

Abstract
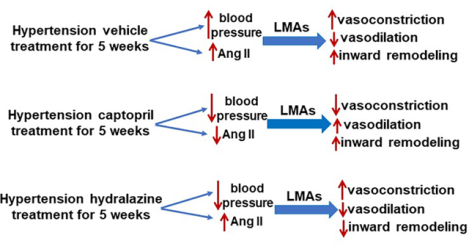
Leptomeningeal anastomoses (LMAs) are pial collaterals that perfuse the penumbra and important for stroke outcome. We previously showed LMAs from spontaneously hypertensive rats (SHRs) were vasoconstricted compared to normotensive Wistar rats. Here, we investigated mechanisms by which hypertension causes LMA vasoconstriction. SHRs were treated with the angiotensin converting enzyme (ACE) inhibitor captopril, an Ang II-independent antihypertensive agent hydralazine, or vehicle for five weeks in drinking water (n=8/group). A group of Wistar rats (n=8) had regular drinking water served as controls. Blood pressure (BP) was measured twice weekly by tail cuff. LMAs were isolated and studied under pressurized conditions. Vasoreactivity of LMAs, including myogenic responses, reactivity to Rho-kinase inhibitor Y-27632 and nitric oxide (NO) were measured. Both captopril and hydralazine lowered BP in SHRs similar to Wistar. However, only captopril normalized LMA increased tone compared to untreated SHRs (15±2% vs. 50±3%, P<0.01) that was similar to Wistar (16±2%), but not hydralazine (38±6%, P>0.05). Vasodilatory response of LMAs to Y-27632 was impaired in SHRs compared to Wistar (28±3% vs. 81±4%, P<0.01) that was restored by captopril (84±5%, P<0.01) and partially hydralazine (59±4%). LMAs from all groups constricted similarly to NO synthase inhibition; however, the vasodilatory response of LMAs to the NO donor sodium nitroprusside was impaired in SHRs compared to Wistar rats (29±4% vs. 80±2%, P<0.01) that was restored by captopril (84±4%, P<0.01), not hydralazine (38±8%, P>0.05). These results suggest that ACE inhibition during chronic hypertension reversed vascular dysfunction and hyperconstriction of LMAs that could improve stroke outcome by increasing collateral perfusion.
Keywords: hypertension, angiotensin II, leptomeningeal anastomoses, vasoconstriction, collateral circulation, angiotensin II type 1 receptor, angiotensin converting enzyme inhibitor
Graphical Abstract
Introduction
Stroke is the second leading cause of death worldwide and the leading cause of devastating disability.1 Ischemic stroke accounts for ~87% of all strokes and results from a thrombotic or embolic occlusion of large extra- and intracranial arteries.1,2 Corresponding to the severe drop of cerebral blood flow (CBF) in the brain downstream from the occlusion, the cerebral tissue develops irreversible injury and is known as infarct.3 The infarct is adjacent to moderately ischemic tissue with metabolic impairment but maintained neuronal integrity, which is called penumbra.4 The penumbra is perfused by leptomeningeal anastomoses (LMAs), which are pial collaterals in the brain that connect distal branches of major cerebral arteries.5 Good LMA circulation is associated with a larger baseline of penumbral flow, limited infarct expansion and better clinical outcomes in patients with acute ischemic stroke.6
LMAs experience unique hemodynamic forces, including low shear stress and bidirectional flow, and lack perivascular innervation.7,8 Zhang et al. reported that endothelial and smooth muscle cells of LMAs have distinct features compared to similarly-sized nonanastomotic pial arterioles, such as fewer cilia and higher expression of pro- and anti-inflammatory genes.9 Our lab has identified that LMAs have less basal tone compared to similarly-sized nonanastomotic arterioles in normotensive rats.8 The influence of this unique vascular environment appears to be associated with distinct vascular functions that could considerably impact collateral perfusion, penumbral flow and ischemic stroke outcome.
