

Abstract
Renal cell carcinoma (RCC) subtypes are increasingly being discerned via their molecular underpinnings. Frequently this can be correlated to histologic and immunohistochemical surrogates, such that only simple targeted molecular assays, or none at all, are needed for diagnostic confirmation. In clear cell RCC, VHL mutation and 3p loss are well known; however, other genes with emerging important roles include SETD2, BAP1, and PBRM1, among others. Papillary RCC type 2 is now known to include likely several different molecular entities, such as fumarate hydratase (FH) deficient RCC. In MIT family translocation RCC, an increasing number of gene fusions are now described. Some TFE3 fusion partners, such as NONO, GRIPAP1, RBMX, and RBM10 may show a deceptive FISH result due to the proximity of the genes on the same chromosome. FH and succinate dehydrogenase (SDH) deficient RCC have implications for patient counseling due to heritable syndromes and the aggressiveness of FH-deficient RCC. Immunohistochemistry is increasingly available and helpful for recognizing both. Emerging tumor types with strong evidence for distinct diagnostic entities include eosinophilic solid and cystic RCC and TFEB / VEGFA / 6p21 amplified RCC. Other emerging entities that are less clearly understood include TCEB1 mutated RCC, RCC with ALK rearrangement, renal neoplasms with mutations of TSC2 or MTOR, and RCC with fibromuscular stroma. In metastatic RCC, the role of molecular studies is not entirely defined at present, although there may be an increasing role for genomic analysis related to specific therapy pathways, such as for tyrosine kinase or MTOR inhibitors.
Keywords: renal cell carcinoma, VHL, molecular pathology, TFE3, TFEB, tuberous sclerosis, MTOR
Introduction
With increasing understanding of the genetic underpinnings of renal cancer, multiple novel subtypes of tumors have been identified (1) and our understanding of well-established renal cancer types has grown dramatically. (2) Additionally, our understanding of hereditary renal cancer syndromes has also grown to include recognition of specific tumor histologies associated with tumor syndromes. (3) Nonetheless, there remain significant practice gaps for implementation of this increasing knowledge into clinical treatment paradigms, as only a few select tumor histologies have specific treatment recommendations. (4) In 2019, in conjunction with the United States and Canadian Academy of Pathology Annual Meeting, the International Society of Urological Pathology (ISUP) convened a consensus conference on molecular pathology of genitourinary tumors. This article summarizes the recommendations of the renal cancer working group and reports the results of a survey of ISUP members with respect to molecular pathology practice in renal cancer. Since other articles have summarized in detail many of the pathologic features of renal cancer types, (5) this article focuses on the latest developments in molecular pathology of renal cancer with emphasis on aspects that are practical for the surgical pathologist.
Meeting format
A web-based survey was circulated to the ISUP membership in advance of the meeting, including a series of questions on renal cancer designed by the working group members (Table 1). Results of the survey (Supplemental File) and overviews of some key areas of emerging data in renal cancer molecular pathology were presented by the group members, followed by a question and comment period. This article represents the consensus of the working group members and organizing committee, taking into account the survey data and open comments provided at the meeting. In contrast to some prior ISUP consensus meetings, this meeting did not include open voting by all attendees; however, any comments or concerns raised at the meeting were considered when making recommendations.
Table 1:
Working Group Members and Organizing Committee
| Pedram Argani | Chair |
|---|---|
| Ondrej Hes | Chair |
| Ying-Bei Chen | Member |
| Anthony J. Gill | Member |
| Sean R. Williamson | Member |
| Lars Egevad | Organizing Committee |
| David J. Grignon | Organizing Committee |
| Glen Kristiansen | Organizing Committee |
Clear cell RCC
Clear cell RCC is overwhelmingly the most common subtype of adult renal cancer, making up approximately 65–70% of tumors. (6) Much of our molecular knowledge of renal cancer stems from this type, both in the hereditary and sporadic settings. (2, 7–9) In brief, it is well known that clear cell RCC typically harbors alterations of the VHL gene, either in the form of mutation or promoter methylation, (2) and a “second hit” typically occurs as large deletion that may include the majority of, or the entire, p arm of chromosome 3. The latter serves as a potential diagnostic marker, as it can be detected by FISH or other copy number assessment techniques, (10) although 3p loss alone may not be entirely specific for clear cell RCC, having been identified in select other histologies. (11–13) There is emerging evidence that 3p loss and VHL mutation are so common in clear cell renal cancer (>90%) that some of the rare tumors lacking them may in fact be misclassified. (14) However, bringing to bear all the necessary techniques to detect these abnormalities (mutation analysis, copy number assessment, and methylation studies) are certainly beyond the scope of most diagnostic pathology practices in a routine setting.
Additionally, there is now increasing awareness of a number of other genes that are frequently altered in clear cell RCC, several of which also reside on chromosome 3p (SETD2, BAP1, PBRM1) (15) and several of which are involved in chromatin remodeling. Tumors with BAP1 or SETD2 mutations appear to have more aggressive behavior, whereas PBRM1 mutated tumors may have more favorable behavior. (6, 15) In current practice, it appears that this information is not being used routinely, based on the low rates of survey respondents utilizing these markers and the lack of their inclusion in current clinical guidelines. (4) However, these may have emerging roles in renal cancer as our integration of molecular pathology matures.
Carbonic anhydrase IX immunohistochemistry
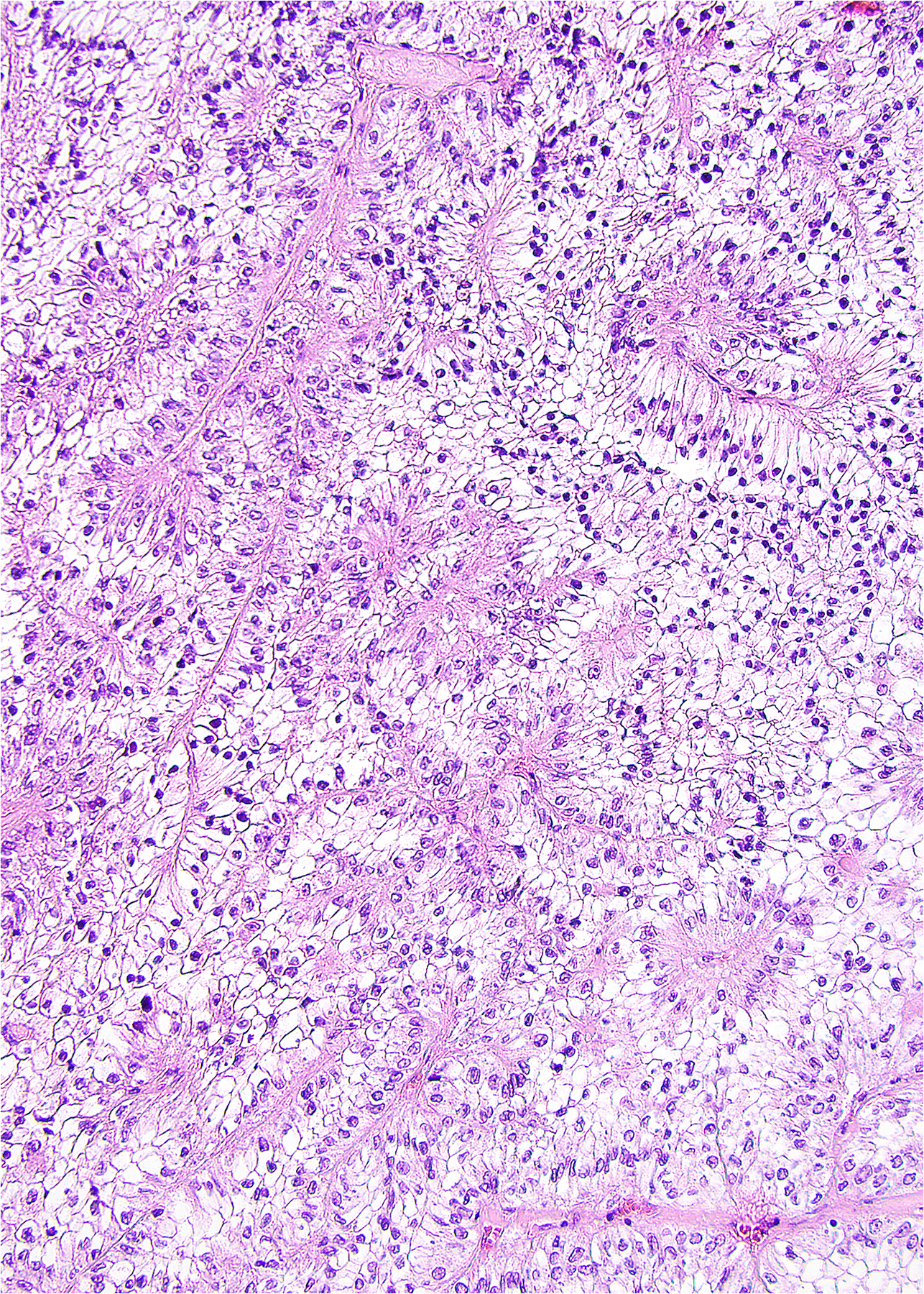
A relatively robust surrogate for pathology practice in supporting clear cell RCC genetics is immunohistochemistry for carbonic anhydrase IX. As part of the downstream hypoxia pathway under VHL, clear cell cancers typically show diffuse membrane staining for this marker, although staining may be decreased in aggressive or poorly-differentiated tumors. (16–21) Several caveats are necessary when using this as a surrogate for molecular pathology. First, positivity can be encountered in non-renal tumors. (18) As such, this should be used cautiously when considering site of origin for a cancer of unknown primary, especially in patients who have no apparent renal mass or history of renal cancer. Secondly, since carbonic anhydrase IX is part of the hypoxia pathway, some degree of positive staining can be encountered in any tumors or tissues with hypoxia or ischemia (Figure 1), which can be misleading in small biopsy samples with limited tissue visualization or necrosis with scant viable cells. Finally, clear cell papillary RCC and potentially related neoplasms, discussed later, have consistent labeling for carbonic anhydrase IX despite usual absence of VHL alterations. A “cup-shaped” pattern of staining has been reported in particular with clear cell papillary RCC. (22)
1.

Although a diffuse membranous pattern of carbonic anhydrase IX staining usually would support a diagnosis of clear cell RCC, focal staining can be encountered in the setting of hypoxic or necrotic tissues. This papillary RCC has focal staining only at the edges of the papillary structures.
Working Group Recommendations, Clear Cell RCC:
In difficult diagnostic cases, molecular evaluation can be used to support a diagnosis of clear cell RCC, such as chromosome 3p loss (FISH, cytogenetics, or copy number analysis) or VHL mutational analysis, with the understanding that 3p loss may not be entirely specific for clear cell RCC in all contexts
Routine use of molecular pathology is not necessary for straightforward cases of clear cell RCC
Carbonic anhydrase IX can be used as a surrogate for molecular pathology in most cases; however, positivity can also be observed in non-renal tumors, hypoxic tissues, and clear cell papillary RCC
Papillary RCC
Papillary RCC is the second most common type of renal cell carcinoma, accounting for approximately 15%−19% of adult renal cancers. (23) It is traditionally classified into type 1 and type 2 tumors; however, there is increasing awareness that in particular type 2 tumors make up likely more than one diagnostic entity. To date, it appears that the most uniform subtype of papillary RCC based on morphologic, immunohistochemical, and molecular features is papillary RCC type 1. (24–26) Recent proposals have attempted to classify papillary RCC into multiple subtypes based on molecular genetic features and/or combined morphologic, immunohistochemical and molecular features. (25–27) As an immunohistochemical surrogate, one finding that appears consistent across papillary RCC subtypes is that staining for alpha-methylacyl-CoA racemase (AMACR) is typically diffuse and strong, with similar intensity that of normal proximal tubules. (21)
Type 1 Papillary RCC
Polysomy or trisomy of chromosomes 7 or 17 are the most common chromosomal changes in type 1 tumors. However, gains of chromosomes 3, 12, 16, and 20 (and less frequently gains of chromosomes 2, 4, 5, 6, 8, 13, and 18) have been also noted in type 1 tumors. Chromosomal losses have also been documented, most commonly of chromosomes 1, 2, 4, 5, 7, 8, 9, 10, 11, 14, 15, 16, 18, 19, 20, 21, and 22. (24) The hereditary papillary RCC syndrome, which manifests as innumerable type 1 tumors, is characterized by germline mutations of MET. (28–30) In sporadic papillary RCCs type 1, MET mutations are also present, although the frequency appears to be lower than in the hereditary setting, (25, 31, 32) contrasting to clear cell RCC, in which VHL alterations are typical in both hereditary and sporadic tumors. Amplifications of MET in some sporadic tumors have also been noted, and potential roles for therapy targeting MET has been reported in tumors with mutations or amplifications. (33–37)
Type 2 Papillary RCC
Type 2 papillary RCC is best considered a histomorphology manifested by multiple specific neoplasms rather than a single specific entity. Although gains of chromosomes 7 and 17 are have been previously noted to be relatively common in type 2 papillary RCC as well, these are in modern studies found in a smaller percentage of cases. Gains of chromosomes 12, 16 and 20 are noted for papillary RCC type 2. (24) Recent works, such as the Cancer Genome Atlas characterization of papillary RCC, have noted that type 2 tumors exhibit CDKN2A silencing, SETD2 mutations, and increased expression of the NRF2–antioxidant response element pathway. (25) The CpG island methylator phenotype (CIMP) has also been noted as a subgroup of type 2 papillary RCC, highly associated with FH gene mutations and decreased expression of the mRNA, suggesting that this represents the emerging category of FH-deficient RCC (often associated with the hereditary leiomyomatosis and renal cell carcinoma syndrome / HLRCC), discussed later.
Oncocytic Papillary RCC / Papillary Renal Cell Neoplasm With Reverse Polarity
A third variant or subtype of papillary RCC is so-called oncocytic papillary RCC. (38–41) Until recently, this has been a poorly understood subcategory of papillary RCC composed of oncocytic cells. Since prior definitions in the literature have been variable, no definitive consensus regarding diagnosis of such tumors has been reached, and therefore this has not been adopted as an official diagnostic entity in the current classification schemes. (5, 23) Previous studies have shown a variable copy number alteration pattern with some showing gains of chromosomes 7 and 17. (39–41) Recent work has suggested that when defined according to strict criteria, there may be a distinct entity within the tumors previously noted as oncocytic papillary RCC. (26, 42, 43) In the classification scheme by Saleeb et al, this subtype was considered type 4 papillary RCC (oncocytic low-grade), (26) and recently, Al-Obaidy et al have proposed this to represent a distinct entity using the nomenclature “papillary renal cell neoplasm with reverse polarity.” (42, 43) These tumors have oncocytic cells, papillary architecture, and nuclei aligned more toward the apex of the cells. In contrast to typical papillary RCC, they show negative immunohistochemistry for vimentin, and in contrast to oncocytoma and chromophobe RCC, they are negative for KIT. (43) These tumors also appear to have consistent positivity for GATA3, contrasting to other papillary RCC subtypes. (26, 43) Very recent studies have found that this tumor is characterized by frequent KRAS mutations, which differs markedly from type 1 and 2 papillary RCC, suggesting that this may be an emerging diagnostic entity in future schemes. (5, 44)
Working Group Recommendations, Papillary RCC
Type 1 papillary RCC is the most uniform subgroup, which can usually be diagnosed by morphology. Ancillary features that may be helpful in difficult cases include common gain of chromosomes 7 and 17 and positive immunohistochemistry for cytokeratin 7
Type 2 papillary RCC is clinically and molecularly heterogeneous. This terminology may still be used at present, but should be used cautiously after consideration of mimics, especially FH-deficient RCC
A definite role for molecular classification schemes in papillary RCC is not yet established for routine diagnostic practice and clinical treatment; however, emerging data suggest that there are more than the historical 2 subtypes
Strong positive staining for AMACR can be used as a surrogate for papillary RCC phenotype in the appropriate context, although not completely specific
Chromophobe renal cell carcinoma
Chromophobe RCC is a generally indolent renal neoplasm with distinct morphologic features including pale cells, sometimes described as resembling plant cells, with prominent cell borders, and smaller eosinophilic cells. Neoplastic cells have accentuated cellular borders, hyperchromatic wrinkled nuclei (raisinoid nuclei), and perinuclear clearing (halos). Despite the longstanding recognition of oncocytoma and chromophobe RCC, there is evidence that this differential diagnosis remains a challenge even today, with incomplete agreement regarding diagnostic markers, use of immunohistochemistry, and need for genetic techniques. (45) Although molecular techniques are used rarely for this diagnosis, analysis of the FLCN (folliculin) gene can be used to support a diagnosis of Birt-Hogg-Dubé syndrome-associated “hybrid” tumors. (46, 47) Renal oncocytosis may also be a consideration for multiple oncocytic neoplasms. (48, 49) Enumeration of the chromosomes, such as by conventional cytogenetics or copy number variation pattern, can be used for routine diagnostic cases, in which multiple chromosome losses (chromosomes Y, 1, 2, 6, 10, 13, 17, 21) are most commonly identified. (50, 51) Nonetheless, other studies have also shown chromosomal gains (chromosomes 4, 7, 15, 19, and 20), losses, and even diploid status, especially in the eosinophilic variant. (52–55) Chromophobe RCC, similar to oncocytoma, has been found to have mutations in mitochondrial genes. (51, 52, 56) TP53 mutations are relatively common in chromophobe RCC, as are alterations of PTEN. (52) TERT gene promoter rearrangements have also been found to occur in a subset of chromophobe tumors. (52)
Working Group Recommendations, Chromophobe RCC
Chromophobe RCC can usually be diagnosed based on typical histologic features, with supportive immunohistochemistry in difficult cases
Chromosomal copy number pattern can be used as a diagnostic adjunct in difficult cases, which often includes losses of multiple chromosomes (chromosomes Y, 1, 2, 6, 10, 13, 17, 21).
