

Abstract
Apolipoprotein E (APOE) ε4 genotype is associated with increased risk of dementia in Parkinson disease (PD) but the mechanism is not clear, as patients often have a mixture of alpha-synuclein (αSyn), amyloid-β (Aβ), and tau pathologies. APOE ε4 exacerbates brain Aβ pathology, as well as tau pathology, but it is not clear whether APOE genotype independently regulates αSyn pathology. In this study we generated A53T αSyn transgenic mice (A53T) on Apoe knockout (A53T/EKO) or human APOE knock-in backgrounds (A53T/E2, E3, E4). At twelve months of age, A53T/E4 mice accumulated higher amounts of brainstem detergent-insoluble phosphorylated αSyn compared to A53T/EKO and A53T/E3; detergent-insoluble αSyn in A53T/E2 mice was undetectable. By immunohistochemistry, A53T/E4 mice displayed a higher burden of phosphorylated αSyn and reactive gliosis compared to A53T/E2. A53T/E2 mice exhibited increased survival and improved motor performance compared to other APOE genotypes. In a complementary model of αSyn spreading, striatal injection of αSyn pre-formed fibrils induced greater accumulation of αSyn pathology in the substantia nigra of A53T/E4 mice compared to A53T/E2 and A53T/EKO. In two separate cohorts of human patients with PD, APOE ε4/ε4 individuals showed the fastest rate of cognitive decline over time. Our results demonstrate that APOE genotype directly regulates αSyn pathology independent of its established effects on Aβ and tau, corroborate the finding that APOE ε4 exacerbates pathology, and suggest that APOE ε2 may protect against αSyn aggregation and neurodegeneration in synucleinopathies.
One Sentence Summary:
APOE genotype regulates disease development in mouse models of alpha-synucleinopathy and affects cognition in patients with Parkinson disease.
INTRODUCTION
Parkinson disease (PD) is a neurodegenerative disorder characterized by a diverse range of motor and non-motor symptoms (1). Of the non-motor symptoms, dementia affects approximately 25–30% of all patients with PD and causes substantial morbidity (2). The risk of dementia in PD increases with age, affecting approximately 80% of patients who survive 20 years (3). Many patients take years to develop dementia, whereas others have a more rapid course, and in some cases dementia precedes motor symptoms. This clinical heterogeneity prompted the classification of patients with PD who develop dementia more than one year after the onset of motor symptoms as having “Parkinson disease dementia” (PDD) whereas patients who develop dementia within one year of, or before, motor symptoms as having “dementia with Lewy bodies” (DLB) (4), but the time to dementia onset in PD is more likely a continuous rather than a categorical variable. The exact basis for this clinical variability is unclear and there are no disease-modifying treatments to slow the course of PD or its associated dementia. Genetic association studies have implicated APOE ε4 as a risk factor for decline in specific cognitive domains in PD (5–7), and APOE ε4 has been linked to rate of cognitive decline in PD (8), but the underlying molecular mechanisms are not well understood.
Pathologically, PD is characterized by the accumulation of insoluble aggregates of alpha-synuclein (αSyn) in Lewy bodies (LB) and Lewy neurites (LN) in multiple brain regions, including limbic areas and the neocortex in advanced cases (9, 10). Additional neuropathologic features classically associated with Alzheimer disease (AD) are also commonly seen in the brains of many patients with PD and dementia, including amyloid plaques composed of Aβ peptides and neurofibrillary tangles (NFT) containing the protein tau (11, 12). This overlapping neuropathology, combined with the well-established role of apoE isoforms in AD pathophysiology (13, 14), is often interpreted as an APOE effect on cognition in PD mediated by co-morbid AD neuropathology (15). However, other studies have demonstrated an effect of APOE genotype on cognition and Lewy pathology in PD that is independent of co-existing AD pathology (16–18), suggesting that APOE may directly influence development and progression of αSyn pathology.
To determine whether human APOE isoforms directly regulate αSyn pathology and associated neurodegeneration, we assessed the effect of human APOE background on pathology in two independent mouse models of pathologic αSyn aggregation. We also examined the effect of APOE genotype and other biomarkers on cognitive function in studies of human patients with PD.
RESULTS
APOE genotypes differentially regulate αSyn aggregation and phosphorylation in A53T mice
To directly assess the effect of APOE genotype on development of pathology in an in vivo model of synucleinopathy, we generated A53T αSyn transgenic (Tg) mice on Apoe knockout (EKO) or human APOE knockin (E2/E3/E4) backgrounds. This αSyn mouse model develops progressive motor deficits and αSyn pathology which ultimately leads to paralysis (19, 20). Brainstem lysates from 12-month-old mice were analyzed by enzyme-linked immunosorbent assay (ELISA) following sequential extraction in a high-salt reassembly (RAB) buffer, RAB containing Triton X-100 (RAB-TX100), radioimmunoprecipitation (RIPA) buffer, and 2% SDS, which contain progressively more insoluble forms of αSyn. A53T mice on all three human APOE backgrounds contained equivalent amounts of total αSyn in the RAB- and RAB + Triton X-100-soluble fractions, which represents the bulk of the αSyn in the brain (Fig. 1, A and B). In the RIPA fraction, A53T/EKO and A53T/E4 mice had significantly higher amounts of total αSyn compared to A53T/E2, with A53T/E3 intermediate to the other human allele backgrounds (Fig. 1C, p<0.01). Total αSyn amount followed a similar pattern in the SDS fraction, with the exception that αSyn in A53T/E2 mice was undetectable (Fig. 1D). To assess the amount of pathologically modified αSyn, we used an antibody that recognizes αSyn phosphorylated on serine 129, which is characteristic of¬¬ LB/LN pathology in human PD brain. Amounts of phosphorylated αSyn (pSyn) followed a similar pattern in the RIPA and SDS fractions, with highest concentrations in A53T/E4, undetectable amounts in A53T/E2, and intermediate concentrations in A53T/E3 and A53T/EKO (Fig. 1, E and F). By 12 months of age, some mice of all genotypes except A53T/E2 had developed endstage paralysis similar to that described in the original report of this strain (19). The mice with paralysis consistently had higher concentrations of RIPA- and SDS-soluble total αSyn than asymptomatic mice (Fig. S1), and pSyn was only detected in symptomatic mice in the RIPA- and SDS-soluble fractions (Fig. 1, E and F).
Fig 1. APOE genotypes differentially regulate αSyn aggregation and phosphorylation in A53T mice.
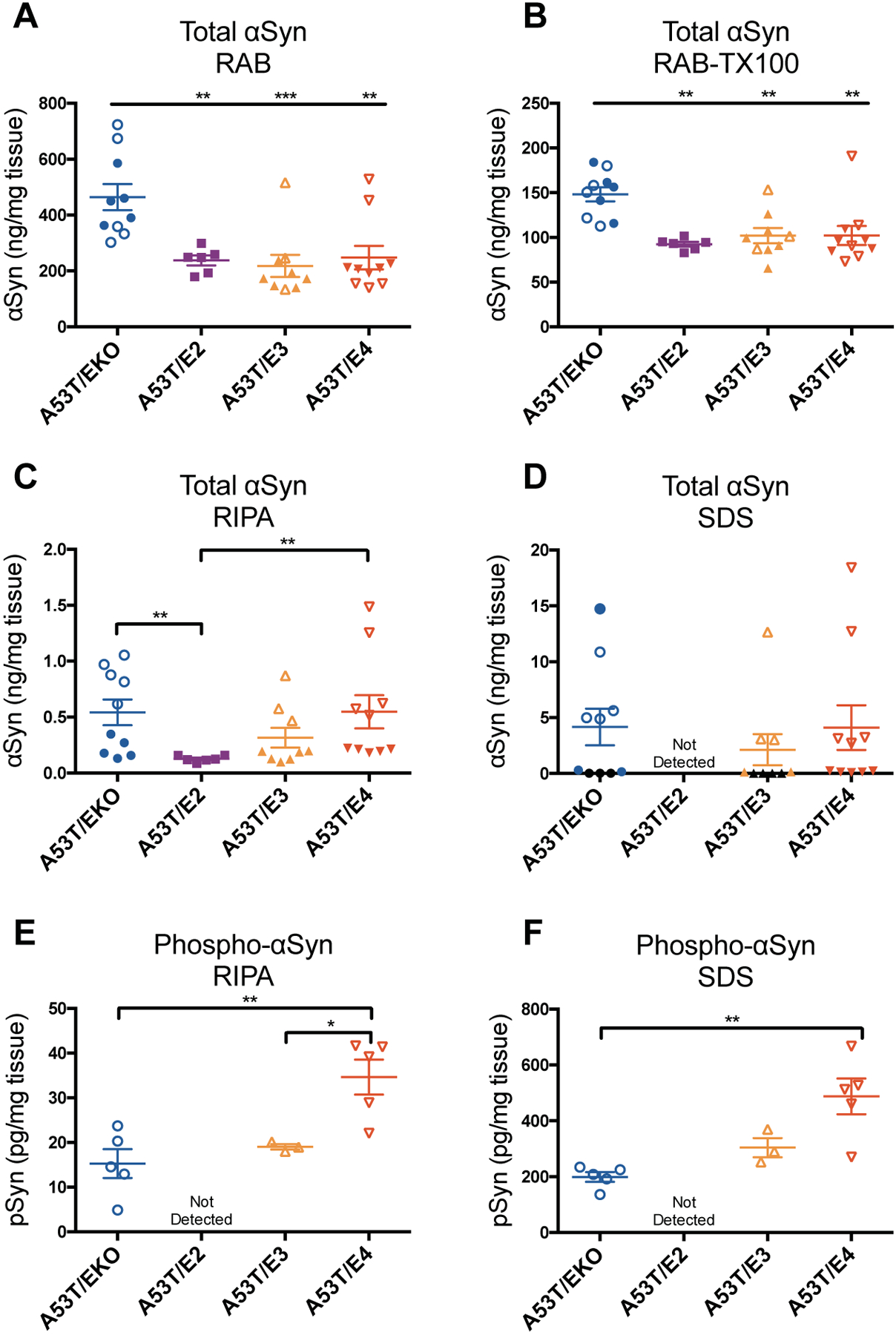
(A-D) Total αSyn concentration was measured by ELISA in RAB, RAB+TX-100, RIPA, and SDS fractions from the brainstem of 12-month old A53T mice. A53T/EKO, n=10; A53T/E2, n=6; A53T/E3, n=9; A53T/E4, n=10. Closed symbols indicate asymptomatic mice; open symbols indicate symptomatic mice with endstage paralysis. Symbols shown in black in (D) indicate A53T/EKO and A53T/E3 samples below the limit of detection. (E-F) Phospho-αSyn concentration was measured by ELISA in RIPA and SDS fractions. Data expressed as mean ± SEM, one-way ANOVA with Tukey’s multiple comparisons test (A, B, E, F) or Kruskal-Wallis test with Dunn’s multiple comparisons test (C). *p<0.05, **p<0.01, ***p<0.001.
To corroborate the ELISA measurement of total and phosphorylated αSyn we performed immunoblot analysis on RAB, RAB+TX-100, RIPA, and SDS-soluble fractions from asymptomatic and symptomatic mice of all APOE genotypes using antibodies that recognize total αSyn or pSyn. We observed enrichment of both total αSyn and pSyn in RIPA and SDS-soluble fractions in symptomatic mice. We also observed high-molecular-weight bands in the SDS-soluble fraction, present in symptomatic mice, that may represent αSyn oligomers (Fig. S2).
