

Summary
The mouse embryo undergoes compaction at the 8-cell stage and its transition to 16 cells generates polarity such that the outer apical cells are trophectoderm (TE) precursors and the inner cell mass (ICM) gives rise to the embryo. We report here, that this first cell fate specification event is controlled by glucose. Glucose does not fuel mitochondrial ATP generation and glycolysis is dispensable for blastocyst formation. Furthermore, glucose does not help synthesize amino acids, fatty acids, and nucleobases. Instead, glucose metabolized by the hexosamine biosynthetic pathway (HBP) allows nuclear localization of YAP1. In addition, glucose dependent nucleotide synthesis by the pentose phosphate pathway (PPP), along with sphingolipid (S1P) signaling, activates mTOR and allows translation of Tfap2c. YAP1, TEAD4 and TFAP2C interact to form a complex that controls TE-specific gene transcription. Glucose signaling has no role in ICM specification, and this process of developmental metabolism specifically controls TE cell fate.
Graphical Abstract
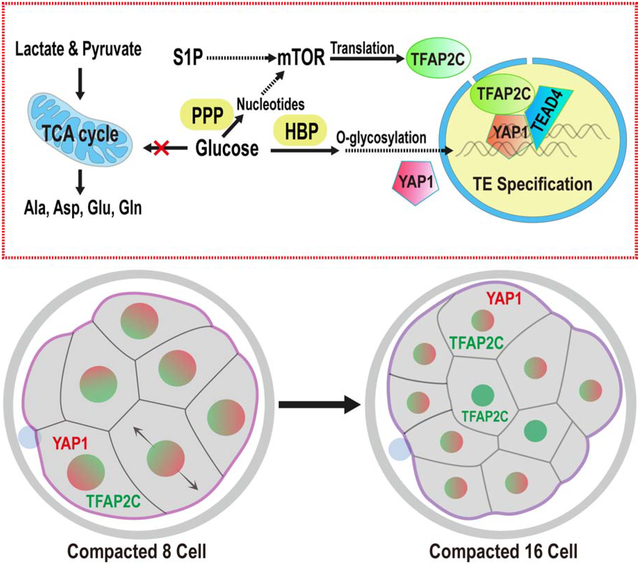
eTOC
Distinct Trophectoderm (TE) and Inner Cell Mass (ICM) precursors are first evident in the compacted morula stage. Chi, Sharpley, and Nagaraj et al. show that glucose metabolism controls this early developmental event. Glucose metabolized by multiple pathways initiates signaling and transcriptional events that control TE but not ICM cell fate.
Introduction
Following fertilization, the single-cell mouse zygote undergoes three cleavages to generate eight totipotent blastomeres. The 8-cell embryo then undergoes compaction, which generates tightly connected cells that have barely distinguishable boundaries. As this “compacted morula” transitions to the 16-cell stage, the outer cells acquire an apical (facing outward) and a basal surface, while the resulting inner cells, surrounded by other cells are non-polar. The first cell-specification event that creates two distinct lineages is now evident and the apical blastomeres ultimately differentiate to form the extraembryonic trophectoderm (TE) while the inner cells remain nonpolar and largely contribute to the pluripotent inner cell mass (ICM) at the blastocyst stage (Chazaud and Yamanaka, 2016; Leung et al., 2016; Rossant, 2018; White et al., 2018).
The nutritional requirements of the preimplantation embryo are minimal and are largely derived from the oviductal fluid in which it floats. An in vitro culture medium with only pyruvate, lactate, and glucose as nutrients, but lacking any amino acids, fats or proteins supports normal development through the 4.5 days of preimplantation stages (Biggers et al., 1997; Nagaraj et al., 2017). Growth factors or cytokines from the local environment are not crucial for development as early embryogenesis occurs normally without any proteins added to the medium. In this manuscript, we demonstrate that developmental cues are instead generated by cooperative interactions between metabolite uptake and internal signaling events. The embryo is self-sufficient in producing all necessary components to sustain all early developmental events. We find that this combined metabolic and developmental strategy is different in many respects, and similar in others, to that seen in differentiated cells, stem cells and cancer tissues.
A growth medium lacking pyruvate is unable to support progression beyond the 2-cell stage (Biggers et al., 1965; Biggers et al., 1967; Brown and Whittingham, 1991; Whittingham and Biggers, 1967). In a previous study we showed that pyruvate is essential for initiating zygotic genome activation (ZGA) and also for the selective translocation of key mitochondrial TCA cycle proteins to the nucleus. This unusual process allows epigenetic remodeling and ZGA (Nagaraj et al., 2017). Glucose is not required during this 2-cell stage. Rather, Whittingham and co-workers established a specific requirement for glucose during the compacted morula to blastocyst transition (Brown and Whittingham, 1991). This requirement for glucose is absolute, and glucose cannot be substituted by pyruvate and lactate that fully support the earlier developmental stages (Martin and Leese, 1995). While this might suggest a bioenergetic role of glucose at this stage, early studies reported minimal oxidation of glucose in the mitochondria (Fridhandler, 1961; Fridhandler et al., 1967). Our data support, extend and unify these empirical observations as we investigate how the three major arms of glucose metabolism, glycolysis, pentose phosphate pathway (PPP) and hexosamine biosynthetic pathway (HBP) control developmental signals that allow transition to the blastocyst stage.
Results
Glucose is Essential for the Morula to Blastocyst Transition
In the absence of glucose, zygotes proceed normally through early cleavage stages, and undergo the compaction process (Figure 1A–M), but then block in their development precisely at the compacted 8-cell morula stage (Figure 1N). These arrested embryos eventually de-compact and fragment. The term “morula” in this paper refers to embryos cultured until 78h post injection of hCG injection, which promotes ovulation. In normal media, 78h embryos are at the post-compaction 8–16 cell stage.
Figure 1. Role of glucose in early embryonic development.
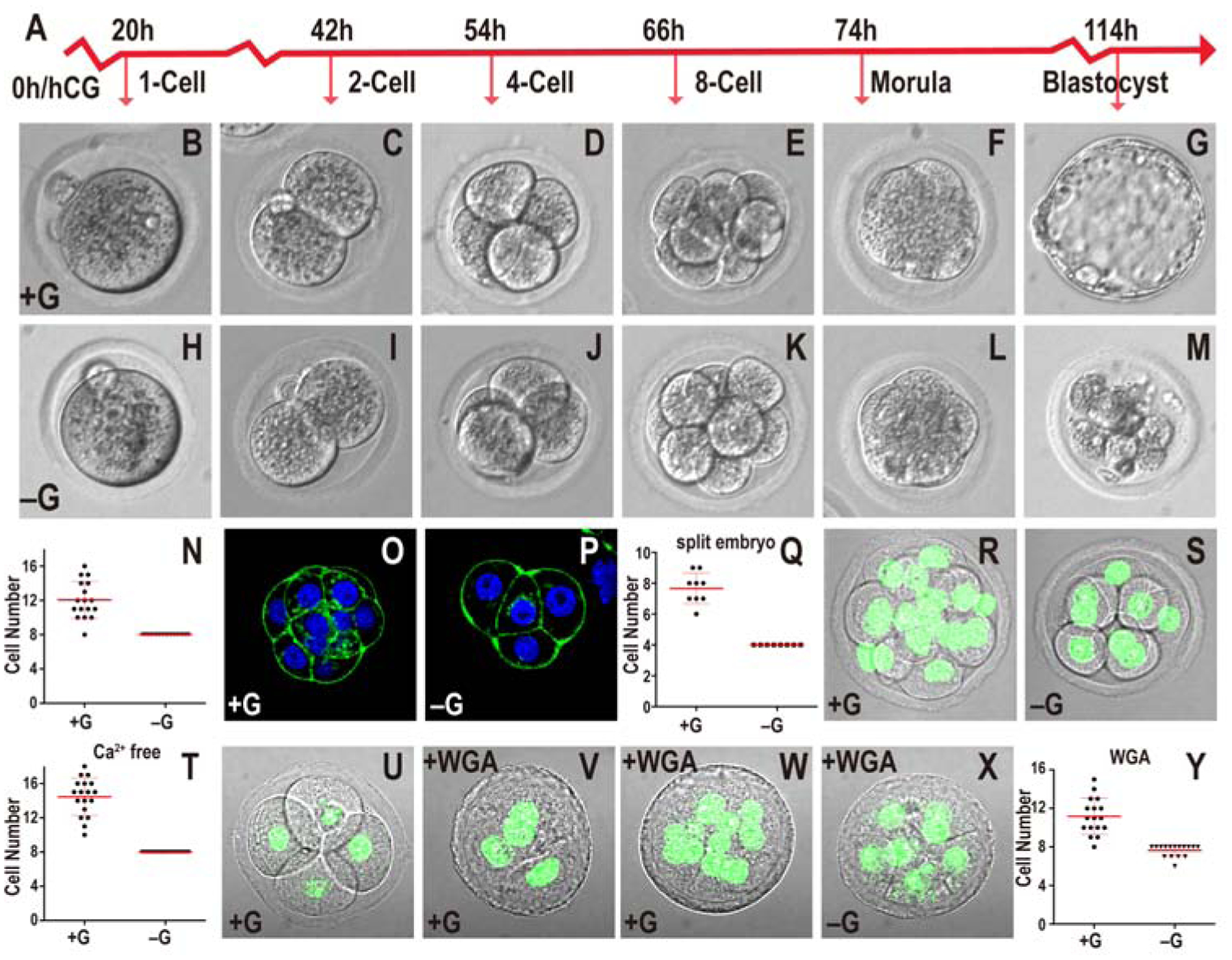
In all figures, hours (h) is following human chorionic gonadotropin (hCG) injection. Growth media including or lacking glucose indicated as +G or −G respectively. Zygotes are isolated at 18h and cultured until the specified hours (h) post hCG. All quantitative data include mean ± SD.
(A) Timeline of preimplantation development. Morula is 78h post hCG that in +G media are at post-compaction 8–16 cell stage with no hint of blastocyst cavity formation
(B-N) Embryos cultured in +G (B-G) or −G (H-M). The times and stages are as indicated in (A). In −G, the embryo fails to make a blastocyst (compare G and M). Every embryo in −G (n=17) is blocked at the 8-cell stage (N).
(O-Q) 2-cell embryos mechanically split into two individual blastomeres and grown in +G (O) or −G (P) until 78h. In both cases the embryos compact at 4-cell. In +G, split embryos proceed to 8-cells but −G embryos are blocked at 4-cell (Q). Note: As 2-cell embryos are split at 46h, they are “4-cell” at 78h.
(R-T) Ca2+ depletion prevents compaction in both +G (R) and −G (S) 78h embryos. Quantitation (T) shows +G embryos contain 10–18 cells (n=18) and −G (n=21) is blocked at 8-cells.
(U-Y) +G grown 4-cell embryos compact when WGA is added (56h; U, 58h; V) proceed to develop beyond 8-cells (n=17) (78h; W, Y). In −G, WGA treated embryos block at 8-cells (n=16) (78h; X, Y).
2-cell embryos that are mechanically split into single cell components and allowed to develop, proceed through all preimplantation development stages and give rise to a smaller than normal blastocyst (Casser et al., 2017). We confirm that split embryos grown with glucose compact at the 4-cell stage, before progressing to the 8-cell and blastocyst stages (Figure 1O, Q). Split embryos grown without glucose also undergo compaction at the 4-cell stage, however they fail to develop beyond 4 cells (Figure 1P, Q). Thus reaching the 8-cell stage per se is not an absolute requirement for the arrest caused by a lack of glucose.
To investigate if the process of compaction itself is causal to the morula block, we utilized earlier observations that showed that embryos fail to compact in a Ca2+ free medium (Shirayoshi et al., 1983), and that WGA (wheat germ agglutinin) confers compaction within 1–2 hours at any developmental stage (Johnson, 1986; Watson and Kidder, 1988). We find that while a calcium-free medium with glucose prevents compaction, the embryos proceed past the 8-cell stage (Figure 1R, T). In contrast, in medium that is both glucose and calcium free, the uncompacted embryos also exhibit an 8-cell developmental block (Figure 1S, T). Similarly, while WGA applied to a 4-cell embryo always causes rapid compaction (Figure 1U, V), such prematurely compacted embryos continue to develop beyond the 8-cell stage only when glucose is added to the medium (Figure 1W–Y). We conclude that compaction does not determine the timing of the glucose requirement. The requirement for glucose seems to follow a counting process that follows ZGA, the mechanism for which is as yet unclear.
Glucose Supports Anabolic Processes but not Energy Production
Based on the observation that glucose uptake increases rapidly from the 8-cell to the blastocyst stage (Leese and Barton, 1984), it is tempting to speculate (Johnson et al., 2003) that the 8-cell block is due to lowered energy levels in the absence of glucose. To track the breakdown products of glucose through metabolomic analysis, we incubated embryos throughout their developmental stages in the presence of uniformly-labeled (U-13C) glucose and unlabeled pyruvate/lactate. In a parallel experiment, the medium contained unlabeled (12C) glucose and labeled, U-13C pyruvate/lactate. The two metabolites, pyruvate and lactate, are labeled together since they interconvert rapidly with the carbon on lactate contributing to pyruvate (Brinster, 1967; Landau and Wahren, 1992; Lane and Gardner, 2005). Embryos are allowed to develop until the compacted morula stage and are then analyzed for metabolites by MS. Two different platforms are utilized to separate the metabolites. When properly optimized, the HPLC-MS method combined with the more sensitive ion chromatography system (ICS-MS) detects approximately 100 metabolites from 250 embryos per sample. Each sample is run in three biological replicates. In spite of the limitations of tissue availability, this technique enables us to map the distribution of glucose and pyruvate/lactate derived carbons with a high degree of confidence. The MS analysis identifies metabolites that derive carbons from glucose as opposed to pyruvate/lactate and also those that are not labeled with either, indicating that they are either maternally derived from the oocyte or endogenously generated from the breakdown of other stored metabolites.
Surprisingly, we find that glucose does not contribute significant levels of carbon to pyruvate as would be expected in cells with active glycolysis (Figure 2A, B). More importantly, metabolites derived from the TCA cycle are not labeled by glucose carbons either. Instead, it is exogenously derived-carbons provided by pyruvate/lactate, that solely populate TCA cycle metabolites (Figure 2B). For instance, 82% of citrate carbons are derived from pyruvate/lactate, the contribution from glucose, less than 0.5%, is close to our limit of detection. The remaining unlabeled citrate is not derived from exogenously added nutrients. Since citrate carbons are not acquired from glucose, citrate-derived fatty acids, cholesterol and several other lipids would not acquire glucose carbons either.
Figure 2. Metabolomics analysis of the compacted morula.