Hypertension is one of the strongest risk factors for stroke,10 and other cardiovascular diseases.11 Though the pathogenesis of hypertension is multifactorial and highly complex,12 the renin-angiotensin system is a major cause in the development and maintenance of hypertension.13 Ang II is generated from angiotensin I by ACE, and elevated Ang II is involved in the pathogenesis of hypertension.14 Increased Ang II promotes vasoconstriction, vascular remodeling and increased expression of proinflammatory factors, as well as inhibiting endothelium-dependent relaxation via activating Ang II type 1 receptor (AT1R).15–17 In addition, local Ang II produces superoxide-mediated vascular dysfunction in cerebral arterioles.18 Treatment with an ACE inhibitor or AT1R blocker has beneficial effects including, normalizing cerebrovascular autoregulation,19,20 increasing cerebral cortical angiogenesis,21 reversing vascular hypertrophy and inward remodeling in cerebral arteries,22 and attenuating brain superoxide levels and inflammation22 that together reduces ischemic injury.20 We previously showed that LMAs from chronically hypertensive rats were significantly vasoconstricted compared to LMAs from normotensive rats that likely limits the amount of salvageable tissue (penumbra) when stroke occurs in chronic hypertension.8,23 In the current study, we investigated the underlying mechanisms by which hypertension causes LMA vasoconstriction. We hypothesized that increased vasoreactivity of LMAs from hypertensive rats is Ang II-dependent that chronic ACE inhibition would reverse the increased vasoreactivity of LMAs.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
Experiments were conducted using adult male SHRs (21–25 weeks) and age-matched male Wistar rats (21–25 weeks, n=8) from Charles River (Wilmington, MA, USA). Sex differences were not studied because we previously showed that LMAs from male and female Wistar rats had similar myogenic tone, structural, and vascular function, and numbers of LMAs.24 All animal procedures were approved by the Institutional Animal Care and Use Committee at University of Vermont and complied with National Institutes of Health guidelines for care and usage of laboratory animals.
SHRs were divided into three groups and treated for five weeks as follows: one group of SHRs were treated with the ACE inhibitor captopril in the drinking water (SHR-Capto, 100 mg/kg/day; n=8), a second group of SHRs were treated with hydralazine in the drinking water (SHR-Hydral, 20 mg/kg/day; n=8) to lower BP without inhibiting Ang II as a control for the BP lowering effects of captopril, a third group of SHRs had regular drinking water thus served as a vehicle control (SHR-Veh; n=8). A group of Wistar rats were included as a normotensive control. The order of treatment groups was randomized using an online randomization tool (https://www.random.org/lists/).
Blood Pressure Measurements
All rats were handled for three days prior to the treatment to acclimate to BP measurements. BP was measured twice every week by a tail-cuff method using the CODA 8 System (Kent Scientific, Torrington, CT, USA).25
Experimental Protocols
LMAs were isolated and studied pressurized as we previously reported.8 Isolated LMAs were equilibrated at 40 mmHg for 1 hour to allow the development of spontaneously myogenic tone, after which intravascular pressure was increased step-wise in 20 mmHg increments to 100 mmHg to measure myogenic reactivity. Lumen diameter and wall thickness were recorded once stable. Then LMA reactivity to various pharmacological reagents were measured: paxilline (10−6 M), a large-conductance Ca2+-activated K+ (BK) channel inhibitor; Y-27632 (10−8 - 10−5 M), a Rho-kinase inhibitor; NG-nitro-L-arginine methyl ester (L-NAME, 10−3 M), a nitric oxide (NO) synthase (NOS) inhibitor; and sodium nitroprusside (SNP, 10−8 - 10−5 M), a NO donor. If myogenic tone did not develop after the wash, a thromboxane A2 agonist U46619 (10−7 M) was given to preconstrict the LMAs in preparation for subsequent dilation.26 Passive structural measurements were obtained in fully relaxed conditions at the end of experiments in zero calcium PSS and the presence of diltiazem and papaverine. To investigate the regional differences of vascular function in chronic hypertension, the MCAs were also isolated and mounted in the arteriograph and myogenic responses and structural characteristics were studied. See the online-only Data Supplement for details.
Expression of AT1R on LMAs and Vasoactivity to Ang II.
To determine whether LMAs had AT1R expression on smooth muscle and/or endothelial cells, cross sections LMAs were immunohistochemically stained with primary antibodies against AT1R, α-smooth muscle Actin (αSMA), a vascular smooth muscle marker, and von Willebrand factor (vWF), an endothelial marker. In another set of experiments, the vasoactivity of isolated and pressurized LMAs to Ang II and AT1R blocker candesartan were done to further confirm the presence of AT1R on LMAs. Please see online-only Data Supplement for details.
Drugs and Solutions
Details on Drugs and Solutions are available in the online-only Data Supplement.