APOE genotype relates to histopathology in A53T mice
To further assess pathology in A53T/APOE mice, we performed immunohistochemistry (IHC) using an anti-pSyn antibody. In mice 9–12 months of age, we observed abundant pSyn staining most notably in the brainstem in A53T/EKO and A53T/E4 mice compared to A53T/E2 mice, which had no detectable pSyn staining. As with ELISA measurements of insoluble and phosphorylated αSyn, pSyn staining appeared to be linked with the presence of endstage paralysis which was frequently seen in A53T/EKO and A53T/E4 mice at this age (Fig. 2A, B, D). This accumulation of pSyn pathology localized to neurons (Fig. S3) and was accompanied by increased glial fibrillary acidic protein (GFAP) immunostaining in the surrounding tissue, indicating that astrogliosis accompanies the development of pSyn pathology. Astrogliosis was most abundant in A53T/EKO and A53T/E4 mice and nearly absent in A53T/E2 mice, again largely in relation to the presence of endstage paralysis (Fig. 2A, C, E). Because there was considerable variation in pSyn and GFAP staining among groups of mice, we performed a linear regression analysis to determine if these histologic findings were related in individual animals. The degree of pSyn pathology was significantly correlated with the extent of GFAP staining across all genotypes, suggesting that these pathologic changes are linked (p<0.0001; Fig. 2F).
Fig 2. APOE genotype relates to pSyn pathology and astrogliosis in A53T mice.
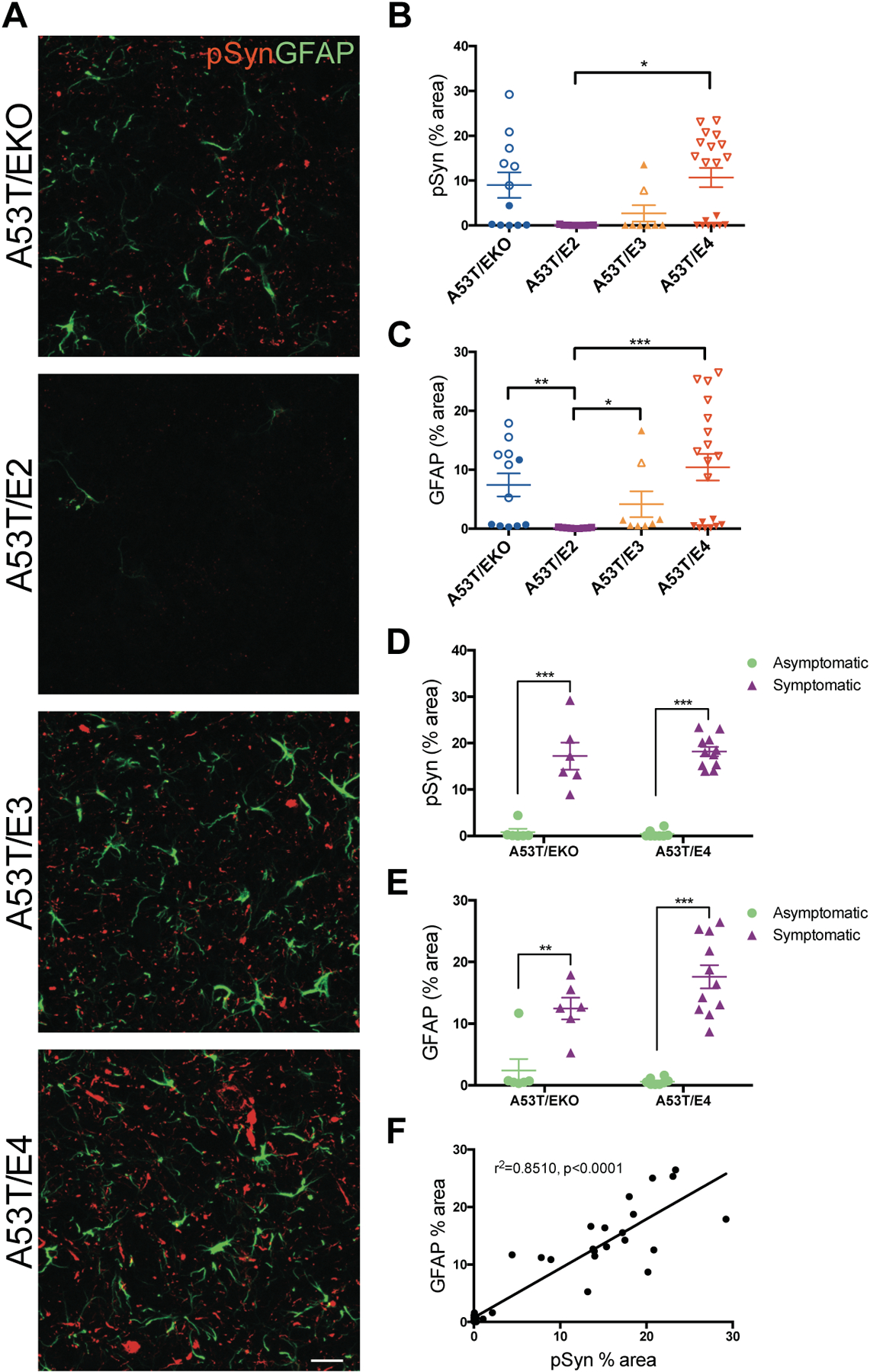
(A) Representative images showing pSyn pathology (b81A) and astrogliosis (GFAP) in the brainstem of 9–12 month old A53T mice. Images represent maximum-intensity projections of z stacks. Scale bar, 50 μm. Quantitation of the percent area covered by (B) pSyn and (C) GFAP staining in the brainstem of A53T/EKO (n=12), A53T/E2 (n=9), A53T/E3 (n=8), and A53T/E4 (n=19) mice. Closed symbols indicate asymptomatic mice; open symbols indicate symptomatic mice with endstage paralysis. Each data point represents the average of 2 adjacent regions of interest from 3 brain sections spaced 300 μm apart. Data expressed as mean ± SEM, Kruskal-Wallis test with Dunn’s multiple comparisons test. *p<0.05, **p<0.01, ***p<0.001. (D) Stratification of pSyn percent area by symptomatic vs asymptomatic status of A53T/EKO (n=12) and A53T/E4 (n=19). Data expressed as mean ± SEM, multiple t-tests, ***p<0.001. (E) Stratification of GFAP percent area by symptomatic vs asymptomatic status of A53T/EKO (n=12) and A53T/E4 (n=19). Data expressed as mean ± SEM, multiple t-tests, **p<0.01, ***p<0.001. (F) Correlation between pSyn and GFAP staining in the brainstem of A53T mice (n=12 A53T/EKO, n=9 A53T/E2, n=8 A53T/E3, n=19 A53T/E4; r2=0.8510, p<0.0001).
To assess microglial reactivity in A53T/APOE mice, we performed IHC using anti-Iba1 and anti-CD68 antibodies. In A53T/E2 mice, which had no detectable pSyn staining, microglia were more extensively ramified and did not express detectable CD68. In contrast, in A53T/EKO, A53T/E3, and A53T/E4 mice with some or extensive pSyn staining, microglia were more ameboid in morphology and were CD68-positive, indicating a reactive state (Fig. S4A). As observed for astrogliosis, microglial reactivity correlated with endstage paralysis and was not clearly stratified primarily on the basis of APOE genotype (Fig. S4 B–E).
Much of the pSyn pathology in A53T mice was found in the brainstem, although we did observe rare pathology in the neocortex. Quantitation of cortical pSyn neuronal inclusions showed no differences between genotypes (Fig. S5).
Inflammatory gene expression in A53T mice correlates with pSyn pathology but not APOE genotype
Our group recently found that APOE4 was associated with microglia and astrocyte activation in a mouse model of tauopathy (21). To further investigate the mechanism of increased pSyn pathology and gliosis we observed in A53T mice, we performed multiplex gene expression analysis using a customized Nanostring nCounter panel enriched for inflammatory genes. We quantified expression of 781 genes from the midbrains of 12-month-old A53T/EKO, A53T/E2, and A53T/E4 mice, which develops similar pSyn pathology compared to the adjacent brainstem (19). We designed this experiment so that approximately half of the A53T/EKO and A53T/E4 mice analyzed had endstage paralysis at the time of tissue collection, whereas none of our A53T/E2 mice showed signs of paralysis by this age. We found that the majority of the sample variance correlated with the presence of pSyn pathology as determined by IHC performed on the accompanying hemibrains of each mouse (Fig. 3A, Fig. S6 for principal component analysis, and Table S1 for gene list). Stratification by APOE genotype did not reveal changes in the majority of genes analyzed (Fig. 3B, C, and Tables S2 and S3). Together these results indicate that APOE-dependent effects on pSyn pathology, rather than APOE genotype, drive downstream inflammatory gene expression, regulating inflammation independently of synucleinopathy in this model system.
Fig 3. Inflammatory gene expression in A53T mice correlates with pSyn pathology but not APOE genotype.
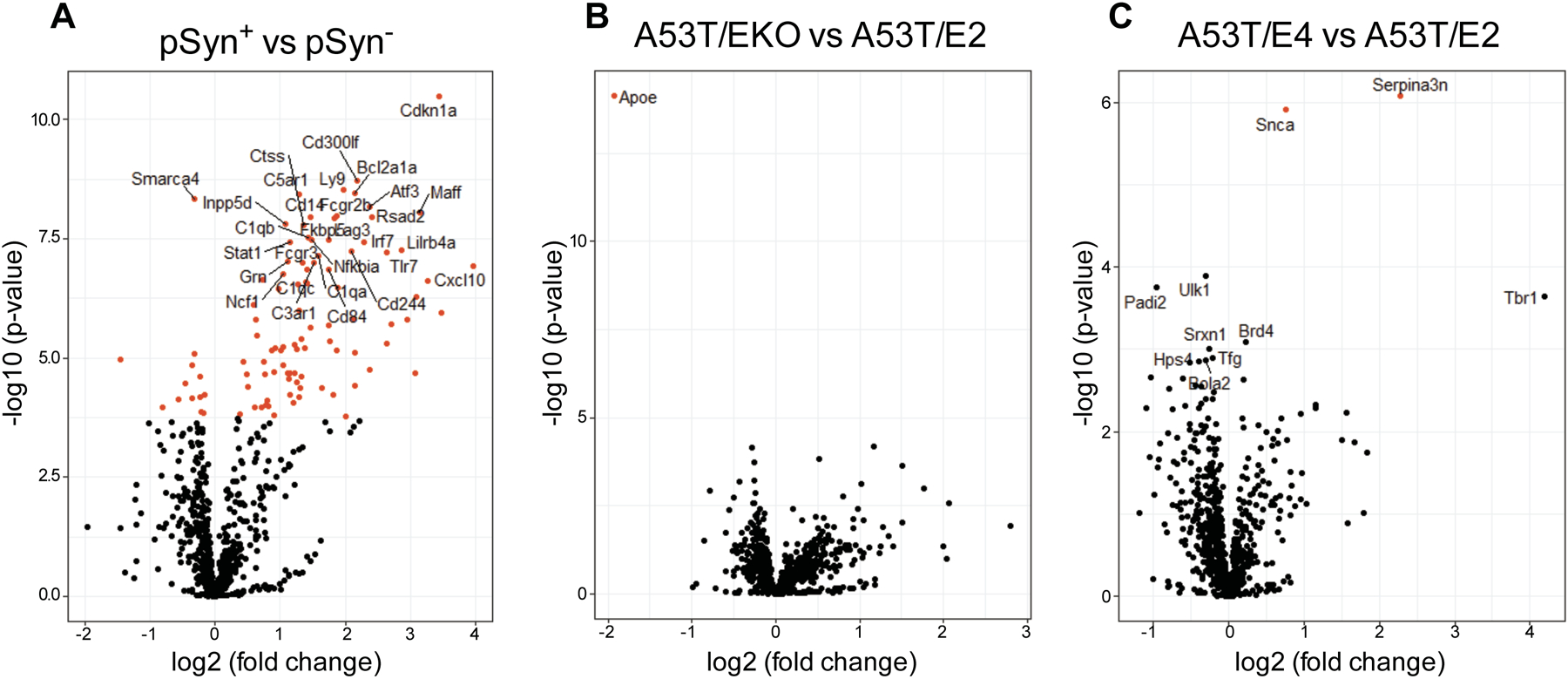
Volcano plot showing differences in gene expression in the midbrain of A53T mice stratified by (A) presence of pSyn pathology in corresponding immunohistochemical analysis, (B) EKO vs. E2 as baseline, (C) E4 vs. E2 as baseline. For each plot, significance is plotted against fold-change. Red symbols denote genes with adjusted significance of p<0.01.
