochondrial DNA instability and release in type I interferon responses.
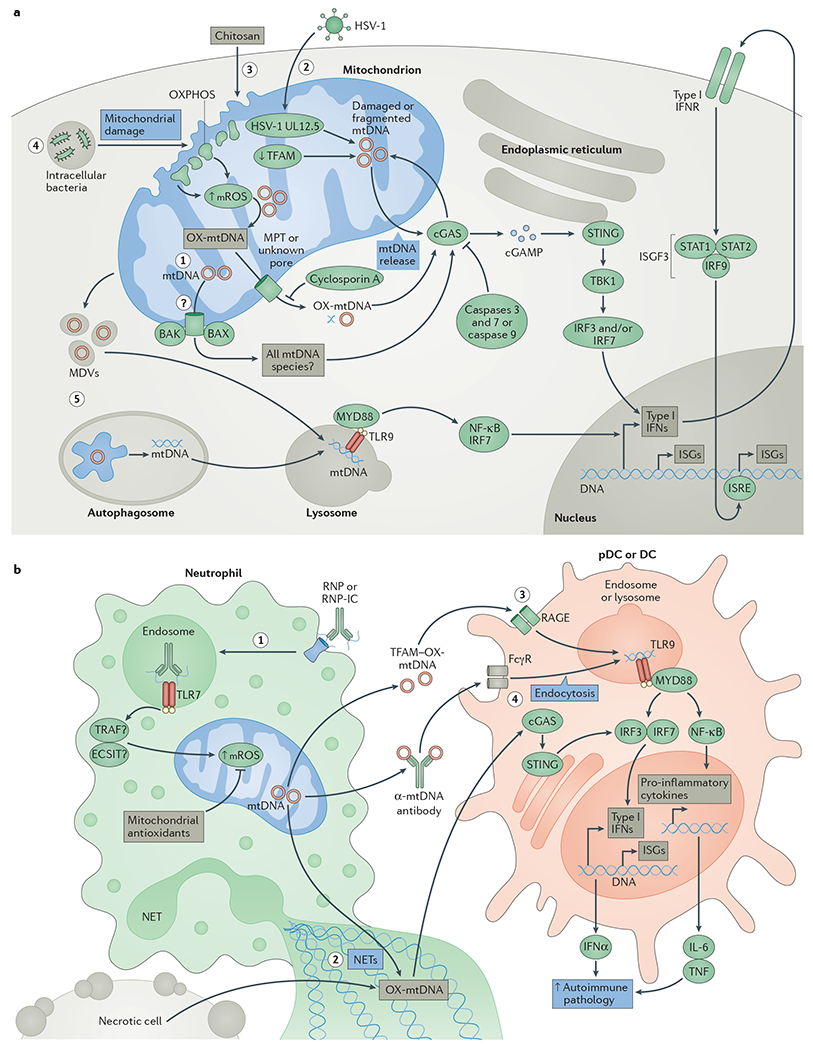
a | Event 1: initiation of apoptosis results in the BCL-2-like protein 4 (BAX)- and BCL-2 homologous antagonist/killer (BAK)-dependent release of mitochondrial DNA (mtDNA), which triggers cyclic GMP–AMP (cGAMP) synthase (cGAS) activation in the absence of apoptotic caspase 9 or both caspase 3 and caspase 7. Event 2: herpes simplex virus 1 (HSV-1) infection and mitochondrial expression of the HSV-1 protein UL12.5, or decreased expression of transcription factor A, mitochondrial (TFAM), results in mtDNA instability and release of fragmented mtDNA into the cytosol to activate cGAS. Events 3 and 4: exposure to the adjuvant chitosan, or infection with intracellular bacteria such as Mycobacterium tuberculosis, results in mitochondrial damage, increased levels of mitochondrial reactive oxygen species (mROS) and release of oxidized mtDNA (OX-mtDNA) into the cytosol to engage cGAS. Cyclosporin A, an inhibitor of the mitochondrial permeability transition (MPT) pore, can decrease cGAS activation by chitosan. On cGAS activation, cGAMP triggers conformational changes of the endoplasmic reticulum-resident protein stimulator of interferon genes (STING), which engages TANK-binding kinase 1 (TBK1) to activate interferon regulatory factor 3 (IRF3) and/or IRF7 to stimulate transcription of type I interferons (IFNs) and interferon-stimulated genes (ISGs). Type I IFNs can then activate the type I IFN receptor (IFNR) in an autocrine and/or paracrine manner to engage the interferon-stimulated gene factor 3 (ISGF3) complex, which consists of signal transducer and activator of transcription 1 (STAT1), STAT2 and IRF9. The ISGF3 complex further enhances ISG expression by binding to interferon-stimulated response elements (ISREs) in the promoters of these genes. Event 5: damaged mitochondria targeted to mitophagy or mtDNA in mitochondria-derived vesicles (MDVs) may also engage Toll-like receptor 9 (TLR9) in lysosomes if the mtDNA they contain is not completely degraded, resulting in engagement of the type I IFN response. b | Event 1: ribonucleoprotein-containing immune complexes (RNP-ICs) are internalized by neutrophils, where they stimulate a TLR7-dependent increase in mROS production, which enhances mtDNA oxidation and mitochondrial re-localization to the plasma membrane. Event 2: neutrophil extracellular traps (NETs) containing OX-mtDNA can be taken up by neighbouring conventional dendritic cells (DCs) or plasmacytoid dendritic cells (pDCs), resulting in engagement of the cGAS–STING axis to increase expression of type I IFNs, ISGs and pro-inflammatory cytokines. Event 3: in addition, TFAM–OX-mtDNA complexes can be endocytosed by DCs in a receptor for advanced glycosylation end products (RAGE)-dependent fashion to engage endosomal TLR9 and enhance type I IFN and inflammatory responses. Event 4: anti-mtDNA immune complexes can also engage Fcγ receptors (FcγRs) to stimulate endosomal TLR9 signalling. All outcomes enhance local and/or systemic type I IFN and inflammatory responses to promote pathology in systemic lupus erythematosus (SLE) or other autoimmune or autoinflammatory diseases. ECSIT, evolutionarily conserved signalling intermediate in Toll pathway, mitochondrial; IL-6, interleukin-6; MYD88, myeloid differentiation primary response protein 88; NF-κB, nuclear factor-κB; OXPHOS, oxidative phosphorylation; TNF, tumour necrosis factor; TRAF, TNF receptor-associated factor.