Data Calculations and Statistical Analysis
Data are presented as mean ± SEM. Data were analyzed using one-way analysis of variance (ANOVA), or mixed model repeated measures ANOVA. For the mixed model repeated measures ANOVA, non-normally distributed data were transformed using Natural Logarithm. The D’Agostino-Pearson omnibus normality test and ANOVA were performed using GraphPad Prism (version 8.0, San Diego, CA, USA); mixed model repeated measures ANOVA was conducted using SAS (SAS Institute, Inc.; Cary, NC, USA). Differences were considered statistically significant at P<0.05. Please see online-only Data Supplement for all data calculations. See figure legends for detailed statistical analysis.
Results
Hydralazine and Captopril Treatment Both Lowered Systolic and Diastolic Blood Pressure in Hypertensive Rats
As shown in Figure 1A, after five weeks of treatment, both hydralazine and captopril decreased systolic BP significantly compared to SHRs with vehicle treatment (SHR-Hydral: 122±3 mmHg, SHR-Capto: 131±3 mmHg vs. SHR-Veh: 197±6 mmHg, P<0.01), and showed no difference compared to Wistar rats (125±3 mmHg, P>0.05). After five weeks of treatment, both hydralazine and captopril significantly decreased diastolic BP (Figure 1B) in comparison to SHRs with vehicle treatment (SHR-Hydral: 85±3 mmHg, SHR-Capto: 93±3 mmHg vs. SHR-Veh: 150±7 mmHg, P<0.01), and showed no difference with Wistar rats (89±1 mmHg, P>0.05). A significant decrease in both systolic and diastolic BP were detected as early as the first day after hydralazine and captopril treatment.
Figure 1.
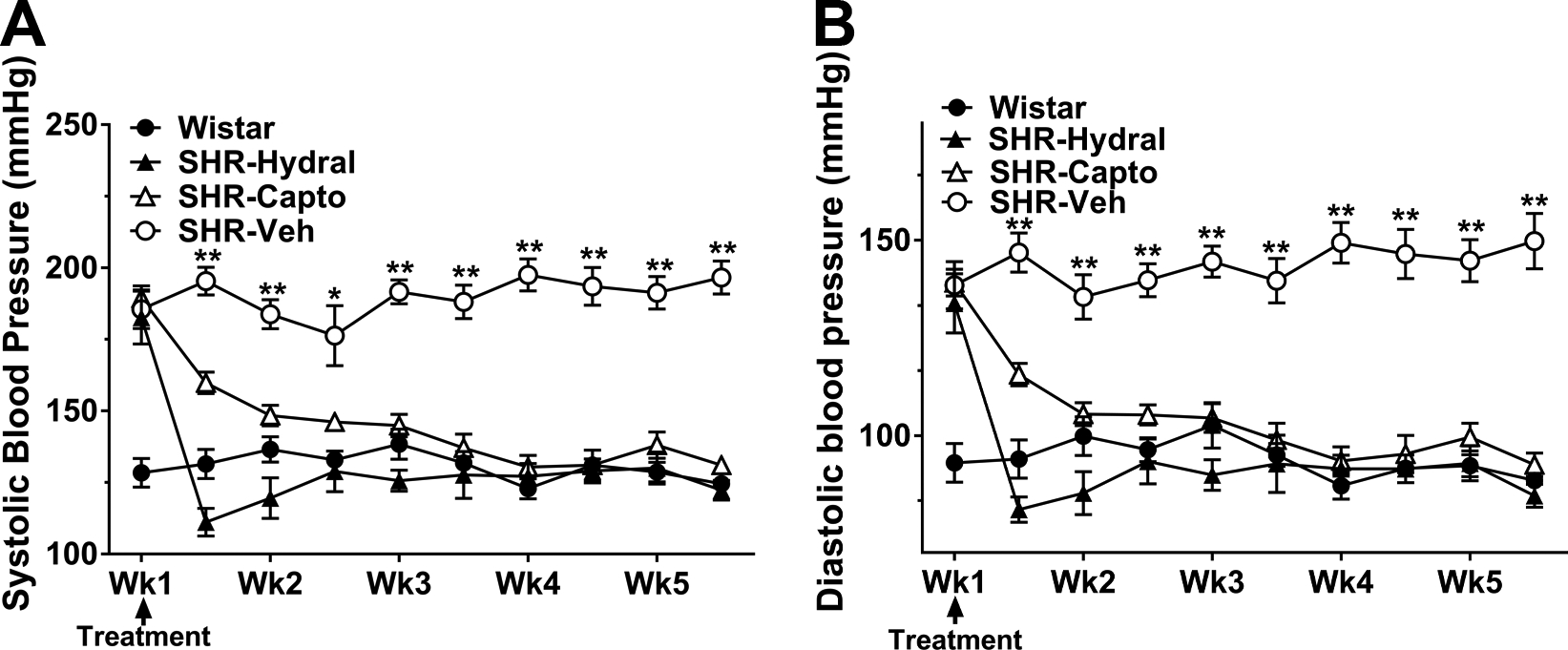
Blood pressure measured in conscious rats before and during treatment. A, Systolic BP of rats before and during treatment. B, Diastolic BP of rats before and during treatment. Values are mean ± SEM for groups of 8 animals each. *P<0.05, **P<0.01 vs. all groups by one-way ANOVA with post-hoc Bonferroni correction for multiple comparisons.