Gene co-expression analysis defines modules associated with pSyn and APOE
To further investigate relationships between APOE genotype and pSyn pathology in A53T mice, we performed weighted gene co-expression network analysis (WGCNA) using the Nanostring nCounter data. We identified five gene co-expression modules, two of which, the turquoise and green modules, exhibited significant (r>0.5, p<0.01) correlations with either APOE genotype or pSyn burden (Fig. 4A, B, Tables S4 and S5). Gene ontology (GO) analysis demonstrated that the turquoise module, comprised of 233 genes, was enriched for genes related to the immune response (Tables S4 and S6). The turquoise module was highly correlated with pSyn burden (Fig. 4B), consistent with our initial analysis that stratification by the presence of pSyn pathology accounted for a large proportion of genes changed at the p<0.01 level (Fig. 3A). Analysis of eigengene expression in the turquoise module by individual animal demonstrated that increased expression in this module was tightly linked to the endstage paralysis phenotype and the presence of pSyn pathology (Fig. 4C–E, Fig. S7A). The green module, comprised of 83 genes, was enriched for genes related to myelination, and was negatively correlated with pSyn burden but positively correlated with APOE2 genotype (Fig. 4B, Tables S4 and S6). Analysis by individual animal corroborated that eigengene expression in this module varied by APOE genotype and also by endstage paralysis phenotype in APOE4 background mice (Fig. 4F, G, Fig. S7B). Although the implications of increased expression of myelination genes in the APOE2 background is not immediately clear, the link between oligodendrocytes and the pathophysiology of synucleinopathies is intriguing, based in part on the fact that oligodendrocytes are the principal cell type in which αSyn aggregates in multiple system atrophy (MSA), a rapidly progressive synucleinopathy. We performed double-label immunohistochemistry with pSyn and 2’, 3’-cyclic nucleotide 3’-phosphodiesterase (CNPase), a myelin-associated enzyme that marks oligodendrocytes. We observed minimal colocalization between pSyn and CNPase in the brainstem where the majority of pSyn pathology develops, indicating that pSyn aggregates do not substantially accumulate in oligodendrocytes in A53T mice at this age (Fig. S8). We also observed a modest negative correlation (r=−0.46, p=0.02) of the blue module with pSyn pathology (Fig. 4B, Tables S4 and S6). GO analysis found that the blue module, comprised of 228 genes, was enriched for genes associated with synaptic transmission and thus the negative correlation with pSyn may reflect a neurodegenerative association in this model system. Finally, we observed a modest positive correlation (r=0.47, p=0.02) of the yellow module with APOE2 genotype (Fig. 4B, Tables S4 and S6). GO analysis found that the yellow module contained 94 genes including multiple anti-apoptotic genes. Whether this anti-apoptotic module is mechanistically related to the delay in progression to endstage paralysis we observed in A53T/E2 mice is unclear given the modest statistical correlation we observed.
Fig 4. Gene co-expression analysis in A53T mice defines modules associated with pSyn and APOE.
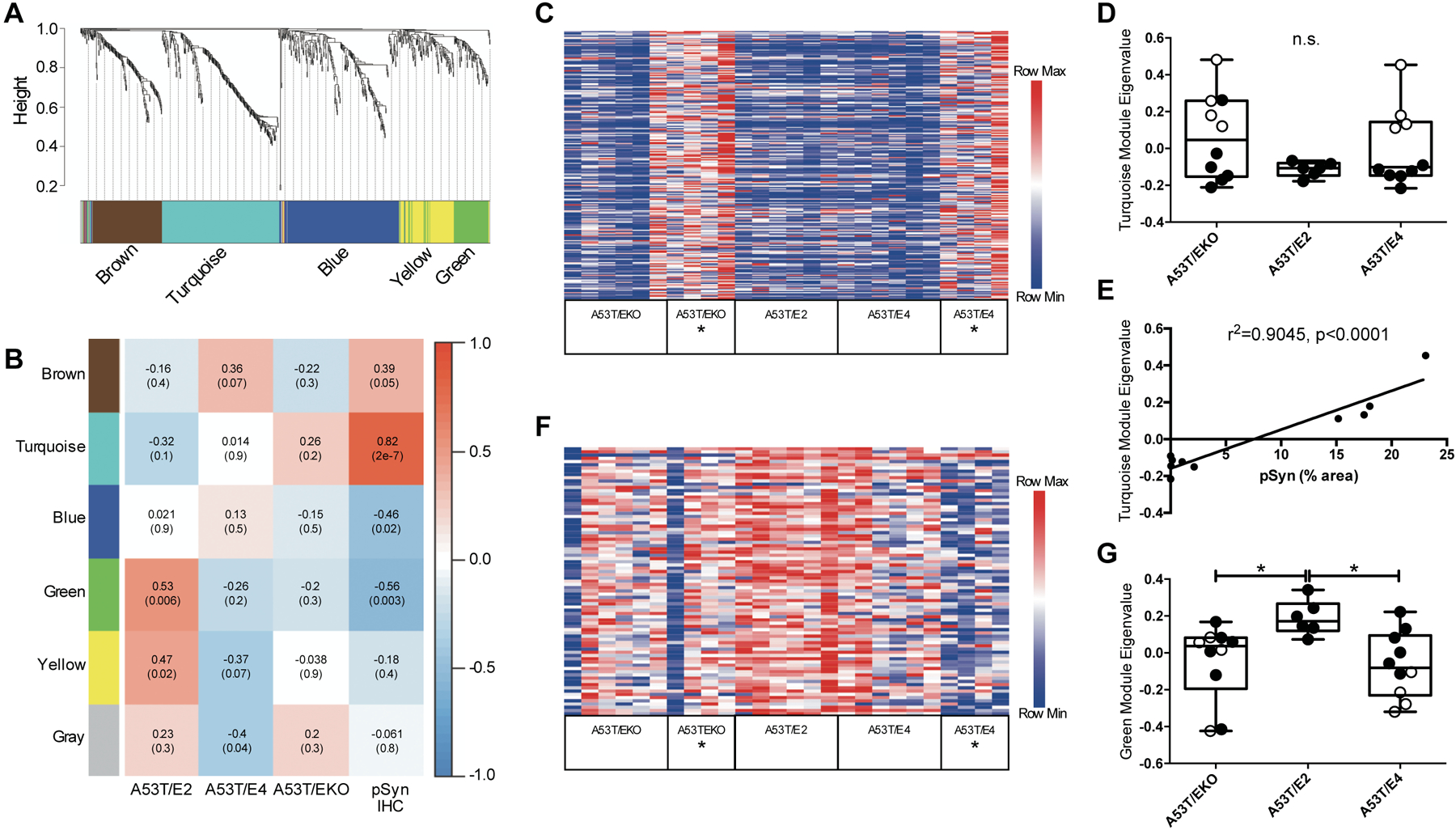
(A) WGCNA dendrogram groups genes measured in the midbrain of 12 month-old A53T mice into distinct modules defined by dendrogram branch clustering, enriched for gene ontologies linked to specific cell type or cellular function. (B) Module-trait analysis between gene modules defined by WGCNA and APOE genotype or pSyn IHC. Data are shown as correlation co-efficient (p-value). (C) Heatmap of relative expression of turquoise module genes in A53T mice stratified by APOE genotype and endstage paralysis (denoted with *). (D) Eigengene analysis for turquoise module by APOE genotype. Open symbols indicate mice with endstage paralysis. Data are expressed as mean ± SEM, Kruskal-Wallis test. (E) Linear regression between pSyn IHC % area and turquoise module eigenvalue among A53T/E4 mice (n=10; r2=0.9045, p<0.0001). (F) Heatmap of relative expression of green module genes stratified by APOE genotype and endstage paralysis (denoted with *). (G) Eigengene analysis for green module by APOE genotype. Open symbols indicate mice with endstage paralysis. Data are expressed as mean ± SEM, Kruskal-Wallis test with Dunn’s multiple comparisons test. *p<0.05
APOE genotype regulates motor phenotype and survival in A53T mice
Given the sharp decline we observed in A53T mice once they began to develop endstage paralysis, we measured latency to fall in an inverted wire screen test to assess subtle motor dysfunction that might develop ahead of this precipitous decline, despite the lack of obvious abnormalities observed in these mice during routine ambulation. In longitudinal measurements, A53T/E2 mice consistently reached a criterion of 60 seconds through 12 months of age, whereas other genotypes displayed a decline in motor function beginning around 4–6 months, with A53T/EKO and A53T/E4 trending worse than A53T/E3 (Fig. 5A). Survival analysis showed that A53T/E2 mice lived longer (median survival 18.4 months) than A53T/EKO (median survival 11.6 months) and A53T/E4 (median survival 11.7 months) (Fig. 5B). After correction for multiple comparisons (Bonferroni corrected threshold p=0.0083), differences in survival remained significant between A53T/E2 and A53T/E4 (p=0.0008) as well as between A53T/E2 and A53T/EKO (p=0.0013). These two findings set APOE2 apart as the strongest genetic influence on motor dysfunction and neurodegeneration in A53T αSyn-Tg mice and suggest there may be a protective gain of function associated with APOE2 in this model system.
Fig 5. APOE2 genotype protects against motor deficits and prolongs survival in A53T mice.