White et al.65 and Rongvaux et al.66 showed that mtDNA released during BCL-2-like protein 4 (BAX)- and BCL-2 homologous antagonist/killer (BAK)-mediated apoptosis can engage cGAS–STING–IRF3 signalling and trigger type I IFN responses and expression of ISGs. In the absence of caspase 9 or of both caspase 3 and caspase 7, mtDNA activates cGAS to enhance type I IFN responses and ISG expression, suggesting that the apoptotic caspase cascade functions to suppress cell-intrinsic, mtDNA-dependent type I IFN responses during programmed cell death. Using immunoprecipitation assays, White et al.65 reported that cGAS binds indiscriminately to mtDNA after permeabilization of the mitochondrial outer membrane by BAX and BAK. This indicates that the entire mitochondrial genome is exposed during apoptosis, which raises interesting questions regarding the mechanisms of release and the possible accessibility of cGAS to inner mitochondrial compartments during mitochondrial permeabilization.
Activation of the mitochondrial permeability transition (MPT) pore is associated with release of linear mtDNA fragments up to 700 bp in length; thus, under certain circumstances, it is possible that MPT is responsible for the liberation of mtDNA fragments during apoptosis and/or mitochondrial stress67,68. In agreement with an MPT-dependent mechanism of mtDNA release, the vaccine adjuvant chitosan stimulates mitochondrial stress, production of mROS and accumulation of cytosolic DNA to activate cGAS–STING signalling and type I IFN responses in DCs69. Although this report does not definitively demonstrate that mtDNA is the activating cGAS ligand, type I IFN and ISG expression induced by chitosan exposure was inhibited by the addition of cyclosporin A, a compound that was previously shown to block MPT and the release of fragmented mtDNA from isolated mitochondria in vitro68 (FIG. 3a). However, these findings must be interpreted carefully as cyclosporin A can directly inhibit PRR and NF-κB signalling and thus its modulation of type I IFN responses in this system could be independent of alterations to mitochondrial permeability70,71. The hypothesized role of the MPT pore as a direct conduit for mtDNA release has been challenged recently and certainly requires more investigation72. The mitochondrial ATP synthase seems to be a major component of the MPT pore, and might even be the actual pore73–75. Interestingly, TrwB, a member of the conserved FtsK/SpoIIIE family of bacterial DNA transporters, is a DNA-dependent F1-ATPase that is involved in transfer of DNA between bacteria during conjugation76,77. TrwB is structurally related to mitochondrial F1-ATPase, and in light of findings that implicate mitochondrial ATP synthase in MPT and MPT in mtDNA release, it is intriguing to consider that regulated mtDNA release might be an evolutionary vestige of the bacterial origin of this organelle. Future studies should clarify the role of MPT in mtDNA release and, perhaps more importantly, define the mtDNA species that uniquely engage cGAS over other DNA sensors.