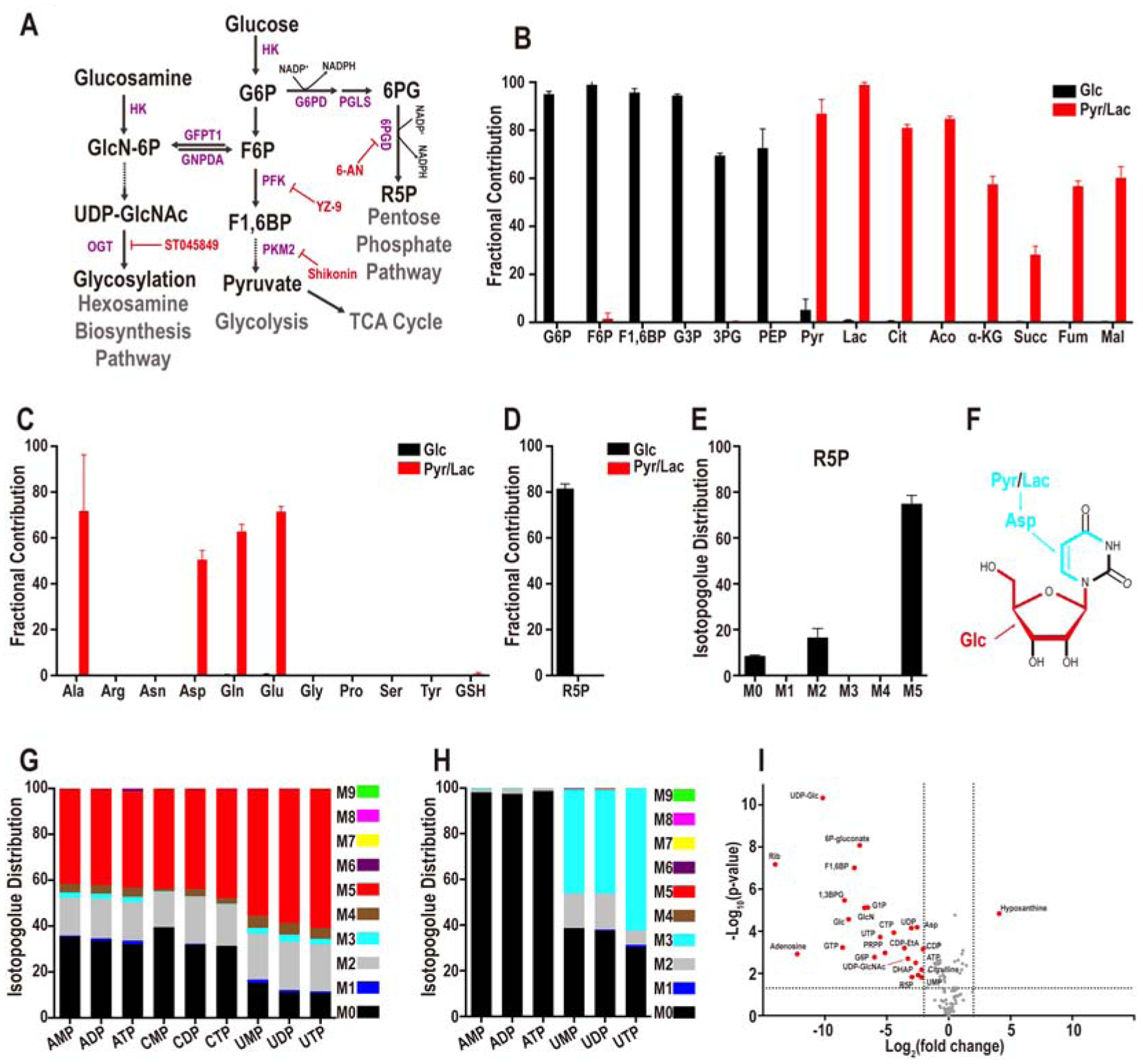
(A) Representative steps in glucose metabolism.
(B) U-13C glucose (black bars) contributes to glycolytic intermediates but not to TCA cycle metabolites while U-13C pyruvate+lactate (red bars) contribute to TCA intermediates.
(C) Glucose (black) does not contribute carbons to amino acids and contribution of pyruvate/lactate (red) carbons is limited to the four amino acids, Ala, Asp, Gln, and Glu.
(D, E) Fractional contribution of U-13C glucose (black) and U-13C pyruvate/lactate (red) (D) to the PPP metabolite ribose-5-phosphate (R5P), and glucose isotopologue distribution of R5P (E). The most abundant isotopologue is M5 in which 5 of the donated carbons are from exogenous U-13C-glucose (black). M0 peak corresponds to unlabeled metabolites from internal resources. The origin of the M2 peak is unknown.
(F-H) Structural representation of uridine (F). Glucose contributes 5 carbons to the ribose sugar (red), while pyruvate/lactate contributes 3 carbons to the base (light blue), via aspartate for de novo pyrimidine biosynthesis. (G) U-13C glucose contributes carbons to all nucleotides. No isotopologues greater than M5 are detected suggesting that only the ribose ring of the nucleotides is synthesized from glucose. (H) U-13C pyruvate/lactate contributes 3 carbons (light blue) to pyrimidine based nucleotides (e.g. UMP, UDP, UTP), and no carbons (M0) from pyruvate/lactate are used to populate purine nucleotides (e.g. AMP, ADP, ATP).
(I) Identification of glucose sensitive metabolites. 25 metabolites (red) differ by more than 4-fold (P< 0.05). Metabolites that do not meet this criterion are marked in grey.
The TCA cycle metabolite alpha-ketoglutarate (α-KG) shows a labeling pattern that is similar to citrate with 57% of α-KG carbons obtained from pyruvate/lactate, but < 0.05% from glucose (Figure 2B). Pyruvate and TCA cycle intermediates, such as α-KG, are the precursors of the four abundant amino acids, alanine, glutamate, glutamine, and aspartate that are extensively labeled by carbons from pyruvate/lactate, but not by carbons from glucose (Figure 2C). The rest of the amino acids are endogenous as they are not labeled by either glucose or pyruvate/lactate (Figure 2C). Similarly, neither glucose nor pyruvate/lactate contributes to glutathione, and thus, the entire pool of glutathione in the compacted morula stage is maternally derived. (Figure 2C).
The labeling data show that PPP is one of the limited numbers of metabolic pathways that is sourced by glucose at this stage of development. Labeling experiments, including isotopologue analyses, provide evidence that ribose-5-phosphate (R5P) is generated from glucose, but not from pyruvate/ lactate (Figure 2D, E). R5P is generated by either the oxidative (via G6P) or the non-oxidative (via F6P) arm of the PPP. The metabolic data suggest no significant contribution of glucose to the base components of purine nucleotides (Figure 2F, G; Figure S1A). Pyruvate and lactate make some contribution to the carbons used to synthesize pyrimidine bases (uracil 37%, cytosine 19%), while glucose does not label any of the detectable nucleobases (uracil < 0.02%, adenine < 0.05%, cytosine < 0.01% and thymine < 0.03%) (Figure 2G, H; Figure S1A). The lack of glucose contribution to purine bases is consistent with amino acid labeling data that finds glycine and serine are neither labeled by glucose (both < 0.2%) nor by pyruvate/lactate (both < 0.3%) (Figure 2C). In systems with proven contribution of glucose carbons to serine, this amino acid is converted to glycine which in turn contributes to purine bases (Mattaini et al., 2016).
The contribution of exogenous glucose to the ribose group is extensive. For instance, 89% of the UTP in the embryo contains ribose that is derived from glucose, while pyruvate/lactate do not provide any carbon to UTP ribose (< 0.1%) (Figure 2F–H). Thus, much of the UTP, and other uridine containing nucleotides acquire carbons from glucose (Figure 2F, G). Adenine and cytidine nucleotides have a larger unlabeled component (indicating maternal origin) compared to the uridine nucleotides (~35 % unlabeled vs ~10 % for uridine, Figure 2G). The low maternal contribution of uridine likely reflects a combination of its small pool size and increased turnover (Clegg and Piko, 1977; Hsieh et al., 1979).
Pyrimidine nucleotides contain carbons derived from pyruvate/lactate (Figure 2F, H). Given that pyruvate/lactate do not contribute to R5P, we infer that the donated carbons belong to the base and not the ribose sugar. Thus, both glucose and pyruvate/lactate contribute to nucleotide formation at this stage, but they do so by very different pathways. In addition to the PPP, glucose derived carbons are incorporated into HBP related metabolites such as GlcNAc-1P that is labeled by U-13C glucose (Figure S1B). To identify the metabolic pathways that are most sensitive to glucose omission, embryos are cultured with and without glucose present in the medium. Metabolites are extracted at the compacted morula stage and analyzed by MS following separation by the two platforms described above. Twenty-four metabolites (of total 106) are highly glucose sensitive and their levels decrease by more than 4-fold in the absence of glucose (Figure 2I). Notable examples include direct or indirect derivatives of the upper arm of glycolysis (e.g. G6P), or PPP (e.g. R5P, UTP), or the HBP (e.g. glucosamine and UDP-GlcNAc). Also notable is the observation that pyruvate and lactate remain unaffected in the metabolomic experiments that are discussed above (Figure 2I). In summary, the compacted morula does not use glucose to fuel mitochondrial ATP generation, or provide carbon to synthesize amino acids, fatty acids, and nucleobases. These processes are fueled by pyruvate and lactate or by endogenous sources. Glucose contributes to the synthesis of metabolites derived from HBP and PPP.
Glycolysis, PPP and HBP Play Distinct Roles in Developmental Progression
Loss of function analysis of metabolic enzymes poses a significant challenge in the context of the preimplantation embryo since enzymes involved in glucose metabolism are maternally deposited as proteins and RNA based knockdowns are therefore ineffective. Zygotic null alleles (such as for PDH) often bypass early phenotypes (Johnson et al., 2001), and as essential genes, we expect that maternal nulls in metabolic enzymes will affect prezygotic stages (Johnson et al., 2007). We use two independent strategies to generate loss of function. The first is to use multiple enzyme inhibitors for each pathway. The second and more definitive method is to use the “Trim-Away” (Clift et al., 2017) method, that directly eliminates both maternally deposited and newly synthesized proteins (see STAR methods).
Glycolysis and generation of energy:
The inhibitor YZ9 (Figure 2A) blocks glycolysis at the level of PFK and therefore inhibits the cascade that generates pyruvate. PPP and HBP are not affected by YZ9 (Seo et al., 2011). Strikingly, continuous inhibition of glycolysis from the 1-cell stage onward has no adverse effect on progress to the blastocyst stage (Figure 3A). While counterintuitive and surprising, this finding of glucose independence of bioenergetics is completely consistent with the 13C-glucose labeling results (Figure 2B). In those experiments, we find that it is exogenous pyruvate and not glucose that contributes to the TCA cycle. Consistent with these requirements, addition of YZ9 blocks blastocyst formation if the growth medium lacks pyruvate (Figure 3A). A mere lack of pyruvate with no added YZ9 has no effect on blastocyst formation (not shown). Thus, YZ9 indeed functions as a specific inhibitor of glycolysis, but its block at the level of PFK is bypassed when pyruvate feeds into the pathway further downstream and provides carbons to the TCA cycle. We also used a second inhibitor, shikonin, which selectively blocks the function of the embryonic pyruvate kinase M2 (PKM2) (Chen et al., 2011). Similar to the results with YZ9, shikonin has no phenotypic consequence for blastocyst development, but a medium lacking pyruvate but with shikonin added is unable to sustain growth beyond the morula stage even when glucose is present (Figure S2A).
Figure 3. Metabolic contribution of glycolysis, PPP and HBP.
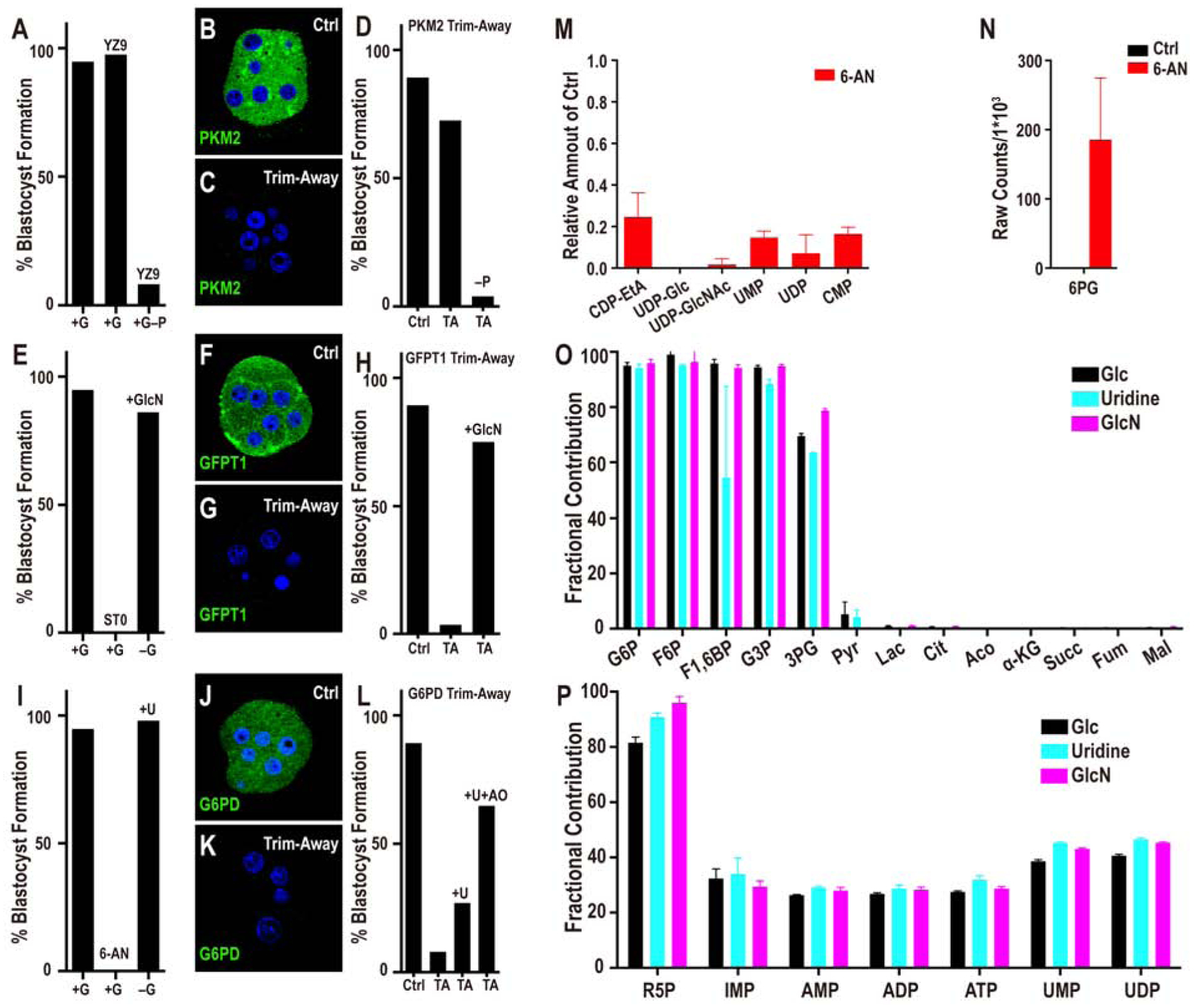
(A) Glycolysis inhibition by YZ9 has no adverse effect on blastocyst formation unless the medium lacks pyruvate (+G–P +YZ9). (B-C) PKM2 staining at the morula stage. (B) Control injected with unrelated antibody and mCherry-Trim21 mRNA shows robust expression of PKM2. (C) Injection of mCherry-Trim21 mRNA along with an antibody against PKM2 leads to complete loss of the protein. (D) PKM2 depletion does not affect blastocyst formation unless the growth medium lacks pyruvate.