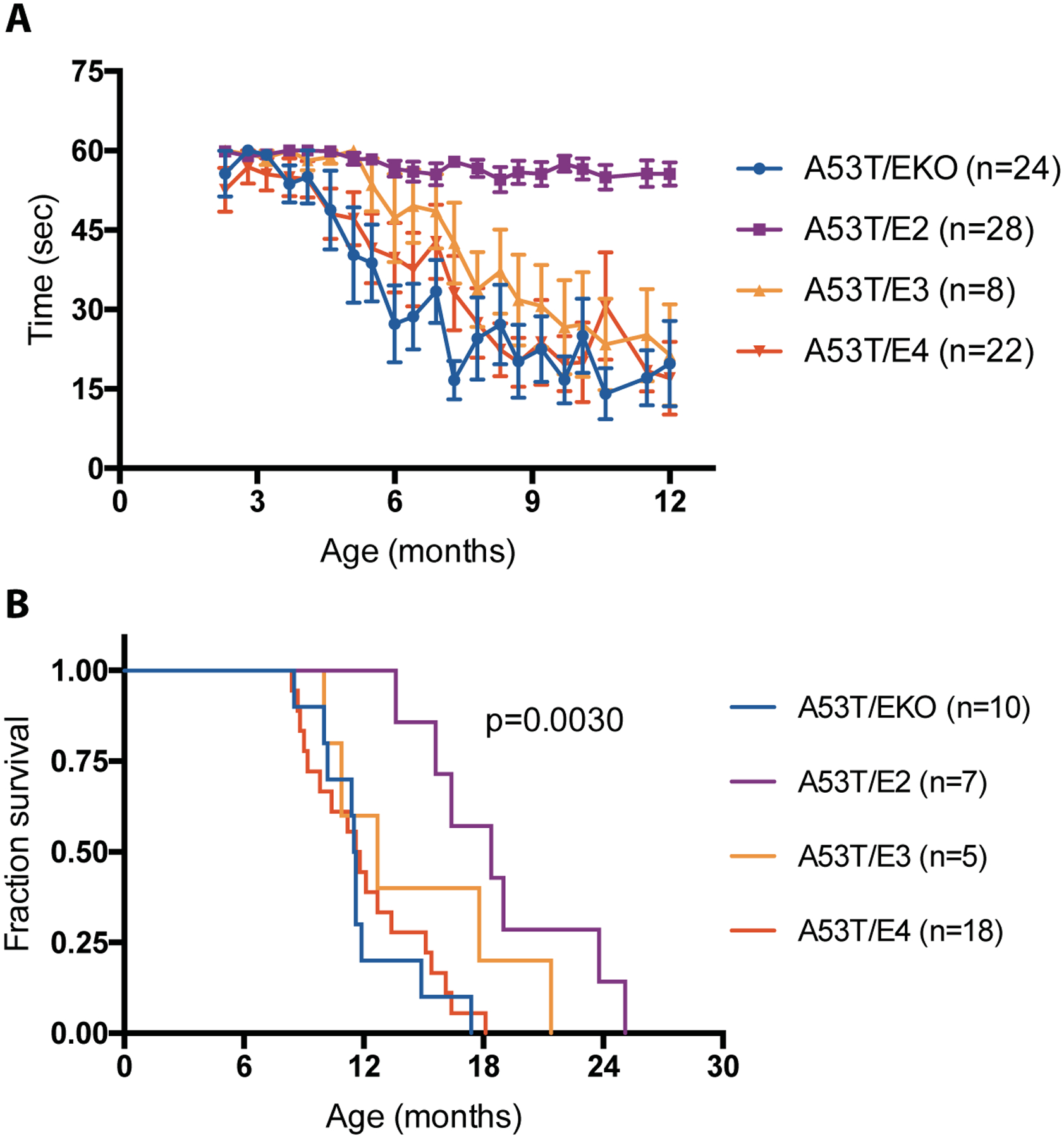
(A) Assessment of motor function in A53T mice. Latency to fall in the inverted wire screen test was measured for A53T/EKO (n=24), A53T/E2 (n=28), A53T/E3 (n=8), and A53T/E4 (n=22) mice. (B) Kaplan-Meier survival analysis of A53T mice by APOE genotype for A53T/EKO (n=10, median survival 11.6 months), A53T/E2 (n=7, median survival 18.4 months), A53T/E3 (n=5, median survival 12.7 months), A53T/E4 (n=18, median survival 11.7 months) mice. Overall log-rank (Mantel-Cox) p=0.0030.
APOE4 potentiates spreading of αSyn pathology
To determine if APOE genotype regulates αSyn pathology in a complementary model system, we performed stereotaxic injection of αSyn pre-formed fibrils (PFFs) into the dorsal striatum of Apoe knockout (KO) and human APOE2, APOE3, or APOE4 knockin mice. This paradigm initiates robust spreading of αSyn pathology from the striatum to connected regions including substantia nigra, and causes degeneration of dopaminergic neurons in the substantia nigra (22). Importantly, this model does not require overexpression of transgenic αSyn, but rather the αSyn PFFs induce aggregation of endogenous mouse αSyn. Following unilateral injection of αSyn PFFs into the striatum, APOE4 mice accumulated more pSyn pathology in the ipsilateral substantia nigra pars compacta (SNpc) than APOE2 or Apoe KO mice, indicating that APOE genotype regulates spreading of pSyn pathology (Fig. 6A and B). pSyn pathology in the striatum was not changed across APOE genotypes (Fig. S8). αSyn PFF injection also decreased the number of tyrosine hydroxylase (TH)-positive dopaminergic neurons in the ipsilateral SNpc in APOE2, APOE3, and APOE4 mice compared to the contralateral side, suggesting that human apoE isoforms may potentiate the toxic effect of αSyn PFFs on dopaminergic neurons in this model system (Fig. 6A and C).
Fig 6. APOE4 exacerbates spreading of αSyn pathology.
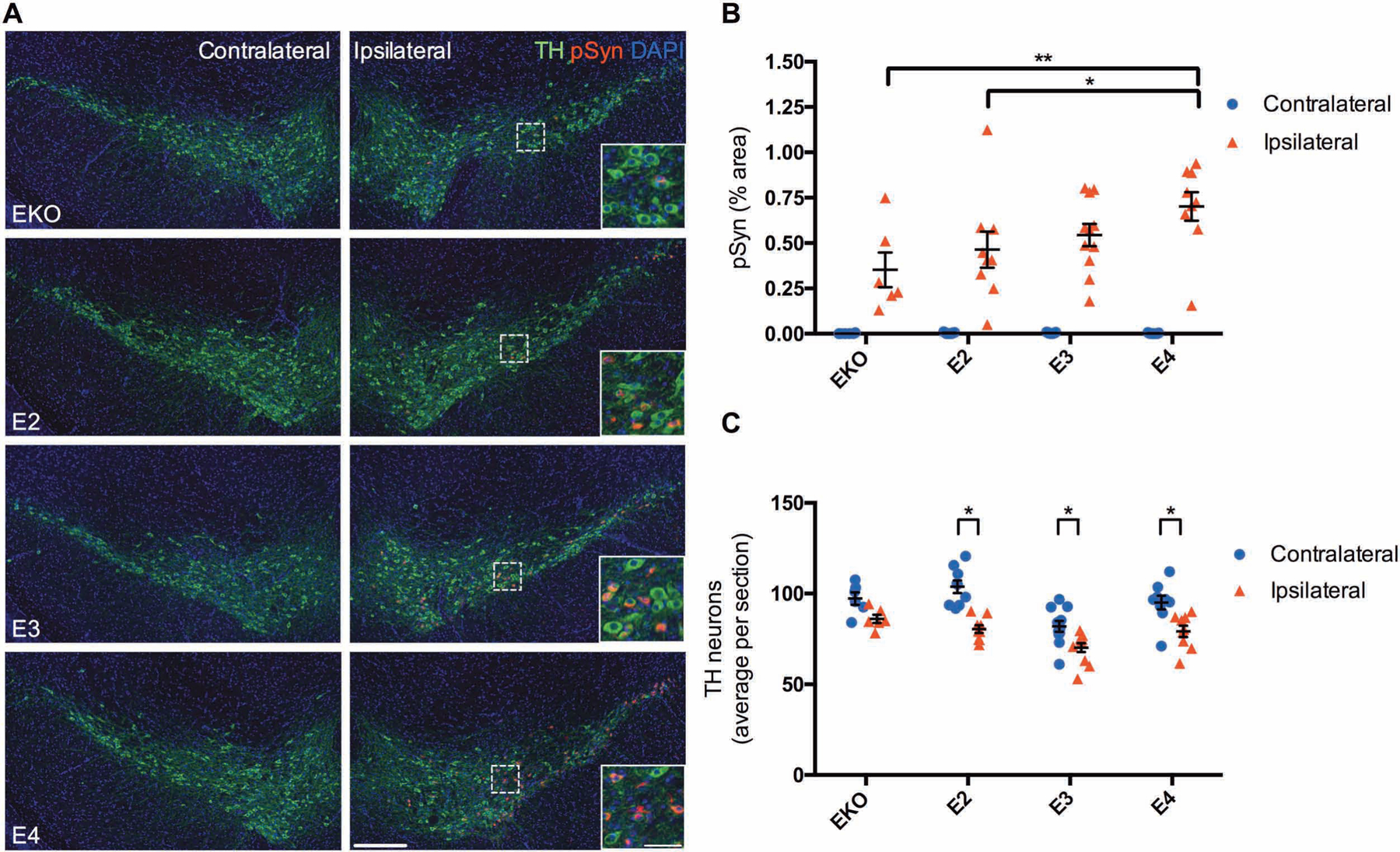
(A) Representative images showing pSyn pathology within the SNpc three months after unilateral injection of αSyn PFFs into the striatum of EKO (n=6), E2 (n=9), E3 (n=11), and E4 (n=9) mice. Scale bar, 250 μm; inset scale bar, 50 μm. (B) Quantitation of the percent area covered by pSyn staining in the SNpc. Data are expressed as mean ± SEM, two-way ANOVA with Tukey’s multiple comparisons test *p<0.05, **p<0.01. (C) Cell counts of TH-positive neurons from 4 sections spaced 150 μm apart. Data are expressed as mean ± SEM, multiple t-tests with correction for multiple comparisons using the Holm-Sidak method.
APOE4 accelerates cognitive decline in human patients with PD
Given our findings in mouse models that APOE4 exacerbated pSyn pathology whereas APOE2 exerted a protective effect, we examined cohorts of patients with PD to determine if a similar effect is present in humans. Given that previous studies have described an APOE genotype effect on dementia in PD, but not on overall disease risk or motor symptoms (23), we focused our attention on cognition. Several studies have reported that APOE ε4 is a risk factor for dementia related to PD (5–8, 16, 24–26), but less is known about the risk associated with APOE ε2. We analyzed data from 251 patients enrolled in the Parkinson’s Progression Markers Initiative (PPMI), a longitudinal study of patients with PD with clinical, genetic, and biomarker data that is publicly available (27). We used a linear mixed model incorporating genetic, demographic, and the last available cerebrospinal fluid (CSF) biomarker data from the PPMI study to determine the impact of APOE genotype on the rate of decline of MoCA scores over time. We found that after accounting for all variables in the model, the presence of APOE ε4 was associated with a faster rate of decline in MoCA score over time (Table 1). Our model included CSF measures of Aβ42 and phosphorylated tau (pTau) from the last available sample which related to MoCA decline in an expected manner, with individuals with lower CSF Aβ42 or higher CSF pTau having a faster rate of decline in MoCA score over time. Similarly, we observed an expected effect of education level, with fewer years of education being associated with a faster rate of decline. The fact that APOE ε4 remained significant (p=0.0119) in this multivariate model indicates that it affects cognitive decline independently of other contributors including CSF Aβ42 and pTau. We did not find any association between CSF αSyn concentration and rate of decline of MoCA score, possibly because this association lacked sufficient strength and did not meet threshold for significance after statistical correction in our multivariate model, or possibly due to technical and biological challenges that are recognized as limitations of CSF αSyn as a biomarker in PD (28).