Our recent work also provides support for mtDNA-dependent activation of the cGAS–STING pathway (FIG. 3a). We found that cells and tissues from TFAM heterozygous (Tfam+/−) mice — which have altered nucleoid morphology, presumably due to altered mtDNA packaging — exhibit increased expression of ISGs and antiviral factors and are markedly resistant to viral infection78. The antiviral ‘priming’ phenotype in Tfam+/− cells is driven by constitutive cGAS–STING–TBKl signalling to IRF3 and depends on the presence of abnormal nucleoid structures and the stress that they impart. Treatment of Tfam+/− cells with dideoxycytosine — which specifically inhibits mtDNA replication and promotes mtDNA depletion — markedly decreased ISG expression and viral resistance, thus implicating mtDNA-dependent cGAS activation in this genetic background. Moreover, we observed enrichment of specific mtDNA fragments in the cytosol of TFAM-deficient fibroblasts and macrophages. That is, we found that mtDNA sequences corresponding to the D-loop regulatory region (BOX 1) were more prevalent in the cytoplasm of Tfam+/− cells. It is thus tempting to speculate that specific sequences derived from this three-stranded region of mtDNA (BOX 1) might resist nuclease degradation and/or bind with higher affinity to cGAS, thus rendering them more immunostimulatory. Moreover, mtDNA, and perhaps the D-loop region in particular, is closely associated with the inner mitochondrial membrane and inner–outer membrane contact sites, and thus this region of mtDNA may be more readily released during mitochondrial membrane breakage, fission or fusion79–81. It is also noteworthy that the 7S DNA strand that forms the D-loop is constantly made and degraded, which may be relevant to its role in innate immune or mitochondrial stress signalling82. Although our work does not reveal the precise mechanism by which mtDNA fragments are liberated from mitochondria, Tfam+/− cells have elongated mitochondria and the ISG response is dampened by knockdown of mitofusin 1 (MFN1), suggesting that altered mitochondrial morphology or dynamics also contributes to mtDNA release. Altogether, our findings further support the idea that mitochondrial stress can liberate cytosolic mtDNA that serves as an endogenous cGAS ligand.
The physiological relevance of cGAS activation in Tfam+/− mice is not completely clear, but our results suggest that mtDNA stress may contribute to innate immune activation and type I IFN responses in various pathological states, from infectious diseases to cancer, neurodegeneration and other mitochondria-related illnesses. With regard to pathogen-mediated mtDNA stress, we and others have shown that cellular infection by herpes simplex virus 1 (HSV-1) and HSV-2 causes mtDNA stress and a swift decline in mtDNA copy number78,83,84. A mutant HSV-1 strain lacking the gene responsible for mtDNA targeting is less efficient at triggering antiviral responses and ISG expression, possibly suggesting that cellular monitoring of mtDNA homeostasis is an evolutionarily beneficial mechanism that cooperates with canonical sensing of viral nucleic acids to fully engage antiviral innate immunity78,85. Conversely, host type I IFN responses can enhance the pathogenesis of certain microorganisms, such as Mycobacterium tuberculosis, and tuberculosis infection triggers both cGAS–STING signalling and mitochondrial stress57,59,86–88. These findings raise the interesting possibility that mitochondrial damage and mtDNA release may be a strategy used by M. tuberculosis or other microorganisms to boost cGAS activation, increase type I IFN responses and enhance intracellular survival (FIG. 3a).
Extracellular mtDNA in type I IFN responses.
In addition to an intracellular role for mtDNA in triggering type I IFN responses, mtDNA released from activated neutrophils can engage either the cGAS–STING pathway or the endosomal TLR9 pathway on neighbouring immune cells89–91 (FIG. 3b; TABLE 1). Neutrophil extracellular trap (NET) formation — a process implicated in bacterial clearance and sterile inflammatory diseases such as systemic lupus erythematosus (SLE) — results in cell death and extrusion of neutrophil DNA and/or protein complexes into the extracellular space. NETs have recently been shown to contain mtDNA, thus raising the possibility that extracellular mtDNA could contribute to type I IFN-mediated pathology observed in individuals with SLE89,92. Consistent with this hypothesis, Lood et al.90 have shown that triggering of neutrophils by ribonucleoprotein-containing immune complexes (RNP-ICs) increases mROS generation and mitochondrial translocation to the cellular surface to support the formation of NETs that are enriched in oxidized mtDNA. The degree of oxidation of NET DNA is a crucial determinant of its immunostimulatory potential. The expression of type I IFNs and pro-inflammatory cytokines by human peripheral blood mononuclear cells and splenic cells is potentiated by oxidation, and lupus-like disease is reduced in mice treated with mROS scavengers. Interestingly, the response to mtDNA-enriched NETs requires STING but not TLR9–MYD88, indicating a crucial role for cGAS in this response90. It is unclear how RNP-IC stimulation increases mROS production to oxidize mtDNA, although one possibility may involve TLR7 activation of a signalling cascade dependent on MYD88, TNF receptor-associated factor 6 (TRAF6) and evolutionarily conserved signalling intermediate in Toll pathway, mitochondrial (ECSIT), which has been shown to trigger mROS production from oxidative phosphorylation complex I93.