FLCN gene analysis or patient genetic counseling can be undertaken for tumors with so-called “hybrid” (chromophobe-oncocytic) morphology and in suspect clinical situations, such as multiple oncocytic neoplasms
Oncocytoma
Renal oncocytoma is a tumor composed of cells with granular eosinophilic cytoplasm and round, regular nuclei, arranged in solid or alveolar architecture. Although a number of morphologic and architectural variants have been described, such as tubular, cystic, and telangiectatic patterns (57, 58) or so-called small cell variant, (59–63) it appears that the morphology, immunohistochemistry, and genetics remain fairly consistent with a few recurring genetic findings. (64, 65) The most used immunohistochemical markers include cytokeratin 7 (showing rare cells positive, except in scar areas), KIT (CD117, showing membranous positivity, sometimes weak), and vimentin (negative, except in scar areas). (45, 66) A specific threshold for cytokeratin 7 staining remains incompletely agreed upon. (45) Three genetic patterns are usually noted: 1) loss of chromosome 1 (in whole or in part) and loss of chromosome Y, 2) rearrangements of 11q13 (mostly translocation t(5;11)(q35;q13)), chromosome 14 deletion, and 3) a normal karyotype. (67–72) These patterns have led some to propose two or three dominant subtypes of oncocytoma. (56, 72) Recently it has been recognized that the 11q13 locus, being the site of CCND1 gene (cyclin D1), typically represents rearrangement of CCND1 in this subset of oncocytomas. (56, 72, 73) As such, recent series have divided oncocytomas into 2–3 types or classes, separating those with CCND1 rearrangement from those with other copy number alterations. (56, 72) Some data suggest that cyclin D1-positive oncocytomas are more often solitary, whereas there may be more multifocality in the cyclin D1-negative patients. (73) There does appear to be some correlation of immunohistochemistry for cyclin D1 with the presence of rearrangement; (73) however, evaluation of cyclin D1 has not gained substantial traction in pathology practice at present. Other oncocytoma tumors have been noted to have loss of chromosome 1, X, Y, 14, or 21. Since this is more similar to the genetic pattern expected of chromophobe RCC, it has been speculated that this group may be a precursor to eosinophilic variant chromophobe renal cell carcinoma, which can be difficult to distinguish from oncocytoma. (56) As with chromophobe RCC, mutations of mitochondrial genes have also been found in oncocytoma. (56) Again, as with chromophobe RCC, patients with multiple oncocytic neoplasms or “hybrid” tumors may be candidates for assessment of the FLCN gene to evaluate for Birt-Hogg-Dubé syndrome. Otherwise, multiple oncocytic tumors may indicate renal oncocytosis. (48, 49)
Working Group Recommendations, Oncocytoma
For the most part, oncocytoma is diagnosed based on typical histologic and immunohistochemical features
In diagnostically challenging cases, copy number or cytogenetic techniques can be used, with which oncocytoma often shows loss of chromosome 1 or Y, rearrangements of 11q13 (CCND1), or a normal karyotype. There is emerging evidence that CCND1 rearranged tumors may be a distinctive subset
FLCN gene analysis or patient genetic counseling can be undertaken for tumors with so-called “hybrid” (chromophobe-oncocytic) morphology and in suspect clinical situations, such as multiple oncocytic neoplasms
Clear cell papillary renal cell carcinoma
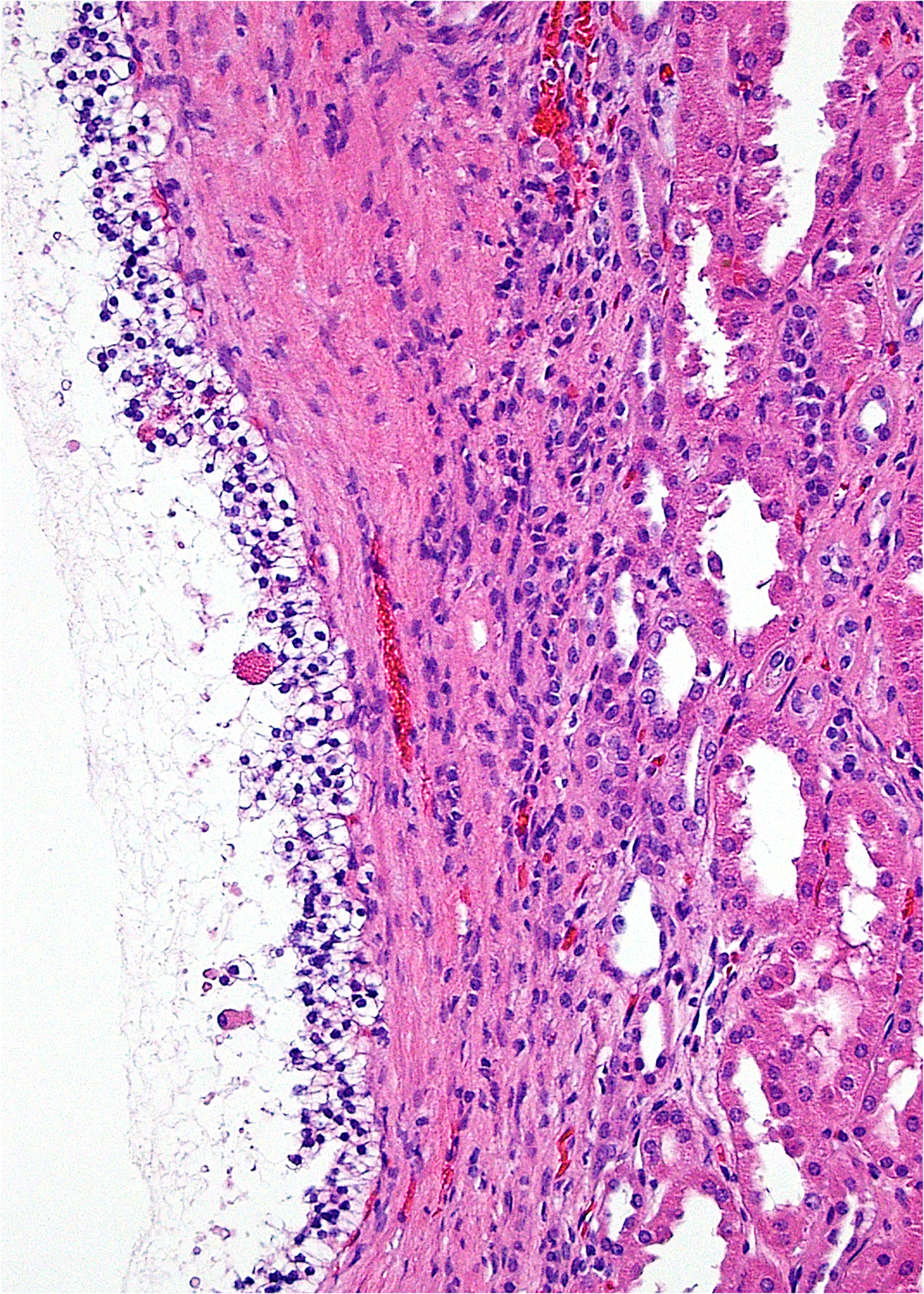
Despite being recognized only in 2006, (74) clear cell papillary RCC has now been accepted as a well-defined diagnostic entity that likely makes up as much as 4% of RCC, making it likely the 4th most common RCC subtype. (22, 75–83) Although these tumors have been historically most often mistaken for clear cell RCC, (82) they have a characteristic histology including branched glandular structures, nuclear alignment above the basement membrane (Figure 2), and variable papillary structures protruding into cystic spaces. (5, 82) Using immunohistochemistry, they have a characteristic staining pattern with diffuse cytokeratin 7 positivity, common high molecular weight cytokeratin and GATA3 positivity, consistent carbonic anhydrase IX staining (often in a “cup-shaped” distribution with the apical cell membrane being negative), (22) and negative results for AMACR and CD10, contrasting to typical papillary RCC and clear cell RCC, respectively. (82, 84, 85) Despite the resemblance of this entity to clear cell RCC, chromosome 3p25 loss and VHL gene alterations are lacking in almost all tumors. A few rare cases have been reported to have such alterations, (86) the significance of which is debatable. In general, these tumors do not have a defining pattern of recurrent genetic alterations or copy number changes. (75, 87–91) Recent work has found it to be a genomically stable tumor with severe depletion of mtDNA and a distinct metabolic phenotype.(87)
2.

Clear cell papillary RCC is composed most often of branched glandular structures with cells possessing clear cytoplasm. The nuclei are often aligned away from the basement membrane.
Recognition of this entity is important, as it appears to have highly favorable behavior, (92) although it may be multifocal or bilateral. Previously, no definite examples of aggressive behavior have been published; however, a recent case of a metastatic lesion with a compelling morphologic, immunohistochemical, and molecular phenotype has been reported (although the primary tumor was not resected). (93) There are occasional tumors that have mixed features of clear cell and clear cell papillary RCC, which have behavior and genetics closer to those of a clear cell RCC. Thus, when encountering a case with borderline features, it is worthwhile to evaluate with immunohistochemistry. If the staining results are imperfect, such as with positive AMACR or CD10, or with less than diffuse cytokeratin 7 staining, it is likely best to classify such tumors as clear cell RCC. (94–96)
Working Group Recommendations, Clear Cell Papillary RCC
The expected immunohistochemical pattern of clear cell papillary RCC (cytokeratin 7, high molecular weight cytokeratin, GATA3 positive; AMACR and CD10 negative) is a relatively robust surrogate for genetics and can be used to support the diagnosis
It is not totally clear at present whether immunohistochemical confirmation is needed even in morphologically typical cases; however, 77.4% of respondents noted using do so at present (Supplemental File)
Immunohistochemistry should be used for any tumor with borderline features of clear cell vs clear cell papillary RCC; an imperfect staining pattern should warrant classification as clear cell RCC
In cases that remain equivocal, genetic studies may be helpful; VHL mutation or chromosome 3p loss should preclude diagnosis of clear cell papillary RCC
MIT family translocation-associated RCC
In the current classification of renal cell neoplasms, tumors with TFE3, TFEB, and more recently MITF rearrangements are now grouped under the heading of MIT family translocation RCC. (97) Most common are TFE3 rearrangements, located at Xp11.2, which has led to the designation Xp11 translocation RCC for this tumor. Other works have described the recurring histologic patterns in translocation RCC tumors. (97) Common recurring features of translocation tumors include a mixture of clear and eosinophilic cells, a mixture of papillary and nested architecture, psammoma bodies, and hyalinized stroma. (98) Currently described fusion partners of TFE3 include ASPSCR1, PRCC, NONO, SFPQ, CLTC, PARP14, LUC7L3, KHSRP, DVL2, MED15, NEAT1, RBM10, KAT6A, GRIPAP1, as well as some unknown genes. (97–105) Tumors with ASPSCR1-TFE3 fusion tend to have more papillary architecture and psammoma bodies, whereas those with PRCC-TFE3 fusion tend to have less abundant cytoplasm, more compact architecture, and fewer psammoma bodies. (98, 102)
Much less common than TFE3 rearrangement is TFEB rearrangement in renal cancer, also known as t(6;11) RCC for the recurring translocation that fuses MALAT1 and TFEB. More recently a few alternative partners have been recognized, including COL21A1, CADM2, and KHDRBS2. (25, 106) The prototypical TFEB rearrangement tumor has a unique histologic pattern of nested structures formed by cells with clear cytoplasm surrounding smaller cells with less cytoplasm and associated hyaline globules, yielding a rosette-like formation. (107) However, this is not a requirement nor entirely specific for the diagnosis, as it is sometimes not well visualized, and a similar finding can occasionally be seen in TFE3 rearrangement tumors. (98) Tumors with TFEB amplification have been recently recognized, discussed later under emerging RCC types. (11, 108–111)
Although for many years TFE3 and TFEB were considered the only members of the MITF gene family that participated in rearrangements in RCC, recent work suggests that the MITF gene itself can be rearranged, with one study reporting an ACTG1-MITF fusion (112) and another finding PRCC-MITF. (113) The detailed pathologic characterization of the tumor with PRCC-MITF fusion by Xia et al noted similar findings to translocation RCC in general, including rosette-like architecture, psammoma bodies, with positive cathepsin K but negative TFE3, TFEB, and melanocytic marker staining. (113)
Surrogates available to the pathologist to recognize translocation RCC prior to, or in lieu of, genetic studies include several immunohistochemical markers (Table 2). In contrast to clear cell RCC, translocation tumors show negative or minimal carbonic anhydrase IX staining, and often some degree of melanocytic marker positivity is present. (114) Immunohistochemical staining for TFE3 and TFEB proteins is also of value, although this can be technically difficult and dependent on the laboratory staining conditions. (98) Classically translocation carcinomas show less cytokeratin staining than RCC in general; however, again this is not an unbreakable rule, as cytokeratin positivity can also be observed. (114) Staining for cathepsin K is also useful in recognizing translocation tumors, although positivity varies depending on the specific gene fusion. (115, 116) Therefore, a positive result is highly supportive of translocation RCC, but a negative result does not exclude it. FISH studies are a mainstay of diagnosis. (117–119) However, recent work has found that several specific fusions may show a false-negative or subtle positive FISH result, due to fusion of genes located close to each other on the X chromosome, in particular NONO (Figure 3), GRIPAP1, RBMX, and RBM10. (120–125) Therefore, other molecular studies, such as RNA sequencing or other techniques may be necessary to confirm difficult cases.
Table 2:
Tools for recognition of MIT family translocation-associated RCC
|
3.
This translocation-associated RCC has NONO-TFE3 fusion, which may exhibit nuclear alignment, similar to that of clear cell papillary RCC, although often with higher nuclear grade. FISH can show a subtle rearrangement pattern or it can be false-negative due to the close proximity of these genes on the X chromosome.