Table 1. Multivariate regression analysis of rate of change of MoCA scores over time in PPMI participants.
Covariates included age at onset of PD (Age at Onset), age at baseline MoCA test (Age at Baseline), sex, years of education (Education), baseline MoCA score (Baseline MoCA), principal component analysis of genetic ancestry (PC1–4), APOE genotype, last available CSF Aβ42, tau, and αSyn concentrations, and the time interval between MoCA assessments. Interaction of covariates with the rate of change of MoCA score over time are listed as “Covariate : Time.” Data are presented as beta coefficients with 95% confidence interval and p-value, after accounting for the effect of covariates.
| Variable | Beta | 95% CI | p-value | |
|---|---|---|---|---|
| Lower | Upper | |||
| Age at Onset | −5.22E−02 | −1.46E−01 | 4.15E−02 | 2.79E−01 |
| Age at Baseline | −1.20E−02 | −8.28E−02 | 1.07E−01 | 8.06E−01 |
| Sex | 3.81E−01 | 5.04E−02 | 7.07E−01 | 2.39E−02 |
| Education | 1.45E−02 | −4.02E−02 | 6.98E−02 | 6.07E−01 |
| Baseline MoCA | 6.29E−01 | 5.11E−01 | 7.49E−01 | 1.20E−22 |
| PC1 | −1.45E+02 | −4.35E+02 | 1.46E+02 | 3.33E−01 |
| PC2 | −1.07E+01 | −1.87E+02 | 1.62E+02 | 9.04E−01 |
| PC3 | −9.10E+00 | −2.47E+01 | 6.52E+00 | 2.57E−01 |
| PC4 | 1.18E+01 | −2.26E+01 | 4.62E+01 | 5.05E−01 |
| APOE ε2 | 4.34E−02 | −3.92E−01 | 4.84E−01 | 8.47E−01 |
| APOE ε4 | −3.51E−02 | −4.07E−01 | 3.32E−01 | 8.53E−01 |
| CSF Aβ42 | −6.70E−05 | −5.76E−04 | 4.45E−04 | 7.98E−01 |
| CSF pTau | 2.23E−03 | −3.18E−02 | 3.58E−02 | 8.98E−01 |
| CSF αSyn | −6.61E−02 | −3.27E−01 | 1.89E−01 | 6.18E−01 |
| Time Interval | 5.78E−02 | −2.03E+00 | 2.15E+00 | 9.58E−01 |
| Age at onset : Time | 1.63E−02 | −2.75E−02 | 6.06E−02 | 4.80E−01 |
| Age at baseline : Time | −2.91E−02 | −7.39E−02 | 1.52E−02 | 2.14E−01 |
| Sex : Time | −8.07E−02 | −2.30E−01 | 7.21E−02 | 3.09E−01 |
| Education : Time | 2.99E−02 | 4.60E−03 | 5.48E−02 | 2.40E−02 |
| Baseline MoCA : Time | −3.24E−02 | −8.69E−02 | 2.11E−02 | 2.54E−01 |
| PC1 : Time | −1.05E+02 | −2.36E+02 | 2.52E+01 | 1.27E−01 |
| PC2 : Time | −7.63E+00 | −8.61E+01 | 7.28E+01 | 8.54E−01 |
| PC3 : Time | −5.18E+00 | −1.20E+01 | 1.64E+00 | 1.51E−01 |
| PC4 : Time | 4.55E+00 | −1.09E+01 | 2.01E+01 | 5.78E−01 |
| APOE ε2 : Time | −4.02E−02 | −2.41E−01 | 1.57E−01 | 7.00E−01 |
| APOE ε4 : Time | −2.26E−01 | −3.95E−01 | −5.43E−02 | 1.19E−02 |
| CSF Aβ42 : Time | 5.19E−04 | 2.81E−04 | 7.57E−04 | 4.77E−05 |
| CSF pTau : Time | −3.85E−02 | −5.43E−02 | −2.27E−02 | 5.87E−06 |
| CSF αSyn : Time | −4.42E−02 | −1.63E−01 | 7.69E−02 | 4.82E−01 |
| APOE2 Count | 0 | 1 | 2 | |
| Number of Individuals: | 217 | 31 | 3 | |
| APOE4 Count | 0 | 1 | 2 | |
| Number of Individuals: | 182 | 61 | 8 | |
In a similar analysis of an independent cohort of 177 patients with PD at Washington University in St Louis Movement Disorders Center (WUSTLPD), we found that APOE ε4 was significantly associated with a faster rate of decline of Mini-Mental State Exam (MMSE) score (Table 2, p=0.0369). In this cohort, we also identified younger age at first evaluation as associated with a faster rate of MMSE decline, and older age at onset of PD as associated with a slower rate of MMSE decline.
Table 2. Multivariate regression analysis of rate of change of MMSE scores over time in WUSTLPD participants.
Covariates included age at onset of PD (Age at Onset), age at baseline MMSE test (Age at Baseline), sex, baseline MMSE score (Baseline MMSE), principal component analysis of genetic ancestry (PC1–4), APOE genotype, and the time interval between MMSE assessments. Interaction of covariates with the rate of change of MMSE score over time are listed as “Covariate : Time.” Data are presented as beta coefficients with 95% confidence interval and p-value, after accounting for the effect of covariates.
| Variable | Beta | 95% CI | p-value | |
|---|---|---|---|---|
| Lower | Upper | |||
| Age at Onset | 1.89E−02 | −2.02E−02 | 5.84E−02 | 3.48E−01 |
| Age at Baseline | −4.33E−02 | −9.05E−02 | 3.56E−03 | 7.31E−02 |
| Sex | 1.49E−01 | −2.73E−01 | 5.71E−01 | 4.91E−01 |
| Baseline MMSE | 6.97E−01 | 5.59E−01 | 8.36E−01 | 3.81E−22 |
| PC1 | 3.22E+01 | −1.74E+02 | 2.39E+02 | 7.61E−01 |
| PC2 | 1.49E+01 | −1.53E+02 | 1.84E+02 | 8.63E−01 |
| PC3 | −3.28E+00 | −1.21E+01 | 5.59E+00 | 4.68E−01 |
| PC4 | −1.24E+00 | −2.61E+01 | 2.37E+01 | 9.22E−01 |
| APOE ε2 | 3.90E−01 | −3.51E−01 | 1.12E+00 | 3.03E−01 |
| APOE ε4 | 1.46E−01 | −3.14E−01 | 6.08E−01 | 5.36E−01 |
| Time Interval | 1.51E+00 | −1.13E+00 | 4.16E+00 | 2.77E−01 |
| Age at Onset : Time | 2.10E−02 | 2.97E−03 | 3.88E−02 | 2.59E−02 |
| Age at Baseline : Time | −4.29E−02 | −6.39E−02 | −2.14E−02 | 1.68E−04 |
| Sex : Time | 6.88E−02 | −1.17E−01 | 2.55E−01 | 4.82E−01 |
| Baseline MMSE : Time | 7.08E−04 | −6.13E−02 | 6.17E−02 | 9.82E−01 |
| PC1 : Time | −2.66E+01 | −1.18E+02 | 6.46E+01 | 5.78E−01 |
| PC2 : Time | 6.99E−01 | −7.19E+01 | 7.35E+01 | 9.85E−01 |
| PC3 : Time | 3.78E−01 | −3.43E+00 | 4.18E+00 | 8.50E−01 |
| PC4 : Time | −3.28E+00 | −1.42E+01 | 7.67E+00 | 5.69E−01 |
| APOE ε2 : Time | −1.82E−01 | −4.93E−01 | 1.32E−01 | 2.69E−01 |
| APOE ε4 : Time | −2.21E−01 | −4.23E−01 | −2.18E−02 | 3.69E−02 |
| APOE2 Count | 0 | 1 | 2 | |
| Number of Individuals: | 162 | 15 | 0 | |
| APOE4 Count | 0 | 1 | 2 | |
| Number of Individuals: | 127 | 48 | 2 | |
In a cross-sectional analysis of a separate cohort of 1030 patients with PD enrolled in the NeuroGenetics Research Consortium study (dbGaP: phs000196.v3.p1), we found that APOE ε4 was associated with lower MMSE score, as was older age, male sex, and younger age at onset (Table S7). Taken together, these data corroborate the finding that APOE ε4 is associated with cognitive impairment and a faster rate of cognitive decline in PD and indicate that this effect is independent of amyloid and tau pathology.
DISCUSSION
Dementia is a major cause of morbidity and caregiver burden in PD, with no currently available disease-modifying treatments or markedly effective symptomatic therapies. The molecular etiology of dementia in PD is unclear. Although there is some evidence that the presence of cortical αSyn pathology correlates at least to some degree with cognitive symptoms (29–31), other studies clearly show that Lewy pathology does not fully account for the dementia phenotype, and biomarker studies have shown that PD dementia has a complex landscape (10, 12, 32–35). The presence of amyloid plaques and neurofibrillary tangles in a large proportion of patients with PD dementia has focused attention on the role that AD co-pathology may play in the pathophysiology of PD dementia (15). In this context, the strong genetic association of APOE4 with PD dementia could be assumed to relate to the well-established effect of apoE4 on Aβ plaques (14, 36) and the more recent link demonstrated between apoE and tau pathology (21). Although there may be a strong contribution of AD-related pathology in PD dementia, either mediated through an APOE-dependent or separate mechanism, several careful studies have demonstrated a direct relationship between APOE genotype, LB/LN pathology, and risk of dementia related to PD, implying that apoE may regulate αSyn pathophysiology directly, independent of its effects on Aβ or tau (16–18). Here we found that A53T αSyn-Tg mice expressing human APOE4 accumulate higher amounts of insoluble phosphorylated αSyn and have reduced survival compared to A53T mice expressing APOE2. APOE4 mice exhibited more spreading of αSyn pathology compared to APOE2 mice following injection with fibrillar αSyn seeds. Corroborating this finding, we observed that cognitive function in APOE ε4/ε4 patients with PD declined more rapidly than other APOE genotypes, and that a small number of APOE ε2/ε2 patients remained cognitively stable. These results support a direct role for APOE4 in exacerbating αSyn pathology and suggest that APOE2 may exert a protective effect.