Oxidized mtDNA has also been shown to engage TLR9 on plasmacytoid DCs (pDCs)94, and Caielli et al.91 further implicated the release of oxidized mtDNA from neutrophils as a driver of TLR9-dependent IFNα secretion by pDCs. They showed that neutrophils constitutively extrude mtDNA–TFAM complexes (and selectively extrude other mitochondrial cargo) into the extracellular space as a result of an attenuated mitophagy pathway, although they argue that MDVs normally direct oxidized mtDNA to lysosomes for degradation91. However, this process is disrupted in neutrophils taken from individuals with SLE in a way that leads to extrusion of extracellular oxidized mtDNA–TFAM complexes91. These complexes then activate the receptor for advanced glycosylation end products (RAGE; also known as ACER)- and TLR9-dependent production of IFNα from pDCs to increase SLE pathology (FIG. 3b). Although these observations raise the interesting possibility of neutrophil-specific mechanisms for mitophagy and mtDNA turnover, the mechanisms governing vesicular trafficking and packaging of oxidized mtDNA remain unclear. Nonetheless, these reports strongly implicate extracellular mtDNA in the inflammatory and type I IFN-mediated pathology of SLE and perhaps other autoimmune diseases (TABLE 2).
Conclusion and future perspectives
Mitochondria are multifaceted organelles that are key hubs of cellular metabolism and signalling, but increasingly they are also being documented as important participants in innate immune responses to pathogens and cellular damage. Mitochondria not only house machinery that is necessary for antiviral and inflammasome signalling but are also important sources of endogenous DAMPs. Specific characteristics of mtDNA, such as its relative hypomethylation, unique structural features and heightened susceptibility to oxidative damage, make it a potent DAMP that activates TLR9, NLRs, cGAS and perhaps other innate sensors to trigger pro-inflammatory processes and type I IFN responses.
The evidence that immunostimulatory mtDNA can enter the cytoplasm, the extracellular space and even the circulation, is robust, and it is perhaps straightforward to envision how damaged or dying cells may release mtDNA into the extracellular milieu. However, regulated and non-necrotic mtDNA release mechanisms have also been suggested, and therefore additional studies are warranted to clarify the physiological and pathological relevance of these pathways. For example, depletion of the mitochondrial fusion machinery can decrease antiviral signalling and mtDNA stress-induced ISG expression, suggesting that altered mitochondrial membrane fusion may promote the release of mtDNA and perhaps other DAMPs4,78. In addition, the intersection of autophagy and mitophagy pathways and mtDNA release should be examined more closely. It is possible that incomplete mitochondrial degradation by these pathways leads to mtDNA fragmentation and cytoplasmic release, and substantial evidence supports the notion that macroautophagy is the predominant mechanism for preventing cytoplasmic mtDNA accumulation38,42. Last, several reports suggest that mtDNA release is controlled by various mitochondrial pores or associated regulatory proteins such as the MPT pore, the voltage-dependent anion channel, hexokinase, BAX and BAK38,65,66,95. However, so far, no definitive genetic or biochemical experiments have demonstrated the direct release of mtDNA by a mitochondrial pore. Moreover, many of the proteins implicated thus far primarily regulate mitochondrial outer membrane permeability, which in isolation does not provide a complete route for mtDNA to transit from the matrix to the cytoplasm72. Hopefully, genetic studies coupled with super-resolution, live-cell imaging modalities will clarify the open mechanistic questions regarding gated mtDNA release.
It is also curious why mtDNA release does not uniformly activate both pro-inflammatory and type I IFN responses. Cytosolic mtDNA accumulation after activation of BAX and BAK or mtDNA stress preferentially activates cGAS-STING signalling and type I IFN responses, with no observed or reported effects on inflammasome activation, IL-1β production or pro-inflammatory cytokine expression65,66,78. This is somewhat surprising, as extra-mitochondrial mtDNA should be accessible to bind to NLRP3 or AIM2, and could also enter the endolysosomal compartment through autophagy to engage TLR9. It is therefore likely that unique aspects of mtDNA, such as its length, conformation, sequence, degree of oxidation or the precise location of its release, govern its differential agonist activities. More research is required to define the unique mechanisms by which mtDNA engages each of these sensors, and these efforts will be key to understanding how mtDNA activates specific inflammatory profiles in various disease contexts.