(E) Inhibition of O-glycosylation (by ST045849) blocks morula to blastocyst transition. Supplementation with GlcN fully rescues the morula block phenotype seen in −G. (F) Control embryo shows robust expression of GFPT1 that is lost upon Trim-Away depletion (G). This causes a morula block that is fully rescued by GlcN (H).
(I) Inhibition of PPP by 6-AN blocks the transition from morula to blastocyst. Supplementation with uridine (U) fully rescues the morula block phenotype seen in −G and restores blastocyst formation. (J-K) G6PD staining at the morula stage. (J) Control IgG shows robust expression of G6PD. (K) G6PD Trim-Away leads to complete loss of the protein. (L) Depletion of G6PD (PPP) by Trim-Away causes a morula block and prevents blastocyst formation. Uridine partially rescues G6PD loss, and this rescue is significantly better in the presence of an antioxidant (AO).
Note: experiments shown in (A, E, I) and (D, H, L) were performed together, and so the respective controls are shared between the groups.
(M) Blocking the oxidative branch of the PPP pathway with 6-AN causes a significant reduction in the levels of nucleotides and nucleotide derived metabolites. The data are normalized to the corresponding controls set at 1.0. (N) 6-PG accumulation following 6-AN treatment.
(O, P) Embryos cultured in U-13C glucose (black), or in U-13C GlcN (without glucose, light blue) or in 13C uridine (without glucose, purple). U-13C GlcN and 13C uridine show a virtually identical metabolic labeling pattern as U-13C glucose.
Depletion of PKM2 using Trim-Away both establishes the efficacy of the method and validates the inhibitor experiment. We find that the procedure eliminates all of the detectable PKM2 protein (Figure 3B, C), and yet PKM2-depleted embryos form blastocysts with the same efficiency as in controls (Figure 3D). However, as predicted by the inhibitor analysis, in a medium lacking pyruvate, PKM2 depleted embryos are unable to transition to blastocysts (Figure 3D). Taken together, the metabolomic, inhibitor and protein loss data unambiguously establish that core glycolysis, the conversion of glucose to pyruvate, is not essential for the morula to blastocyst transition. The insensitivity of embryos to the inhibition of PFK or PKM contrasts with the morula block that is caused by treatment with 2-deoxyglucose (2-DG), an inhibitor that blocks early enzymes in glycolysis, including hexokinase (Chen and Gueron, 1992). In addition to glycolysis, 2-DG treatment is expected to affect HBP and the PPP, which are not affected by PFK or PKM inhibition.
Hexosamine biosynthetic pathway (HBP)
Treatment of embryos with the commonly used inhibitors of the HBP, such as azaserine and DON that target GFPT1, results in a developmental arrest at the compacted morula stage (Figure S2A). However, these inhibitors can, under some circumstances, have off-target effects. More convincingly, we recall from the metabolomic data that UDP-GlcNAc, the critical product generated by HBP, is labeled by 13C-glucose and that its levels decrease in the absence of exogenously added glucose (Figure 2I). UDP-GlcNAc facilitates both N- and O-linked glycosylation. Experiments with tunicamycin that blocks N-glycosylation have suggested a role for this protein modification in the morula to blastocyst transition (Surani, 1979). However, we later show that although N-glycosylation plays a role in the proper localization of apical-domain proteins, its role in cell fate determination is minor, especially in comparison to that of O-glycosylation in this process. ST045849 is a widely used, and very specific inhibitor of OGT (O-linked N-acetyl glucosamine transferase) function (Gross et al., 2005). When added to post ZGA 2-cell embryos, ST045849 causes a developmental arrest at the compacted morula stage (Figure 3E). The morphological phenotype of O-glycosylation inhibited embryos is indistinguishable from those cultured without glucose. Also, glucosamine (GlcN) is able to bypass and rescue the morula block caused by lack of glucose (Figure 3E) (Pantaleon et al., 2008). These data convincingly show that glucose metabolism by HBP and its role in protein O-glycosylation are critically important for the progression to the blastocyst stage.
To be absolutely sure of this important conclusion, we used the Trim-Away method with a specific GFPT1 antibody. This process completely eliminates the protein and also causes a compacted morula block (Figure 3F–H). Importantly, GFPT1 protein depleted embryos are rescued to form blastocysts if the growth medium is supplemented with GlcN (Figure 3H) that directly generates glucosamine-6-phosphate (GlcN-6P), bypassing the need for an active GFPT1 enzyme.
Pentose phosphate pathway (PPP):
13C labeling by glucose demonstrates that carbons from glucose contribute extensively towards nucleotide ribose sugar biosynthesis suggesting an influx of glucose into the PPP at this stage. Furthermore, the level of nucleotides and PPP metabolites decrease substantially in embryos that are cultured without glucose. We assessed the role of PPP by inhibiting the pathway using 6-aminonicotinamide (6-AN) that is a well-established competitive inhibitor of 6-phosphogluconate dehydrogenase (PGD), the last enzyme of the oxidative PPP (Downs SM, 1998; Kohler et al., 1970; Lange and Proft, 1970). With PPP thus blocked, embryos proceed normally to the 8-cell stage, undergo compaction, but arrest in development at the compacted morula stage and fail to form blastocysts (Figure 3I). The terminal product of the PPP pathway includes ribose sugars that contribute to nucleotide formation. We find that uridine, which is a source of ribose for all nucleotides, added to a medium lacking glucose restores blastocyst formation (Figure 3I).
Phenotypically, 6-AN treatment closely recapitulates the developmental block caused by glucose omission. Nevertheless, 6-AN activity could, in principle, extend to other NADP+ dependent enzymes, although within the bounds of glucose metabolism, the PPP arm is heavily impacted. 6-phosphogluconate is an upstream metabolite of 6-AN inhibition in the PPP. We find that it is undetectable in control embryos, but accumulates to very high levels when 6-AN is added (Figure 3N).
In order to verify results from inhibitor and metabolomic analyses, we specifically depleted G6PD, the first enzyme in the oxidative branch of the PPP using Trim-Away. A monoclonal antibody that specifically recognizes G6PD is used in these experiments and we find that the G6PD protein becomes undetectable following the procedure (Figure 3J, K). Importantly, this manipulation also causes a specific developmental block at the compacted morula stage. To test the specificity of the depletion, we asked if restoring PPP activity downstream of G6PD rescues the phenotype. Partial rescue is observed when the metabolite uridine is used to supplement the culture medium of embryos with depleted G6PD (Figure 3L). This rescue is much improved when antioxidants are used along with uridine to remove the extra ROS accumulated when this enzyme function is blocked (Figure 3L).
Remarkably, of the metabolites detected in our assay, only 6 decrease substantially (more than 4-fold with P < 0.05) upon 6-AN treatment, and these metabolites all contain a nucleotide group (Figure 3M). 6-AN treatment almost completely abolishes the synthesis of nucleotide ribose. And also, pyrimidine nucleotides and their derivatives are particularly affected, which may reflect the smaller sizes of these pools and/or increased turnover at this stage of development (Clegg and Piko, 1977; Hsieh et al., 1979). The high sensitivity of UTP production to 6-AN explains why the HBP component UDP-GlcNAc is eliminated when PPP is blocked, while other metabolites belonging to the HBP pathway remain unaffected. This result highlights one of the important mechanisms of cross-talk between PPP and HBP (see below). In summary, the PPP is absolutely critical for the transition from the morula to the blastocyst stage and additionally, these data provide the first hints of a cross-talk between the different arms of the glucose metabolism pathways.
Cross-talk Between PPP and HBP:
In the experiments described above we show that exogenously added GlcN or uridine overcomes the morula block seen upon HBP or PPP inhibition respectively (Figure 3H, L). However, we also find that both GlcN and uridine can independently overcome the phenotype resulting from glucose depletion (Figure 3E, I). Without glucose, both PPP and HBP are nonfunctional and it is puzzling how such an overall deficit in glucose metabolism is rescued by separately adding either GlcN or uridine to the medium.
To investigate this question, we started with a glucose-free medium and supplemented it with fully 13C-labeled glucosamine (U-13C-GlcN). Embryos are allowed to grow to the compacted morula stage, and metabolites are then extracted and the flow of labeled carbons to other metabolites is tracked using MS. We find that GlcN carbons not only populate HBP metabolites (such as GlcN-6P, GlcNAc-P, UDP-GlcNAc), but interestingly, they also populate metabolites associated with the PPP (such as ribose-phosphate) and glycolytic intermediates (such as G6P) in a manner similar to that seen upon glucose labeling (Figure 3O, P). Also, similar to glucose, GlcN does not contribute carbons to the TCA cycle, to amino acid synthesis, or to nucleobases (Figure 3O, S2B–C).
In a similar vein, when uridine whose 5 ribose-ring carbons are labeled with 13C [1’,2’,3’,4’,5’−13C5] uridine (the base is not labeled and is 12C) is used instead in the above labeling experiment, carbons from the ribose ring also populate metabolites in a pattern that is virtually identical to that seen when glucose is used for isotope labeling (Figure 3O, P). For instance, as with glucose, nucleotide containing metabolites, derived from the PPP, are labeled with uridine, and importantly, so are the intermediates of glycolysis and the HBP (Figure 3O, P). We conclude that uridine is metabolized and fed into glycolysis by non-oxidative PPP. As with glucose, TCA cycle intermediates, amino acids and nucleobases are not labeled by uridine (Figure 3O, Figure S2B–C). In summary, the labeling data show that either uridine or GlcN can supply carbons to all the metabolites that G6P generates and provides evidence for extensive cross-talk between these two arms of the glucose metabolism pathways.
Glucose Metabolism and TE Specification
The first lineage specification event in the developing embryo is later than but not far in time from when glucose becomes essential for further development leading us to investigate whether lineage specification might be causally linked to glucose utilization. We find that the canonical TE specification and maturation transcription factor CDX2 (Strumpf et al., 2005), is not expressed in embryos cultured in a glucose deprived medium or those in which either the HBP or the PPP pathway is specifically inhibited (Figure 4A–E). To our surprise, the expression of ICM markers such as OCT4 and NANOG remain entirely unaffected in the absence of glucose, and their expression also remains normal when HBP derived glycosylation or PPP is inhibited (Figure 4F–O).
Figure 4. Glucose is required for TE fate initiation and specification.
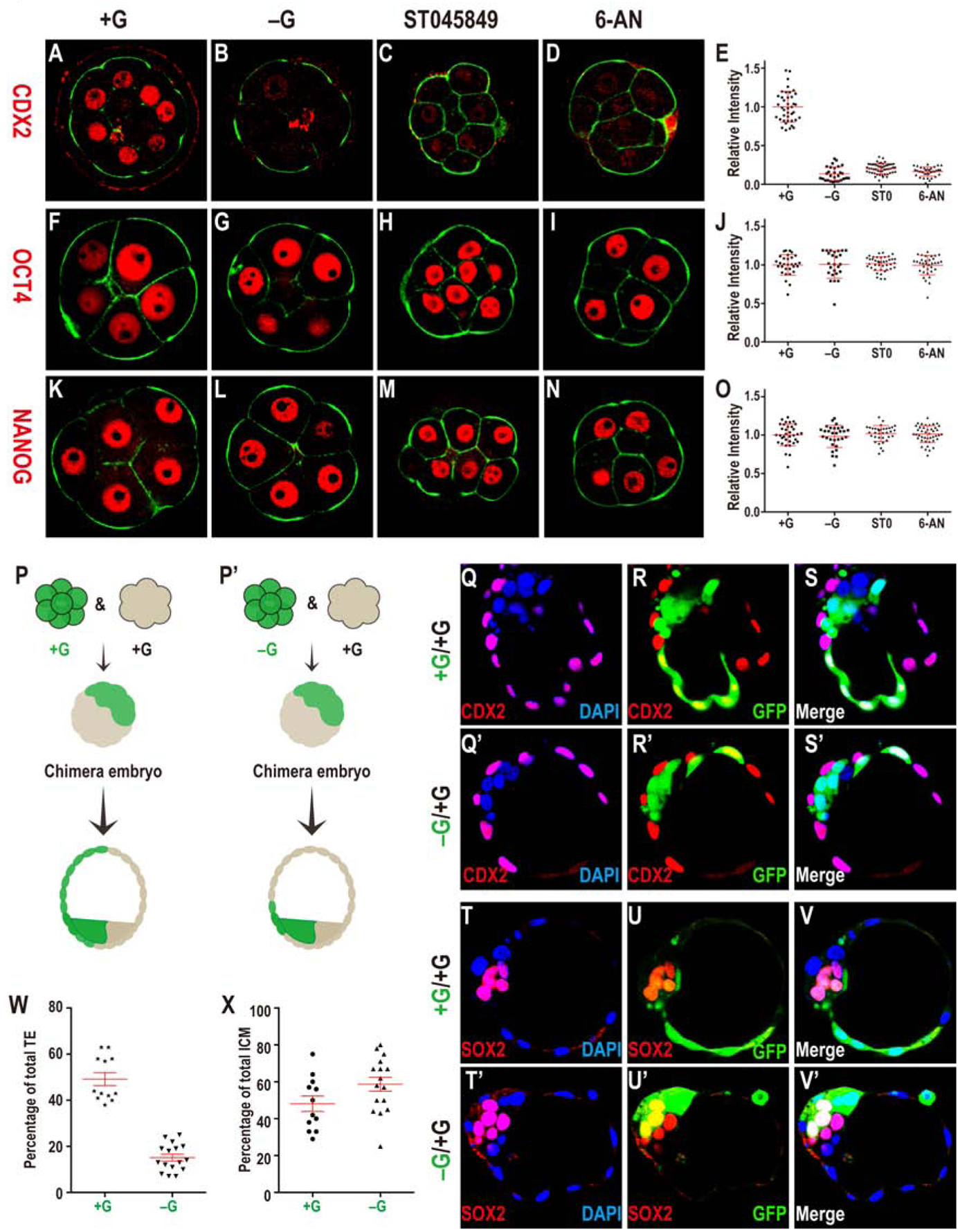
CDX2 (A-E) expression is observed in outer blastomeres in +G (A), but not in −G (B), and not when O-glycosylation (HBP, C) or PPP (D) is blocked. Quantitation of CDX2 data (E).
OCT4 (F-J) and NANOG (K-O) expression in +G (F, K), −G (G, L), ST045849 (H, M), or 6-AN (I, N). Quantitation of OCT4 (J) and NANOG (O) shows that their levels are unchanged without glucose, or when PPP or O-linked glycosylation is blocked.
(P, P’) Schematic representation of control (+G; P) and experimental (-G; P’) derived chimeras. A GFP marked embryo (green) is cultured either in +G (P) or −G (P’). An unmarked embryo (not green) is separately grown in +G to the 8-cell stage. The green and non-green embryo are fused to make a chimera that is grown without glucose (for both P, P’) to the blastocyst stage. In (P) green cells distribute equally between TE and ICM. In (P’) green cells rarely take on TE fate.
(Q-X) Data supporting the scheme in (P, P’). (Q-S’) CDX2 (red, purple in merge with DAPI showing nuclei, GFP green) expression in TE cells in control (Q-S) and experimental (Q’-S’) chimeras. In the control, green cells populate both ICM and TE while in the experimental chimera, they are largely restricted to the ICM.