Our biochemical and histologic data suggest that APOE4 may have a direct effect on αSyn aggregation. This hypothesis is supported by several findings including that αSyn contains multiple repeats harboring an amphipathic alpha-helical motif typical of apolipoproteins (37, 38), human APOE isoforms influence αSyn aggregation in vitro (39), and APOE has been shown to co-localize with Lewy bodies in human brain (40, 41). It is not immediately clear how APOE, which is primarily an extracellular protein secreted by astrocytes and activated microglia, would interact pathologically with αSyn which is localized mainly to cytosolic and membrane-associated compartments in presynaptic nerve terminals, although several possibilities exist including apoE isoform-dependent alterations in cholesterol homeostasis which could disrupt αSyn association with lipid membranes (42). An alternative mechanism could involve a direct interaction of apoE in the extracellular space with a monomeric or oligomeric form of αSyn (43–46). The simplest explanation for our data showing that APOE4 exacerbates spreading of pSyn pathology following αSyn PFF injection is that apoE isoforms regulate a cell autonomous mechanism, given that the uptake of αSyn PFFs by striatal dopaminergic terminals and aggregation of endogenous αSyn in their corresponding soma in the SNpc involves a single neuron projecting from the SNpc to the striatum. This scenario may be less likely though, given that neurons do not produce apoE under most conditions, implying that a more complex, non-cell autonomous process is involved. Further work is needed to better characterize the mechanism of apoE isoform-dependent effects on αSyn aggregation and spreading, the cell types involved, and the implications for TH neuron toxicity and motor phenotype. At three months following αSyn PFF injection, we found a modest increase in pSyn pathology in the SNpc in E4 mice compared to E2 mice, but the E3 mice displayed an intermediate phenotype and were not different compared to the other genotypes. It is possible that with longer incubation times that these differences may be more pronounced. Also, in this study, we observed that the increased pSyn pathology burden in the SNpc in E4 mice did not correlate with a greater magnitude of TH neuron loss. One possible reason for this apparent discrepancy is that these events are not temporally synchronized and that with additional time, the increased pSyn pathology in E4 mice may translate into greater neuron loss. Additional experiments with a range of incubation times following αSyn PFF injection will be needed to test this hypothesis.
Given the astrogliosis and microgliosis we observed in A53T/APOE mice with pSyn pathology, we anticipated that inflammatory gene expression might correlate strongly with the presence of pSyn histopathology. However, given the relationship between APOE and inflammation described in multiple other models of brain disease (21, 47, 48), we were surprised to see few if any differences in gene expression in A53T mice when stratified by APOE genotype. This likely reflects that approximately half of the mice in the A53T/EKO and A53T/E4 groups had substantial amounts of pSyn pathology, whereas the other half of these groups, as well as the A53T/E2 group, had no measurable pSyn pathology. One of the most striking phenotypic differences between the current experiments in A53T αSyn-Tg mice and those that our group reported with tau-Tg mice is that the absence of APOE actually did not change the amount of insoluble tau (21). One of the main effects of deletion of APOE in the tau model was reducing neurodegeneration in the context of tauopathy, presumably by affecting the innate immune response to tau pathology (21, 49). We recently showed that the protective effect of loss of APOE in the tauopathy model is mediated through a reduction in microglia-induced neurodegeneration (50). Our findings in A53T αSyn-Tg mice are distinct and suggest that the main effect of APOE genotype in this model is on aggregation of αSyn and not on the inflammatory response to αSyn pathology. One possible mechanism for this difference could be that human APOE isoforms interact differently with αSyn than with tau, possibly because of the lipid-binding properties of αSyn. Another possibility is that pathogenic tau aggregates stimulate the innate immune system in a different manner than pathogenic αSyn aggregates.
The finding that a myelination-enriched gene module was positively associated with APOE2 genotype and negatively correlated with pSyn pathology is intriguing and may indicate that oligodendrocytes regulate either the pathologic aggregation of αSyn or a compensatory response, in an APOE2-dependent manner. In the context of synucleinopathies, this is of particular interest given that MSA, an aggressive illness characterized by aggregates with high potency for pathologic seeding and spreading, is characterized by αSyn accumulation in oligodendrocytes (51, 52). One study of 44 patients with MSA found no difference in the frequency of the APOE ε2 allele in patients with MSA compared to healthy controls (53), and another recent study of 168 autopsy-confirmed patients with MSA found no difference in disease risk or αSyn burden when stratified by APOE ε2 or APOE ε4 (54). The role of oligodendrocytes in the pathogenesis of MSA is not fully appreciated and is even less clear in PD and PD dementia, but deserves more attention especially considering the strong associations we observed for APOE2 and pSyn pathology. We did not observe localization of pSyn pathology in oligodendrocytes in the brainstem of A53T mice in this study, but a recent report described pSyn pathology in oligodendrocytes 18 months following injection of αSyn fibrils, suggesting that oligodendrocytes may play a role in other synucleinopathies besides MSA (55). Further work is needed to explore the role of oligodendrocytes in human synucleinopathies.
Our analysis of APOE genotype effect on cognitive impairment in human patients with PD confirms several previous reports that APOE ε4 exacerbates or accelerates dementia in PD, including several studies in large cohorts (7, 8, 25, 26, 34, 56, 57). Given that the studies we examined are ongoing and many patients are still living, we were not statistically powered to perform a comprehensive analysis incorporating neuropathology data stratified by APOE genotype. Similar to previous studies (26, 58–62), we found that lower CSF Aβ42 concentration and higher CSF pTau concentration were associated with a faster rate of cognitive decline in PD. However, our finding that the relationship between APOE ε4 and cognitive decline is independent of CSF Aβ42 and pTau indicates that amyloid and tau pathology do not completely account for the APOE ε4 effect on dementia in PD, supporting the possibility of a direct link between apoE and αSyn. Additional longitudinal analyses, especially those incorporating neuropathology data to anchor findings to a gold standard, will be critical to interpret genetic and biomarker signatures and develop protocols for clinical assessment and response to disease-modifying treatment trials. Additional longitudinal follow up will be important to further delineate the role of specific APOE alleles, including whether APOE ε2 may be protective in synucleinopathies in addition to the deleterious effect of APOE ε4 relative to APOE ε2 and APOE ε3 backgrounds.
Our study has several limitations. First, although the mouse models utilized in these experiments exhibit pathological αSyn aggregation and motor dysfunction, they do not develop robust cognitive phenotypes and therefore do not perfectly recapitulate the full spectrum of symptoms seen in humans with PD dementia. Recently, injection of αSyn PFFs into the gut was shown to result in extensive spreading of αSyn pathology to cortical and limbic areas, accompanied by deficits in multiple cognitive tests (63). Additional experiments are needed in model systems that will allow examination of risk factors including APOE genotype on cortical/limbic αSyn pathology and cognitive behavioral phenotypes. Second, the A53T mouse model that we employed relies on overexpression of a mutant transgene, which raises certain issues with respect to expression amount and molecular specificity of interaction with other proteins. This may contribute to the pattern we observed with respect to αSyn pathology in Apoe KO mice, which consistently displayed higher burden of αSyn pathology compared to APOE2. Multiple other models of protein aggregation and neurodegeneration that have examined an effect of APOE genotype found the lowest amounts of pathology in Apoe KO mice, including a study in a different αSyn-Tg model (21, 64–67). Third, the three-month incubation time we used for the αSyn PFF injection experiment was sufficient to detect modest loss of TH-positive dopaminergic neurons in human APOE genotype mice, but we did not observe any difference in the magnitude of TH-neuron loss across APOE genotypes. Other studies that utilized a longer duration of incubation following αSyn PFF injection have reported a larger magnitude of TH-neuron loss, which may afford a larger dynamic range and facilitate detection of differences among groups. This will be important to incorporate in future studies to investigate the role of specific genetic manipulations on αSyn pathology and dopaminergic neurodegeneration. Fourth, our analysis of human studies would benefit from larger sample sizes and having neuropathology data as a gold standard for diagnosis and co-pathology stratification. This is a common issue with studies of chronic neurodegenerative illnesses and a shared challenge for multiple ongoing studies.
Our findings indicate that apoE isoforms independently regulate αSyn pathology and contribute to disease progression in synucleinopathies. It will be important to further elucidate the molecular and cellular mechanisms of this effect to determine if discrete steps in this cascade may represent therapeutic targets for intervention. Whether this could yield generalizable approaches to treat multiple synucleinopathies is not clear, but it is intriguing to speculate whether APOE and other potential genetic risk or resilience genes could be useful as screening tools to stratify risk for individual patients and tailor preventative or interventional treatments for PD and other neurodegenerative diseases.
MATERIALS AND METHODS
Study Design
The objective of this study was to determine if APOE genotype regulates αSyn pathology and progression of disease in mouse models of synucleinopathy and in human patients with PD. After analysis was underway, we also sought to determine if APOE genotype affected gene expression in A53T αSyn-Tg mice. The study design included controlled laboratory experiments and cohort studies of human patients. The sample sizes for A53T αSyn-Tg mouse and αSyn PFF stereotaxic injection experiments were initially selected based on power analyses of published data (19, 20, 22). Sample sizes and genotypes for Nanostring gene expression analysis were selected based on αSyn pathology and biochemistry results. Assessment and quantitation of data from mouse experiments was performed by individuals blinded to genotype of the mice. Replication numbers for experiments are listed in the figure legends.
Animals
A53T αSyn-Tg mice (The Jackson Laboratory, 006823) were crossed with human APOE2, APOE3, or APOE4 knock-in mice (provided by P.M. Sullivan, Duke University) or Apoe knockout mice (The Jackson Laboratory, 002052) to generate A53T mice homozygous for one of the three human alleles or completely null for Apoe. Male and female mice were used in this study. All mice were on a C57BL/6 background. Animal procedures were performed in accordance with protocols approved by the Animal Studies Committee at Washington University School of Medicine.
Brain Extraction and Tissue Homogenzation
Mice were anesthetized with an intraperitoneal injection of pentobarbital and perfused with cold heparinized phosphate buffered saline (PBS). The brains were carefully removed and separated into two hemispheres along the mid-sagittal plane. The left hemisphere was fixed in 4% paraformaldehyde in phosphate buffer overnight and then transferred to 30% sucrose in PBS and allowed to sink at 4°C before sectioning. The right hemisphere was dissected into regions and frozen at −80°C until use. Brainstems were weighed and homogenized using a Dounce tissue grinder (Kimble Kontes) in 10 mL/g cold reassembly buffer (RAB), composed of 750 mM NaCl, 50 mM Tris-HCl pH 7.4, 5 mM EDTA. Before use, protease inhibitor cocktail (Roche Complete) and phosphatase inhibitors (Roche PhosSTOP) were added at 1x recommended concentrations, hereafter referred to as “PIC/PI.” Homogenates were centrifuged at 100,000 × g for 20 minutes at 4°C and the supernatants taken as the RAB-soluble fraction. The pellets were homogenized in RAB plus 1% Triton X-100 plus PIC/PI and centrifuged at 100,000 × g for 20 minutes at 4°C, with the supernatants saved as the RAB-TX-100-soluble fraction. The pellets were homogenized in RAB-TX-100 plus 1 M sucrose, centrifuged at 100,000 × g for 20 minutes at 4°C, and the supernatants with associated myelin were removed with a cotton swab. The pellet was resuspended in radioimmunoprecipitation assay (RIPA) buffer composed of 150 mM NaCl, 50 mM Tris pH 8.0, 5 mM EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, plus PIC/PI, then centrifuged at 100,000 × g for 20 minutes at 4°C, with the supernatant saved as the RIPA-soluble fraction. The pellets were homogenized in 2% SDS plus PIC/PI and centrifuged at 100,000 × g for 20 minutes at room temperature, with the supernatants saved as the SDS-soluble fraction. All fractions were stored at −80°C until analyzed.
ELISA
To measure total αSyn, half-area 96 well plates were coated overnight at 4°C with Syn1 (BD Transduction Laboratories) and blocked with bovine serum albumin (BSA). Samples and recombinant αSyn standard (Anaspec) were diluted in sample buffer (PBS, 0.1% BSA, 0.1% SDS, 1% TX-100) and incubated overnight at 4°C, followed by detection with biotinylated 13G5 anti-αSyn antibody. To measure phosphorylated αSyn, 81A antibody (BioLegend) was used as the capture antibody and biotinylated Syn1 as the detection antibody, with phospho-serine 129 αSyn peptide (Proteos) as a standard. Both ELISAs were developed using streptavidin-conjugated horseradish peroxidase (Fitzgerald) and super-slow TMB substrate (Sigma) with absorbance read at 650 nm.