Aberrant innate immune responses are being implicated in an ever-growing list of pathologies, including autoimmune disease, metabolic syndrome, neurodegeneration and cancer. Mitochondrial dysfunction and/or damage is a shared feature in nearly all of these diseases, as well as in ageing, and thus it is interesting to speculate that the release of mitochondrial constituents may be a common factor in the propagation, and perhaps initiation, of the inflammatory pathology of these conditions. Following the emergence of mtDNA as an important mitochondrial DAMP (TABLES 1,2), future work to unravel the mechanistic aspects of mtDNA release, sensing and resulting inflammatory pathology will have broad implications for understanding the mitochondrial aetiology of human disease and ageing, perhaps leading to new avenues for therapeutic intervention to improve human health.
Acknowledgements
The authors thank B. Kaufman for helpful comments on the manuscript, and we acknowledge L. Ciaccia West for assistance with the figures. G.S.S. is the Joseph A. and Lucille K. Madri Endowed Professor of Experimental Pathology, and this work was supported by US National Institutes of Health grant R01 AG047632.
Glossary
- Pattern-recognition receptors
(PRRs). Evolutionarily conserved receptors of the innate immune system that detect foreign viral, bacterial and/or fungal constituents, as well as endogenous molecules released from injured cells and tissues
- Damage-associated molecular patterns
(DAMPs). Molecules that are exposed or released by injured, necrotic or dying cells and are recognized by pattern-recognition receptors
- Nucleoids
Functional mitochondrial DNA packaging complexes in the mitochondrial matrix that consist of one or more mitochondrial DNA genomes and associated proteins
- Transcription factor A, mitochondrial
(TFAM). A dual high-mobility-group box protein in mitochondria that promotes packaging of mitochondrial DNA and regulates transcription from mitochondrial DNA promoters
- Inflammasomes
Multi-protein complexes that activate caspase 1 to induce processing of pro-interleukin-1β and pro-interleukin-18 into mature and secreted forms
- Haplogroups
Clusters of single-nucleotide polymorphisms in mitochondrial DNA that define inherited lineages
- Cyclic GMP–AMP synthase
(cGAS). A cytosolic DNA sensor that catalyses the production of the second messenger cyclic GMP–AMP (cGAMP) on binding to DNA
- Stimulator of interferon genes
(STING). An endoplasmic reticulum-resident adaptor protein that binds to cyclic GMP–AMP (cGAMP) to trigger type I interferon production
- Aicardi–Goutières syndrome
A disease in which mutations in the cytosolic enzyme 3 repair exonuclease 1 (TREX1)or other nucleases lead to the intracellular accumulation of endogenous nucleic acids, triggering chronic type I interferon responses that cause debilitating autoinflammatory and neurodegenerative pathology
- D-loop
A stable three-stranded DNA structure in mammalian mitochondrial DNA that is caused by premature termination of replication
- Systemic lupus erythematosus
(SLE). A chronic autoimmune disease that is linked to aberrant type I interferon responses in which autoantibodies specific for DNA, RNA or proteins ass.’sciated with nucleic acids form immune complexes that accumulate in multiple tissues to cause pathology
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Friedman JR & Nunnari J Mitochondrial form and function. Nature 505, 335–343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shadel GS & Clayton DA Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem 66, 409–435 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Shadel GS & Horvath TL Mitochondrial ROS signaling in organismal homeostasis. Cell 163, 560–569 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.West AP, Shadel GS & Ghosh S Mitochondria in innate immune responses. Nat. Rev. Immunol 11, 389–402 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinberg SE, Sena LA & Chandel NS Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakahira K, Hisata S & Choi AMK The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxid. Redox Signal. 23, 1329–1350 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calvo SE & Mootha VK The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 11, 25–44 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonawitz ND, Clayton DA & Shadel GS Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol. Cell 24, 813–825 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Hong EE, Okitsu CY, Smith AD & Hsieh C-L Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol. Cell. Biol 33, 2683–2690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawid IB 5-Methylcytidylic acid: absence from mitochondrial DNA of frogs and HeLa cells. Science 184, 80–81 (1974). [DOI] [PubMed] [Google Scholar]
- 11.Maekawa M et al. Methylation of mitochondrial DNA is not a useful marker for cancer detection. Clin. Chem 50, 1480–1481 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Bellizzi D et al. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. 20, 537–547 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shock LS, Thakkar PV, Peterson EJ, Moran RG & Taylor SM DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl Acad. Sci. USA 108, 3630–3635 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shmookler Reis RJ & Goldstein S Mitochondrial DNA in mortal and immortal human cells. Genome number, integrity, and methylation. J. Biol. Chem 258, 9078–9085 (1983). [PubMed] [Google Scholar]
- 15.Pollack Y, Kasir J, Shemer R, Metzger S & Szyf M Methylation pattern of mouse mitochondrial DNA. Nucleic Acids Res. 12, 4811–4824 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nass MM Differential methylation of mitochondrial and nuclear DNA in cultured mouse, hamster and virus-transformed hamster cells. In vivo and in vitro methylation. J. Mol. Biol 80, 155–175 (1973). [DOI] [PubMed] [Google Scholar]