Captopril, Not Hydralazine, Lowered Myogenic Reactivity and Tone of LMAs in Hypertensive Rats
Figure 2A shows the change in active lumen diameter of LMAs to increased intravascular pressure. LMAs from SHR-Veh and SHR-Hydral constricted significantly [F(3, 81)=13.67, F(3, 81)=17.57; P<0.01] with increasing intravascular pressure, indicating considerable myogenic response. In contrast, this pressure effect was absent in LMAs from Wistar and SHR-Capto rats [F(3, 81)=0.05, F(3, 81)=0.18; P>0.05], demonstrating pressure had less effect on LMAs from those groups. Thus, only captopril treatment prevented vasoconstriction in response to pressure in SHR. However, LMAs from SHR-Veh and SHR-Capto rats both had significantly smaller lumen diameters compared to LMAs from Wistar rats (P<0.01).
Figure 2.
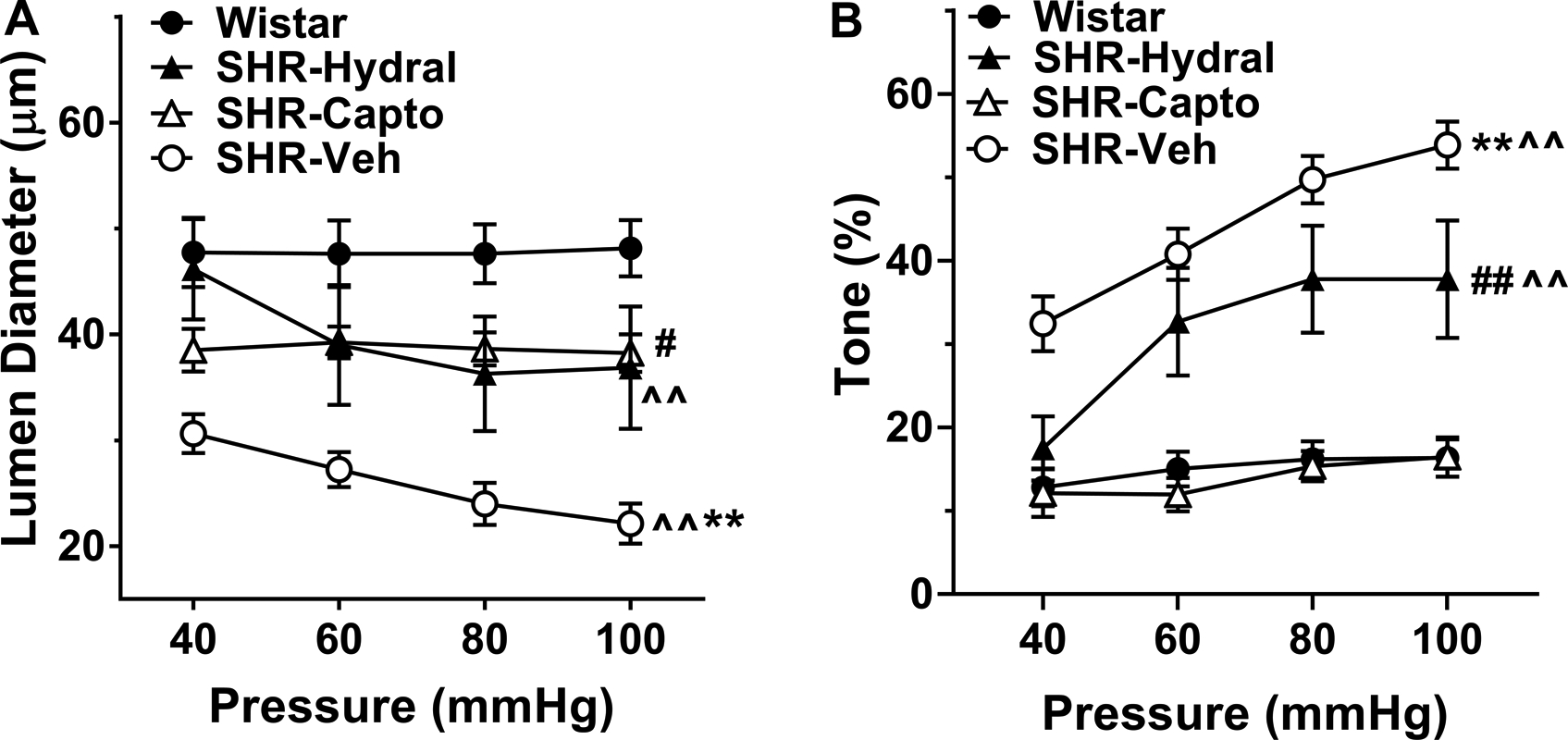
Myogenic reactivity and percent myogenic tone to increased intravascular pressure in LMAs isolated from Wistar rats, and SHRs with or without treatment. A, Change in active lumen diameter in response to increased intravascular pressure of LMAs from Wistar rats, SHRs treated with hydralazine, captopril or vehicle. There was a significant group by pressure interaction, a group effect and pressure effect [F(9, 81)=5.71, F(3, 27)=9.07, F(3, 81)=15.46; P<0.01)]. ^^P<0.01, indicating significant pressure effect in SHR-Veh and SHR-Hydral groups; **P<0.01 vs. all groups, #P<0.05 vs. Wistar rats by mixed model repeated measures ANOVA. B, Percent myogenic tone at different intravascular pressures of LMAs from Wistar rats, SHRs treated with hydralazine, captopril or vehicle. There was a significant group by pressure interaction, as well as a group effect and pressure effect [F(9, 81)=4.75, F(3, 27)=23.71, F(3, 81)=27.17; P<0.01]. ^^P<0.01, indicating significant pressure effect in SHR-Veh and SHR-Hydral groups; **P<0.01 vs. all groups, ## P<0.01 vs. Wistar and SHR-Capto by mixed model repeated measures ANOVA. Wistar: n=8; SHR-Hydral: n=7; SHR-Capto: n=8; SHR-Veh: n=8.