Working Group Recommendations, Translocation RCC:
Translocation RCC should be considered when encountering RCC with a mixture of clear cell and papillary features, psammoma bodies, abnormally voluminous cytoplasm, or hyalinized stroma, or in a patient of unexpectedly young age
Helpful pathologic surrogates for the diagnosis include positive immunohistochemical staining for TFE3 or TFEB proteins, melanocytic markers, or cathepsin K, with negative or minimal staining for carbonic anhydrase IX
FISH for TFE3 or TFEB rearrangement is a helpful diagnostic tool; however, recent work has shown that some fusions resulting from X chromosome inversion (particularly RBM10, RBMX, GRIPAP1, and NONO fusions) may show a false-negative FISH result due to the proximity of the genes involved in the rearrangement, in which case sequencing studies may be used to verify rearrangement
Renal medullary carcinoma
Renal medullary carcinoma is an aggressive renal adenocarcinoma classically found in the setting of sickle cell trait or rarely with other hemoglobinopathies. These tumors can show an infiltrative glandular architecture, often with necrosis and inflammation. (126) Recent studies have shown that medullary carcinoma is characterized by loss of the SMARCB1 (INI1) gene, with mechanisms including hemizygous loss and balanced translocation of the gene, homozygous loss, or pathogenic somatic mutation. (127–129) As such, immunohistochemistry for the SMARCB1 (INI1) protein has emerged as a helpful diagnostic tool for this entity, showing abnormal negative staining of the tumor cells. (126, 130–132) Of note, OCT3/4, often used in diagnosis of germ cell tumors, also may show positivity in medullary carcinoma. (133) Interestingly, rare tumors have also recently been recognized to have alterations of SMARCB1 or abnormal negative staining for the protein in the absence of sickle trait. The term “RCC unclassified with medullary phenotype” has been proposed for this scenario, until it becomes better understood. (134, 135)
Working Group Recommendations, Renal Medullary Carcinoma:
When encountering an aggressive renal carcinoma with tubular, papillary, or infiltrative architecture, correlation with clinical history for sickle trait or other hemoglobinopathy and evaluation of SMARCB1 protein staining can be used to support a diagnosis of medullary carcinoma
Rare carcinomas resembling medullary carcinoma with SMARCB1 loss in the absence of sickle trait or other hemoglobinopathies are currently recommended to be classified as RCC unclassified with medullary phenotype, pending further study of this rare phenomenon
Collecting duct carcinoma
Once considered one of the major subtypes of renal cancer, collecting duct carcinoma is now considered quite rare and essentially a diagnosis of exclusion after other subtypes are argued against, particularly FH-deficient renal cancer, urothelial carcinoma, metastatic carcinoma from another organ, and renal medullary carcinoma. A recent study proposed an algorithm from discrimination between these entities, all of which can be composed of infiltrative glands, papillary structures, and cribriform structures, among other patterns. (126) Such a histology, in combination with sickle trait and abnormal negative SMARCB1 staining would be diagnostic of medullary carcinoma, whereas abnormal negative staining for FH, positive staining for 2-succino-cysteine (2SC), or FH mutation would support FH-deficient RCC. Careful exclusion of urothelial carcinoma invading the kidney or metastatic carcinoma from another origin is also necessary before arriving at such a diagnosis. Nonetheless, there do remain a subset of tumors that would fall into the category of collecting duct carcinoma using such a system. Recent molecular characterization of collecting duct carcinoma has found most common genomic alterations in NF2, SETD2, and CDKN2A. A subset of tumors was noted to have alterations of SMARCB1 or FH homozygous loss, (136) suggesting that these likely represent either medullary carcinoma or RCC unclassified with medullary phenotype, or FH-deficient renal cancer, respectively.
Working Group Recommendations, Collecting Duct Carcinoma:
Collecting duct carcinoma is a diagnosis of exclusion for a tumor that has been proven to be of primary renal cell lineage (not urothelial or metastatic) and for which FH-deficient and medullary carcinoma have been argued against
Other RCC types (mucinous tubular and spindle cell carcinoma, tubulocystic carcinoma, and acquired cystic kidney disease RCC)
Mucinous tubular and spindle cell carcinoma, tubulocystic carcinoma, and acquired cystic kidney disease RCC all demonstrate some overlapping features of papillary RCC, yet each are considered currently distinct diagnostic entities, based on some unique pathologic features. (5, 137–143) Although the spindle-shaped cell areas of mucinous tubular and spindle cell carcinoma could be confused with sarcomatoid changes, these tumors are usually nonaggressive, although rare metastatic examples have been reported. (144–146) In pure form, mucinous tubular and spindle cell carcinoma has a recurring copy number variation pattern with multiple chromosomal losses involving chromosomes 1, 4, 6, 8, 9, 13, 14, 15, and 22, without the gains of chromosomes 7 and 17 that is typical of papillary RCC. (147–150) Recent molecular characterization has found that this tumor also demonstrates inactivation of Hippo pathway tumor suppressor genes, with PTPN14 and NF2 being most common alterations. (151, 152) Recently, VSTM2A overexpression with RNA in situ hybridization has been noted as a sensitive and specific biomarker for this tumor. (153) There remains some debate as to whether tubulocystic RCC is closely related to papillary RCC, as some studies have found similar chromosomal copy number patterns (gain of chromosome 7 or 17 and loss of Y) or clustering with papillary RCC, whereas others have found trisomy 7 and 17 to be lacking in pure tubulocystic carcinoma. (140, 141, 154) Loss of chromosome 9 has also been noted. (155, 156) It has recently been shown that some tumors that resemble tubulocystic carcinoma yet which have an abrupt transition to high-grade infiltrative carcinoma (157) are likely best classified as fumarate hydratase (FH)-deficient RCC, discussed additionally later. (158) In a genomic profiling study of tumors in end-stage renal disease, acquired cystic kidney disease RCC tumors were found to cluster more closely with papillary and clear cell papillary RCC than clear cell RCC. (89) Although gains of chromosomes 7 and 17 can be observed, resembling type 1 papillary RCC, these tumors can also show gain of chromosomes 3, 16, and Y. (142, 159–163)
Working Group Recommendations, Other RCC types
Diagnosis of these RCC types can usually be made based predominantly on morphology
Copy number assessment can be used in difficult cases to support diagnosis of mucinous tubular and spindle cell carcinoma, which typically shows losses of chromosomes 1, 4, 6, 8, 9, 13, 14, 15, and 22, and lack of gains of chromosomes 7 and 17
Heterogeneity of patterns in a tumor resembling tubulocystic carcinoma should prompt consideration of FH-deficient carcinoma or hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC)
Hereditary renal cancer syndromes
von Hippel-Lindau disease
von Hippel-Lindau disease is the prototypical hereditary renal cancer syndrome, associated with multiple clear cell RCC tumors, renal cysts (Figure 4), and extrarenal manifestations including: hemangioblastoma of the central nervous system and retina, pheochromocytoma, pancreatic cysts and neuroendocrine tumors, epididymal and broad ligament cystadenomas, and endolymphatic sac tumors of the inner ear. (164, 165) Patients with von Hippel-Lindau disease have a germline mutation of the VHL gene, which is also commonly mutated in sporadic renal cancer. Therefore, only one genetic “hit” is needed for tumor development, in contrast to the typical “two-hit” mechanism expected in the sporadic setting. (3, 165, 166) The findings of the renal cancers in these disease patients generally resemble those in the sporadic setting, except that tumors and cysts can sometimes be numerous and occasionally microscopic incipient tumors can be found in the grossly normal-appearing renal parenchyma. When encountering clear cell RCC in a patient of young age (under age 46) (167) or with this constellation of multiple tumors, it would be appropriate for the pathologist to communicate with clinicians that a hereditary syndrome may be a consideration and that genetic counseling could be considered.
4.
Patients with von Hippel-Lindau disease have multiple clear cell RCC tumors and often renal cysts lined by cells with clear cytoplasm, which are thought to be precursors to neoplasms. This cyst is lined by cells with prominent clear cytoplasm and a slight heaping up of the lining cells.
Of note, the renal cancers in patients with von Hippel-Lindau disease sometimes morphologically closely resemble clear cell papillary RCC, (96) which is counterintuitive since sporadic clear cell papillary RCC tumors rarely if ever harbor alterations of VHL. However, when using immunohistochemistry, these clear cell papillary-like tumors in von Hippel-Lindau patients typically show an atypical staining pattern, such as with incomplete or negative cytokeratin 7 staining and positive CD10 or AMACR reactivity. Most have loss of chromosome 3p using FISH, suggesting that these are best classified instead as clear cell RCC. (96)
Working Group Recommendations, von Hippel-Lindau Disease
When encountering a clear cell RCC in a patient under age 46 or multiple clear RCC tumors, cysts, or microscopic clear cell tumors, it is worthwhile to communicate with clinicians that evaluation for a hereditary renal cancer syndrome may be considered
Tumors that resemble clear cell papillary RCC in von Hippel-Lindau disease patients typically do not show the expected staining pattern and are likely better classified as clear cell RCC due to the known risks of multiple renal cancers in these patients
Succinate dehydrogenase (SDH) deficient neoplasia
Autosomal dominant germline mutations of SDHA, SDHB, SDHC and SDHD cause a hereditary cancer syndrome characterized by paraganglioma/pheochromocytoma, gastrointestinal stromal tumor (GIST), RCC, and pituitary adenoma. (168, 169) Immunohistochemistry for SDHB is abnormally negative (which defines a tumor as being SDH deficient) whenever there is bialleic inactivation of any of the SDH genes. (168, 169) SDH deficiency is almost always associated with germline SDH mutations. (168–171) Care must be taken when interpreting SDHB immunohistochemistry to ensure that there are internal positive controls of non-neoplastic cells demonstrating strong granular cytoplasmic (mitochondrial) staining and to distinguish cases where the neoplastic cells show weak and diffuse staining (which is considered negative) from true positive (granular) staining (Figure 5A).
5.
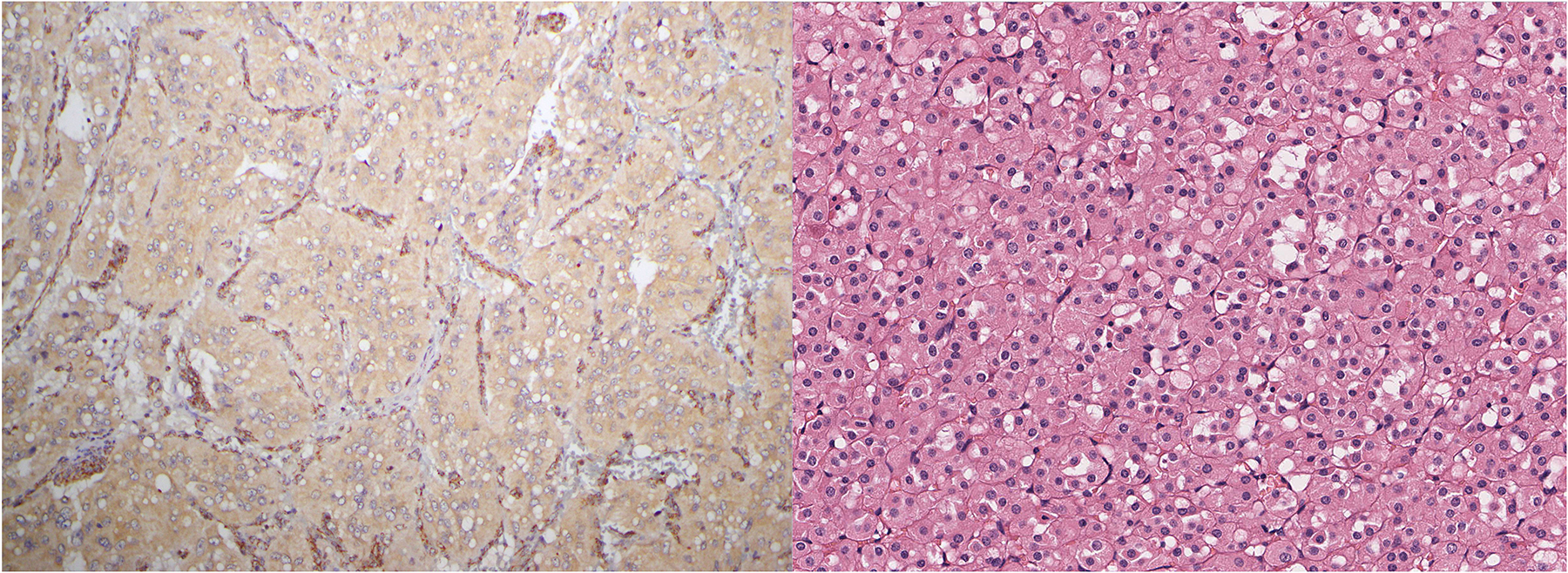
A) Care must be taken in interpreting SDHB immunohistochemistry. In this paraganglioma associated with SDHD mutation, the neoplastic cells show a weak diffuse cytoplasmic blush. This is considered SDHB immunohistochemistry ‘negative,’ as it contrasts strongly with the strong granular cytoplasmic (mitochondrial) staining in the internal positive controls provided by endothelial cells. B) SDH-deficient renal cell carcinoma showing typical features exhibits intracytoplasmic vacuoles/inclusions.
When there is double hit inactivation of SDHA, SDHA immunohistochemistry is also negative. (172, 173) SDHA germline mutations can occur in the general healthy population (up to 0.3% in some studies) (173) with a very low penetrance (currently estimated at 1.7%). (174) Therefore, the significance of SDHA mutations, particularly when discovered incidentally as part of a personalized medicine approach, should always be interpreted in the clinical context.
Succinate dehydrogenase deficient renal carcinoma is considered a distinct type of renal carcinoma under the WHO 2016 classification. (175) The majority of SDH deficient renal cancers demonstrate distinctive morphology illustrated in Figure 5B. (175–180) Briefly, the tumors are relatively circumscribed but entrap tubules and commonly show cystic change. The neoplastic cells demonstrate flocculent eosinophilic cytoplasm. Commonly there are distinctive, intracytoplasmic inclusions containing eosinophilic or wispy pale material, which appear to correspond to altered mitochondria. However, with the more widespread availability of screening immunohistochemistry, variant morphologies are being increasingly recognized.
Although metastasis is rare in low grade tumors, SDH deficient renal carcinomas with sarcomatoid change or coagulative necrosis are considered high risk with a metastatic rate approaching 70%. (175, 179, 180) The overwhelming majority of SDH deficient renal carcinomas reported to date have been associated with germline mutation in one of the SDH genes – usually SDHB or SDHC. Cases associated with SDHA mutation more commonly show variant morphology and are often identified only after molecular testing, where SDHA immunohistochemistry is particularly useful to distinguish between an incidental finding and true pathogenicity. (181–183)
Working Group Recommendations, SDH Deficient RCC
Care is required in interpreting SDHB IHC, particularly to ensure internal positive controls are present and to identify distinguish weak diffuse (considered negative) staining.
The diagnosis of SDH deficient RCC should be strongly considered with the stereotypical eosinophilic vacuolated cell morphology; however, variant morphologies are increasingly being recognized, such that the diagnosis may also be considered for other unusual patterns of RCC
The overwhelming majority of SDH deficient renal cell carcinomas are associated with germline mutation of the SDH subunits (usually SDHB), and therefore clinical genetic counseling should typically be undertaken when this diagnosis is made
Fumarate hydratase (FH) mutation and hereditary leiomyomatosis and renal cell carcinoma (HLRCC)
Autosomal dominant germline FH mutation causes the HLRCC syndrome, characterized by benign leiomyomas of the skin and uterus, RCC and, rarely, pheochromocytoma. (184, 185) Similarly to SDH deficiency, fumarate hydratase deficiency can be identified by abnormal negative immunohistochemical staining for fumarate hydratase (loss). However, in contrast to SDHB, FH loss is not completely sensitive for FH deficiency and it is currently estimated that only approximately 80 to 90% of FH deficient tumors will show negative staining using immunohistochemistry. (126, 158, 184, 186–191) Positive staining for 2SC is a promising more sensitive marker of fumarate hydratase deficiency; however, limited availability has significantly limited its use and validation. (184, 186, 188) The cutaneous and uterine leiomyomas associated with FH deficiency are characterized by greater cytological atypia which commonly has a symplastic quality. (184) Other clues to the diagnosis of fumarate hydratase deficiency, best seen in uterine leiomyomas, include a staghorn vasculature and prominent nucleoli. (184, 192)
Initially the renal carcinomas arising in the setting of HLRCC were characterized as type 2 papillary RCC with prominent inclusion-like nucleoli. (193) Since then several studies have reported that morphologies are more variable and are commonly mixed. (126, 158, 186–188, 190, 191, 194) In addition to the classic type 2 papillary RCC-like appearance, other morphologies including solid, cribriform/sieve-like, tubular, cystic, low grade oncocytic, and sarcomatoid are increasingly being recognized. (188) Given this morphological variability, a low threshold for FH and 2SC immunohistochemistry is indicated for any tumor, particularly in younger patients, that does not neatly fit into other diagnostic categories (Figure 6). Furthermore, given the limited sensitivity of FH immunohistochemistry and the relative lack of availability of 2SC, in cases with suggestive morphology it is still reasonable to proceed to molecular testing even if FH immunohistochemistry is positive.
6.