(T-V’) SOX2 expression (red) is in the ICM. While green cells can be either SOX2 positive or negative in control chimera (T-V), in +G/−G chimera, the green cells are usually SOX2 positive (T’-V’) (see text). (W, X) Quantitation of the chimera data.
The timing of the 8-cell morula block is earlier than that of the first expression of CDX2 (16-cell stage). To determine if the results above merely represent the fact that glucose deprived embryos simply do not reach the stage that corresponds to CDX2 expression, we decided to cause a cell cycle block by alternative means. The drug aphidicolin inhibits DNA replication, and cytochalasin D inhibits the cleavage of blastomeres. When added to early 8-cell embryos they both cause a compacted 8-cell morula block. In contrast to glucose deprived embryos, however, these 8-cell blocked embryos express normal levels of CDX2 at 78h post hCG (Figure S3A, A’, D, D”) showing that CDX2 expression does not require the embryos be at the 16-cell stage or later.
A morula blocked embryo never reaches the blastocyst stage which is when the fates of the TE and ICM cells become obvious, not only by markers but also by their cell position and shape. To address whether glucose is important for this later process of TE determination, rather than simply for the expression of a few isolated markers, we developed a chimera assay for our system. In these experiments, two embryos grown independently until the 8-cell stage are mechanically aggregated together to form one larger embryo that is competent to develop further to form “chimeric” blastocysts (Spindle, 1982; Tarkowski, 1961). For fate mapping purposes, one of the embryos is injected at the 1-cell stage with GFP mRNA that is efficiently translated into the green protein and marks all progeny to the blastocyst stage (Figure 4P, P’). As control, both the green and the non-green embryos are grown in normal medium containing glucose, fused at the 8-cell stage, and then further grown to the blastocyst stage in a medium lacking glucose. As expected, we find in this control that the blastomeres from the GFP and non-GFP embryos are equally competent to generate ICM and TE lineages (Figure 4Q–S, 4T–V). For the experimental group, the GFP labeled embryo is allowed to mature from the 1-cell to the early 8-cell stage in a glucose-free medium while an unlabeled embryo is grown in the normal glucose containing medium. The two embryos are aggregated and the resulting chimera is grown in a glucose-free medium until the blastocyst stage (Figure. 4Q’–S’, 4T’–V’). We find a very significant decrease (P < 0.0001) in the propensity of labeled (glucose deprived) vs the unlabeled (glucose supplemented) cells to be CDX2 positive (Figure 4Q’–S’, W). There is no significant difference in the total number of ICM or TE cells (regardless of GFP status) in chimeric blastocysts (P = 1.0). A few green cells are CDX2 positive and these are often found directly abutted to an unlabeled neighbor (Figure 4Q’–S’), and the simplest explanation is that these cells exchange a glucose derived metabolite with the neighboring cell. The proportion of glucose deprived blastomeres in the TE and ICM are shown in Figure 4W, X. Overall, these results provide a strong basis in favor of the hypothesis that the presence of glucose is required for TE fate. The loss of TE related markers is seen at a stage earlier than the actual establishment of the fate. The chimera allows us to grow the embryo until the blastocyst stage when the fate choices are clearly discernible.
Molecular Mechanisms Linking Glucose Metabolism to CDX2 Expression
The next step in this analysis is to probe the molecular mechanism that links CDX2 expression to PPP and HBP, since expression of CDX2 is critically dependent on the proper functioning of these two arms of glucose metabolism. The transcription factors that control CDX2 expression are well characterized in past studies (Cao et al., 2015; Rayon et al., 2014). Principal amongst these are pre-TE specification factors YAP1 and TFAP2C. The active YAP1 (Sasaki, 2017) is nuclearly localized within the pre-TE, but not in the ICM cells. TFAP2C is widely expressed in the 8-cell morula, but is later an important factor in controlling CDX2 in the TE (Auman et al., 2002; Cao et al., 2015; Kuckenberg et al., 2012). We focused on nuclear YAP1 and TFAP2C since they are dramatically affected in the absence of glucose (Figure 5A–F) and because these two transcription factor complexes are implicated in the direct control of CDX2 (Cao et al., 2015; Nishioka et al., 2009). Interestingly, TEAD4, the binding partner of YAP1, is also reduced in the absence of glucose, and this low TEAD4 level also likely contributes to prevent TE fate (Figure S4D’–G’). Other transcription factors such as GATA3 and SBNO1 have also been linked to TE fate initiation (Home et al., 2009; Ralston et al., 2010; Watanabe et al., 2017), but since they do not show glucose dependent expression (Figure S4A–D), we have not studied these further. The expression of TFAP2C and YAP1 remains high in embryos that are arrested at the 8-cell stage following treatment with aphidicolin or cytochalasin D, and, thus, their expression is not affected by cell cycle arrest at this stage, allowing them to activate CDX2 (Figure S3A–F; D”–F”).
Figure 5. Glucose dependent activation of transcription factors.
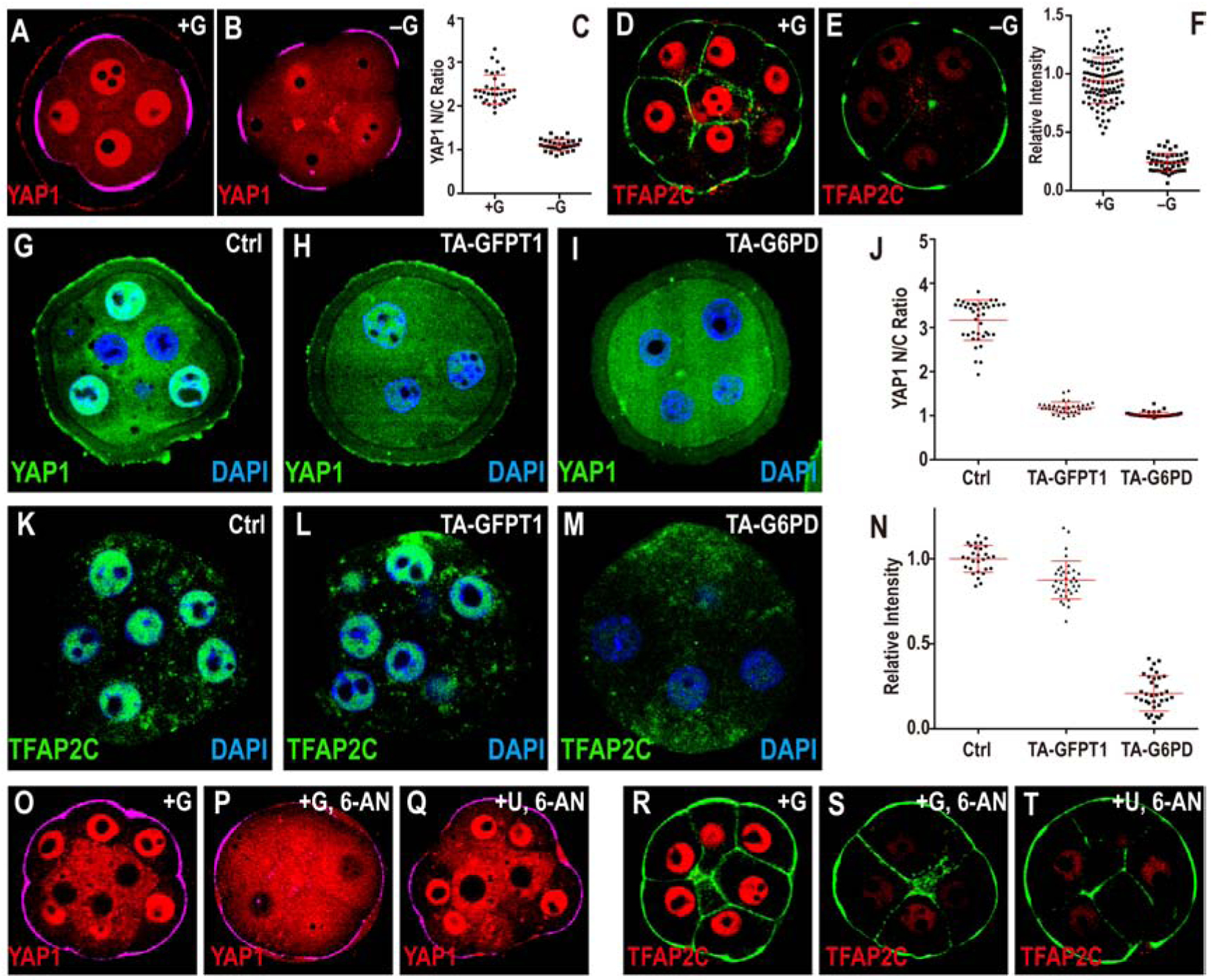
Cell membranes (green) are marked using phalloidin-FITC in (D, E, R, S, T), apical surfaces (purple) are marked by phospho-Ezrin/Radixin/Moesin (pERM) in (A, B, O, P, Q).
In +G control (A) YAP1 is localized to the nucleus in pERM (purple) marked polar cells (74h). This nuclear localization is significantly reduced in −G (B). (C) Quantitation of YAP1 nuclear to cytoplasmic (N/C) ratio.
(D-F) In control media TFAP2C (red) is expressed in the nuclei of all blastomeres (D) at the morula stage (78h). This expression is lost in −G (E) Quantitation shown in (F).
Inhibition of PPP or HBP both give results similar to that in −G (see Figure. S4E–J)
(G-N) Loss of function using Trim-Away (TA).
(G) Control, morula injected with control IgG antibody. Strong nuclear YAP1 staining (green) is seen. Trim-Away (TA) with either anti-GFPT1 (HBP) (H) or anti-G6PD (PPP) (I) results in a significant reduction in nuclear YAP1. (J) Quantitation of YAP1 nuclear to cytoplasmic (N/C) ratio. (K) Control, morula injected with non-specific IgG antibody shows robust nuclear TFAP2C (green). Injection with anti-G6PD (M) causes a significant reduction in TFAP2C expression while injection with anti-GFPT1 (L) does not influence TFAP2C expression. (N) Quantitation of nuclear TFAP2C.
(O-T) Rescue experiments at morula stage (78h). YAP1 nuclear localization seen in control embryos (O) and lost in PPP blocked embryos (P) is rescued by addition of uridine (Q). In contrast, TFAP2C expression (R), also lost upon PPP inhibition (S) is not rescued by uridine (T). Quantitation for YAP1 and TFAP2C are shown in Figure S4G, J.
Loss of HBP and PPP manifest themselves in distinct ways in affecting YAP1 vs TFAP2C. For blocking either pathway, once again, we used the reliable and specific Trim-Away technique. Loss of GFPT1 protein (HBP) using this method blocks nuclear localization of YAP1 (Figure 5G, H) but has no effect on TFAP2C expression (Figure 5K, L). Whereas, G6PD (PPP) loss by Trim-Away causes complete elimination of TFAP2C expression and YAP1 nuclear localization is also affected (Figure 5I, M). We conclude that PPP plays an important role in causing TFAP2C expression, while HBP does not (Figure 5K–M). YAP1 nuclear localization is affected when either HBP or PPP is perturbed (Figure 5G–I). These data are quantified in (Figure 5J, N).
The nuclear localization of YAP1 is severely compromised in embryos in which O-glycosylation is inhibited (Figure S4E, G). Recent studies have shown that nuclear localization of YAP1 requires O-linked glycosylation at 1–4 specific sites on the protein (Peng et al., 2017; Zhang et al., 2017). This is a likely explanation for loss of YAP1 localization when this protein modification is blocked. However, with technology currently available to us, combined with the limited availability of tissue, we cannot rule out the possibility that loss of O-glycosylation affects an unrelated protein that in turn affects YAP1 localization. In either case, it remains clear that HBP related O-glycosylation is essential for producing active nuclear YAP1 which is necessary for CDX2 expression. Phosphorylation at serine-127 also correlates with YAP1 activity (Zhao et al., 2007) but is not altered in response to glucose in our assay (Figure 6E). Consistent with the Trim-Away data, expression of TFAP2C is not at all altered when HBP-related glycosylation is inhibited (Figure S4H, J). The impact of glucose on YAP1, TFAP2C, and CDX2 expression, but not ICM markers, is phenocopied when embryos are treated with 2-DG (Figure S3D–F’), which is expected to inhibit glycolysis, PPP and HBP.
Figure 6. Modulation of TFAP2C Translation.
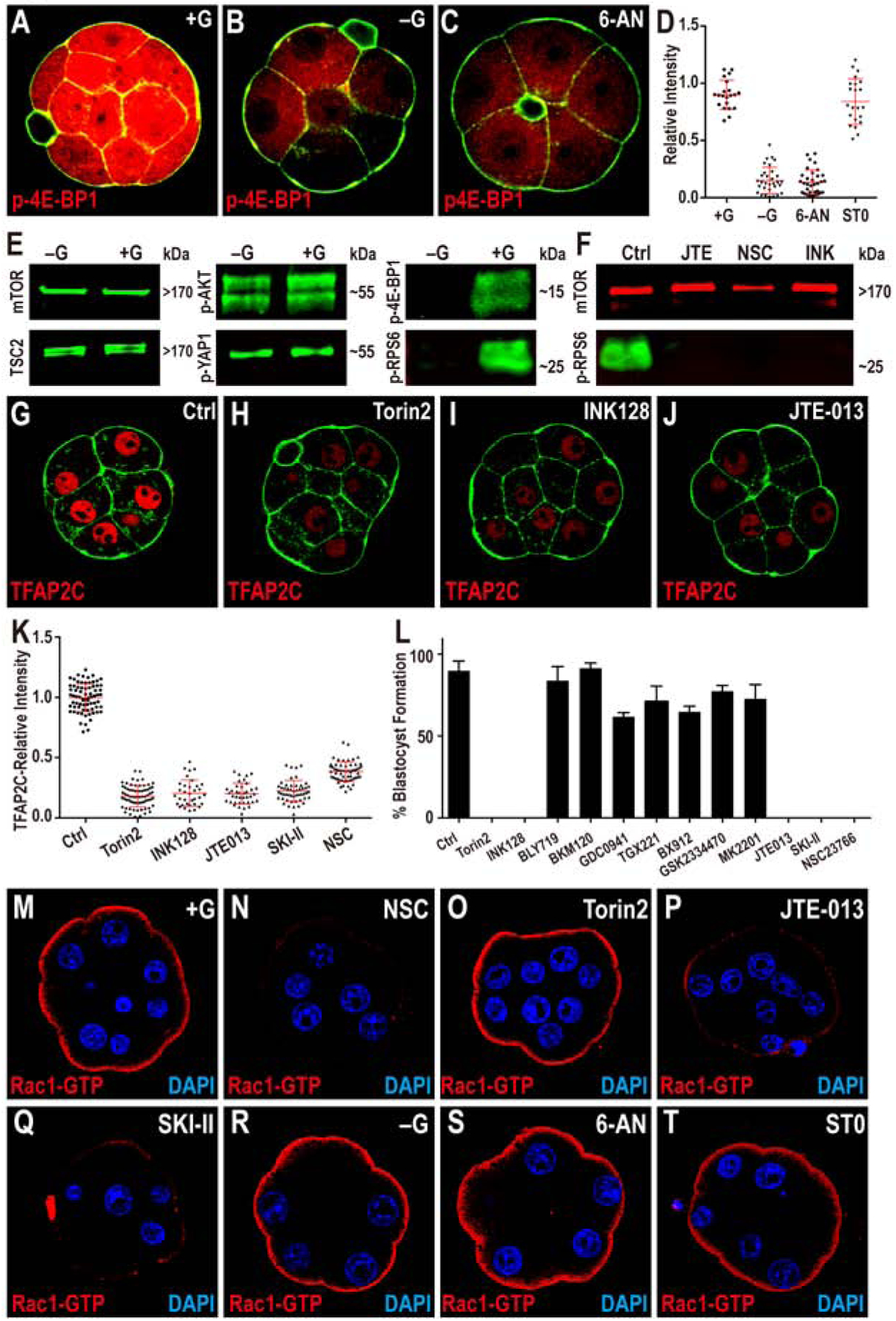
(A-D) TFAP2C translation is controlled by PPP function. Phosphorylation of threonine 37/46 on 4E-BP1 (p-4E-BP1), a target of mTOR is expressed at high levels in the 8-cell embryo (A). This expression is lost in −G (B) or when PPP is inhibited (C). Quantitation in (D) also shows that blocking HBP does not affect p-4E-BP1.