- 17.Rebelo AP, Williams SL & Moraes CT In vivo methylation of mtDNA reveals the dynamics of protein–mtDNA interactions. Nucleic Acids Res. 37, 6701–6715 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Z et al. High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep. 3, 567–576 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh S, Sengupta S & Scaria V Hydroxymethyl cytosine marks in the human mitochondrial genome are dynamic in nature. Mitochondrion 27, 25–31 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Wu TP et al. DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature 532, 329–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parisi MA & Clayton DA Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science 252, 965–969 (1991). [DOI] [PubMed] [Google Scholar]
- 22.Bestwick ML & Shadel GS Accessorizing the human mitochondrial transcription machinery. Trends Biochem. Sci. 38, 283–291 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kazak L, Reyes A & Holt IJ Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 13, 659–671 (2012). [DOI] [PubMed] [Google Scholar]
- 24.O’Rourke TW, Doudican NA, Mackereth MD, Doetsch PW & Shadel GS Mitochondrial dysfunction due to oxidative mitochondrial DNA damage is reduced through cooperative actions of diverse proteins. Mol. Cell. Biol 22, 4086–4093 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheibye-Knudsen M, Fang EF, Croteau DL, Wilson DM & Bohr VA Protecting the mitochondrial powerhouse. Trends Cell Biol. 25, 158–170 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crousei ED et al. Monocyte activation by necrotic cells is promoted by mitochondrial proteins and formyl peptide receptors. Crit. Care Med. 37, 2000–2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Julian MW et al. Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J. Immunol 189, 433–443 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collins LV, Hajizadeh S, Holme E, Jonsson I-M & Tarkowski A Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J. Leukoc. Biol 75, 995–1000 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Barbalat R, Ewald SE, Mouchess ML & Barton GM Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol 29, 185–214 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Zhang Q et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; A seminal report documenting pro-inflammatory roles for mitochondrial DAMPs in sterile injury and trauma.
- 31.Zhang Q, Itagaki K & Hauser CJ Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 34, 55–59 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Oka T et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that the accumulation of mtDNA activates cell-intrinsic, TLR9-dependent inflammation leading to cardiomyopathy.
- 33.Liu Y et al. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J. Hepatol 63, 114–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matheoud D et al. Parkinson’s disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell 166, 314–327 (2016). [DOI] [PubMed] [Google Scholar]
- 35.De Leo MG et al. Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat. Cell Biol. 18, 839–850 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Man SM & Kanneganti T-D Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol 16, 7–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elliott EI & Sutterwala FS Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev 265, 35–52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakahira K et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol 12, 222–230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This important study shows that mROS and mtDNA contribute to macrophage NLRP3 and AIM2 inflammasome activation.
- 39.Shimada K et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes that oxidized mtDNA is detected by the NLRP3 inflammasome during apoptosis.
- 40.Yu J et al. Inflammasome activation leads to caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl Acad. Sci. USA 111, 15514–15519(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bronner DN et al. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity 43, 451–462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong Z et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tumurkhuu G et al. Ogg 1 -dependent DNA repair regulates NLRP3 inflammasome and prevents atherosclerosis. Circ. Res 119, e76–e90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jabii MS et al. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 11, 166–182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dib B et al. Mitochondrial DNA has a pro-inflammatory role in AMD. Biochim. Biophys. Acta 1853,2897–2906(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Burgh R et al. Defects in mitochondrial clearance predispose human monocytes to interleukin-1β hypersecretion. J. Biol. Chem 289, 5000–5012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lawlor KE & Vince JE Ambiguities in NLRP3 inflammasome regulation: is there a role for mitochondria? Biochim. Biophys. Acta 1840, 1433–1440 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Subramanian N, Natarajan K, Clatworthy MR, Wang Z & Germain RN The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wallace DC Mitochondrial DNA variation in human radiation and disease. Cell 163, 33–38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cordero MD et al. Mutation in cytochrome b gene of mitochondrial DNA in a family with fibromyalgia is associated with NLRP3-inflammasome activation. J. Med. Genet 53, 113–122 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Lorente L et al. Severe septic patients with mitochondrial DNA haplogroup JT show higher survival rates: a prospective, multicenter, observational study. PLoS ONE 8, e73320 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kenney MC et al. Inherited mitochondrial DNA variants can affect complement, inflammation and apoptosis pathways: insights into mitochondrial-nuclear interactions. Hum. Mol. Genet 23, 3537–3551 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roers A, Hiller B & Hornung V Recognition of endogenous nucleic acids by the innate immune system. Immunity 44, 739–754 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Chen Q, Sun L & Chen ZJ Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol 17, 1142–1149 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Barber GN STING: infection, inflammation and cancer. Nat. Rev. Immunol 15, 760–770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao D et al. Cyclic GMP–AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341,903–906(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Collins AC et al. Cyclic GMP–AMP synthase is an innate immune DNA sensor for Mycobacterium tuberculosis. Cell Host Microbe 17,820–828(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Storek KM, Gertsvolf NA, Ohlson MB & Monack DM cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J. Immunol 194, 3236–3245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Watson RO et al. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe 17, 811–819 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schoggins JW et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crow YJ & Manel N Aicardi–Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol 15,429–440 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Mackenzie KJ et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 35, 831–844 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gray EE, Treuting PM, Woodward JJ & Stetson DB Cutting edge: cGAS is required for lethal autoimmune disease in the Trex 1-deficient mouse model of Aicardi–Goutières syndrome. J. Immunol 195, 1939–1943 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Q et al. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.White MJ et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159, 1549–1562 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rongvaux A et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.García N & Chávez E Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci. 81, 1160–1166(2007). [DOI] [PubMed] [Google Scholar]
- 68.Patrushev M et al. Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell. Mol. Life Sci. 61,3100–3103 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carroll EC et al. The vaccine adjuvant chitosan promotes cellular immunity via DNA sensor cGAS–STING-dependent induction of type I interferons. Immunity 44, 597–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nishiyama S et al. Cyclosporin A inhibits the early phase of NF-kappaB/RelA activation induced by CD28 costimulatory signaling to reduce the IL-2 expression in human peripheral T cells. Int. Immunopharmacol 5, 699–710 (2005). [DOI] [PubMed] [Google Scholar]
- 71.Howell J et al. Cyclosporine and tacrolimus have inhibitory effects on Toll-like receptor signaling after liver transplantation. Liver Transpl 19, 1099–1107 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Kanneganti T-D, Kundu M & Green DR Innate immune recognition of mtDNA — an undercover signal? Cell Metab. 21,793–794 (2015). [DOI] [PubMed] [Google Scholar]
- 73.Alavian KN et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl Acad. Sci. USA 111, 10580–10585 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bernardi P, Rasola A, Forte M & Lippe G The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev 95, 1111–1155 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jonas EA, Porter GA, Beutner G, Mnatsakanyan N & Alavian KN Cell death disguised: the mitochondrial permeability transition pore as the c-subunit of the F1, Fo ATP synthase. Pharmacol. Res 99, 382–392 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Burton B & Dubnau D Membrane-associated DNA transport machines. Cold Spring Harb. Perspect. Biol. 2, a000406 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matilla I et al. The conjugative DNA translocase TrwB is a structure-specific DNA-binding protein. J. Biol. Chem 285, 17537–17544 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.West AP et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that mtDNA instability induced by TFAM deficiency or herpesvirus infection triggers cGAS activation and type I IFN responses. References 65, 66 and 78, were the first to document that mtDNA engages the cGAS–STING axis.
- 79.Albring M, Griffith J & Attardi G Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc. Natl Acad. Sci. USA 74, 1348–1352 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gerhold JM et al. Human mitochondrial DNA–protein complexes attach to a cholesterol-rich membrane structure. Sci. Rep 5, 15292 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lewis SC, Uchiyama LF & Nunnari J ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353, aaf5549 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Clayton DA Replication of animal mitochondrial DNA. Cell 28, 693–705 (1982). [DOI] [PubMed] [Google Scholar]
- 83.Saffran HA, Pare JM, Corcoran JA, Weller SK & Smiley JR Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep. 8, 188–193 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper was the first to document that alphaherpesviruses target mtDNA through the UL12.5 nuclease.
- 84.Corcoran JA, Saffran HA, Duguay BA & Smiley JR Herpes simplex virus UL12.5 targets mitochondria through a mitochondrial localization sequence proximal to the N terminus. J. Virol 83, 2601–2610 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duguay BA et al. Elimination of mitochondrial DNA is not required for herpes simplex virus 1 replication. J. Virol 88, 2967–2976 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McNab F, Mayer-Barber K, Sher A, Wack A & O’Garra A Type I interferons in infectious disease. Nat. Rev. Immunol 15, 87–103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wassermann R et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe 17, 799–810 (2015). [DOI] [PubMed] [Google Scholar]
- 88.Wiens KE & Ernst JD The mechanism for type I interferon induction by Mycobacterium tuberculosis is bacterial strain-dependent. PLoS Pathog. 12, e1005809 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang H, Li T, Chen S, Gu Y & Ye S Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. 67, 3190–3200(2015). [DOI] [PubMed] [Google Scholar]
- 90.Lood C et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med 22, 146–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper documents a role for mtDNA in NET-mediated type I IFN responses in lupus.