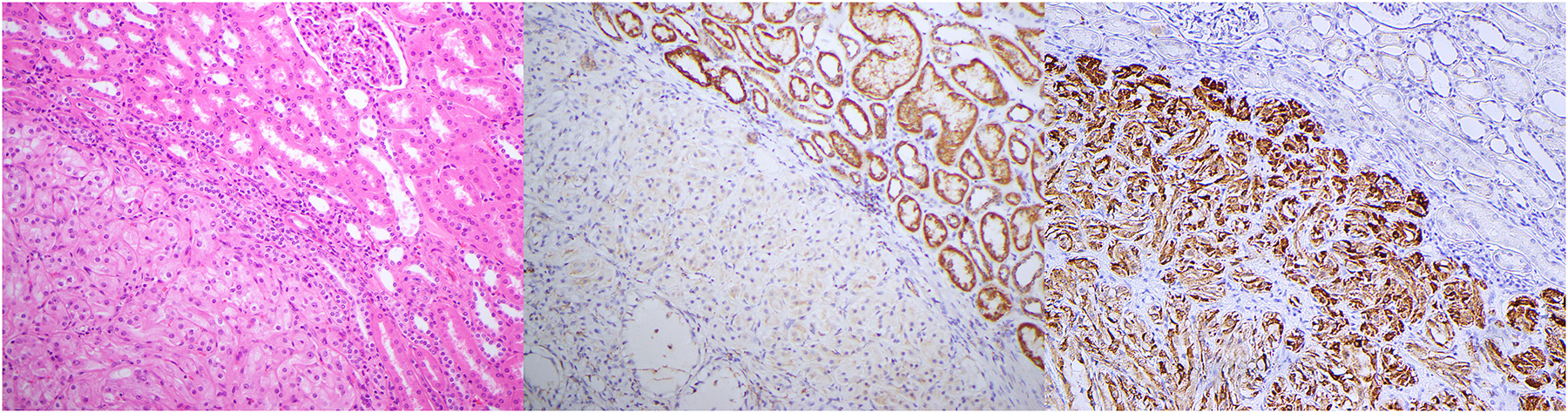
Serial sections of an FH-deficient RCC stained with A) H&E, B) fumarate hydratase, and C) 2SC immunohistochemistry. A) In this case the neoplastic cells have prominent nucleoli but lack the typical papillary or tubulocystic-like architecture of more readily recognized FH-deficient RCC. B) FH immunohistochemistry shows negative staining in all neoplastic cells that contrasts with the positive granular cytoplasmic (mitochondrial) staining in the internal positive controls. C) 2SC immunohistochemistry is positive in a nuclear and cytoplasmic pattern in all the neoplastic cells.
Working Group Recommendations, FH Deficient RCC (HLRCC)
Type 2 papillary RCC-like morphology with prominent nucleoli is helpful to recognize FH-deficient RCC and prompt confirmatory testing
Variant morphologies with heterogeneous patterns (papillary, tubulocystic, infiltrative, or mixed architecture, with or without prominent nucleoli) are increasingly recognized and should also prompt immunohistochemistry or molecular testing
FH negative immunohistochemistry occurs in only 80–90% of FH deficient neoplasms, therefore normal positive staining or equivocal weak positive staining does not exclude FH mutation; molecular testing may be considered
Although somatic only mutations do occur (especially in uterine leiomyomas in older women), most FH deficient RCCs reported to date have been associated with germline FH mutation (HLRCC syndrome) and should prompt genetic counseling
Other hereditary kidney tumor syndromes
Other hereditary syndromes associated with renal tumors include the hereditary papillary RCC syndrome, characterized by mutation of MET and numerous papillary RCC tumors, and Birt-Hogg-Dubé syndrome, characterized by mutation of FLCN and multiple oncocytic neoplasms (oncocytoma and chromophobe RCC or “hybrid” tumors). (195) In addition to the well-known development of angiomyolipomas in patients with tuberous sclerosis complex, these patients can also develop RCC, including some novel subtypes such as eosinophilic solid and cystic RCC, discussed later. (196, 197) In general, there is increasing recognition of the roles of the tuberous sclerosis genes (TSC1 and TSC2) in emerging subtypes of renal cancer, (198–203) discussed in the next section.
Emerging renal cancer types
Eosinophilic solid and cystic RCC
Eosinophilic solid and cystic RCC was first recognized as an unusual pattern of RCC in patients with tuberous sclerosis. (196, 197) These tumors are notable for solid and cystic growth, both composed of cells with voluminous cytoplasm and basophilic stippling of the cytoplasm (Figure 7). (203) Following the recognition of this tumor in the setting of tuberous sclerosis, it was shown that it can occur sporadically, predominantly in women, and that the tumors often have positive immunohistochemistry for cytokeratin 20 (ranging from focal to diffuse, with rare tumors negative). (203) Later it was found by several groups simultaneously that even the sporadic tumors have molecular alterations of TSC1 or TSC2. (199–201) Although initial reports suggested that these neoplasms are non-aggressive, a few recent studies have reported metastases, supporting their classification as carcinomas. (204, 205) The recurring constellation of pathologic features and molecular pathology strongly support this tumor as a novel diagnostic entity.
7.
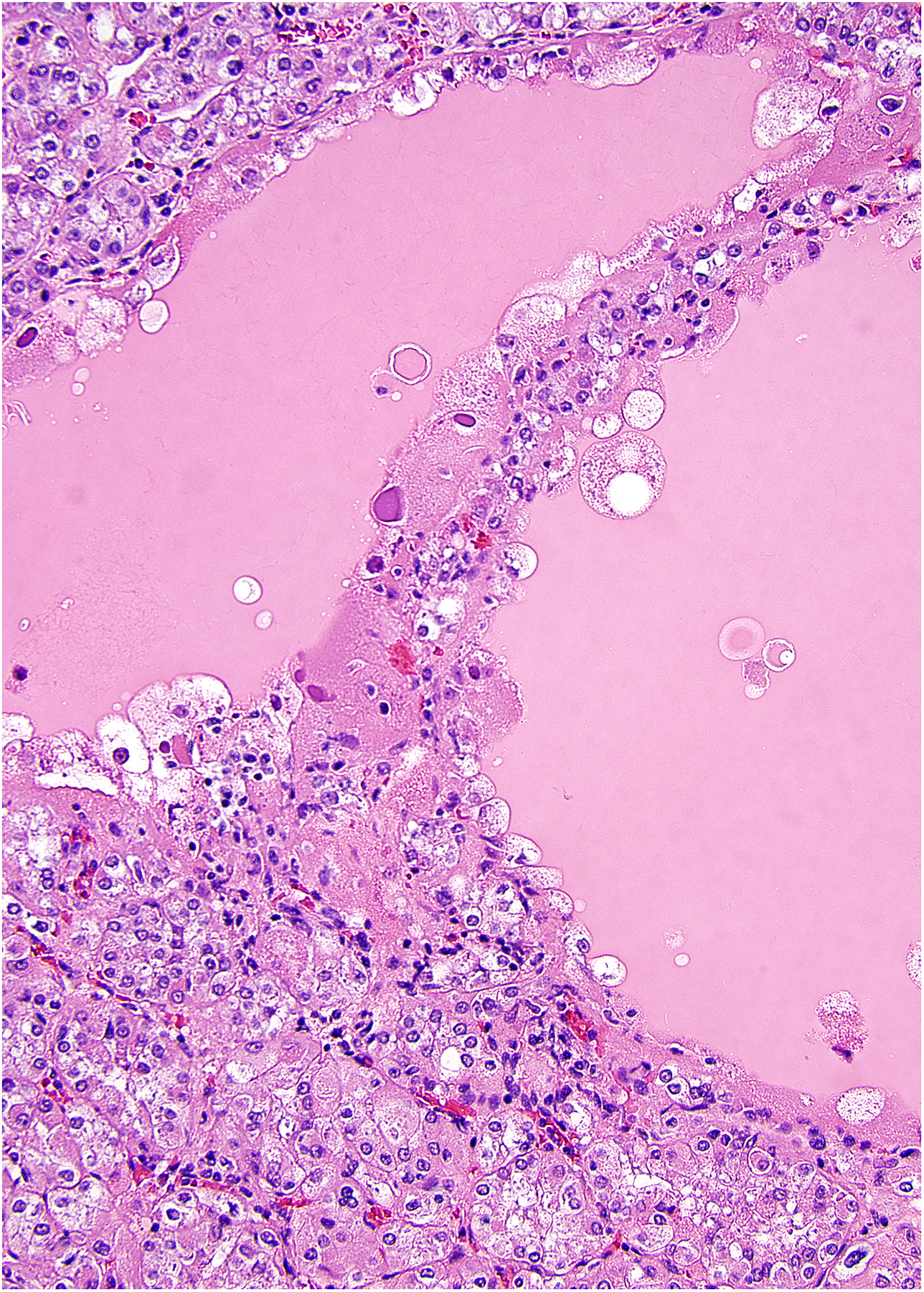
Eosinophilic solid and cystic RCC has been recently recognized to be composed of cells with voluminous eosinophilic cytoplasm, often containing granular basophilic stippling of the cytoplasm. Cysts are lined by cells with similar cytology.
RCC with TSC / MTOR gene mutations
In addition to eosinophilic solid and cystic RCC, a few other subtypes of renal cancer are now being recognized to have mutations of TSC1, TSC2, or MTOR, again likely corresponding to some of the unique histologic patterns that were recognized in patients with tuberous sclerosis complex. (196, 197) For example, one pattern resembled so-called renal angiomyoadenomatous tumor or RCC with smooth muscle or angioleiomyoma-like stroma, (196, 206) in which there are clear cells forming glandular structures dispersed in muscular stroma or within the cores of papillary structures. (206) A subset of tumors with such morphology has been recently found to harbor mutations in TSC1, TSC2, or MTOR. (207, 208) Since only some of these patients have clinical stigmata of tuberous sclerosis complex, (207–210) it seems that this tumor may again have both sporadic and inherited forms associated with alterations of TSC1, TSC2, or MTOR.
Secondly, it has been recently found that some oncocytic neoplasms with vacuolated cytoplasm have TSC2 or MTOR mutations, (198) which appears to correspond to an entity described by another group as high-grade oncocytic tumor. (211, 212) The clinical behavior described for these tumors thus far appears indolent. In general, these tumors exhibit a chromophobe-like histology with prominent nucleoli and eosinophilic cytoplasm with areas of cytoplasmic clearing or vacuoles. (198, 211) Some of these tumors may show histologic features overlapping with eosinophilic solid and cystic RCC (e.g. basophilic stippling of eosinophilic cytoplasm, nested or solid architecture), which may reflect their shared molecular alterations in TSC1 or TSC2. Taken together, these appear to be emerging subcategories of renal neoplasms with alterations in the MTOR pathway.
TCEB1 mutated renal cell carcinoma
Although some of the renal cancers with fibromuscular stroma appear to be associated with TSC1 or TSC2 mutations, recent work has recognized tumors that resemble clear cell RCC with fibromuscular stroma that harbor mutations of TCEB1 rather than VHL, likely accompanied by loss of chromosome 8 (often in the form of monosomy). (14, 206, 207, 213–216) It is not entirely clear at present if these tumors can be readily discriminated from clear cell RCC prospectively, as they are also positive for carbonic anhydrase IX. However, cytokeratin 7 positivity appears to be increased in this tumor type. Although initial data on this subset suggested that they are non-aggressive, recent reports of aggressive behavior have been published. (213, 217)
RCC with TFEB / 6p21 / VEGFA amplification
Although RCC with TFEB rearrangement has been recognized for many years, very recent work has found that occasional renal tumors exhibit amplification of chromosome 6p21 including the TFEB and VEGFA genes. (11, 108–112, 218, 219) These tumors thus far appear to be highly aggressive, with a mixture of histologic patterns predominantly resembling papillary RCC (Figure 8), although sometimes with areas suggesting clear cell or chromophobe RCC. Like translocation RCC, these tumors have been found to have some positivity for melanocytic immunohistochemical markers, particularly melan-A (more often than HMB45), and cathepsin K is often positive. (11, 218) If break-apart FISH for TFEB is used, it typically reveals numerous copies of the probes (at least 10), although low-level amplification has been reported in some cases, the significance of which is less clear. (109) Most of these tumors have shown amplification in the absence of rearrangement of TFEB; however, both rearrangement and amplification has been reported. (111) Recent work shows that TFEB gene expression is increased in these tumors, although not as much as in TFEB translocation tumors, raising the possibility that other genes at the 6p21 locus, such as VEGFA or CCND3 or other genes, may be responsible for the aggressive behavior. (108) The independent confirmation of this phenomenon by multiple groups strongly supports consideration of this tumor type as a significant diagnostic entity.
8.
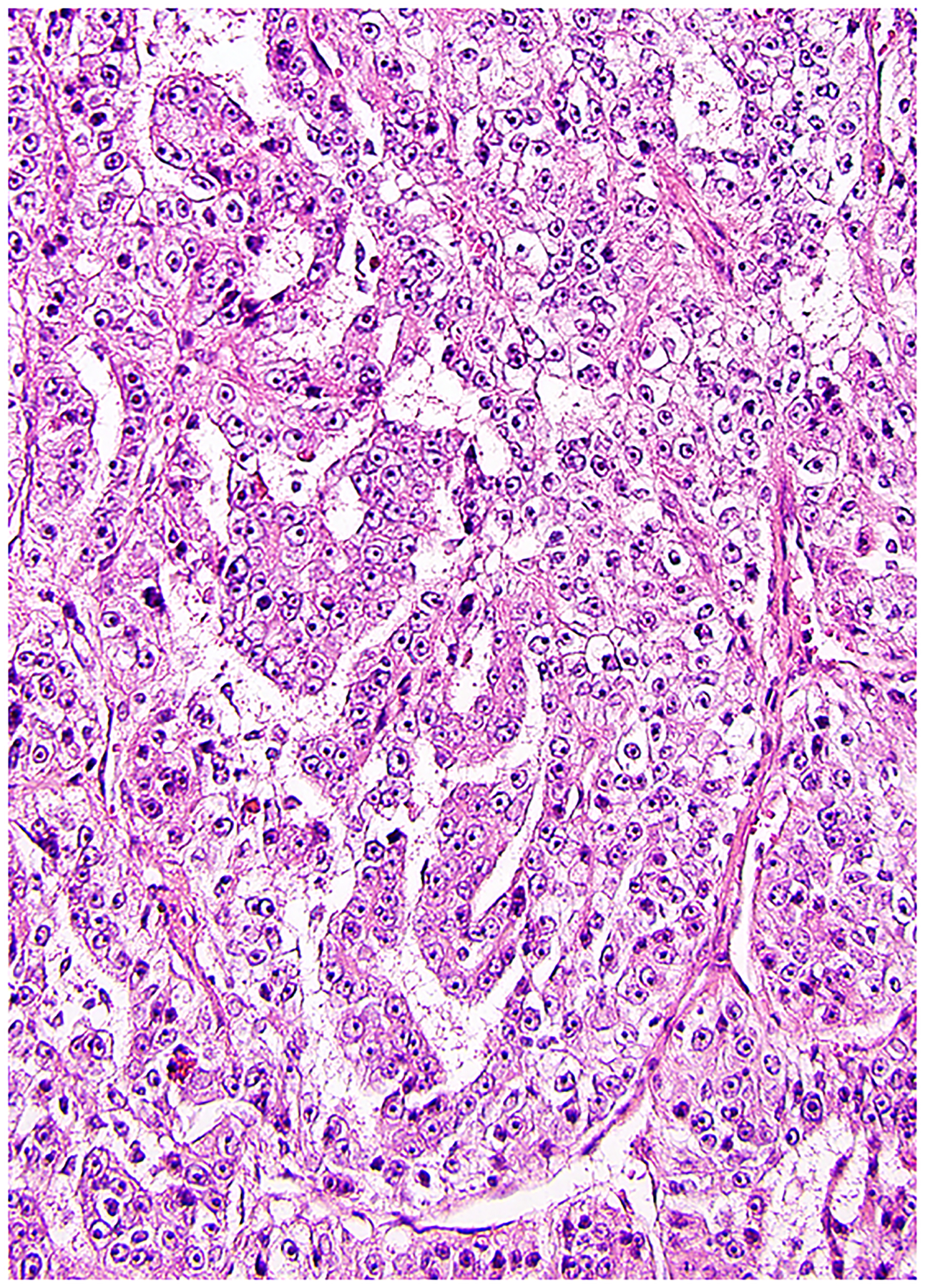
RCC with amplification of TFEB / 6p21 / VEGFA often has a papillary-like morphology, composed of clear or eosinophilic cells with prominent nucleoli, although it can exhibit multiple histologic patterns. These tumors appear to be highly aggressive.
ALK rearranged RCC
Rearrangement of ALK has been described in various tumors. However, an increasing number of renal cancers with ALK rearrangement have been recently reported. (164, 220–236) ALK rearranged renal cancers have been reported to be mostly papillary or cribriform, some having mucin production (Figure 9) or myxoid changes. Cases with the VCL-ALK fusion have been associated with sickle trait and have demonstrated prominent cytoplasmic vacuolization. Several fusion partners have been identified in ALK rearranged RCC, including TPM3, STRN, VCL, HOOK1. Novel partners CLIP1 and KIF5B have been identified recently. Tumors with unusual morphology have been recently noted, including resembling metanephric adenoma or mucinous tubular and spindle cell carcinoma. (228, 237) Of note, clinical response to ALK inhibitor alectinib has been reported in patients with ALK-rearranged tumors, implying that this may be a targetable therapeutic option. (233) For detection of ALK rearranged renal cancer, immunohistochemical screening with ALK antibody and confirmation by FISH or sequencing methods is generally recommended.
9.