(E, F) Western blot analysis of members of mTOR signaling pathway and its activation. (E) Total mTOR, TSC2, phosphorylation level of serine 473 on AKT1/2 (p-AKT) and serine 127 on YAP1 (p-YAP1) all remain unchanged in −G embryos, but phosphorylated mTOR targets 4E-BP1 and RPS6 (p-RPS6) levels are significantly reduced. (F) p-RPS6 is eliminated when S1PR2, Rac1 or mTOR is inhibited.
(G-K) TFAP2C expression requires mTOR and S1P signaling. The expression of TFAP2C (G) is eliminated in the presence of mTOR inhibitors (H, I) and S1PR2 inhibitor JTE (J). (K) Quantitation of TFAP2C expression in embryos in which the mTOR and S1P pathways are inhibited.
(L) Quantitation shows that the morula to blastocyst transition is acutely sensitive to mTOR inhibitors, inhibitors of the S1P pathway and its downstream component Rac1, but is not affected by PI3K inhibitors.
(M-T) Role of glucose and S1P signaling in controlling Rac1 expression. In control, Rac1-GTP staining is observed only on the apical surface of the polar cells (M). Inhibition of Rac1 (N), S1P receptor (P), and S1P biosynthesis (Q) cause a significant reduction in Rac1-GTP staining. In contrast, inhibition of mTOR (O), lack of glucose during culture (R), or inhibition of specific arms of glucose metabolism (PPP and HBP) (S-T) has no effect on Rac1-GTP levels or localization.
Tunicamycin, a drug that blocks the HBP-related process of N-linked glycosylation affects the localization of apical membrane proteins (Olden et al., 1979) such as PARD6B (Figure S4K, L). However, this perturbation has no effect on either YAP1 nuclear localization or TFAP2C expression (Figure S4K–N) suggesting parallel and perhaps a more minor contribution of N-glycosylation compared to its O-glycosylation counterpart in the process of TE fate determination. Finally, it is interesting that YAP1 nuclear localization is affected by PPP. Our metabolomic analysis shows a high sensitivity of UDP-GlcNAc levels to PPP inhibition (Figure 3B). UTP is one of the most strongly affected metabolites upon PPP loss and is critically important for the generation of UDP-GlcNAc. We propose that the link between PPP and O-glycosylation is through this pathway’s ability to generate precursors for UDP-GlcNAc. We are able to demonstrate this to be the likely scenario by supplementing PPP inhibited embryos with uridine, a source of ribose that bypasses the PPP block to replenish UTP levels. However, we note that uridine can also rescue the levels of other nucleotides (Figure 3P). Addition of uridine to PPP inhibited embryos fully restores YAP1 nuclear localization (Figure 5O–Q), but not TFAP2C expression (Figure 5R–T). This further demonstrates the independent regulation of TFAP2C and YAP1. Finally, although we are not yet able to measure the level of O-glycosylation on any individual protein, we can demonstrate that bulk O-glycosylation levels in the embryo are not only sensitive to HBP, but also to PPP inhibition (Figure S4O–R). This supports our hypothesis that UDP-GlcNAc generation is critically dependent on the PPP.
We next investigated the mechanism for the control of TFAP2C by PPP. From ongoing RNA-seq profiling across developmental stages we find that Tfap2c mRNA level reaches a relatively high plateau that remains virtually unchanged between the 4-cell and blastocyst stages (not shown). Importantly, this mRNA expression pattern is not dependent on the presence of glucose in the medium (Figure S4A’). Moreover, EU incorporation results demonstrate that global transcription is not abolished in embryos cultured without glucose (Figure S4B’, C’). In contrast, the TFAP2C protein, barely detectable until the early 8-cell stage, rises abruptly and dramatically at the compacted morula stage (Figure S4S–V). Also, the TFAP2C protein is eliminated upon deprivation of glucose (Figure 5D–F). These results first suggested to us that the control of TFAP2C expression is likely to be post-transcriptional. To test this hypothesis, early 8-cell embryos were treated with α-amanitin to inhibit transcription or with cycloheximide to inhibit translation. We find that TFAP2C protein expression remains unaffected in the presence of α-amanitin but is eliminated by cycloheximide (Figure S4W–Z). This suggests that glucose metabolism is involved in translational control of Tfap2c at the compacted morula stage.
In search of a mechanism, we turned to an analysis of the mTOR pathway that regulates protein translation in response to nutrients (Kim and Guan, 2019). To determine if mTOR activity is sensitive to glucose availability, embryos are cultured in a glucose deprived medium from the zygote stage and the level of several mTOR targets assayed in the compacted morulae. In addition to immunofluorescence analysis, we also used western blots against several components of the mTOR pathway. A key phosphorylation target of mTOR is 4E-BP1, which enables CAP-dependent translation (Sonenberg and Hinnebusch, 2009). In embryos cultured without glucose, phospho-4E-BP1 expression is abolished (Figure 6A–E) as is a second target, phospho-ULK1 (Kim et al., 2011b) (Figure S5A, B). Finally, another critical target in the mTOR-related translational cascade is the ribosomal protein S6 (RPS6), and we find that its phosphorylation level is also highly glucose sensitive (Figure 6E, S5C, D). The sensitivity of mTOR activity to glucose is not related to HBP and is traced entirely to the PPP arm of glucose metabolism (Figure 6D).
Four independent mTOR inhibitors, Torin2 and INK128 (Figure 6L), as well as rapamycin and PP242 (not shown) cause a block in the transition from the compacted morula to the blastocyst (Figure 6L). Importantly, these mTOR inhibitors abolish TFAP2C (Figure 6G–I, K) and CDX2 (Figure S5E–G) expression, but OCT4 (Figure S5I–K) and YAP1 (Figure S5M–O) remain unaffected. The fact that YAP1 nuclear localization is not mTOR dependent lends further support to the idea that the PPP dependence of YAP1 is secondarily related to its role in UDP-GlcNAc generation.
Together, our analysis shows that PPP, but not HBP, is necessary to maintain TFAP2C protein levels, and glucose metabolism does not control Tfap2c transcription. PPP inhibition also causes a decrease in the activity of mTOR. Given the well described role of mTOR in translational control, and the data showing that mTOR inhibition also decreases TFAP2C protein levels, we propose a model in which glucose is metabolized through PPP and generates an as yet unidentified metabolite that activates mTOR and thereby translation of Tfap2c. Such a metabolite would represent a nutritional signal for mTOR activation. In this context, we note that amino acids that are typically associated with mTOR activation, or the metabolite SAM, are not glucose dependent (Figure 2I) and also that the PI3K/RTK/AKT dependent regulation of mTOR is not involved in this pathway at the 8-cell stage (Figure 6L).
The most sensitive set of metabolites to PPP inhibition are nucleotides, and there is precedence in the published literature on unrelated systems of mTOR inhibition upon attenuation of de novo nucleotide biosynthesis in general (Hoxhaj et al., 2017), and guanine nucleotides in particular (Emmanuel et al., 2017). De novo purine synthesis inhibitors (LTX, MTX, Miz, or AVN944), added to the culture medium at the 2-cell stage cause developmental arrest at the compacted morula stage (8–16 cells). LTX and MTX block early stages of purine synthesis and are expected to decrease both adenine and guanine nucleotide pools. Miz and AVN944 block only the synthesis of guanine nucleotides, and so will specifically deplete the guanine nucleotide pool (GMP, GDP, GTP) (Hoxhaj et al., 2017).
Importantly, the pan-purine nucleotide synthesis inhibitors and the specific inhibitors of guanine nucleotide synthesis also reduce mTOR activity and TFAP2C expression, but do not influence YAP1 nuclear localization (Figure S5Q–Y). The pyrimidine synthesis inhibitor leflunomide also causes a morula block, and a modest decrease in TFAP2C expression. Taken together the most parsimonious explanation is that glucose metabolism, via the PPP, is necessary for nucleotide synthesis, and that a decrease in purine nucleotides is sensed by the mTOR pathway that controls the translation of Tfap2c.
Activation of mTOR by a Signaling Lipid
The mechanism linking depletion in GTP and other purines to mTOR activity is likely to be complex and subject to a variety of stress response systems. We focused on defining other mechanisms of mTOR activation during normal development. A non-RTK driven mechanism for mTOR activation involves G-protein coupled receptors (GPCRs) (Puertollano, 2019; Wauson et al., 2013). Expression analysis reveals that a small but specific class of GPCRs that bind the signaling lipid sphingosine-1-phosphate (S1P) is highly represented in the transcriptome across preimplantation stages. These attracted our attention since S1P receptors are involved in mTOR activation (Liu et al., 2009; Liu et al., 2010; Maeurer et al., 2009; Taniguchi et al., 2012). Of the several S1P receptors expressed in the embryo, the receptor S1PR2 follows a profile that reaches peak expression level at the morula stage.
S1P binds the extracellular domain of S1PR2 and activates its intracellular signaling cascade (Pyne and Pyne, 2010). The compound JTE-013, is a specific antagonist that exclusively prevents S1P binding to S1PR2 (Ohmori et al., 2003; Osada et al., 2002; Parrill et al., 2004). When included in the culture medium at the late 2-cell stage and beyond, JTE-013 causes a compacted morula block, and a marked decrease in phospho-4E-BP1 (Figure S5Z) and phospho-RPS6 levels (Figure 6F). Moreover, S1PR2 inhibition causes loss of TFAP2C (Figure 6J, K) and CDX2 expression (Figure S5H), without affecting ICM markers (OCT4 shown in Figure S5L), or the nuclear localization of YAP1 (Figure S5P). We conclude that in addition to being downstream of the PPP, the mTOR/4E-BP1/RPS6/TFAP2C cascade is also downstream of the S1P/S1PR2 pathway.
Although S1P binds the extracellular domain of the receptor, no lipid is exogenously provided in the medium to initiate such a signal. We therefore reasoned that S1P is synthesized within the embryo, and locally transported out of the cell. Once extruded, this lipid is able to bind the cell-surface receptor. Beyond a threshold ligand concentration, this complex would engage an intracellular signal. Several genes involved in salvage or de novo synthesis of sphingosine as well as sphingosine kinase (SPHK that phosphorylates sphingosine to form S1P) are all very well represented in the transcriptome at this stage of development. Inhibition of SPHK causes phenotypes that are identical to those seen upon loss of S1PR2 and includes a morula block (Figure 6L), loss of p-4E-BP1 (Figure S5Z), and TFAP2C (Figure 6K). Thus, like glucose, the sphingolipid pathway also regulates mTOR to control TFAP2C.
The molecular details of the link between GPCRs and mTOR are not fully understood, although several past studies have placed Rac1 as an actin cytoskeleton based downstream target of S1P/S1PRs (Gonzalez et al., 2006; Kim et al., 2011a; Reinhard et al., 2017; Zhao et al., 2009). Furthermore, a biochemical analysis has proposed that Rac1 activates mTOR by altering its localization within the cell (Saci et al., 2011). The exact activation mechanism is not the central goal of our study and we focus here instead on testing whether Rac1 is a possible link between lipid signaling and mTOR activation in our system. Following our standard assays we determined that inactivation of Rac1 function (using the Rac1-GEF inhibitor NSC23766) leads to loss of p-RPS6 (Figure 6F), p-4E-BP1 (Figure S5Z), and TFAP2C (Figure 6K) and a failure to make the compact morula to blastocyst transition (Figure 6L).
The active, GTP bound form of Rac1 is detected at the apical surface of the 8-cell embryo cultured in the presence of glucose (Figure 6M) and as a control we demonstrate that Rac1-GTP staining is lost when the interaction between Rac1 and its GEF is inhibited (Gao et al., 2004) (Figure 6N). Rac1-GTP localization is unchanged when mTOR is inhibited (Figure 6O). Importantly however, expression of Rac1-GTP is completely lost when either S1PR2 or SPHK is inhibited (Figure 6P, Q). These results place Rac1 upstream of mTOR and as a downstream target of S1P/S1PR2.
Interestingly, in sharp contrast to the dependence of Rac1 on lipid signaling, absence of glucose, or inhibition of either PPP or HBP has no effect on Rac1-GTP expression (Figure 6R–T). In other words, Rac1-GTP expression is S1P signaling dependent and glucose signaling independent. Yet, loss of either S1P or glucose (PPP) generated signal causes loss of mTOR function and prevents mTOR-induced downstream translational control. We conclude that S1P/S1PR2/Rac1 and glucose/PPP provide parallel inputs into mTOR. Dual inputs usually regulate mTOR (Gonzalez and Hall, 2017). For the 8-cell embryo, these are the signals generated by a lipid and an unidentified glucose (PPP) metabolism byproduct.
Functional Integration of HBP and PPP Inputs through YAP1/TEAD4 and TFAP2C
The TE specific marker CDX2 is downstream of YAP1 and TFAP2C and published literature has shown that these two proteins participate in the direct transcriptional activation of Cdx2.
Consistent with these results, the proposed promoter and enhancer regions of Cdx2 contain both TEAD4 and TFAP2C binding sites (Cao et al., 2015; Rayon et al., 2014). TEAD4 is the DNA binding component of the YAP1/TEAD4 transcription complex (Li et al., 2010), and we wondered if TFAP2C functions separately or as part of a larger complex with YAP1/TEAD4. This question was addressed using co-immunoprecipitation assays. Due to the very limited availability of embryos, these experiments are performed in extracts of HEK293T cells that are transfected with and express uniquely tagged YAP1, TFAP2C and TEAD4 proteins. The results from the co-immunoprecipitation assays support a physical association between YAP1/TEAD4 and TFAP2C that would result in the formation of a larger transcription complex (Figure 7A–C). Interactions between proteins over-expressed in HEK293T cells are not always physiologically relevant. Therefore, we used a proximity ligation assay (PLA) as a reporter ((Bedzhov and Stemmler, 2015); see STAR methods) for an in situ interaction inside the normal embryo. As a control, we establish that the PLA assay can detect known heterodimer formation between YAP1 and TEAD4 and that this interaction is restricted to the TE but not the ICM cell population (Figure 7D–F). In the experimental set up, a PLA assay using antibodies against YAP1 and TFAP2C demonstrates that a similar protein level interaction is seen between YAP1 and TFAP2C, also restricted to the TE cells (Figure 7G–I).