Immunoblot
Total protein concentrations were determined by BCA assay (Thermo). 15 μg (RAB, RAB-TX100) or 1.5 μg (RIPA, SDS) from each sample was separated on 4–12% gradient gels (Invitrogen), transferred to 0.2 μm PVDF membranes (Millipore), blocked with 2% BSA in TBS, and incubated overnight at 4°C with Syn1 and MJF-R13. Membranes were washed in TBS + 0.1% Tween-20, incubated for 2 hours at room temperature with goat-anti-mouse Alexa-488 and goat-anti-rabbit Alexa-647 secondary antibodies (Invitrogen), washed thoroughly with TBS+Tween, rinsed with TBS, and scanned using a ChemiDoc MP imaging system (BioRad).
Immunohistochemistry
Free-floating A53T mouse brain sections (50 μm) were stained with primary and fluorophore-conjugated antibodies, counterstained with DAPI, and images acquired using either a Zeiss LSM 880 Airyscan confocal microscope, an Olympus FV1200 confocal microscope, or a Hamamatsu NanoZoomer 2.0-HT slide scanner. Images were processed and quantified using Imaris 8.1 (Bitplane), NDP.view2 (Hamamatsu), ImageJ version 2.0.0 (National Institutes of Health), and Photoshop CS5 (Adobe) software. Detailed Materials and Methods available in the Supplementary Material.
Nanostring gene expression analysis
In coordination with Canopy Biosciences, RNA was isolated from 12 month-old A53T/EKO (n=10), A53T/E2 (n=6), and A53T/E4 (n=10) mouse midbrain and 793 transcripts were quantified with the Nanostring nCounter multiplexed target platform using a customized chip based on the Mouse Neuroinflammation panel (https://www.nanostring.com). The geometric mean of negative control lanes was subtracted from gene transcript counts and gene expression data were normalized to the geometric mean of positive control lanes and the following housekeeping genes: Aars, Asb10, Ccdc127, Cnot10, Csnk2a2, Fam104a, Gusb, Lars, Mto1, Supt7l, Tada2b, Tbp, and Xpnpep1. Differential gene expression and principal component analysis were performed using nSolver 4.0 and the Advanced Analysis 2.0 plugin (Nanostring). For differential expression analysis, pSyn pathology was expressed as low (<5% coverage by IHC) or high (>5% coverage by IHC) for each animal and APOE genotype and pSyn pathology were selected as predictor covariates. Fold-change expression and p-values were calculated by linear regression analysis using negative binomial or log-linear models. P-values were corrected for multiple comparisons using the Benjamini-Yekutieli method. Volcano plots of differential expression data were plotted using the −log10 (uncorrected p-value) and log2 fold-change using the Ggplot2 package in R. Gene co-expression analysis was performed using the WGCNA package in R as described previously (68). A soft thresholding power of 4 resulted in a scale-free topology fit index of 0.90 and was selected to calculate the signed-hybrid adjacency for normalized gene counts. Hierarchical clustering of the topological overlap matrix dissimilarity was used to produce a gene dendrogram. Gene modules were identified using a dynamic tree cut with a minimum module size of 30 genes. Eigengenes for each module were calculated and correlations with dummy-coded APOE genotype or the percent pSyn pathology were calculated using biweight midcorrelation. Gene significance and module membership were also calculated using biweight midcorrelation. Gene ontology enrichment analysis was performed using Panther (geneontology.org) (69–71) Heatmaps were constructed using Phantasus (https://bioconductor.org/packages/release/bioc/html/phantasus.html).
Inverted Wire Screen Test
The inverted wire screen test of motor function was performed by placing the mouse upright on a platform covered with a wire screen and then inverting the platform. The latency of the mouse to fall off of the wire screen was measured and the better time was taken from two trials. Trials were stopped if a mouse reached a criterion of 60 seconds.
Recombinant αSyn Pre-formed Fibrils
Purification of recombinant mouse sequence αSyn monomer and in vitro fibril assembly was performed as described (72) with minor modifications. Fibril assembly reactions were carried out in an Eppendorf Thermomixer at 1000 rpm for 3 days. An aliquot of the resulting suspension was centrifuged at 15,000 × g for 20 min and the PFF concentration was estimated by subtracting the concentration of αSyn monomer in the resulting supernatant from the starting monomer concentration. PFFs were aliquoted and stored at −80°C until use. Prior to use, aliquots were thawed and sonicated briefly in a water bath sonicator (QSonica).
Stereotaxic Injection
Apoe knockout and APOE2, APOE3, or APOE4 knock-in mice (3–5 months of age) were anesthetized with isoflurane and stereotaxically injected with 2.5 μL of αSyn PFF suspension (approximately 4 μg) into the left dorsal striatum (0.2 mm anterior and 2.0 mm lateral to Bregma, and 3.5 mm below the surface of the skull) using a Hamilton microsyringe attached to a motorized injector (Stoelting). At three months post-injection, animals were anesthetized with pentobarbital and transcardially perfused with heparinized PBS. The brains were carefully removed, fixed in 4% paraformaldehyde overnight, then transferred to 30% sucrose in PBS and allowed to sink at 4°C before sectioning. Whole brains were cut in the coronal plane at 50 μm and processed as described above for fluorescence immunohistochemistry. For manual neuron counts, TH-positive cell bodies in the substantia nigra pars compacta were counted in four sections spaced 300 μm apart.
Cognitive Assessment in Human Patients with PD
Clinical, biomarker, and genetic data from the Parkinson’s Progression Markers Initiative (PPMI) were obtained from the PPMI database repository (www.ppmi-info.org), accessed most recently on April 1, 2019. Cognition was assessed per PPMI study protocol using the Montreal Cognitive Assessment (MoCA). We generated principal components from the genetic data to infer genetic ancestry and filtered to include only individuals of European ancestry. We further filtered the dataset to analyze only those individuals with a clinical diagnosis of PD who did not have a known mutation in LRRK2, SNCA, or GBA genes. Prior to analysis, we excluded all individuals without data for APOE genotype or CSF biomarkers, those with APOE ε2/ε4 genotype, those with less than three cognitive assessments, and those with baseline MoCA score of 25 or less. 251 individuals met inclusion criteria, with a mean follow up time of three years.
We identified individuals from the Washington University in St Louis Movement Disorder Center (WUSTLPD) with a clinical diagnosis of PD according to United Kingdom Brain Bank criteria (73) modified for genetic studies (74) who had longitudinal Mini-Mental State Examination (MMSE) data. As with the PPMI cohort, we applied inclusion criteria of clinical diagnosis of PD, European ancestry, at least three cognitive assessments, non-APOE ε2/ε4 genotype, and baseline MMSE score of 24 or greater. 177 individuals met inclusion criteria with a mean follow up time of five years.
Detailed Materials and Methods available in the Supplementary Material.
Statistical Analysis
Statistical tests included one-way ANOVA (or Kruskal-Wallis test for nonparametric data), multiple t-test, linear regression, Kaplan-Meier survival analysis, two-way ANOVA, and a linear mixed effects model. Data were expressed as mean ± SEM. Significance level (alpha) of 0.05 and two-sided tests were used. Tukey’s multiple comparisons test (one-way ANOVA), Dunn’s multiple comparisons test (Kruskal-Wallis test), Bonferroni’s correction (Kaplan-Meier), and the Benjamini-Yekutieli method were used to correct for multiple comparisons.
Supplementary Material
Fig. S1. Insoluble total αSyn correlates with endstage paralysis in A53T mice.
Fig. S2. Immunoblot analysis of total and phosphorylated αSyn
Fig. S3. pSyn pathology A53T mice localizes to neurons
Fig. S4. Microglial reactivity in A53T mice with pSyn pathology
Fig. S5. pSyn pathology in the cortex in A53T mice
Fig. S6. Sample variance of inflammatory gene expression correlates with pSyn pathology in A53T mice.
Fig. S7. Gene expression modules correlate with endstage paralysis in A53T mice.
Fig. S8. Minimal colocalization of pSyn pathology and oligodendrocytes in A53T mice.
Fig. S9. pSyn pathology in the striatum following αSyn PFF injection
Table S7. Linear regression analysis of MMSE scores in dbGaP participants.
Table S1. pSyn IHC differential expression data (see separate Excel file).
Table S3. E4 vs E2 differential expression data (see separate Excel file).
Table S2. EKO vs E2 differential expression data (see separate Excel file).
Table S5. Gene-trait significance (see separate Excel file).
Table S4. Gene module membership (see separate Excel file).
Table S6. Gene ontology analysis for selected modules (see separate Excel file).
ACKNOWLEDGEMENTS
We thank the patients who participated in this research, their families, and the investigators and staff at the Washington University Movement Disorders Center; Drs. John R. Cirrito, Sarah Fritschi, Gilbert Gallardo, Erik Musiek, and Laura A. Volpicelli-Daley for helpful discussions; Drs. Kelvin C. Luk and Virginia M.-Y. Lee for providing the mouse αSyn plasmid and for helpful discussions; and Mary Beth Finn, Javier Remolina Serrano, and Nathan Scott for expert technical assistance. Canopy Biosciences performed RNA extraction and Nanostring nCounter assays. Digital immunohistochemistry images were captured using instruments in the Alafi Neuroimaging Laboratory and the Washington University Center for Cellular Imaging, with expert assistance from Gary London, Michael Shih, and Dennis Oakley.
Data used in the preparation of this article were obtained from the Parkinson’s Progression Markers Initiative (PPMI) database (www.ppmi-info.org/data). For up-to-date information on the study, visit www.ppmi-info.org. PPMI, a public-private partnership, is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners, including Abbvie, Allergan, Avid, Biogen, BioLegend, Bristol-Myers Squibb, Celgene, Denali, GE Healthcare, Genentech, GlaxoSmithKline, Lilly, Lundbeck, Merck, Meso Scale Discovery, Pfizer, Piramal, Prevail, Roche, Sanofi Genzyme, Servier, Takeda, Teva, UCB, Verily, Voyager, and Golub Capital.
FUNDING
This work was supported by an American Academy of Neurology / American Brain Foundation Clinical Research Training Fellowship (A.A.D.), the BrightFocus Foundation (A.A.D.), the Mary E. Groff Charitable Trust (A.A.D.), the Dobbins Family Fund (A.A.D.), Barnes-Jewish Hospital Foundation (Elliot Stein Family Fund; J.S.P.), the Riney Foundation (J.S.P.), the American Parkinson Disease Association (APDA) (J.S.P.), the Greater St. Louis Chapter of the APDA (J.S.P.), The JPB Foundation (D.M.H.), NIH K08NS101118 (A.A.D.), R01AG044546 (C.C.), RF1AG053303 (C.C.), RF1AG058501 (C.C.), U01AG052411 (C.C.), U01AG058922 (C.C.), NS075321 (J.S.P.), NS097799 (P.T.K.), R01NS090934 (D.M.H.), and R01 AG047644 (D.M.H.).
Footnotes
* This manuscript has been accepted for publication in Science Translational Medicine. This version has not undergone final editing. Please refer to the complete version of record at www.sciencetranslationalmedicine.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior written permission of AAAS.