RCC with ALK gene rearrangement has been noted to contain mucin or myxoid material in a subset of cases. This tumor was found to have rearrangement between ALK and TPM3.
Working Group Recommendations, Emerging Renal Cancer Types:
Consistent morphology and molecular findings support eosinophilic solid and cystic RCC as a distinct tumor type
There is growing, strong evidence supports RCC with TFEB / 6p21 / VEGFA amplification as a distinct entity in renal cancer with aggressive behavior
Other RCC types with TSC1, TSC2, or MTOR alterations are emerging renal cancer types that may be considered distinctive entities in future classification schemes, including RCC with smooth muscle stroma and eosinophilic neoplasms recently reported
There is insufficient evidence for recognition of TCEB1 mutated RCC as a definitive tumor type at present, although this may change with acquisition of more data regarding this emerging tumor type
There is some evidence for ALK rearranged RCC as a distinct tumor type, particularly in view of potential targeted therapy; however, multiple histologic patterns have been recognized
Metastatic renal cancer
Currently, a specific role for molecular pathology in metastatic renal cancer has not been definitively established. (4) However, at the experimental level, genetic profiling of metastatic renal cancer may be considered by clinicians when formulating treatment plans. The most relevant decision for the pathologist in metastatic renal cancer is to attempt to determine clear cell vs non-clear cell renal cancer, which has different preferred treatment regimens. (4) As noted previously, a helpful surrogate for the surgical pathologist is immunohistochemical staining for carbonic anhydrase IX; (16–21) however, the limitations of this marker must be kept in mind, as staining can be decreased or absent in poorly-differentiated clear cell RCC tumors (238) and non-renal tumors can also be positive. (18) Of course, molecular pathology can be utilized in attempting to confirm clear cell vs non-clear cell metastatic renal cancer, such as sequencing studies evaluating VHL gene alterations. Although FISH for chromosome 3p deletion may be used as a surrogate, (239, 240) some studies have found 3p loss to be not entirely specific for clear cell RCC, since they can be seen in 6p21 / TFEB amplified tumors, papillary RCC with clear cell changes, and unclassified RCC. (11–13) As found in the survey data, it appears that few pathologists are using such molecular techniques extensively in current diagnostic practice.
If comprehensive genomic profiling studies are requested by the oncologist in the setting of metastatic renal cancer, genes that may be relevant to modifying or confirming the treatment plan could include clear cell RCC-associated genes, such as VHL, BAP1, ARID1A, PBRM1, and SETD2, or genes involved in other pathways, such as the MTOR pathway, like TSC1, TSC2, PIK3CA, and MTOR. (241, 242) Secondly, immune checkpoint inhibitors have begun to establish a role in renal cancer. (4, 243) However, since multiple antibody clones currently exist with varying scoring systems for different cancers, the role of pathologic assessment for PD-L1 status in renal cancer remains incompletely understood at present. (243) Nonetheless, treatment with checkpoint inhibitor therapy is gaining traction as a therapeutic option, particularly in clear cell RCC, including a role in national guidelines, but also in some scenarios for non-clear cell RCC. (4, 244–247) Despite the currently limited role of molecular pathology in metastatic RCC, this is an area of tremendous exploration and it is possible that this role will expand significantly in the future.
Summary
Molecular pathology has dramatically influenced our understanding of renal cancer and continues to reshape and elucidate new diagnostic entities. However, knowledge of the genetics of renal cancer subtypes gained from the research setting can often be translated to the diagnostic setting through relatively simple surrogates, including histologic pattern, immunohistochemistry, and copy number analyses, precluding the need for extensive molecular evaluation in diagnostic practice. With the continued explosion of knowledge in molecular pathology of cancer, genetics will doubtlessly have a major impact in classification and possibly in prognostication and treatment selection for RCC going forward.
Supplementary Material
Footnotes
Conflict of Interest: None
Contributor Information
Sean R Williamson, Department of Pathology and Laboratory Medicine and Henry Ford Cancer Institute, Henry Ford Health System, Detroit, MI, USA; and Department of Pathology, Wayne State University School of Medicine, Detroit, MI, USA;.
Anthony J Gill, NSW Health Pathology, Department of Anatomical Pathology, Royal North Shore Hospital, St Leonards, NSW, Australia 2065; Cancer Diagnosis and Pathology Research Group, Kolling Institute of Medical Research, Royal North Shore Hospital NSW Australia 2065; University of Sydney, Sydney, NSW, Australia 2006.
Pedram Argani, Department of Pathology, Johns Hopkins University School of Medicine, Baltimore, Maryland, USA..
Ying-Bei Chen, Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, New York, USA..
Lars Egevad, Department of Oncology and Pathology, Karolinska Institutet, Stockholm, Sweden..
Glen Kristiansen, Institute of Pathology, University Hospital Bonn, Bonn 53127, Germany..
David J. Grignon, Department of Pathology, Indiana University School of Medicine, Indianapolis, Indiana, USA..
Ondrej Hes, Department of Pathology, Charles University, Medical Faculty and Charles University Hospital Plzen..
References
- 1.Moch H, Cubilla AL, Humphrey PA, et al. The 2016 WHO classification of tumours of the urinary system and male genital organs-part A: renal, penile, and testicular tumours. Eur Urol. 2016;70:93–105. [DOI] [PubMed] [Google Scholar]
- 2.Brugarolas J Molecular genetics of clear-cell renal cell carcinoma. J Clin Oncol. 2014;32:1968–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carlo MI, Hakimi AA, Stewart GD, et al. Familial Kidney Cancer: Implications of New Syndromes and Molecular Insights. Eur Urol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Motzer RJ, Jonasch E, Agarwal N, et al. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines) Kidney Cancer. 2019. Available at: https://www.nccn.org/professionals/physician_gls/PDF/kidney.pdf. Accessed 8/28/2019.
- 5.Srigley JR, Delahunt B, Eble JN, et al. The International Society of Urological Pathology (ISUP) Vancouver classification of renal neoplasia. Am J Surg Pathol. 2013;37:1469–1489. [DOI] [PubMed] [Google Scholar]
- 6.Moch H, Bonsib SM, Delahunt B, et al. Clear cell renal cell carcinoma In: Moch H, Humphrey PA, Ulbright TM., et al. eds. WHO Classification of Tumours of the Urinary System and Male Genital Organs. Lyon: International Agency for Research on Cancer; 2016:18–21. [Google Scholar]
- 7.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato Y, Yoshizato T, Shiraishi Y, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45:860–867. [DOI] [PubMed] [Google Scholar]
- 9.Chen F, Zhang Y, Senbabaoglu Y, et al. Multilevel Genomics-Based Taxonomy of Renal Cell Carcinoma. Cell Rep. 2016;14:2476–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Magers MJ, Cheng L. Practical Molecular Testing in a Clinical Genitourinary Service. Arch Pathol Lab Med. 2019. [DOI] [PubMed] [Google Scholar]
- 11.Williamson SR, Grignon DJ, Cheng L, et al. Renal cell carcinoma with chromosome 6p amplification including the TFEB gene: a novel mechanism of tumor pathogenesis? Am J Surg Pathol. 2017;41:287–298. [DOI] [PubMed] [Google Scholar]
- 12.Klatte T, Said JW, Seligson DB, et al. Pathological, immunohistochemical and cytogenetic features of papillary renal cell carcinoma with clear cell features. J Urol. 2011;185:30–35. [DOI] [PubMed] [Google Scholar]
- 13.Chen YB, Xu J, Skanderup AJ, et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun. 2016;7:13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Favazza L, Chitale DA, Barod R, et al. Renal cell tumors with clear cell histology and intact VHL and chromosome 3p: a histological review of tumors from the Cancer Genome Atlas database. Mod Pathol. 2017;30:1603–1612. [DOI] [PubMed] [Google Scholar]
- 15.Piva F, Santoni M, Matrana MR, et al. BAP1, PBRM1 and SETD2 in clear-cell renal cell carcinoma: molecular diagnostics and possible targets for personalized therapies. Expert Rev Mol Diagn. 2015;15:1201–1210. [DOI] [PubMed] [Google Scholar]
- 16.Al-Ahmadie HA, Alden D, Fine SW, et al. Role of immunohistochemistry in the evaluation of needle core biopsies in adult renal cortical tumors: an ex vivo study. Am J Surg Pathol. 2011;35:949–961. [DOI] [PubMed] [Google Scholar]
- 17.Al-Ahmadie HA, Alden D, Qin LX, et al. Carbonic anhydrase IX expression in clear cell renal cell carcinoma: an immunohistochemical study comparing 2 antibodies. Am J Surg Pathol. 2008;32:377–382. [DOI] [PubMed] [Google Scholar]
- 18.Donato DP, Johnson MT, Yang XJ, et al. Expression of carbonic anhydrase IX in genitourinary and adrenal tumours. Histopathology. 2011;59:1229–1239. [DOI] [PubMed] [Google Scholar]
- 19.Genega EM, Ghebremichael M, Najarian R, et al. Carbonic anhydrase IX expression in renal neoplasms: correlation with tumor type and grade. Am J Clin Pathol. 2010;134:873–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stillebroer AB, Mulders PF, Boerman OC, et al. Carbonic anhydrase IX in renal cell carcinoma: implications for prognosis, diagnosis, and therapy. Eur Urol. 2010;58:75–83. [DOI] [PubMed] [Google Scholar]
- 21.Feldman DR, Iyer G, Van Alstine L, et al. Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant germ cell tumors. Clin Cancer Res. 2014;20:3712–3720. [DOI] [PubMed] [Google Scholar]
- 22.Rohan SM, Xiao Y, Liang Y, et al. Clear-cell papillary renal cell carcinoma: molecular and immunohistochemical analysis with emphasis on the von Hippel-Lindau gene and hypoxia-inducible factor pathway-related proteins. Mod Pathol. 2011;24:1207–1220. [DOI] [PubMed] [Google Scholar]
- 23.Delahunt B, Algaba F, Eble J, et al. Papillary renal cell carcinoma In: Moch H, Humphrey PA, Ulbright TM., et al. eds. WHO Classification of Tumours of the Urinary System and Male Genital Organs. Lyon: International Agency for Research on Cancer; 2016:23–25. [Google Scholar]
- 24.Pitra T, Pivovarcikova K, Alaghehbandan R, et al. Chromosomal numerical aberration pattern in papillary renal cell carcinoma: Review article. Ann Diagn Pathol. 2019;40:189–199. [DOI] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas Research Network, Linehan WM, Spellman PT, et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N Engl J Med. 2016;374:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saleeb RM, Brimo F, Farag M, et al. Toward Biological Subtyping of Papillary Renal Cell Carcinoma With Clinical Implications Through Histologic, Immunohistochemical, and Molecular Analysis. Am J Surg Pathol. 2017;41:1618–1629. [DOI] [PubMed] [Google Scholar]
- 27.Lee BH. Commentary on: “Comprehensive molecular characterization of papillary renal-cell carcinoma.” Cancer Genome Atlas Research Network.: N Engl J Med. 2016. January 14;374(2):135–45. Urol Oncol. 2017;35:578–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zbar B, Tory K, Merino M, et al. Hereditary papillary renal cell carcinoma. J Urol. 1994;151:561–566. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68–73. [DOI] [PubMed] [Google Scholar]
- 30.Dharmawardana PG, Giubellino A, Bottaro DP. Hereditary papillary renal carcinoma type I. Curr Mol Med. 2004;4:855–868. [DOI] [PubMed] [Google Scholar]
- 31.Akhtar M, Al-Bozom IA, Al Hussain T. Papillary Renal Cell Carcinoma (PRCC): An Update. Adv Anat Pathol. 2019;26:124–132. [DOI] [PubMed] [Google Scholar]
- 32.Lubensky IA, Schmidt L, Zhuang Z, et al. Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol. 1999;155:517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhoades Smith KE, Bilen MA. A Review of Papillary Renal Cell Carcinoma and MET Inhibitors. Kidney Cancer. 2019;3:151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schoffski P, Wozniak A, Escudier B, et al. Crizotinib achieves long-lasting disease control in advanced papillary renal-cell carcinoma type 1 patients with MET mutations or amplification. EORTC 90101 CREATE trial. Eur J Cancer. 2017;87:147–163. [DOI] [PubMed] [Google Scholar]
- 35.Gilbert JA. Savolitinib for MET-driven papillary renal cell carcinoma. Lancet Oncol. 2017;18:e440. [DOI] [PubMed] [Google Scholar]
- 36.Fay AP, Signoretti S, Choueiri TK. MET as a target in papillary renal cell carcinoma. Clin Cancer Res. 2014;20:3361–3363. [DOI] [PubMed] [Google Scholar]
- 37.Choueiri TK, Vaishampayan U, Rosenberg JE, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lefevre M, Couturier J, Sibony M, et al. Adult papillary renal tumor with oncocytic cells: clinicopathologic, immunohistochemical, and cytogenetic features of 10 cases. Am J Surg Pathol. 2005;29:1576–1581. [DOI] [PubMed] [Google Scholar]
- 39.Han G, Yu W, Chu J, et al. Oncocytic papillary renal cell carcinoma: A clinicopathological and genetic analysis and indolent clinical course in 14 cases. Pathol Res Pract. 2017;213:1–6. [DOI] [PubMed] [Google Scholar]
- 40.Kunju LP, Wojno K, Wolf JS Jr., et al. Papillary renal cell carcinoma with oncocytic cells and nonoverlapping low grade nuclei: expanding the morphologic spectrum with emphasis on clinicopathologic, immunohistochemical and molecular features. Hum Pathol. 2008;39:96–101. [DOI] [PubMed] [Google Scholar]
- 41.Hes O, Brunelli M, Michal M, et al. Oncocytic papillary renal cell carcinoma: a clinicopathologic, immunohistochemical, ultrastructural, and interphase cytogenetic study of 12 cases. Ann Diagn Pathol. 2006;10:133–139. [DOI] [PubMed] [Google Scholar]
- 42.Al-Obaidy KI, Eble JN, Nassiri M, et al. Recurrent KRAS mutations in papillary renal neoplasm with reverse polarity. Mod Pathol. 2019:epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 43.Al-Obaidy KI, Eble JN, Cheng L, et al. Papillary Renal Neoplasm With Reverse Polarity: A Morphologic, Immunohistochemical, and Molecular Study. Am J Surg Pathol 2019;43:1099–1111. [DOI] [PubMed] [Google Scholar]
- 44.Tong K, Zhu W, Fu H, et al. Frequent KRAS Mutations in Oncocytic Papillary Renal Neoplasm with Inverted Nuclei. Histopathology. 2020. [DOI] [PubMed] [Google Scholar]
- 45.Williamson SR, Gadde R, Trpkov K, et al. Diagnostic criteria for oncocytic renal neoplasms: a survey of urologic pathologists. Hum Pathol. 2017;63:149–156. [DOI] [PubMed] [Google Scholar]
- 46.Delongchamps NB, Galmiche L, Eiss D, et al. Hybrid tumour ‘oncocytoma-chromophobe renal cell carcinoma’ of the kidney: a report of seven sporadic cases. BJU Int. 2009;103:1381–1384. [DOI] [PubMed] [Google Scholar]
- 47.Petersson F, Gatalica Z, Grossmann P, et al. Sporadic hybrid oncocytic/chromophobe tumor of the kidney: a clinicopathologic, histomorphologic, immunohistochemical, ultrastructural, and molecular cytogenetic study of 14 cases. Virchows Arch. 2010;456:355–365. [DOI] [PubMed] [Google Scholar]