Figure 7. YAP1/TEAD4 and TFAP2C form a complex.
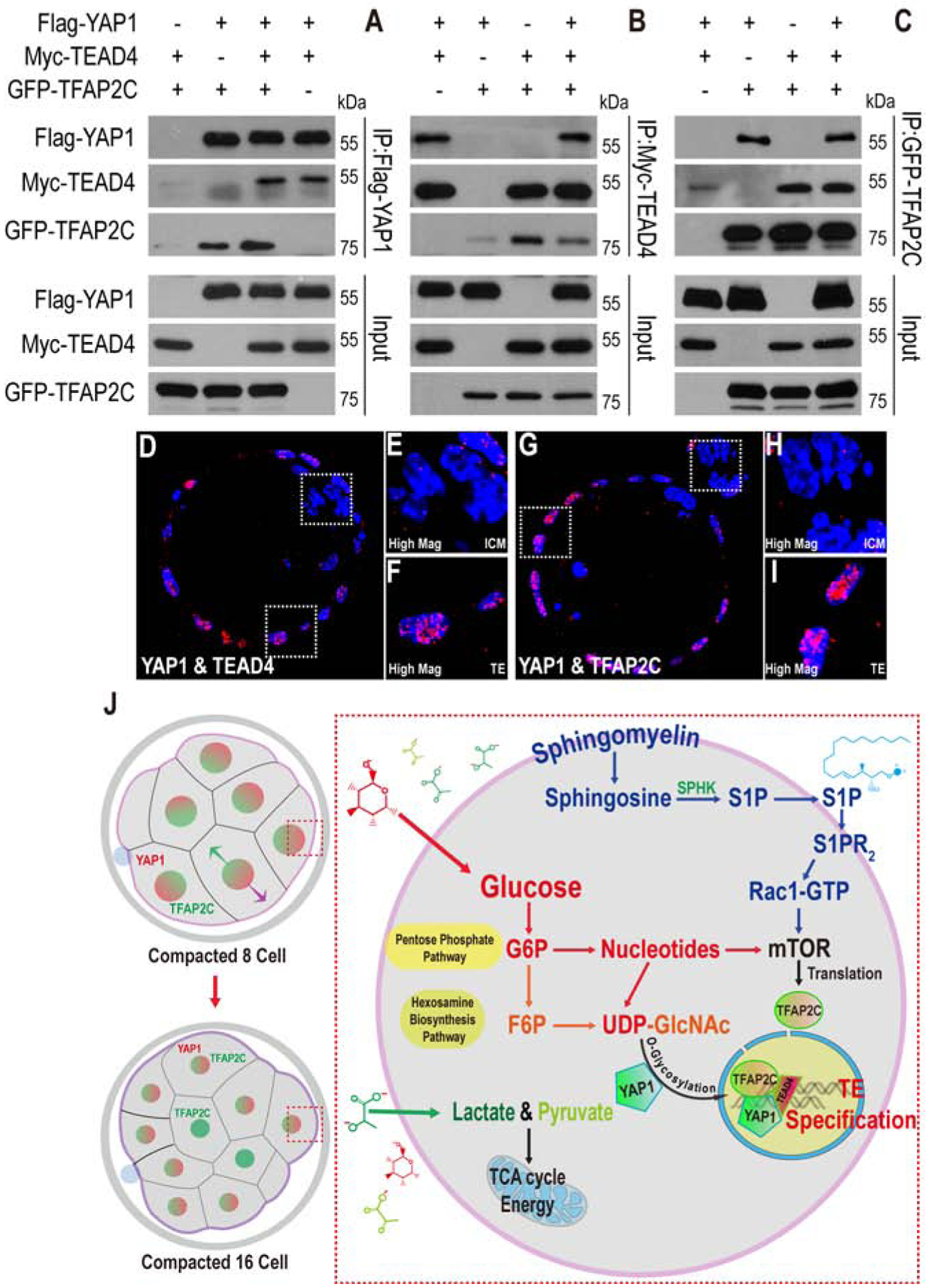
(A-C) Flag-YAP1, Myc-TEAD4, and GFP-TFAP2C are over-expressed in HEK293T cells for immunoprecipitation (IP) assay using specific antibodies as indicated. Anti-Flag antibody immunoprecipitates YAP1 (as expected) but also co-immunoprecipitates TFAP2C and TEAD4 (A). Likewise, immunoprecipitation with anti-Myc also results in the co-precipitation of TFAP2C and YAP1 (B) and anti-GFP co-precipitates both YAP1 and TEAD4 (C).
(D-I) Blastocyst embryos were harvested for Duolink PLA. (D-F) YAP1 and TEAD4 form complex in TE (E) but not in the ICM (F). (G-I) YAP1 and TFAP2C form complex in TE (H), but not in the ICM (I).
(J) A model for metabolic control of preimplantation development by glucose.
Discussion
A model that summarizes the analysis presented in this paper is presented in Figure 7J. Following formation of the 8-cell compacted morula, the next cell division generates apical and basal cells (Sasaki, 2017). This polarity arises independently of glucose and relies in part on mechanical forces (White et al., 2018). YAP1 is sequestered and activated in apical cells and its inactive form is degraded in basal cells (Rossant, 2018) by mechanisms that too are independent of any input from glucose. The basal cells, devoid of YAP1 function will eventually adopt an ICM fate.
In the apical cells, active YAP1 needs to be localized to the nucleus in order for it to be functional as a transcription factor, and this event of nuclear localization is glucose/HBP/O-linked glycosylation dependent. TFAP2C expression is translationally controlled by mTOR in all cells at the 8-cell stage. This process requires a dual input into the mTOR pathway by a signaling lipid, S1P, and by an unidentified PPP generated metabolite. The HBP plays no role in TFAP2C regulation. Although TFAP2C expression is ubiquitous at this stage, it forms a complex with YAP1/TEAD4 in only the apical cells that have all three proteins in the nucleus. The heterotrimer functions as a transcription factor, and activates TE-specific markers such as CDX2. Loss of Tead4 causes a similar loss of TE but not ICM markers (Cao et al., 2015; Yagi et al., 2007).
Amongst the metabolic curiosities of the system, we find that glucose and glycolysis do not function to generate energy since glucose does not fuel the TCA cycle and does not support anabolic pathways that are TCA cycle dependent. Pyruvate takes over these functions and the prominence of the lactate/pyruvate system over glucose for fulfilling bioenergetic needs is a metabolic signature of the preimplantation embryo. The importance of pyruvate has been emphasized in many classical studies (Biggers et al., 1965; Biggers et al., 1967; Brown and Whittingham, 1991; Whittingham and Biggers, 1967; Leese and Barton, 1984; Lane and Gardner, 2000; Barbehenn et al., 1974; O’Fallon and Wright, 1987). Combined with this current analysis, we conclude that the embryo, unlike cancer cells, utilizes only a small amount of glucose that is not used for bioenergetic functions. However, this small amount is necessary and sufficient for the signaling functions that drive an important developmental transition. Also in stark contrast with cancer cells (Locasale et al., 2011; Snell, 1984), glucose does not generate fatty acids and cholesterol, amino acids including glycine and serine or purine nucleobases.
Bioenergetic needs could be met by a plethora of metabolites, for example, pyruvate in the embryo and glutamine in many cancer cells. But only glucose provides anabolic and protein modification functions that are essential for lineage specification, differentiation and morphogenesis during the morula to blastocyst transition. Keeping the flux through glycolysis at a minimum allows glucose derivatives to be used for the PPP and the HBP that are otherwise minor arms of the pathway. The relatively free exchange of carbon between the metabolites generated in PPP, HBP and glycolysis in spite of the divergent function of each arm is a surprising finding of this study. It is worth investigating, if this extent of cross-talk occurs in other systems and whether low activity through the PFK-step of glycolysis is especially conducive to exchange of metabolite carbons between the arms.
Several unique features of metabolic control make the preimplantation mammalian embryo a superb system in which to study “Developmental Metabolism” or the metabolic control of normal development. The relative solitude of the pre-implantation embryo demands self-sufficiency and requires built-in metabolic mechanisms that compensate for the paucity of extracellularly triggered signal transduction events. The interplay between metabolic pathways and proliferation or differentiation has been studied extensively in the context of cancer cell expansion and tumor growth (Pavlova and Thompson, 2016; Kang et al., 2018) or in model systems (Mandal et al., 2005; Owusu-Ansah and Banerjee, 2009). As in cancer, metabolic and signaling pathways are closely intertwined during development, with the role of intrinsic metabolic pathways difficult to dissect away from interactions between cells and their environment. Any imbalance between these two fundamentally important sets of pathways would lead to developmental disorders and malignancies suggesting that “Developmental Metabolism” would provide valuable insights for “Cancer Metabolism”.
Experimental Model and Subject Details
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Utpal Banerjee (banerjee@mbi.ucla.edu).
Mouse embryo culture
All animal care and procedures used in this study are approved by the Animal Regulatory Committee (ARC) of the University of California at Los Angeles (UCLA).
Mouse zygotes and preimplantation embryos were collected from super-ovulated 4-week old C57BL/6J X C3He (Jackson Labs) F1 females. Mice were super-ovulated by peritoneal injection of 7.5 IU of PMSG (Pregnant Mare Serum Gonadotropin) to stimulate egg production, followed by 7.5 IU of hCG (human Chorionic Gonadotropin) 48h after PMSG. Embryos were obtained by mating the super-ovulated females with C57BL/6 X C3He F1 males. Mating was confirmed by the presence of the vaginal plug. For isolation of fertilized 1-cell zygotes, super-ovulated females were euthanized 18h post hCG and zygotes were dissected out of the ampulla in the oviduct. The embryo cumulus complexes were treated with 300μg/ml of hyaluronidase to disperse the cumulus cells, washed in mKSOM medium without pyruvate/glucose and transferred to the appropriate culture medium (+G or −G) and cultured at 37°C in 5% CO2. All mouse embryos used in this study were cultured in a modified KSOM medium whose composition is identical to KSOM in salts, glucose, lactate and pyruvate (95mM NaCl, 2.5mM KCl, 0.35mM KH2PO4, 0.20mM MgSO4, 25mM NaHCO3, 1.71mM CaCl2, 0.01 mM EDTA, 0.20mM glucose, 10mM lactate, 0.20mM pyruvate) but was devoid of all amino acids and BSA. The medium also contained 0.01% PVA (polyvinyl alcohol). At the 72h post hCG, an embryos growing in our mKSOM medium compacts and contains 8 cells. When grown in −G, the embryos are blocked at 8-cell stage. Most of the staining were performed at 78h post hCG, at which stage the embryos contains 12–16 cells.
Method Details
Antibody staining for immunofluorescence
Embryos were fixed in 4% paraformaldehyde for 30 min at room temperature, permeabilized for 30 min in PBS with 0.4% Triton (PBST), blocked in PBST with 3% albumin (PBSTA) for 30 min and incubated with the desired primary antibody in PBSTA overnight at 4°C. The following day the embryos were washed in PBST 4 times for 10 min each, blocked with PBSTA, incubated with the appropriate secondary antibody (1:500 dilution) and DAPI overnight at 4°C. Embryos were washed again 3 times for 10 min each in PBST, deposited on glass slides and mounted in Vectashield (Vector Laboratories) medium. Images were captured using Zeiss LSM700 or LSM880 confocal microscopes.
Inhibitor experiments
Most of the inhibitors were used to treat the embryos in the oil-free condition to avoid the oil absorption at the late 2-cell stage (at 50h post hCG). Early 4-cell stage embryos (56h post hCG) were treated with 20μg/ml WGA for 2 hours to induce premature compaction. Early 8-cell stage embryos (66h post hCG) were treated with 10μM of aphidicolin, 100μg/ml of α-amanitin, or 10μg/ml of cycloheximide for 12 hours (until 78h post hCG) in the oil-free condition. Cytochalasin D (CCD, 500nM) was added to the embryos at the early 8-cell stage (66h) for 8 hours to inhibit cleavage, then transferred to normal medium, allowing them compaction for 4 hours. The embryos treated with different drugs were fixed in 4% paraformaldehyde for 30 min for immunofluorescence.
Measurement of metabolite levels
The metabolites are measured using the procedure described previously (Nagaraj et al., 2017; Sullivan et al., 2018). For samples analyzed under different conditions, embryos were very briefly washed in ice-cold 150mM ammonium acetate, and transferred into 80% methanol. Dried metabolites were re-suspended in 20μl 50% ACN and 5μl was injected for chromatographic separation on the UltiMate 3000 RSLC (Thermo Scientific) UHPLC system which is coupled to a Thermo Scientific Q Exactive that is run in polarity switching mode. For the UHPLC separation, the mobile phase comprised (A) 5mM NH4AcO, pH 9.9, and (B) ACN. A Luna 3mm NH2 100A (150 × 2.0mm) (Phenomenex) column was used with a 300 μL/min flow-rate. The gradient ran from 15% A to 95% A in 18 min, followed by an isocratic step for 9 min and re-equilibration for 7 min. The remainder of the sample was diluted 2.67-fold and 10μl were applied to a Thermo Scientific Ion Chromatography System (ICS) 5000 that is also coupled to a Thermo Scientific Q Exactive run in negative polarity mode. The gradient ran from 10mM to 90mM KOH over 25 min with a flow rate of 350μl/min. The settings for the HESI-II source were: S-lens 50, Sheath Gas 18, Aux Gas 4, spray heater 320°C, and spray voltag e −3.2 kV. Metabolites were identified based on accurate mass (±3 ppm) and retention times of pure standards. Relative amounts of metabolites and contribution of 13C labeled nutrients (following correction for natural abundance) were quantified using TraceFinder 3.3. The data were analyzed using the Prism software package.
Trim-Away Experiment
The Trim-Away procedure was performed as previously described in the published literature with minor modifications (Clift et al., 2018). mCherry-Trim21 mRNA was in vitro transcribed using the HiScribe™ T7 ARCA mRNA Kit according to the manufacturer’s instructions. Specific antibodies in carrier-free solution (PBS) were purchased from CST or Abcam as listed above, with the stock concentration 2mg/ml. Then the mCherry-Trim21 mRNA (final concentration: 400ng/μl) and specific antibody (final concentration: 1mg/ml) were mixed with each other, and the detergent NP-40 so that the final concentration of NP-40 is 0.01%. The mixture is then microinjected into 1-cell (20h hCG) and 2-cell stage (46h hCG) embryos using a FemtoJet microinjector. A negative capacitance is generated using micro-ePORE (World Precision Instruments) to assist the 2-cell microinjection (Gu et al., 2018; Zernicka-Goetz et al., 1997). The developmental progression of these Trim-Away embryos was monitored until 114h post hCG or the embryos were fixed at the morula stage for further immunofluorescence analysis.
Aggregation of chimeric embryos
Embryos were labeled by microinjecting 100ng/μl GFP mRNA at 1-cell stage (20h hCG), and then cultured in +G or −G medium until the morula stage (76h hCG). Unlabeled embryos were cultured in +G medium until the compacted morula stage (76h hCG) as aggregation partners. The zona pellucida was removed by incubating each group of embryos in pre-warmed acidic tyrode’s solution for 30 seconds at 37°C. Zona-free embryos from GFP labeled (−G, or +G in the control group) and unlabeled (+G in both control and experimental) groups were immediately aggregated and cultured in small drops (10μl) of −G medium. The drops were covered with mineral oil. Each small drop of the −G medium contains a tiny well that is created by pressing and rotating the tip of a ball-point pen on the surface of the tissue culture dishes. The significance of the difference in the propensity to form TE and ICM cells was determined using a 2×2 contingency table that contained the number GFP labeled and unlabeled cells in each population and was analyzed using Fisher’s exact test.