COMPETING INTERESTS
C.C. receives research support from: Biogen, EISAI, Alector and Parabon, none of which had any role in the collection, analysis, or interpretation of data; in the writing of the report; or in the decision to submit the paper for publication. C.C. is a member of the advisory board of ADx Healthcare, Halia Terapeutics and Vivid Genomics. D.M.H. co-founded and is on the scientific advisory board of C2N Diagnostics, LLC. D.M.H. is on the scientific advisory board of Denali and consults for Genentech and Idorsia. All other authors declare that they have no competing interests.
DATA AND MATERIALS AVAILABILITY
All the data are present in the main text or in the supplementary material.
REFERENCES
- 1.Obeso JA et al. , Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Movement disorders : official journal of the Movement Disorder Society 32, 1264–1310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willis AW et al. , Predictors of survival in patients with Parkinson disease. Archives of neurology 69, 601–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG, The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Movement disorders : official journal of the Movement Disorder Society 23, 837–844 (2008). [DOI] [PubMed] [Google Scholar]
- 4.McKeith IG et al. , Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89, 88–100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bras J et al. , Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Human molecular genetics, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guerreiro R et al. , Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet neurology 17, 64–74 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mata IF et al. , APOE, MAPT, and SNCA genes and cognitive performance in Parkinson disease. JAMA neurology 71, 1405–1412 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morley JF et al. , Genetic influences on cognitive decline in Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society 27, 512–518 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braak H et al. , Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of aging 24, 197–211 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Halliday G, Hely M, Reid W, Morris J, The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta neuropathologica 115, 409–415 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Irwin DJ, Lee VM, Trojanowski JQ, Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nature reviews. Neuroscience 14, 626–636 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotzbauer PT et al. , Pathologic accumulation of alpha-synuclein and Abeta in Parkinson disease patients with dementia. Archives of neurology 69, 1326–1331 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim J, Basak JM, Holtzman DM, The role of apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holtzman DM, Herz J, Bu G, Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harbor perspectives in medicine 2, a006312 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irwin DJ et al. , Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet neurology 16, 55–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuang D et al. , APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA neurology 70, 223–228 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dickson DW et al. , APOE epsilon4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 91, e1182–e1195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabir MS et al. , Assessment of APOE in atypical parkinsonism syndromes. Neurobiology of disease 127, 142–146 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee MK et al. , Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 99, 8968–8973 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin LJ et al. , Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. The Journal of neuroscience : the official journal of the Society for Neuroscience 26, 41–50 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi Y et al. , ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luk KC et al. , Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science (New York, N.Y.) 338, 949–953 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Federoff M, Jimenez-Rolando B, Nalls MA, Singleton AB, A large study reveals no association between APOE and Parkinson’s disease. Neurobiology of disease 46, 389–392 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paul KC et al. , APOE, MAPT, and COMT and Parkinson’s Disease Susceptibility and Cognitive Symptom Progression. Journal of Parkinson’s disease, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tropea TF et al. , APOE, thought disorder, and SPARE-AD predict cognitive decline in established Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society 33, 289–297 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shahid M et al. , An increased rate of longitudinal cognitive decline is observed in Parkinson’s disease patients with low CSF Ass42 and an APOE epsilon4 allele. Neurobiology of disease 127, 278–286 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marek K. e. a., for the Parkinson’s Progression Markers Initiative, The Parkinson Progression Marker Initiative (PPMI). Progress in neurobiology 95, 629–635 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eusebi P et al. , Diagnostic utility of cerebrospinal fluid alpha-synuclein in Parkinson’s disease: A systematic review and meta-analysis. Movement disorders : official journal of the Movement Disorder Society 32, 1389–1400 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Braak H, Rub U, Jansen Steur EN, Del Tredici K, de Vos RA, Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 64, 1404–1410 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Galvin JE, Pollack J, Morris JC, Clinical phenotype of Parkinson disease dementia. Neurology 67, 1605–1611 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Hurtig HI et al. , Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 54, 1916–1921 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Parnetti L et al. , Cerebrospinal fluid biomarkers in Parkinson’s disease with dementia and dementia with Lewy bodies. Biological psychiatry 64, 850–855 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Jellinger KA, A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochimica et biophysica acta 1792, 730–740 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Irwin DJ et al. , Neuropathologic substrates of Parkinson disease dementia. Annals of neurology 72, 587–598 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao L et al. , Cerebrospinal fluid alpha-synuclein as a biomarker for Parkinson’s disease diagnosis: a systematic review and meta-analysis. The International journal of neuroscience 125, 645–654 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Huynh TV, Davis AA, Ulrich JD, Holtzman DM, Apolipoprotein E and Alzheimer’s disease: the influence of apolipoprotein E on amyloid-beta and other amyloidogenic proteins. Journal of lipid research 58, 824–836 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davidson WS, Jonas A, Clayton DF, George JM, Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. The Journal of biological chemistry 273, 9443–9449 (1998). [DOI] [PubMed] [Google Scholar]
- 38.George JM, Jin H, Woods WS, Clayton DF, Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 15, 361–372 (1995). [DOI] [PubMed] [Google Scholar]
- 39.Emamzadeh FN, Aojula H, McHugh P, Allsop D, Effects of different isoforms of apoE on aggregation of the alpha-synuclein protein implicated in Parkinson’s disease. Neuroscience letters, (2016). [DOI] [PubMed] [Google Scholar]
- 40.Rohn TT, Mack JM, Apolipoprotein E Fragmentation within Lewy Bodies of the Human Parkinson’s Disease Brain. International journal of neurodegenerative disorders 1, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilhelmus MM et al. , Apolipoprotein E and LRP1 Increase Early in Parkinson’s Disease Pathogenesis. The American journal of pathology 179, 2152–2156 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fantini J, Yahi N, The driving force of alpha-synuclein insertion and amyloid channel formation in the plasma membrane of neural cells: key role of ganglioside- and cholesterol-binding domains. Advances in experimental medicine and biology 991, 15–26 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Danzer KM et al. , Exosomal cell-to-cell transmission of alpha synuclein oligomers. Molecular neurodegeneration 7, 42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Danzer KM et al. , Heat-shock protein 70 modulates toxic extracellular alpha-synuclein oligomers and rescues trans-synaptic toxicity. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 25, 326–336 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emmanouilidou E et al. , Assessment of alpha-synuclein secretion in mouse and human brain parenchyma. PloS one 6, e22225 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamada K, Iwatsubo T, Extracellular alpha-synuclein levels are regulated by neuronal activity. Molecular neurodegeneration 13, 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin S et al. , Apolipoprotein E mediation of neuro-inflammation in a murine model of multiple sclerosis. Journal of neuroimmunology 271, 8–17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ulrich JD et al. , ApoE facilitates the microglial response to amyloid plaque pathology. The Journal of experimental medicine 215, 1047–1058 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krasemann S et al. , The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581.e569 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi Y et al. , Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. The Journal of experimental medicine 216, 2546–2561 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jellinger KA, Neuropathology of multiple system atrophy: new thoughts about pathogenesis. Movement disorders : official journal of the Movement Disorder Society 29, 1720–1741 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Peng C et al. , Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 557, 558–563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cairns NJ et al. , Apolipoprotein E e4 allele frequency in patients with multiple system atrophy. Neuroscience letters 221, 161–164 (1997). [DOI] [PubMed] [Google Scholar]
- 54.Ogaki K et al. , Multiple system atrophy and apolipoprotein E. Movement disorders : official journal of the Movement Disorder Society 33, 647–650 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uemura N et al. , Slow Progressive Accumulation of Oligodendroglial Alpha-Synuclein (alpha-Syn) Pathology in Synthetic alpha-Syn Fibril-Induced Mouse Models of Synucleinopathy. Journal of neuropathology and experimental neurology 78, 877–890 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ibanez L et al. , Pleiotropic Effects of Variants in Dementia Genes in Parkinson Disease. Frontiers in neuroscience 12, 230 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parsian A, Racette B, Goldsmith LJ, Perlmutter JS, Parkinson’s disease and apolipoprotein E: possible association with dementia but not age at onset. Genomics 79, 458–461 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Liu C et al. , CSF tau and tau/Abeta42 predict cognitive decline in Parkinson’s disease. Parkinsonism & related disorders 21, 271–276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hall S et al. , CSF biomarkers and clinical progression of Parkinson disease. Neurology 84, 57–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Backstrom DC et al. , Cerebrospinal Fluid Patterns and the Risk of Future Dementia in Early, Incident Parkinson Disease. JAMA neurology 72, 1175–1182 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Siderowf A et al. , CSF amyloid {beta} 1–42 predicts cognitive decline in Parkinson disease. Neurology 75, 1055–1061 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Montine TJ et al. , CSF Abeta(42) and tau in Parkinson’s disease with cognitive impairment. Movement disorders : official journal of the Movement Disorder Society 25, 2682–2685 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim S et al. , Transneuronal Propagation of Pathologic alpha-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holtzman DM et al. , Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America 97, 2892–2897 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fagan AM et al. , Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiology of disease 9, 305–318 (2002). [DOI] [PubMed] [Google Scholar]
- 66.Gallardo G, Schluter OM, Sudhof TC, A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nature neuroscience 11, 301–308 (2008). [DOI] [PubMed] [Google Scholar]
- 67.Bales KR et al. , Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 6771–6779 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langfelder P, Horvath S, WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.The Gene Ontology Resource: 20 years and still GOing strong. Nucleic acids research 47, D330–d338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ashburner M et al. , Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mi H et al. , PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic acids research 45, D183–d189 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Volpicelli-Daley LA, Luk KC, Lee VM, Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nature protocols 9, 2135–2146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hughes AJ, Daniel SE, Kilford L, Lees AJ, Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. Journal of neurology, neurosurgery, and psychiatry 55, 181–184 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Racette BA, Rundle M, Parsian A, Perlmutter JS, Evaluation of a screening questionnaire for genetic studies of Parkinson’s disease. American journal of medical genetics 88, 539–543 (1999). [PubMed] [Google Scholar]
- 75.Factor SA et al. , Postural instability/gait disturbance in Parkinson’s disease has distinct subtypes: an exploratory analysis. Journal of neurology, neurosurgery, and psychiatry 82, 564–568 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Insoluble total αSyn correlates with endstage paralysis in A53T mice.
Fig. S2. Immunoblot analysis of total and phosphorylated αSyn
Fig. S3. pSyn pathology A53T mice localizes to neurons
Fig. S4. Microglial reactivity in A53T mice with pSyn pathology
Fig. S5. pSyn pathology in the cortex in A53T mice
Fig. S6. Sample variance of inflammatory gene expression correlates with pSyn pathology in A53T mice.
Fig. S7. Gene expression modules correlate with endstage paralysis in A53T mice.
Fig. S8. Minimal colocalization of pSyn pathology and oligodendrocytes in A53T mice.
Fig. S9. pSyn pathology in the striatum following αSyn PFF injection
Table S7. Linear regression analysis of MMSE scores in dbGaP participants.
Table S1. pSyn IHC differential expression data (see separate Excel file).
Table S3. E4 vs E2 differential expression data (see separate Excel file).
Table S2. EKO vs E2 differential expression data (see separate Excel file).
Table S5. Gene-trait significance (see separate Excel file).
Table S4. Gene module membership (see separate Excel file).
Table S6. Gene ontology analysis for selected modules (see separate Excel file).