- 48.Tickoo SK, Reuter VE, Amin MB, et al. Renal oncocytosis: a morphologic study of fourteen cases. Am J Surg Pathol. 1999;23:1094–1101. [DOI] [PubMed] [Google Scholar]
- 49.Gobbo S, Eble JN, Delahunt B, et al. Renal cell neoplasms of oncocytosis have distinct morphologic, immunohistochemical, and cytogenetic profiles. Am J Surg Pathol. 2010;34:620–626. [DOI] [PubMed] [Google Scholar]
- 50.Speicher MR, Schoell B, du Manoir S, et al. Specific loss of chromosomes 1, 2, 6, 10, 13, 17, and 21 in chromophobe renal cell carcinomas revealed by comparative genomic hybridization. Am J Pathol. 1994;145:356–364. [PMC free article] [PubMed] [Google Scholar]
- 51.Paner G, Amin MB, Moch H, et al. Chromophobe renal cell carcinoma In: Moch H, Humphrey PA, Ulbright TM., et al. eds. WHO Classification of Tumours of the Urinary System and Male Genital Organs. Lyon: International Agency for Research on Cancer; 2016:27–28. [Google Scholar]
- 52.Davis CF, Ricketts CJ, Wang M, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vieira J, Henrique R, Ribeiro FR, et al. Feasibility of differential diagnosis of kidney tumors by comparative genomic hybridization of fine needle aspiration biopsies. Genes Chromosomes Cancer. 2010;49:935–947. [DOI] [PubMed] [Google Scholar]
- 54.Sperga M, Martinek P, Vanecek T, et al. Chromophobe renal cell carcinoma--chromosomal aberration variability and its relation to Paner grading system: an array CGH and FISH analysis of 37 cases. Virchows Arch. 2013;463:563–573. [DOI] [PubMed] [Google Scholar]
- 55.Tan MH, Wong CF, Tan HL, et al. Genomic expression and single-nucleotide polymorphism profiling discriminates chromophobe renal cell carcinoma and oncocytoma. BMC Cancer. 2010;10:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joshi S, Tolkunov D, Aviv H, et al. The Genomic Landscape of Renal Oncocytoma Identifies a Metabolic Barrier to Tumorigenesis. Cell Rep. 2015;13:1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xiao GQ, Ko HB, Unger P. Telangiectatic oncocytoma: a previously undescribed variant of renal oncocytoma. Am J Clin Pathol. 2013;140:103–108. [DOI] [PubMed] [Google Scholar]
- 58.Skenderi F, Ulamec M, Vranic S, et al. Cystic Renal Oncocytoma and Tubulocystic Renal Cell Carcinoma: Morphologic and Immunohistochemical Comparative Study. Appl Immunohistochem Mol Morphol. 2016;24:112–119. [DOI] [PubMed] [Google Scholar]
- 59.Hes O, Michal M, Boudova L, et al. Small cell variant of renal oncocytoma--a rare and misleading type of benign renal tumor. Int J Surg Pathol. 2001;9:215–222. [DOI] [PubMed] [Google Scholar]
- 60.Kuroda N, Yorita K, Naroda T, et al. Renal oncocytoma, small cell variant, with pseudorosettes, showing cyclin D1 expression and tubulovesicular cristae of mitochondria. Pathol Int. 2016;66:409–410. [DOI] [PubMed] [Google Scholar]
- 61.Magro G, Gardiman MP, Lopes MR, et al. Small-cell variant of renal oncocytoma with dominating solid growth pattern: a potential diagnostic pitfall. Virchows Arch. 2006;448:379–380. [DOI] [PubMed] [Google Scholar]
- 62.Petersson F, Sima R, Grossmann P, et al. Renal small cell oncocytoma with pseudorosettes A histomorphologic, immunohistochemical, and molecular genetic study of 10 cases. Hum Pathol. 2011;42:1751–1760. [DOI] [PubMed] [Google Scholar]
- 63.Zhang W, Yu W, Wang Q, et al. The clinicopathological, ultrastructural, genetic features and diagnosis of small cell variant renal oncocytoma. Acta Histochem. 2015;117:505–511. [DOI] [PubMed] [Google Scholar]
- 64.Wobker SE, Williamson SR. Modern Pathologic Diagnosis of Renal Oncocytoma. J Kidney Cancer VHL. 2017;4:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hes O, Moch H, Reuter V. Oncocytoma In: Moch H, Humphrey PA, Ulbright TM., et al. eds. WHO Classification of Tumours of the Urinary System and Male Genital Organs. Lyon: International Agency for Research on Cancer; 2016:43–44. [Google Scholar]
- 66.Hes O, Michal M, Kuroda N, et al. Vimentin reactivity in renal oncocytoma: immunohistochemical study of 234 cases. Arch Pathol Lab Med. 2007;131:1782–1788. [DOI] [PubMed] [Google Scholar]
- 67.Crotty TB, Lawrence KM, Moertel CA, et al. Cytogenetic analysis of six renal oncocytomas and a chromophobe cell renal carcinoma. Evidence that -Y, −1 may be a characteristic anomaly in renal oncocytomas. Cancer Genet Cytogenet. 1992;61:61–66. [DOI] [PubMed] [Google Scholar]
- 68.Fuzesi L, Gunawan B, Braun S, et al. Renal oncocytoma with a translocation t(9;11)(p23;q13). J Urol. 1994;152:471–472. [DOI] [PubMed] [Google Scholar]
- 69.Paner GP, Lindgren V, Jacobson K, et al. High incidence of chromosome 1 abnormalities in a series of 27 renal oncocytomas: cytogenetic and fluorescence in situ hybridization studies. Arch Pathol Lab Med. 2007;131:81–85. [DOI] [PubMed] [Google Scholar]
- 70.Lindgren V, Paner GP, Omeroglu A, et al. Cytogenetic analysis of a series of 13 renal oncocytomas. J Urol. 2004;171:602–604. [DOI] [PubMed] [Google Scholar]
- 71.Picken MM, Chyna B, Flanigan RC, et al. Analysis of chromosome 1p abnormalities in renal oncocytomas by loss of heterozygosity studies: correlation with conventional cytogenetics and fluorescence in situ hybridization. Am J Clin Pathol. 2008;129:377–382. [DOI] [PubMed] [Google Scholar]
- 72.Anderson CB, Lipsky M, Nandula SV, et al. Cytogenetic analysis of 130 renal oncocytomas identify three distinct and mutually exclusive diagnostic classes of chromosome aberrations. Genes Chromosomes Cancer. 2019. [DOI] [PubMed] [Google Scholar]
- 73.Sukov WR, Ketterling RP, Lager DJ, et al. CCND1 rearrangements and cyclin D1 overexpression in renal oncocytomas: frequency, clinicopathologic features, and utility in differentiation from chromophobe renal cell carcinoma. Hum Pathol. 2009;40:1296–1303. [DOI] [PubMed] [Google Scholar]
- 74.Tickoo SK, dePeralta-Venturina MN, Harik LR, et al. Spectrum of epithelial neoplasms in end-stage renal disease: an experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am J Surg Pathol. 2006;30:141–153. [DOI] [PubMed] [Google Scholar]
- 75.Adam J, Couturier J, Molinie V, et al. Clear-cell papillary renal cell carcinoma: 24 cases of a distinct low-grade renal tumour and a comparative genomic hybridization array study of seven cases. Histopathology. 2011;58:1064–1071. [DOI] [PubMed] [Google Scholar]
- 76.Aron M, Chang E, Herrera L, et al. Clear cell-papillary renal cell carcinoma of the kidney not associated with end-stage renal disease: clinicopathologic correlation with expanded immunophenotypic and molecular characterization of a large cohort with emphasis on relationship with renal angiomyoadenomatous tumor. Am J Surg Pathol. 2015;39:873–888. [DOI] [PubMed] [Google Scholar]
- 77.Aydin H, Chen L, Cheng L, et al. Clear cell tubulopapillary renal cell carcinoma: a study of 36 distinctive low-grade epithelial tumors of the kidney. Am J Surg Pathol. 2010;34:1608–1621. [DOI] [PubMed] [Google Scholar]
- 78.Deml KF, Schildhaus HU, Comperat E, et al. Clear cell papillary renal cell carcinoma and renal angiomyoadenomatous tumor: two variants of a morphologic, immunohistochemical, and genetic distinct entity of renal cell carcinoma. Am J Surg Pathol. 2015;39:889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Diolombi ML, Cheng L, Argani P, et al. Do clear cell papillary renal cell carcinomas have malignant potential? Am J Surg Pathol. 2015;39:1621–1634. [DOI] [PubMed] [Google Scholar]
- 80.Leroy X, Camparo P, Gnemmi V, et al. Clear cell papillary renal cell carcinoma is an indolent and low-grade neoplasm with overexpression of cyclin-D1. Histopathology. 2014;64:1032–1036. [DOI] [PubMed] [Google Scholar]
- 81.Srigley JR, Cheng L, Grignon DJ, et al. Clear cell papillary renal cell carcinoma In: Moch H, Humphrey PA, Ulbright TM., et al. eds. WHO Classification of Tumours of the Urinary System and Male Genital Organs. Lyon: International Agency for Research on Cancer; 2016:40–41. [Google Scholar]
- 82.Williamson SR, Eble JN, Cheng L, et al. Clear cell papillary renal cell carcinoma: differential diagnosis and extended immunohistochemical profile. Mod Pathol. 2013;26:697–708. [DOI] [PubMed] [Google Scholar]
- 83.Zhou H, Zheng S, Truong LD, et al. Clear cell papillary renal cell carcinoma is the fourth most common histologic type of renal cell carcinoma in 290 consecutive nephrectomies for renal cell carcinoma. Hum Pathol. 2014;45:59–64. [DOI] [PubMed] [Google Scholar]
- 84.Mantilla JG, Antic T, tretiakova MS. GATA-3 Is a Specific Marker for Clear Cell Papillary Renal Cell Carcinoma. Mod Pathol. 2017;30:241A (abstract). [DOI] [PubMed] [Google Scholar]
- 85.Martignoni G, Brunelli M, Segala D, et al. Validation of 34betaE12 immunoexpression in clear cell papillary renal cell carcinoma as a sensitive biomarker. Pathology. 2017;49:10–18. [DOI] [PubMed] [Google Scholar]
- 86.Morlote D, Rais-Bahrami S, Harada S, et al. A Molecular Profile of Clear Cell Papillary Renal Cell Carcinoma by Next Generation Sequencing. Mod Pathol. 2018;31:370 (abstract). [Google Scholar]
- 87.Xu J, Reznik E, Lee HJ, et al. Abnormal oxidative metabolism in a quiet genomic background underlies clear cell papillary renal cell carcinoma. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fisher KE, Yin-Goen Q, Alexis D, et al. Gene expression profiling of clear cell papillary renal cell carcinoma: comparison with clear cell renal cell carcinoma and papillary renal cell carcinoma. Mod Pathol. 2014;27:222–230. [DOI] [PubMed] [Google Scholar]
- 89.Inoue T, Matsuura K, Yoshimoto T, et al. Genomic profiling of renal cell carcinoma in patients with end-stage renal disease. Cancer Sci. 2012;103:569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Munari E, Marchionni L, Chitre A, et al. Clear cell papillary renal cell carcinoma: micro-RNA expression profiling and comparison with clear cell renal cell carcinoma and papillary renal cell carcinoma. Hum Pathol. 2014;45:1130–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wolfe A, Dobin SM, Grossmann P, et al. Clonal trisomies 7,10 and 12, normal 3p and absence of VHL gene mutation in a clear cell tubulopapillary carcinoma of the kidney. Virchows Arch. 2011;459:457–463. [DOI] [PubMed] [Google Scholar]
- 92.Williamson SR, Cheng L. Clear cell renal cell tumors: not all that is “clear” is cancer. Urol Oncol. 2016;34:292 e217–222. [DOI] [PubMed] [Google Scholar]
- 93.Gupta S, Inwards CY, Van Dyke DL, et al. Defining Clear Cell Papillary Renal Cell Carcinoma in Routine Clinical Practice. Histopathology. 2020. [DOI] [PubMed] [Google Scholar]
- 94.Williamson SR, Gupta NS, Eble JN, et al. Clear cell renal cell carcinoma with borderline features of clear cell papillary renal cell carcinoma: combined morphologic, immunohistochemical, and cytogenetic analysis. Am J Surg Pathol. 2015;39:1502–1510. [DOI] [PubMed] [Google Scholar]
- 95.Dhakal HP, McKenney JK, Khor LY, et al. Renal Neoplasms With Overlapping Features of Clear Cell Renal Cell Carcinoma and Clear Cell Papillary Renal Cell Carcinoma: A Clinicopathologic Study of 37 Cases From a Single Institution. Am J Surg Pathol. 2016;40:141–154. [DOI] [PubMed] [Google Scholar]
- 96.Williamson SR, Zhang S, Eble JN, et al. Clear cell papillary renal cell carcinoma-like tumors in patients with von Hippel-Lindau disease are unrelated to sporadic clear cell papillary renal cell carcinoma. Am J Surg Pathol. 2013;37:1131–1139. [DOI] [PubMed] [Google Scholar]
- 97.Argani P MiT family translocation renal cell carcinoma. Semin Diagn Pathol. 2015;32:103–113. [DOI] [PubMed] [Google Scholar]
- 98.Gandhi JS, Malik F, Amin MB, et al. MiT family translocation renal cell carcinomas: A 15th anniversary update. Histol Histopathol. 2019:18159. [DOI] [PubMed] [Google Scholar]
- 99.Argani P, Antonescu CR, Illei PB, et al. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma: a distinctive tumor entity previously included among renal cell carcinomas of children and adolescents. Am J Pathol. 2001;159:179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Argani P, Lui MY, Couturier J, et al. A novel CLTC-TFE3 gene fusion in pediatric renal adenocarcinoma with t(X;17)(p11.2;q23). Oncogene. 2003;22:5374–5378. [DOI] [PubMed] [Google Scholar]
- 101.Argani P, Ladanyi M. Renal carcinomas associated with Xp11.2 translocations / TFE3 gene fusions In: Eble JN, Sauter G, Epstein JI, et al. eds. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. Lyon: IARC Press; 2004:37–38. [Google Scholar]
- 102.Argani P, Olgac S, Tickoo SK, et al. Xp11 translocation renal cell carcinoma in adults: expanded clinical, pathologic, and genetic spectrum. Am J Surg Pathol. 2007;31:1149–1160. [DOI] [PubMed] [Google Scholar]
- 103.Argani P, Hicks J, De Marzo AM, et al. Xp11 translocation renal cell carcinoma (RCC): extended immunohistochemical profile emphasizing novel RCC markers. Am J Surg Pathol. 2010;34:1295–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ellis CL, Eble JN, Subhawong AP, et al. Clinical heterogeneity of Xp11 translocation renal cell carcinoma: impact of fusion subtype, age, and stage. Mod Pathol. 2014;27:875–886. [DOI] [PubMed] [Google Scholar]
- 105.Argani P, Zhong M, Reuter VE, et al. TFE3-Fusion Variant Analysis Defines Specific Clinicopathologic Associations Among Xp11 Translocation Cancers. Am J Surg Pathol. 2016;40:723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Malouf GG, Su X, Yao H, et al. Next-generation sequencing of translocation renal cell carcinoma reveals novel RNA splicing partners and frequent mutations of chromatin-remodeling genes. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:4129–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Argani P, Hawkins A, Griffin CA, et al. A distinctive pediatric renal neoplasm characterized by epithelioid morphology, basement membrane production, focal HMB45 immunoreactivity, and t(6;11)(p21.1;q12) chromosome translocation. Am J Pathol. 2001;158:2089–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gupta S, Argani P, Jungbluth AA, et al. TFEB Expression Profiling in Renal Cell Carcinomas: Clinicopathologic Correlations. Am J Surg Pathol. 2019;43:1445–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gupta S, Johnson SH, Vasmatzis G, et al. TFEB-VEGFA (6p21.1) co-amplified renal cell carcinoma: a distinct entity with potential implications for clinical management. Mod Pathol. 2017;30:998–1012. [DOI] [PubMed] [Google Scholar]
- 110.Skala SL, Xiao H, Udager AM, et al. Detection of 6 TFEB-amplified renal cell carcinomas and 25 renal cell carcinomas with MITF translocations: systematic morphologic analysis of 85 cases evaluated by clinical TFE3 and TFEB FISH assays. Mod Pathol. 2018;31:179–197. [DOI] [PubMed] [Google Scholar]
- 111.Peckova K, Vanecek T, Martinek P, et al. Aggressive and nonaggressive translocation t(6;11) renal cell carcinoma: comparative study of 6 cases and review of the literature. Ann Diagn Pathol. 2014;18:351–357. [DOI] [PubMed] [Google Scholar]
- 112.Durinck S, Stawiski EW, Pavia-Jimenez A, et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet. 2015;47:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xia QY, Wang XT, Ye SB, et al. Novel gene fusion of PRCC-MITF defines a new member of MiT family translocation renal cell carcinoma: clinicopathological analysis and detection of the gene fusion by RNA sequencing and FISH. Histopathology. 2018;72:786–794. [DOI] [PubMed] [Google Scholar]