Cell culture, transfection, immunoprecipitation
HEK293T cells were cultured in DMEM (Invitrogen) containing 10% FBS (Invitrogen) and 50 μg/ml penicillin/streptomycin (P/S). Transfection with Lipofectamine (Invitrogen) was performed according to the manufacturer’s instructions.
For immunoprecipitation, HEK293T cells were transfected with the indicated plasmids when cells reached 60%–80% confluence. 48h post-transfection, cells were lysed with mild lysis buffer (50mM HEPES at pH 7.5, 150mM NaCl, 1mM EDTA, 1% NP-40, 10mM pyrophosphate, 10mM glycerophosphate, 50mM NaF, 1.5mM Na3VO4, protease inhibitor mixture (Roche), 1mM DTT, 1mM PMSF). Anti-Flag or anti-Myc antibodies were added to the cell lysate and incubated at 4°C for 2h to bind Flag-YAP1 or Myc-TEAD4, and t hen Pierce Protein A/G agarose beads were added to precipitate the antibody-substrate complex at 4°C for 2h. GFP-Trap agarose beads were directly added to the cell lysis to precipitate GFP-TFAP2C. The immuno-precipitates were washed three times with mild lysis buffer and boiled with SDS sample buffer before analysis by western blot. The size of the products on the western blot were determined by reference to a protein molecular weight ladder on the PVDF membrane.
Duolink Proximity Ligation Assay (PLA)
A proximity ligation assay was performed by using the Duolink In Situ Red Starter Kit (Millipore Sigma) to detect the binding between YAP1 and TFAP2C proteins according to the manufacturer’s instructions. YAP1/TEAD4 antibodies were used as a positive control. Blastocyst embryos were fixed with 4% paraformaldehyde for 30 min at room temperature, permeabilized for 30 min in PBST, and blocked in pre-heated Duolink blocking solution for 60 min at 37°C. YAP1/TEAD4 or YAP1/ TFAP2C primary antibodies were diluted at 1:50 in the Duolink antibody diluent, and embryos were incubated in these primary antibodies solution at overnight at 4°C. Then, PLA probe incubation, ligation, amplification processes were performed following the manufacturer’s protocol. Finally, embryos were deposited on glass slides and mounted in Vectashield (Vector Laboratories) medium. Images were captured using a Zeiss LSM880 confocal microscope.
Quantification and Statistical Analysis
Statistical parameters are reported in the figures and figure legends. Data is considered significant if p < 0.05. Statistical analysis was performed using GraphPad Prism software.
Data and Code Availability
This study did not generate datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-PKM2 (D78A4) | Cell Signaling | Cat #4053 |
| Rabbit monoclonal anti-G6PD (D5D2) | Cell Signaling | Cat #12263 |
| Rabbit monoclonal anti-GFPT1 (EPR4854) | Abcam | Cat #ab236053 |
| Mouse IgG1 monoclonal isotype control (MG1–45) | Abcam | Cat #ab18447 |
| Rabbit IgG monoclonal isotype control (EPR25A) | Abcam | Cat #ab199376 |
| Mouse monoclonal anti-CDX2 (CDX2–88) | Biogenex | Cat #MU392A-UC |
| Goat anti-OCT4 | Santa Cruz | Cat #sc-8628 |
| Rabbit anti-NANOG | ReproCELL | Cat #RCAB0002P-F |
| Goat anti-SOX2 | R&D Systems | Cat #AF2018 |
| Mouse monoclonal anti-YAP (2F12) | Abnova | Cat #H00010413-M01 |
| Mouse monoclonal anti-TFAP2C (6E4/4) | Santa Cruz | Cat #sc-12762 |
| Rabbit anti-TFAP2C | Santa Cruz | Cat #sc-8977 |
| Rabbit anti-p-Ezrin (T567)/Radixin (T564)/Moesin (T558) | Cell Signaling | Cat #3141 |
| Rabbit anti-SBNO1 | Abcam | Cat #ab122789 |
| Rabbit anti-GATA3 | Santa Cruz | Cat #sc-9009 |
| Rabbit anti-PARD6B | Santa Cruz | Cat #sc-67393 |
| Mouse monoclonal anti-O-Linked N-Acetylglucosamine (RL2) | Abcam | Cat #ab2739 |
| Rabbit monoclonal anti-pT37/46–4E-BP1 (236B4) | Cell Signaling | Cat #2855S |
| Rabbit monoclonal anti-pS235/236-RPS6 (D57.2.2E) | Cell Signaling | Cat #4858 |
| Rabbit monoclonal anti-mTOR (7C10) | Cell Signaling | Cat #2983 |
| Rabbit monoclonal anti-Tuberin/TSC2 (D93F12) | Cell Signaling | Cat #4308 |
| Rabbit monoclonal anti-pS473-AKT (Ser473) (D9E) | Cell Signaling | Cat #4060 |
| Rabbit anti-pS127-YAP | Cell Signaling | Cat #4911 |
| Rabbit anti-pS757-ULK1 | Cell Signaling | Cat #6888 |
| Mouse monoclonal anti-active Rac1-GTP | NewEast | Cat #26903 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lectin from Triticum vulgaris (wheat)–WGA (20μg/ml) | Sigma-Aldrich | Cat # L0636 |
| YZ9 (1μM) | Cayman Chemical | Cat #15352 |
| ST045849 (0.8μM) | TimTec | Cat #ST045849 |
| Tunicamycin (5μM) | Sigma-Aldrich | Cat #T7765 |
| 6-Aminonicotinamide (6-AN) (2μM) | Sigma-Aldrich | Cat #A68203 |
| N-Acetylcysteine (NAC) (1mM) | Sigma-Aldrich | Cat #A7250 |
| Hypotaurine (1mM) | Sigma-Aldrich | Cat #H1384 |
| Shikonin (1μM) | Cayman Chemical | Cat #14751 |
| Azaserine (5μM) | Sigma-Aldrich | Cat #A4142 |
| 6-Diazo-5-oxo-L-norleucine (DON) (20μM) | Sigma-Aldrich | Cat #D2141 |
| 2-Deoxy-D-glucose (1μM) | Sigma-Aldrich | Cat #D8375 |
| Aphidicolin (10μM) | Cayman Chemical | Cat #14007 |
| Cytochalasin D (500nM) | Cayman Chemical | Cat #11330 |
| α-Amanitin (100μg/ml) | Cayman Chemical | Cat #17898 |
| Cycloheximide (10μg/ml) | Cayman Chemical | Cat #14126 |
| Torin2 (10nM) | Cayman Chemical | Cat #14185 |
| INK128 (100nM) | Cayman Chemical | Cat #11811 |
| JTE013 (1μM) | Tocris | Cat #2392 |
| SKI-II (0.8μM) | Echelon | Cat #B-0024 |
| NSC26722 (100μM) | Tocris | Cat #2161 |
| BYL719 (1μM) | Cayman Chemical | Cat #16986 |
| BKM120 (0.5μM) | Cayman Chemical | Cat #11587 |
| GDC0941 (0.5μM) | Cayman Chemical | Cat #11600 |
| TGX221 (0.5μM) | Cayman Chemical | Cat #10007349 |
| BX912 (100nM) | Cayman Chemical | Cat #14708 |
| GSK2334470 (50nM) | Cayman Chemical | Cat #18095 |
| MK2206 (200nM) | Cayman Chemical | Cat #11593 |
| Lometrexol (250–500nM) | Cayman Chemical | Cat #18049 |
| Methotrexate (200nM) | Cayman Chemical | Cat #13960 |
| Mizoribine (1μM) | Cayman Chemical | Cat #23128 |
| AVN-944 (50nM) | Cayman Chemical | Cat #21284 |
| Leflunomide (5μM) | Cayman Chemical | Cat #14860 |
| U-13C6-D-Glucose (200μM) | Cambridge Isotope Laboratories | Cat #CLM-1396-PK |
| U-13C3-Pyruvic Acid Sodium Salt (200μM) | Toronto Research Chemicals | Cat #P998904 |
| U-13C3-L-(+)-Lactic Acid Sodium Salt (5mM) | Toronto Research Chemicals | Cat #L113507 |
| [1', 2', 3', 4', 5'-13C5]-Uridine (200μM) | Omicron | Cat #NUC-034 |
| U-13C6-D-Glucosamine hydrochloride | Omicron | Cat #GLC-091 |
| Pregnant Mare Serum Gonadotropin (PMSG) | ProSpec | Cat #hor-272 |
| Chorionic gonadotropin human (hCG) | Sigma-Aldrich | Cat #CG10 |
| Critical Commercial Assays | ||
| Click-iT™ RNA Alexa Fluor™ 594 Imaging Kit | Thermo Scientific | Cat #C10330 |
| Duolink® In Situ PLA® Probe Anti-Rabbit PLUS | Sigma-Aldrich | Cat #DUO92002 |
| Duolink® In Situ PLA® Probe Anti-Mouse MINUS | Sigma-Aldrich | Cat #DUO92004 |
| Duolink® In Situ Detection Reagents Red | Sigma-Aldrich | Cat #DUO92008 |
| Duolink® In Situ Wash Buffers, Fluorescence | Sigma-Aldrich | Cat #DUO82049 |
| Experimental Models: Cell Lines | ||
| HEK293T | This lab | N/A |
| Mouse: embryo culture | This lab | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6C3F1/J | Jackson Laboratory | Stock No: 100010 |
| Oligonucleotides | ||
| CleanCap EGFP mRNA (5moU) | TriLink | Cat #L-7201 |
| mCherry-mTrim21 mRNA | This paper | N/A |
| Recombinant DNA | ||
| pGEMHE-mCherry-mTrim21 | Addgene | Cat #105522 |
| pCMV6-Yap1 | OriGene | Cat #MR226049 |
| pCMV6-Tead4 (ATT) | OriGene | Cat #MR219506 |
| pCMV6-Tfap2c | OriGene | Cat #MR207174 |
| pCMV6-Flag-Yap1 | This paper | N/A |
| pCMV6-Myc-Tead4 (ATG) | This paper | N/A |
| pQCMV-GFP-Tfap2c | This paper | N/A |
| Software and Algorithms | ||
| Graphpad Prism 7 | GraphPad | http://www.graphpad.com/ |
| TraceFinder 3.3 | Thermo Scientific | N/A |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| MetaboAnalyst | (Xia and Wishart, 2011) | https://www.metaboanalyst.ca/ |
| Other | ||
| WPI MICRO-ePORE | World Precision Instruments | N/A |
Highlights.
TE formation in mouse embryos is controlled by glucose metabolism activating CDX2.
Glycolysis is dispensible and glucose is not required for amino acid or lipid synthesis.
Nucleotides synthesized from glucose, along with S1P, activate mTOR and TFAP2C.
Glucose metabolism by the HBP controls YAP1 which, together with TFAP2C, activates CDX2.
Acknowledgments
We thank members of our laboratory for suggestions and support. Yonggang Zhou for RNA seq and Daniel Braas, Johanna ten Hoeve-Scott, Tom Graeber and the UCLA Metabolomics Center for help in metabolomic analysis. Heather Christofk and Hilary Coller provided reagents and advice. We thank Huachun Liu, Wei Liao, Qin An and Wanlu Liu for help with molecular techniques, sequencing and bioinformatic analyses. Bin Gu at Sick Kids, Toronto and Bin Zhao at Zhejiang University provided valuable input.