- 91.Caielli S et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med 213, 697–713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that oxidized mtDNA nucleoids are released from lupus neutrophils to trigger type I IFN responses.
- 92.McIlroy DJ et al. Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J. Crit Care 29, 1133.e1–1133.e5 (2014). [DOI] [PubMed] [Google Scholar]
- 93.West AP et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pazmandi K et al. Oxidative modification enhances the immunostimulatory effects of extracellular mitochondrial DNA on plasmacytoid dendritic cells. Free Radio. Biol. Med. 77, 281–290 (2014). [DOI] [PubMed] [Google Scholar]
- 95.Wolf AJ et al. Hexokinase is an innate immune receptor for the detection of bacterial Peptidoglycan. Cell 166, 624–636(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nicholls TJ & Minczuk M In D-loop: 40 yearn of mitochondrial 7S DNA. Exp. Gerontol 56, 175–181 (2014). [DOI] [PubMed] [Google Scholar]
- 97.Reyes A et al. Mitochondrial DNA replication proceeds via a ‘bootlace’ mechanism involving the incorporation of processed transcripts. Nucleic Acids Res. 41,5837–5850 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dayama G, Emery SB, Kidd JM & Mills RE The genomic landscape of polymorphic human nuclear mitochondrial insertions. Nucleic Acids Res. 42, 12640–12649 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li M, Schroeder R, Ko A & Stoneking M Fidelity of capture-enrichment for mtDNA genome sequencing: influence of NUMTs. Nucleic Acids Res. 40, e137 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wolff JN Targeted and robust amplification of mitochondrial DNA in the presence of nuclear-encoded mitochondrial pseudogenes using Φ29 DNA polymerases. Methods Mol. Biol. 1167, 255–263 (2014). [DOI] [PubMed] [Google Scholar]
- 101.Calvignac S, Konecny L, Malard F & Douady CJ Preventing the pollution of mitochondrial datasets with nuclear mitochondrial paralogs (numts). Mitochondrion 11, 246–254 (2011). [DOI] [PubMed] [Google Scholar]
- 102.Mankan AK et al. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J. 33, 2937–2946 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Garcia-Martinez I et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Invest 126, 859–864 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Itagaki K et al. Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PLoS ONE 10, e0120549 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schäfer ST et al. Mitochondrial DNA: an endogenous trigger for immune paralysis. Anesthesiology 124, 923–933 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Schiller M et al. Induction of type I IFN is a physiological immune reaction to apoptotic cell-derived membrane microparticles. J. Immunol 189, 1747–1756 (2012). [DOI] [PubMed] [Google Scholar]
- 107.Ries M et al. Identification of novel oligonucleotides from mitochondrial DNA that spontaneously induce plasmacytoid dendritic cell activation. J. Leukoc. Biol 94, 123–135 (2013). [DOI] [PubMed] [Google Scholar]
- 108.Pouwels SD et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am. J. Physiol. Lung Cell. Mol. Physiol 310, L377–L386 (2016). [DOI] [PubMed] [Google Scholar]
- 109.Hajizadeh S, DeGroot J, TeKoppele JM, Tarkowski A & Collins LV Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res. Ther. 5, R234–R240 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tsuji N et al. Role of mitochondrial DNA in septic AKI via Toll-like receptor 9. J. Am. Soc. Nephrol 27, 2009–2020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yao X et al. Mitochondrial ROS induces cardiac inflammation via a pathway through mtDNA damage in a pneumonia-related sepsis model. PLoS ONE 10, e0139416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kuck JL et al. Mitochondrial DNA damage-associated molecular patterns mediate a feed-forward cycle of bacteria-induced vascular injury in perfused rat lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, L1078–L1085 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McCarthy CG et al. Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc. Res 107, 119–130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ding Z et al. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci. Rep 3, 1077 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang Z et al. Mitochondrial DNA–LL-37 complex promotes atherosclerosis by escaping from autophagic recognition. Immunity 43, 1137–1147 (2015). [DOI] [PubMed] [Google Scholar]
- 116.Terluk MR et al. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci 35, 7304–7311 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Karunadharma PP, Nordgaard CL, Olsen TW & Ferrington DA Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 51, 5470–5479 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pinti M et al. Circulating mitochondrial DNA increases with age and is a familiar trait: implications for “inflamm-aging”. Eur. J. Immunol 44, 1552–1562 (2014). [DOI] [PubMed] [Google Scholar]
- 119.Verschoor CP et al. Circulating TNF and mitochondrial DNA are major determinants of neutrophil phenotype in the advanced-age, frail elderly. Mol. Immunol 65, 148–156(2015). [DOI] [PubMed] [Google Scholar]
- 120.Marques PE et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 56, 1971–1982 (2012). [DOI] [PubMed] [Google Scholar]