- 114.Camparo P, Vasiliu V, Molinie V, et al. Renal translocation carcinomas: clinicopathologic, immunohistochemical, and gene expression profiling analysis of 31 cases with a review of the literature. Am J Surg Pathol. 2008;32:656–670. [DOI] [PubMed] [Google Scholar]
- 115.Martignoni G, Gobbo S, Camparo P, et al. Differential expression of cathepsin K in neoplasms harboring TFE3 gene fusions. Mod Pathol. 2011;24:1313–1319. [DOI] [PubMed] [Google Scholar]
- 116.Martignoni G, Pea M, Gobbo S, et al. Cathepsin-K immunoreactivity distinguishes MiTF/TFE family renal translocation carcinomas from other renal carcinomas. Mod Pathol. 2009;22:1016–1022. [DOI] [PubMed] [Google Scholar]
- 117.Argani P, Yonescu R, Morsberger L, et al. Molecular confirmation of t(6;11)(p21;q12) renal cell carcinoma in archival paraffin-embedded material using a break-apart TFEB FISH assay expands its clinicopathologic spectrum. Am J Surg Pathol. 2012;36:1516–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Green WM, Yonescu R, Morsberger L, et al. Utilization of a TFE3 break-apart FISH assay in a renal tumor consultation service. Am J Surg Pathol. 2013;37:1150–1163. [DOI] [PubMed] [Google Scholar]
- 119.Rao Q, Williamson SR, Zhang S, et al. TFE3 break-apart FISH has a higher sensitivity for Xp11.2 translocation-associated renal cell carcinoma compared with TFE3 or cathepsin K immunohistochemical staining alone: expanding the morphologic spectrum. Am J Surg Pathol. 2013;37:804–815. [DOI] [PubMed] [Google Scholar]
- 120.Xia QY, Wang Z, Chen N, et al. Xp11.2 translocation renal cell carcinoma with NONO-TFE3 gene fusion: morphology, prognosis, and potential pitfall in detecting TFE3 gene rearrangement. Mod Pathol. 2017;30:416–426. [DOI] [PubMed] [Google Scholar]
- 121.Argani P, Zhang L, Reuter VE, et al. RBM10-TFE3 Renal Cell Carcinoma: A Potential Diagnostic Pitfall Due to Cryptic Intrachromosomal Xp11.2 Inversion Resulting in False-negative TFE3 FISH. Am J Surg Pathol. 2017;41:655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Just PA, Letourneur F, Pouliquen C, et al. Identification by FFPE RNA-Seq of a new recurrent inversion leading to RBM10-TFE3 fusion in renal cell carcinoma with subtle TFE3 break-apart FISH pattern. Genes Chromosomes Cancer. 2016;55:541–548. [DOI] [PubMed] [Google Scholar]
- 123.Xia QY, Wang XT, Zhan XM, et al. Xp11 Translocation Renal Cell Carcinomas (RCCs) With RBM10-TFE3 Gene Fusion Demonstrating Melanotic Features and Overlapping Morphology With t(6;11) RCC: Interest and Diagnostic Pitfall in Detecting a Paracentric Inversion of TFE3. Am J Surg Pathol. 2017;41:663–676. [DOI] [PubMed] [Google Scholar]
- 124.Classe M, Malouf GG, Su X, et al. Incidence, clinicopathological features and fusion transcript landscape of translocation renal cell carcinomas. Histopathology. 2017;70:1089–1097. [DOI] [PubMed] [Google Scholar]
- 125.Argani P, Zhang L, Sung YS, et al. A novel RBMX-TFE3 gene fusion in a highly aggressive pediatric renal perivascular epithelioid cell tumor. Genes Chromosomes Cancer. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ohe C, Smith SC, Sirohi D, et al. Reappraisal of Morphologic Differences Between Renal Medullary Carcinoma, Collecting Duct Carcinoma, and Fumarate Hydratase-deficient Renal Cell Carcinoma. Am J Surg Pathol. 2018;42:279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jia L, Carlo MI, Khan H, et al. Distinctive mechanisms underlie the loss of SMARCB1 protein expression in renal medullary carcinoma: morphologic and molecular analysis of 20 cases. Mod Pathol. 2019;32:1329–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Calderaro J, Masliah-Planchon J, Richer W, et al. Balanced Translocations Disrupting SMARCB1 Are Hallmark Recurrent Genetic Alterations in Renal Medullary Carcinomas. Eur Urol. 2016;69:1055–1061. [DOI] [PubMed] [Google Scholar]
- 129.Carlo MI, Chaim J, Patil S, et al. Genomic Characterization of Renal Medullary Carcinoma and Treatment Outcomes. Clin Genitourin Cancer. 2017;15:e987–e994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Calderaro J, Moroch J, Pierron G, et al. SMARCB1/INI1 inactivation in renal medullary carcinoma. Histopathology. 2012;61:428–435. [DOI] [PubMed] [Google Scholar]
- 131.Liu Q, Galli S, Srinivasan R, et al. Renal medullary carcinoma: molecular, immunohistochemistry, and morphologic correlation. Am J Surg Pathol. 2013;37:368–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cheng JX, Tretiakova M, Gong C, et al. Renal medullary carcinoma: rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod Pathol. 2008;21:647–652. [DOI] [PubMed] [Google Scholar]
- 133.Rao P, Tannir NM, Tamboli P. Expression of OCT3/4 in renal medullary carcinoma represents a potential diagnostic pitfall. Am J Surg Pathol. 2012;36:583–588. [DOI] [PubMed] [Google Scholar]
- 134.Sirohi D, Smith SC, Ohe C, et al. Renal cell carcinoma, unclassified with medullary phenotype: poorly differentiated adenocarcinomas overlapping with renal medullary carcinoma. Hum Pathol. 2017;67:134–145. [DOI] [PubMed] [Google Scholar]
- 135.Amin MB, Smith SC, Agaimy A, et al. Collecting duct carcinoma versus renal medullary carcinoma: an appeal for nosologic and biological clarity. Am J Surg Pathol. 2014;38:871–874. [DOI] [PubMed] [Google Scholar]
- 136.Pal SK, Choueiri TK, Wang K, et al. Characterization of Clinical Cases of Collecting Duct Carcinoma of the Kidney Assessed by Comprehensive Genomic Profiling. Eur Urol. 2015. [DOI] [PubMed] [Google Scholar]
- 137.Eble JN. Mucinous tubular and spindle cell carcinoma and post-neuroblastoma carcinoma: newly recognised entities in the renal cell carcinoma family. Pathology. 2003;35:499–504. [DOI] [PubMed] [Google Scholar]
- 138.Paner GP, Srigley JR, Radhakrishnan A, et al. Immunohistochemical analysis of mucinous tubular and spindle cell carcinoma and papillary renal cell carcinoma of the kidney: significant immunophenotypic overlap warrants diagnostic caution. Am J Surg Pathol. 2006;30:13–19. [DOI] [PubMed] [Google Scholar]
- 139.Amin MB, MacLennan GT, Gupta R, et al. Tubulocystic carcinoma of the kidney: clinicopathologic analysis of 31 cases of a distinctive rare subtype of renal cell carcinoma. Am J Surg Pathol. 2009;33:384–392. [DOI] [PubMed] [Google Scholar]
- 140.Zhou M, Yang XJ, Lopez JI, et al. Renal tubulocystic carcinoma is closely related to papillary renal cell carcinoma: implications for pathologic classification. Am J Surg Pathol. 2009;33:1840–1849. [DOI] [PubMed] [Google Scholar]
- 141.Tran T, Jones CL, Williamson SR, et al. Tubulocystic renal cell carcinoma is an entity that is immunohistochemically and genetically distinct from papillary renal cell carcinoma. Histopathology. 2016;68:850–857. [DOI] [PubMed] [Google Scholar]
- 142.Kuroda N, Ohe C, Mikami S, et al. Review of acquired cystic disease-associated renal cell carcinoma with focus on pathobiological aspects. Histol Histopathol. 2011;26:1215–1218. [DOI] [PubMed] [Google Scholar]
- 143.Tickoo SK, Kuroda N. Acquired cystic disease-associated renal cell carcinoma In: Moch H, Humphrey PA, Ulbright TM., et al. eds. WHO Classification of Tumours of the Urinary System and Male Genital Organs. Lyon: International Agency for Research on Cancer; 2016:39. [Google Scholar]
- 144.Thway K, du Parcq J, Larkin JM, et al. Metastatic renal mucinous tubular and spindle cell carcinoma. Atypical behavior of a rare, morphologically bland tumor. Ann Diagn Pathol. 2012;16:407–410. [DOI] [PubMed] [Google Scholar]
- 145.Dhillon J, Amin MB, Selbs E, et al. Mucinous tubular and spindle cell carcinoma of the kidney with sarcomatoid change. Am J Surg Pathol. 2009;33:44–49. [DOI] [PubMed] [Google Scholar]
- 146.Bulimbasic S, Ljubanovic D, Sima R, et al. Aggressive high-grade mucinous tubular and spindle cell carcinoma. Hum Pathol. 2009;40:906–907. [DOI] [PubMed] [Google Scholar]
- 147.Ren Q, Wang L, Al-Ahmadie HA, et al. Distinct Genomic Copy Number Alterations Distinguish Mucinous Tubular and Spindle Cell Carcinoma of the Kidney From Papillary Renal Cell Carcinoma With Overlapping Histologic Features. Am J Surg Pathol. 2018;42:767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Peckova K, Martinek P, Sperga M, et al. Mucinous spindle and tubular renal cell carcinoma: analysis of chromosomal aberration pattern of low-grade, high-grade, and overlapping morphologic variant with papillary renal cell carcinoma. Ann Diagn Pathol. 2015;19:226–231. [DOI] [PubMed] [Google Scholar]
- 149.Sadimin ET, Chen YB, Wang L, et al. Chromosomal abnormalities of high-grade mucinous tubular and spindle cell carcinoma of the kidney. Histopathology. 2017;71:719–724. [DOI] [PubMed] [Google Scholar]
- 150.Cossu-Rocca P, Eble JN, Delahunt B, et al. Renal mucinous tubular and spindle carcinoma lacks the gains of chromosomes 7 and 17 and losses of chromosome Y that are prevalent in papillary renal cell carcinoma. Mod Pathol. 2006;19:488–493. [DOI] [PubMed] [Google Scholar]
- 151.Ged Y, Chen YB, Knezevic A, et al. Mucinous Tubular and Spindle-Cell Carcinoma of the Kidney: Clinical Features, Genomic Profiles, and Treatment Outcomes. Clin Genitourin Cancer. 2019;17:268–274 e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mehra R, Vats P, Cieslik M, et al. Biallelic Alteration and Dysregulation of the Hippo Pathway in Mucinous Tubular and Spindle Cell Carcinoma of the Kidney. Cancer Discov. 2016;6:1258–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wang L, Zhang Y, Chen YB, et al. VSTM2A Overexpression Is a Sensitive and Specific Biomarker for Mucinous Tubular and Spindle Cell Carcinoma (MTSCC) of the Kidney. Am J Surg Pathol. 2018;42:1571–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yang XJ, Zhou M, Hes O, et al. Tubulocystic carcinoma of the kidney: clinicopathologic and molecular characterization. Am J Surg Pathol. 2008;32:177–187. [DOI] [PubMed] [Google Scholar]
- 155.Sarungbam J, Mehra R, Tomlins SA, et al. Tubulocystic renal cell carcinoma: a distinct clinicopathologic entity with a characteristic genomic profile. Mod Pathol. 2019;32:701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Osunkoya AO, Young AN, Wang W, et al. Comparison of gene expression profiles in tubulocystic carcinoma and collecting duct carcinoma of the kidney. Am J Surg Pathol. 2009;33:1103–1106. [DOI] [PubMed] [Google Scholar]
- 157.Al-Hussain TO, Cheng L, Zhang S, et al. Tubulocystic carcinoma of the kidney with poorly differentiated foci: a series of 3 cases with fluorescence in situ hybridization analysis. Hum Pathol. 2013;44:1406–1411. [DOI] [PubMed] [Google Scholar]
- 158.Smith SC, Trpkov K, Chen YB, et al. Tubulocystic Carcinoma of the Kidney With Poorly Differentiated Foci: A Frequent Morphologic Pattern of Fumarate Hydratase-deficient Renal Cell Carcinoma. Am J Surg Pathol. 2016;40:1457–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kuroda N, Shiotsu T, Hes O, et al. Acquired cystic disease-associated renal cell carcinoma with gain of chromosomes 3, 7, and 16, gain of chromosome X, and loss of chromosome Y. Med Mol Morphol. 2010;43:231–234. [DOI] [PubMed] [Google Scholar]
- 160.Kuroda N, Tamura M, Hamaguchi N, et al. Acquired cystic disease-associated renal cell carcinoma with sarcomatoid change and rhabdoid features. Ann Diagn Pathol. 2011;15:462–466. [DOI] [PubMed] [Google Scholar]
- 161.Kuroda N, Yamashita M, Kakehi Y, et al. Acquired cystic disease-associated renal cell carcinoma: an immunohistochemical and fluorescence in situ hybridization study. Med Mol Morphol. 2011;44:228–232. [DOI] [PubMed] [Google Scholar]
- 162.Cossu-Rocca P, Eble JN, Zhang S, et al. Acquired cystic disease-associated renal tumors: an immunohistochemical and fluorescence in situ hybridization study. Mod Pathol. 2006;19:780–787. [DOI] [PubMed] [Google Scholar]
- 163.Kuntz E, Yusenko MV, Nagy A, et al. Oligoarray comparative genomic hybridization of renal cell tumors that developed in patients with acquired cystic renal disease. Hum Pathol. 2010;41:1345–1349. [DOI] [PubMed] [Google Scholar]
- 164.Moch H, Amin M, Argani P, et al. Renal cell tumors In: Moch H, Humphrey P, Ulbright T, et al. eds. WHO classification of tumours of the urinary system and male genital organs. Lyon: International Agency for Research on Cancer; 2016:14–17. [Google Scholar]
- 165.Guo J, McKenney JK. The Pathology of von Hippel-Lindau Disease. Pathol Case Rev. 2014;19:49–56. [Google Scholar]
- 166.Cheng L, Williamson SR, Zhang S, et al. Understanding the molecular genetics of renal cell neoplasia: implications for diagnosis, prognosis and therapy. Expert Rev Anticancer Ther. 2010;10:843–864. [DOI] [PubMed] [Google Scholar]
- 167.Shuch B, Vourganti S, Ricketts CJ, et al. Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol. 2014;32:431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gill AJ. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology. 2012;44:285–292. [DOI] [PubMed] [Google Scholar]
- 169.Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology. 2018;72:106–116. [DOI] [PubMed] [Google Scholar]
- 170.Gill AJ, Benn DE, Chou A, et al. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol. 2010;41:805–814. [DOI] [PubMed] [Google Scholar]
- 171.van Nederveen FH, Gaal J, Favier J, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009;10:764–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Burnichon N, Briere JJ, Libe R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Korpershoek E, Favier J, Gaal J, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab. 2011;96:E1472–1476. [DOI] [PubMed] [Google Scholar]
- 174.Benn DE, Zhu Y, Andrews KA, et al. Bayesian approach to determining penetrance of pathogenic SDH variants. J Med Genet. 2018;55:729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Gill AJ, Amin MB, Smith S, et al. Succinate dehydrogenase-deficient renal carcinoma In: Moch H, Humphrey PA, Ulbright TM., et al. eds. WHO Classification of Tumours of the Urinary System and Male Genital Organs. Lyon: International Agency for Research on Cancer; 2016:35–36. [Google Scholar]
- 176.Gill AJ, Pachter NS, Clarkson A, et al. Renal tumors and hereditary pheochromocytoma-paraganglioma syndrome type 4. N Engl J Med. 2011;364:885–886. [DOI] [PubMed] [Google Scholar]
- 177.Gill AJ, Pachter NS, Chou A, et al. Renal tumors associated with germline SDHB mutation show distinctive morphology. Am J Surg Pathol. 2011;35:1578–1585. [DOI] [PubMed] [Google Scholar]
- 178.Paik JY, Toon CW, Benn DE, et al. Renal carcinoma associated with succinate dehydrogenase B mutation: a new and unique subtype of renal carcinoma. J Clin Oncol. 2014;32:e10–13. [DOI] [PubMed] [Google Scholar]
- 179.Gill AJ, Hes O, Papathomas T, et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol. 2014;38:1588–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Williamson SR, Eble JN, Amin MB, et al. Succinate dehydrogenase-deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol. 2015;28:80–94. [DOI] [PubMed] [Google Scholar]
- 181.Gill AJ, Lipton L, Taylor J, et al. Germline SDHC mutation presenting as recurrent SDH deficient GIST and renal carcinoma. Pathology. 2013;45:689–691. [DOI] [PubMed] [Google Scholar]