This work was supported by the NIH Director’s Pioneer Award to U.B. (DP1DK098059). U.B. is supported by MOD (1-FY17-788). and NCI (R01CA217608). F.C. by a China Scholarship Council Award and a California Institute for Regenerative Medicine pre-doctoral fellowship. We are grateful to Owen Witte and the Broad Stem Cell Research Center for several innovation awards and for continued support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Auman HJ, Nottoli T, Lakiza O, Winger Q, Donaldson S, and Williams T (2002). Transcription factor AP-2γ is essential in the extra-embryonic lineages for early postimplantation development. Development 129, 2733. [DOI] [PubMed] [Google Scholar]
- Barbehenn EK, Wales RG, and Lowry OH (1974). The explanation for the blockade of glycolysis in early mouse embryos. Proc Natl Acad Sci U S A 71, 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedzhov I, and Stemmler MP (2015). Applying the proximity ligation assay (PLA) to mouse preimplantation embryos for identifying protein-protein interactions in situ. Methods Mol Biol 1233, 57–64. [DOI] [PubMed] [Google Scholar]
- Biggers JD, Moore BD, and Whittingham DG (1965). Development of mouse embryos in vivo after cultivation from two-cell ova to blastocysts in vitro. Nature 206, 734–735. [DOI] [PubMed] [Google Scholar]
- Biggers JD, Summers MC, and McGinnis LK (1997). Polyvinyl alcohol and amino acids as substitutes for bovine serum albumin in culture media for mouse preimplantation embryos. Hum Reprod Update 3, 125–135. [DOI] [PubMed] [Google Scholar]
- Biggers JD, Whittingham DG, and Donahue RP (1967). The pattern of energy metabolism in the mouse oocyte and zygote. Proc Natl Acad Sci U S A 58, 560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL (1967). Carbon dioxide production from glucose by the preimplantation mouse embryo. Experimental cell research 47, 271–277. [DOI] [PubMed] [Google Scholar]
- Brown JJ, and Whittingham DG (1991). The roles of pyruvate, lactate and glucose during preimplantation development of embryos from F1 hybrid mice in vitro. Development 112, 99–105. [DOI] [PubMed] [Google Scholar]
- Cao Z, Carey TS, Ganguly A, Wilson CA, Paul S, and Knott JG (2015a). Transcription factor AP-2gamma induces early Cdx2 expression and represses HIPPO signaling to specify the trophectoderm lineage. Development 142, 1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casser E, Israel S, Witten A, Schulte K, Schlatt S, Nordhoff V, and Boiani M (2017). Totipotency segregates between the sister blastomeres of two-cell stage mouse embryos. Scientific reports 7, 8299–8299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazaud C, and Yamanaka Y (2016). Lineage specification in the mouse preimplantation embryo. Development 143, 1063–1074. [DOI] [PubMed] [Google Scholar]
- Chen J, Xie J, Jiang Z, Wang B, Wang Y, and Hu X (2011). Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 30, 4297–4306. [DOI] [PubMed] [Google Scholar]
- Chen W, and Gueron M (1992). The inhibition of bovine heart hexokinase by 2-deoxy-D-glucose-6-phosphate: characterization by 31P NMR and metabolic implications. Biochimie 74, 867–873. [DOI] [PubMed] [Google Scholar]
- Clegg KB, and Piko L (1977). Size and specific activity of the UTP pool and overall rates of RNA synthesis in early mouse embryos. Dev Biol 58, 76–95. [DOI] [PubMed] [Google Scholar]
- Clift D, McEwan WA, Labzin LI, Konieczny V, Mogessie B, James LC, and Schuh M (2017). A Method for the Acute and Rapid Degradation of Endogenous Proteins. Cell 171, 1692–1706.e1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clough JR, and Whittingham DG (1983). Metabolism of [14C]glucose by postimplantation mouse embryos in vitro. J Embryol Exp Morphol 74, 133–142. [PubMed] [Google Scholar]
- Downs SM HP, Leese HJ (1998). Meiotic induction in cumulus cell-enclosed mouse oocytes: involvement of the pentose phosphate pathway. Biol Reprod 58, 1084–1094. [DOI] [PubMed] [Google Scholar]
- Emmanuel N, Ragunathan S, Shan Q, Wang F, Giannakou A, Huser N, Jin G, Myers J, Abraham RT, and Unsal-Kacmaz K (2017). Purine Nucleotide Availability Regulates mTORC1 Activity through the Rheb GTPase. Cell Reports 19, 2665–2680. [DOI] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371 e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridhandler L (1961). Pathways of glucose metabolism in fertilized rabbit ova at various pre-implantation stages. Experimental cell research 22, 303–316. [DOI] [PubMed] [Google Scholar]
- Fridhandler L, Wastila WB, and Palmer WM (1967). The Role of Glucose in Metabolism of the Developing Mammalian Preimplantation Conceptus. Fertility and Sterility 18, 819–830. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, and Zheng Y (2004). Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A 101, 7618–7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A, and Hall MN (2017). Nutrient sensing and TOR signaling in yeast and mammals. EMBO J 36, 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, Kou R, and Michel T (2006). Rac1 modulates sphingosine 1-phosphate-mediated activation of phosphoinositide 3-kinase/Akt signaling pathways in vascular endothelial cells. The Journal of biological chemistry 281, 3210–3216. [DOI] [PubMed] [Google Scholar]
- Gross BJ, Kraybill BC, and Walker S (2005). Discovery of O-GlcNAc transferase inhibitors. J Am Chem Soc 127, 14588–14589. [DOI] [PubMed] [Google Scholar]
- Home P, Ray S, Dutta D, Bronshteyn I, Larson M, and Paul S (2009). GATA3 is selectively expressed in the trophectoderm of peri-implantation embryo and directly regulates Cdx2 gene expression. The Journal of biological chemistry 284, 28729–28737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoxhaj G, Hughes-Hallett J, Timson RC, Ilagan E, Yuan M, Asara JM, Ben-Sahra I, and Manning BD (2017). The mTORC1 Signaling Network Senses Changes in Cellular Purine Nucleotide Levels. Cell reports 21, 1331–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh B, Chi MM, Knor J, and Lowry OH (1979). Enzymes of glycogen metabolism and related metabolites in preimplantation mouse embryos. Dev Biol 72, 342–349. [DOI] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Z, Yanxiang Guo J, et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LV (1986). Wheat germ agglutinin induces compaction- and cavitation-like events in two-cell mouse embryos. Dev Biol 113, 1–9. [DOI] [PubMed] [Google Scholar]
- Johnson MT, Freeman EA, Gardner DK, and Hunt PA (2007). Oxidative Metabolism of Pyruvate Is Required for Meiotic Maturation of Murine Oocytes In Vivo1. Biology of Reproduction 77, 2–8. [DOI] [PubMed] [Google Scholar]
- Johnson MT, Mahmood S, Hyatt SL, Yang H-S, Soloway PD, Hanson RW, and Patel MS (2001). Inactivation of the Murine Pyruvate Dehydrogenase (Pdha1) Gene and Its Effect on Early Embryonic Development. Molecular Genetics and Metabolism 74, 293–302. [DOI] [PubMed] [Google Scholar]
- Johnson MT, Mahmood S, and Patel MS (2003). Intermediary metabolism and energetics during murine early embryogenesis. The Journal of biological chemistry 278, 31457–31460. [DOI] [PubMed] [Google Scholar]
- Kang YP, Ward NP, and DeNicola GM (2018). Recent advances in cancer metabolism: a technological perspective. Experimental & Molecular Medicine 50, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ES, Kim JS, Kim SG, Hwang S, Lee CH, and Moon A (2011a). Sphingosine 1-phosphate regulates matrix metalloproteinase-9 expression and breast cell invasion through S1P3-Galphaq coupling. J Cell Sci 124, 2220–2230. [DOI] [PubMed] [Google Scholar]
- Kim J, and Guan K-L (2019). mTOR as a central hub of nutrient signalling and cell growth. Nature Cell Biology 21, 63–71. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, and Guan KL (2011b). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler E, Barrach H, and Neubert D (1970). Inhibition of NADP dependent oxidoreductases by the 6-aminonicotinamide analogue of NADP. FEBS Lett 6, 225–228. [DOI] [PubMed] [Google Scholar]
- Kuckenberg P, Kubaczka C, and Schorle H (2012). The role of transcription factor Tcfap2c/TFAP2C in trophectoderm development. Reproductive BioMedicine Online 25, 12–20. [DOI] [PubMed] [Google Scholar]
- Landau BR, and Wahren J (1992). Nonproductive exchanges: the use of isotopes gone astray. Metabolism 41, 457–459. [DOI] [PubMed] [Google Scholar]
- Lane M, and Gardner DK (2000). Lactate regulates pyruvate uptake and metabolism in the preimplantation mouse embryo. Biol Reprod 62, 16–22. [DOI] [PubMed] [Google Scholar]
- Lane M, and Gardner DK (2005). Mitochondrial malate-aspartate shuttle regulates mouse embryo nutrient consumption. The Journal of biological chemistry 280, 18361–18367. [DOI] [PubMed] [Google Scholar]
- Lange K, and Proft ER (1970). Inhibition of the 6-phosphogluconate dehydrogenase in the rat kidney by 6-aminonicotinamide. Naunyn Schmiedebergs Arch Pharmakol 267, 177–180. [DOI] [PubMed] [Google Scholar]
- Leese HJ, and Barton AM (1984). Pyruvate and glucose uptake by mouse ova and preimplantation embryos. Journal of reproduction and fertility 72, 9–13. [DOI] [PubMed] [Google Scholar]
- Leung CY, Zhu M, and Zernicka-Goetz M (2016). Polarity in Cell-Fate Acquisition in the Early Mouse Embryo. Curr Top Dev Biol 120, 203–234. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, Guan KL, and Xu Y (2010). Structural insights into the YAP and TEAD complex. Genes Dev 24, 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Burns S, Huang G, Boyd K, Proia RL, Flavell RA, and Chi H (2009). The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol 10, 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Yang K, Burns S, Shrestha S, and Chi H (2010). The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nat Immunol 11, 1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. (2011). Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 43, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeurer C, Holland S, Pierre S, Potstada W, and Scholich K (2009). Sphingosine-1-phosphate induced mTOR-activation is mediated by the E3-ubiquitin ligase PAM. Cell Signal 21, 293–300. [DOI] [PubMed] [Google Scholar]
- Mandal S, Guptan P, Owusu-Ansah E, and Banerjee U (2005). Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev Cell 9, 843–854. [DOI] [PubMed] [Google Scholar]
- Martin KL, and Leese HJ (1995). Role of glucose in mouse preimplantation embryo development. 40, 436–443. [DOI] [PubMed] [Google Scholar]
- Mattaini KR, Sullivan MR, and Vander Heiden MG (2016). The importance of serine metabolism in cancer. The Journal of cell biology 214, 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj R, Sharpley MS, Chi F, Braas D, Zhou Y, Kim R, Clark AT, and Banerjee U (2017). Nuclear Localization of Mitochondrial TCA Cycle Enzymes as a Critical Step in Mammalian Zygotic Genome Activation. Cell 168, 210–223 e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka N, Inoue K. i., Adachi K, Kiyonari H, Ota M, Ralston A, Yabuta N, Hirahara S, Stephenson RO, Ogonuki N, et al. (2009). The Hippo Signaling Pathway Components Lats and Yap Pattern Tead4 Activity to Distinguish Mouse Trophectoderm from Inner Cell Mass. Developmental Cell 16, 398–410. [DOI] [PubMed] [Google Scholar]
- O’Fallon JV, and Wright RW Jr. (1987). Calculation of the pentose phosphate and Embden-Myerhoff pathways from a single incubation with [U-14C]- and [5–3H]glucose. Anal Biochem 162, 33–38. [DOI] [PubMed] [Google Scholar]
- Ohmori T, Yatomi Y, Osada M, Kazama F, Takafuta T, Ikeda H, and Ozaki Y (2003). Sphingosine 1-phosphate induces contraction of coronary artery smooth muscle cells via S1P2. Cardiovasc Res 58, 170–177. [DOI] [PubMed] [Google Scholar]
- Olden K, Pratt RM, Jaworski C, and Yamada KM (1979). Evidence for role of glycoprotein carbohydrates in membrane transport: specific inhibition by tunicamycin. Proc Natl Acad Sci U S A 76, 791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osada M, Yatomi Y, Ohmori T, Ikeda H, and Ozaki Y (2002). Enhancement of sphingosine 1-phosphate-induced migration of vascular endothelial cells and smooth muscle cells by an EDG-5 antagonist. Biochem Biophys Res Commun 299, 483–487. [DOI] [PubMed] [Google Scholar]
- Owusu-Ansah E, and Banerjee U (2009). Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature 461, 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaleon M, Scott J, and Kaye P (2008). Nutrient Sensing by the Early Mouse Embryo: Hexosamine Biosynthesis and Glucose Signaling During Preimplantation Development, Vol 78. [DOI] [PubMed] [Google Scholar]
- Parrill AL, Sardar VM, and Yuan H (2004). Sphingosine 1-phosphate and lysophosphatidic acid receptors: agonist and antagonist binding and progress toward development of receptor-specific ligands. Semin Cell Dev Biol 15, 467–476. [DOI] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C, Zhu Y, Zhang W, Liao Q, Chen Y, Zhao X, Guo Q, Shen P, Zhen B, Qian X, et al. (2017). Regulation of the Hippo-YAP Pathway by Glucose Sensor O-GlcNAcylation. Molecular Cell 68, 591–604.e595. [DOI] [PubMed] [Google Scholar]
- Puertollano R (2019). GPCRs join the mTORC1 regulatory network. Nat Cell Biol 21, 538–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne NJ, and Pyne S (2010). Sphingosine 1-phosphate and cancer. Nat Rev Cancer 10, 489–503. [DOI] [PubMed] [Google Scholar]
- Ralston A, Cox BJ, Nishioka N, Sasaki H, Chea E, Rugg-Gunn P, Guo G, Robson P, Draper JS, and Rossant J (2010). Gata3 regulates trophoblast development downstream of Tead4 and in parallel to Cdx2. Development 137, 395–403. [DOI] [PubMed] [Google Scholar]
- Rayon T, Menchero S, Nieto A, Xenopoulos P, Crespo M, Cockburn K, Canon S, Sasaki H, Hadjantonakis AK, de la Pompa JL, et al. (2014). Notch and hippo converge on Cdx2 to specify the trophectoderm lineage in the mouse blastocyst. Dev Cell 30, 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard NR, Mastop M, Yin T, Wu Y, Bosma EK, Gadella TWJ Jr., Goedhart J, and Hordijk PL (2017). The balance between Galphai-Cdc42/Rac and Galpha12/13-RhoA pathways determines endothelial barrier regulation by sphingosine-1-phosphate. Mol Biol Cell 28, 3371–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossant J (2018). Genetic Control of Early Cell Lineages in the Mammalian Embryo. Annu Rev Genet 52, 185–201. [DOI] [PubMed] [Google Scholar]
- Saci A, Cantley LC, and Carpenter CL (2011). Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Molecular cell 42, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H (2017). Roles and regulations of Hippo signaling during preimplantation mouse development. Development, Growth & Differentiation 59, 12–20. [DOI] [PubMed] [Google Scholar]
- Seo M, Kim J-D, Neau D, Sehgal I, and Lee Y-H (2011). Structure-based development of small molecule PFKFB3 inhibitors: a framework for potential cancer therapeutic agents targeting the Warburg effect. PLoS One 6, e24179–e24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayoshi Y, Okada TS, and Takeichi M (1983). The calcium-dependent cell-cell adhesion system regulates inner cell mass formation and cell surface polarization in early mouse development. Cell 35, 631–638. [DOI] [PubMed] [Google Scholar]
- Snell K (1984). Enzymes of serine metabolism in normal, developing and neoplastic rat tissues. Adv Enzyme Regul 22, 325–400. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, and Hinnebusch AG (2009). Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindle A (1982). Cell allocation in preimplantation mouse chimeras. J Exp Zool 219, 361–367. [DOI] [PubMed] [Google Scholar]
- Strumpf D, Mao CA, Yamanaka Y, Ralston A, Chawengsaksophak K, Beck F, and Rossant J (2005). Cdx2 is required for correct cell fate specification and differentiation of trophectoderm in the mouse blastocyst. Development 132, 2093–2102. [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Kitatani K, Kondo T, Hashimoto-Nishimura M, Asano S, Hayashi A, Mitsutake S, Igarashi Y, Umehara H, Takeya H, et al. (2012). Regulation of autophagy and its associated cell death by “sphingolipid rheostat”: reciprocal role of ceramide and sphingosine 1-phosphate in the mammalian target of rapamycin pathway. The Journal of biological chemistry 287, 39898–39910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski AK (1961). Mouse chimaeras developed from fused eggs. Nature 190, 857–860. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Miyasaka KY, Kubo A, Kida YS, Nakagawa O, Hirate Y, Sasaki H, and Ogura T (2017). Notch and Hippo signaling converge on Strawberry Notch 1 (Sbno1) to synergistically activate Cdx2 during specification of the trophectoderm. Scientific reports 7, 46135–46135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson AJ, and Kidder GM (1988). Immunofluorescence assessment of the timing of appearance and cellular distribution of Na/K-ATPase during mouse embryogenesis. Developmental Biology 126, 80–90. [DOI] [PubMed] [Google Scholar]
- Wauson EM, Lorente-Rodríguez A, and Cobb MH (2013). Minireview: Nutrient sensing by G protein-coupled receptors. Molecular endocrinology (Baltimore, Md) 27, 1188–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MD, Zenker J, Bissiere S, and Plachta N (2018). Instructions for Assembling the Early Mammalian Embryo. Dev Cell 45, 667–679. [DOI] [PubMed] [Google Scholar]
- Whittingham DG, and Biggers JD (1967). Fallopian tube and early cleavage in the mouse. Nature 213, 942–943. [DOI] [PubMed] [Google Scholar]
- Yagi R, Kohn MJ, Karavanova I, Kaneko KJ, Vullhorst D, DePamphilis ML, and Buonanno A (2007). Transcription factor TEAD4 specifies the trophectoderm lineage at the beginning of mammalian development. Development 134, 3827–3836. [DOI] [PubMed] [Google Scholar]
- Zhang X, Qiao Y, Wu Q, Chen Y, Zou S, Liu X, Zhu G, Zhao Y, Chen Y, Yu Y, et al. (2017). The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nature Communications 8, 15280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. (2007). Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21, 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YD, Ohkawara H, Rehman J, Wary KK, Vogel SM, Minshall RD, Zhao YY, and Malik AB (2009). Bone marrow progenitor cells induce endothelial adherens junction integrity by sphingosine-1-phosphate-mediated Rac1 and Cdc42 signaling. Circ Res 105, 696–704, 698 p following 704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets or code.