- 182.Ozluk Y, Taheri D, Matoso A, et al. Renal carcinoma associated with a novel succinate dehydrogenase A mutation: a case report and review of literature of a rare subtype of renal carcinoma. Hum Pathol. 2015;46:1951–1955. [DOI] [PubMed] [Google Scholar]
- 183.Yakirevich E, Ali SM, Mega A, et al. A Novel SDHA-deficient Renal Cell Carcinoma Revealed by Comprehensive Genomic Profiling. Am J Surg Pathol. 2015;39:858–863. [DOI] [PubMed] [Google Scholar]
- 184.Harrison WJ, Andrici J, Maclean F, et al. Fumarate Hydratase-deficient Uterine Leiomyomas Occur in Both the Syndromic and Sporadic Settings. Am J Surg Pathol. 2016;40:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Turchini J, Cheung VKY, Tischler AS, et al. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology. 2018;72:97–105. [DOI] [PubMed] [Google Scholar]
- 186.Trpkov K, Hes O, Agaimy A, et al. Fumarate Hydratase-deficient Renal Cell Carcinoma Is Strongly Correlated With Fumarate Hydratase Mutation and Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome. Am J Surg Pathol. 2016;40:865–875. [DOI] [PubMed] [Google Scholar]
- 187.Pivovarcikova K, Martinek P, Grossmann P, et al. Fumarate hydratase deficient renal cell carcinoma: Chromosomal numerical aberration analysis of 12 cases. Ann Diagn Pathol. 2019;39:63–68. [DOI] [PubMed] [Google Scholar]
- 188.Lau HD, Chan E, Fan AC, et al. A Clinicopathologic and Molecular Analysis of Fumarate Hydratase-Deficient Renal Cell Carcinoma in 32 Patients. Am J Surg Pathol. 2019. [DOI] [PubMed] [Google Scholar]
- 189.Shyu I, Mirsadraei L, Wang X, et al. Clues to recognition of fumarate hydratase-deficient renal cell carcinoma: Findings from cytologic and limited biopsy samples. Cancer Cytopathol. 2018;126:992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chen YB, Brannon AR, Toubaji A, et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol. 2014;38:627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Muller M, Guillaud-Bataille M, Salleron J, et al. Pattern multiplicity and fumarate hydratase (FH)/S-(2-succino)-cysteine (2SC) staining but not eosinophilic nucleoli with perinucleolar halos differentiate hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinomas from kidney tumors without FH gene alteration. Mod Pathol. 2018;31:974–983. [DOI] [PubMed] [Google Scholar]
- 192.Miettinen M, Felisiak-Golabek A, Wasag B, et al. Fumarase-deficient Uterine Leiomyomas: An Immunohistochemical, Molecular Genetic, and Clinicopathologic Study of 86 Cases. Am J Surg Pathol. 2016;40:1661–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Merino MJ, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma (HLRCC)-associated renal cell carcinoma In: Moch H, Humphrey PA, Ulbright TM., et al. eds. WHO Classification of Tumours of the Urinary System and Male Genital Organs. Lyon: International Agency for Research on Cancer; 2016:25–26. [Google Scholar]
- 194.Shyu I, Mirsadraei L, Wang X, et al. Clues to recognition of fumarate hydratase-deficient renal cell carcinoma: Findings from cytologic and limited biopsy samples. Cancer Cytopathol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Adeniran AJ, Shuch B, Humphrey PA. Hereditary Renal Cell Carcinoma Syndromes: Clinical, Pathologic, and Genetic Features. Am J Surg Pathol. 2015;39:e1–e18. [DOI] [PubMed] [Google Scholar]
- 196.Guo J, Tretiakova MS, Troxell ML, et al. Tuberous sclerosis-associated renal cell carcinoma: a clinicopathologic study of 57 separate carcinomas in 18 patients. Am J Surg Pathol. 2014;38:1457–1467. [DOI] [PubMed] [Google Scholar]
- 197.Yang P, Cornejo KM, Sadow PM, et al. Renal cell carcinoma in tuberous sclerosis complex. Am J Surg Pathol. 2014;38:895–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Chen YB, Mirsadraei L, Jayakumaran G, et al. Somatic Mutations of TSC2 or MTOR Characterize a Morphologically Distinct Subset of Sporadic Renal Cell Carcinoma With Eosinophilic and Vacuolated Cytoplasm. Am J Surg Pathol. 2019;43:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mehra R, Vats P, Cao X, et al. Somatic Bi-allelic Loss of TSC Genes in Eosinophilic Solid and Cystic Renal Cell Carcinoma. Eur Urol. 2018;74:483–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Palsgrove DN, Li Y, Pratilas CA, et al. Eosinophilic Solid and Cystic (ESC) Renal Cell Carcinomas Harbor TSC Mutations: Molecular Analysis Supports an Expanding Clinicopathologic Spectrum. Am J Surg Pathol. 2018;42:1166–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Parilla M, Kadri S, Patil SA, et al. Are Sporadic Eosinophilic Solid and Cystic Renal Cell Carcinomas Characterized by Somatic Tuberous Sclerosis Gene Mutations? Am J Surg Pathol. 2018;42:911–917. [DOI] [PubMed] [Google Scholar]
- 202.Trpkov K, Abou-Ouf H, Hes O, et al. Eosinophilic Solid and Cystic Renal Cell Carcinoma (ESC RCC): Further Morphologic and Molecular Characterization of ESC RCC as a Distinct Entity. Am J Surg Pathol. 2017. [DOI] [PubMed] [Google Scholar]
- 203.Trpkov K, Hes O, Bonert M, et al. Eosinophilic, Solid, and Cystic Renal Cell Carcinoma: Clinicopathologic Study of 16 Unique, Sporadic Neoplasms Occurring in Women. Am J Surg Pathol. 2016;40:60–71. [DOI] [PubMed] [Google Scholar]
- 204.Tretiakova MS. Eosinophilic solid and cystic renal cell carcinoma mimicking epithelioid angiomyolipoma: series of 4 primary tumors and 2 metastases. Hum Pathol. 2018;80:65–75. [DOI] [PubMed] [Google Scholar]
- 205.Li Y, Reuter VE, Matoso A, et al. Re-evaluation of 33 ‘unclassified’ eosinophilic renal cell carcinomas in young patients. Histopathology. 2018;72:588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Williamson SR. Renal cell carcinomas with a mesenchymal stromal component: what do we know so far? Pathology. 2019;51:453–462. [DOI] [PubMed] [Google Scholar]
- 207.Parilla M, Alikhan M, Al-Kawaaz M, et al. Genetic Underpinnings of Renal Cell Carcinoma With Leiomyomatous Stroma. Am J Surg Pathol. 2019;43:1135–1144. [DOI] [PubMed] [Google Scholar]
- 208.Jia L, Jayakumaran G, Al-Ahmadie H, et al. Expanding the Morphologic Spectrum of Sporadic Renal Cell Carcinoma (RCC) Harboring Somatic TSC or MTOR Alterations: Analysis of 8 Cases with Clear Cytoplasm and Leiomyomatous Stroma. Mod Pathol. 2019;32:78–79 (abstract). [Google Scholar]
- 209.Williamson SR, Hornick JL, Eble JN, et al. Renal cell carcinoma with angioleiomyoma-like stroma and clear cell papillary renal cell carcinoma: exploring SDHB protein immunohistochemistry and the relationship to tuberous sclerosis complex. Hum Pathol. 2018;75:10–15. [DOI] [PubMed] [Google Scholar]
- 210.Verkarre V, Mensah A, Leroy X, et al. A Clinico-Pathologic Study of 17 Patients With Renal Cell Carcinoma Associated With Leiomyomatous Stroma Identifies a Strong Association With Tuberous Sclerosis. Lab Invest. 2015;95:266A–266A. [Google Scholar]
- 211.He H, Trpkov K, Martinek P, et al. “High-grade oncocytic renal tumor”: morphologic, immunohistochemical, and molecular genetic study of 14 cases. Virchows Arch. 2018;473:725–738. [DOI] [PubMed] [Google Scholar]
- 212.Trpkov K, Bonert M, Gao Y, et al. High-grade oncocytic tumour (HOT) of kidney in a patient with tuberous sclerosis complex. Histopathology. 2019;75:440–442. [DOI] [PubMed] [Google Scholar]
- 213.Hakimi AA, Tickoo SK, Jacobsen A, et al. TCEB1-mutated renal cell carcinoma: a distinct genomic and morphological subtype. Mod Pathol. 2015;28:845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Sato Y, Yoshizato T, Shiraishi Y, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013. [DOI] [PubMed] [Google Scholar]
- 215.Hirsch MS, Barletta JA, Gorman M, et al. Renal Cell Carcinoma With Monosomy 8 and CAIX Expression: A Distinct Entity or Another Member or the Clear Cell Tubulopapillary RCC/RAT Family? Mod Pathol. 2015;28:229A (abstract). [Google Scholar]
- 216.Shah RB, Stohr BA, Tu ZJ, et al. “Renal Cell Carcinoma With Leiomyomatous Stroma” Harbor Somatic Mutations of TSC1, TSC2, MTOR, and/or ELOC (TCEB1): Clinicopathologic and Molecular Characterization of 18 Sporadic Tumors Supports a Distinct Entity. Am J Surg Pathol. 2019. [DOI] [PubMed] [Google Scholar]
- 217.DiNatale RG, Gorelick AN, Makarov V, et al. Putative Drivers of Aggressiveness in TCEB1-mutant Renal Cell Carcinoma: An Emerging Entity with Variable Clinical Course. Eur Urol Focus. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Argani P, Reuter VE, Zhang L, et al. TFEB-amplified Renal Cell Carcinomas: An Aggressive Molecular Subset Demonstrating Variable Melanocytic Marker Expression and Morphologic Heterogeneity. Am J Surg Pathol. 2016;40:1484–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Andeen NK, Qu X, Antic T, et al. Clinical Utility of Chromosome Genomic Array Testing for Unclassified and Advanced-Stage Renal Cell Carcinomas. Arch Pathol Lab Med. 2019;143:494–504. [DOI] [PubMed] [Google Scholar]
- 220.Debelenko LV, Raimondi SC, Daw N, et al. Renal cell carcinoma with novel VCL-ALK fusion: new representative of ALK-associated tumor spectrum. Mod Pathol. 2011;24:430–442. [DOI] [PubMed] [Google Scholar]
- 221.Sugawara E, Togashi Y, Kuroda N, et al. Identification of anaplastic lymphoma kinase fusions in renal cancer: large-scale immunohistochemical screening by the intercalated antibody-enhanced polymer method. Cancer. 2012;118:4427–4436. [DOI] [PubMed] [Google Scholar]
- 222.Sukov WR, Hodge JC, Lohse CM, et al. ALK alterations in adult renal cell carcinoma: frequency, clinicopathologic features and outcome in a large series of consecutively treated patients. Mod Pathol. 2012;25:1516–1525. [DOI] [PubMed] [Google Scholar]
- 223.Bodokh Y, Ambrosetti D, Kubiniek V, et al. ALK-TPM3 rearrangement in adult renal cell carcinoma: Report of a new case showing loss of chromosome 3 and literature review. Cancer Genet. 2018;221:31–37. [DOI] [PubMed] [Google Scholar]
- 224.Cajaiba MM, Jennings LJ, George D, et al. Expanding the spectrum of ALK-rearranged renal cell carcinomas in children: Identification of a novel HOOK1-ALK fusion transcript. Genes Chromosomes Cancer. 2016;55:814–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Cajaiba MM, Jennings LJ, Rohan SM, et al. ALK-rearranged renal cell carcinomas in children. Genes Chromosomes Cancer. 2016;55:442–451. [DOI] [PubMed] [Google Scholar]
- 226.Hodge JC, Pearce KE, Sukov WR. Distinct ALK-rearranged and VCL-negative papillary renal cell carcinoma variant in two adults without sickle cell trait. Mod Pathol. 2013;26:604–605. [DOI] [PubMed] [Google Scholar]
- 227.Jeanneau M, Gregoire V, Desplechain C, et al. ALK rearrangements-associated renal cell carcinoma (RCC) with unique pathological features in an adult. Pathol Res Pract. 2016;212:1064–1066. [DOI] [PubMed] [Google Scholar]
- 228.Kuroda N, Liu Y, Tretiakova M, et al. Clinicopathological Study of Seven Cases of ALK-positive Renal Tumor Identification of New Fusion Partners including CLIP1 and KIF5B Genes. Mod Pathol. 2019;32:85. [Google Scholar]
- 229.Kuroda N, Sugawara E, Kusano H, et al. A review of ALK-rearranged renal cell carcinomas with a focus on clinical and pathobiological aspects. Pol J Pathol. 2018;69:109–113. [DOI] [PubMed] [Google Scholar]
- 230.Kusano H, Togashi Y, Akiba J, et al. Two Cases of Renal Cell Carcinoma Harboring a Novel STRN-ALK Fusion Gene. Am J Surg Pathol. 2016;40:761–769. [DOI] [PubMed] [Google Scholar]
- 231.Lee C, Park JW, Suh JH, et al. ALK-Positive Renal Cell Carcinoma in a Large Series of Consecutively Resected Korean Renal Cell Carcinoma Patients. Korean J Pathol. 2013;47:452–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Marino-Enriquez A, Ou WB, Weldon CB, et al. ALK rearrangement in sickle cell trait-associated renal medullary carcinoma. Genes Chromosomes Cancer. 2011;50:146–153. [DOI] [PubMed] [Google Scholar]
- 233.Pal SK, Bergerot P, Dizman N, et al. Responses to Alectinib in ALK-rearranged Papillary Renal Cell Carcinoma. Eur Urol. 2018;74:124–128. [DOI] [PubMed] [Google Scholar]
- 234.Smith NE, Deyrup AT, Marino-Enriquez A, et al. VCL-ALK renal cell carcinoma in children with sickle-cell trait: the eighth sickle-cell nephropathy? Am J Surg Pathol. 2014;38:858–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Thorner PS, Shago M, Marrano P, et al. TFE3-positive renal cell carcinomas are not always Xp11 translocation carcinomas: Report of a case with a TPM3-ALK translocation. Pathol Res Pract. 2016;212:937–942. [DOI] [PubMed] [Google Scholar]
- 236.Yu W, Wang Y, Jiang Y, et al. Genetic analysis and clinicopathological features of ALK-rearranged renal cell carcinoma in a large series of resected Chinese renal cell carcinoma patients and literature review. Histopathology. 2017;71:53–62. [DOI] [PubMed] [Google Scholar]
- 237.Hang JF, Chung HJ, Pan CC. ALK-rearranged renal cell carcinoma with a novel PLEKHA7-ALK translocation and metanephric adenoma-like morphology. Virchows Arch. 2020. [DOI] [PubMed] [Google Scholar]
- 238.Taneja K, Cheng L, Al-Obaidy K, et al. Clear Cell Renal Cell Carcinoma With a Poorly-Differentiated Component: A Novel Variant Causing Potential Diagnostic Difficulty. Mod Pathol. 2019;32:147–148 (abstract).30171197 [Google Scholar]
- 239.Kouba EJ, Eble JN, Simper N, et al. High fidelity of driver chromosomal alterations among primary and metastatic renal cell carcinomas: implications for tumor clonal evolution and treatment. Mod Pathol. 2016;29:1347–1357. [DOI] [PubMed] [Google Scholar]
- 240.Wang L, Williamson SR, Wang M, et al. Molecular subtyping of metastatic renal cell carcinoma: implications for targeted therapy. Mol Cancer. 2014;13:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.D’Avella C, Abbosh P, Pal SK, et al. Mutations in renal cell carcinoma. Urol Oncol. 2018. [DOI] [PubMed] [Google Scholar]
- 242.Kwiatkowski DJ, Choueiri TK, Fay AP, et al. Mutations in TSC1, TSC2, and MTOR Are Associated with Response to Rapalogs in Patients with Metastatic Renal Cell Carcinoma. Clin Cancer Res. 2016;22:2445–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kammerer-Jacquet SF, Deleuze A, Saout J, et al. Targeting the PD-1/PD-L1 Pathway in Renal Cell Carcinoma. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Grimm MO, Leucht K, Grunwald V, et al. New First Line Treatment Options of Clear Cell Renal Cell Cancer Patients with PD-1 or PD-L1 Immune-Checkpoint Inhibitor-Based Combination Therapies. J Clin Med. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Nunes-Xavier CE, Angulo JC, Pulido R, et al. A Critical Insight into the Clinical Translation of PD-1/PD-L1 Blockade Therapy in Clear Cell Renal Cell Carcinoma. Curr Urol Rep. 2019;20:1. [DOI] [PubMed] [Google Scholar]
- 246.Flaifel A, Xie W, Braun DA, et al. PD-L1 Expression and Clinical Outcomes to Cabozantinib, Everolimus, and Sunitinib in Patients with Metastatic Renal Cell Carcinoma: Analysis of the Randomized Clinical Trials METEOR and CABOSUN. Clin Cancer Res. 2019;25:6080–6088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.McKay RR, Bosse D, Xie W, et al. The Clinical Activity of PD-1/PD-L1 Inhibitors in Metastatic Non-Clear Cell Renal Cell Carcinoma. Cancer Immunol Res. 2018;6:758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.