Figure 2B shows the myogenic tone of LMAs at different pressures. LMAs from SHR-Veh had significantly more myogenic tone compared to all other groups (P<0.01). Captopril treatment prevented the increased myogenic tone in LMAs that had similar tone as from Wistar rats (P>0.05). However, SHR-Hydral rats had LMAs with significantly increased myogenic tone compared to Wistar rats (P<0.01). Thus, only captopril treatment reversed the increased LMA myogenic tone in SHRs.
Figure S1A shows the active lumen diameter of MCAs from the same animals. MCAs from Wistar rats had significantly larger lumen diameter compared to MCAs from SHR-Hydral, SHR-Capto and SHR-Veh (P<0.01 for all groups). MCAs from SHR-Veh maintained lumen diameter with increased intravascular pressure (P>0.05); however, MCAs from Wistar rats, SHR-Hydral and SHR-Capto increased lumen diameter with the increasing of intravascular pressure (P<0.01 for all groups). Figure S1B shows myogenic tone of MCAs at various pressures. MCAs from SHR-Veh (P<0.05) and SHR-Hydral (P<0.05) had significantly more myogenic tone compared to MCAs from Wistar rats; MCAs from SHR-Capto showed similar tone compared to Wistar rats (P>0.05). These results indicate that only captopril treatment lowered MCA myogenic tone that was similar to Wistar rats.
Captopril Did Not Restore Structural Properties of LMAs
To understand if chronic hypertension changed LMA structural properties, and if changes were modified by captopril or hydralazine, we measured inner diameter and wall thickness of LMAs under passive conditions and calculated outer diameter and distensibility. As shown in Figure 3A and 3B, passive inner and outer diameters of LMAs from SHR-veh were structurally smaller than Wistar, but this did not reach statistical significance (P=0.07). Treatment with hydralazine reversed these structural changes and showed similar inner (P>0.05) and outer (P>0.05) diameters compared to Wistar rats. Interestingly, captopril did not prevent inward remodeling and showed similar inner (P>0.05) and outer (P>0.05) diameters compared to SHR-veh rats. Thus, while captopril prevented hypertension-induced vasoconstriction in response to pressure, it did not prevent structural inward remodeling whereas hydralazine had the opposite effect and prevented remodeling but not vasoconstriction. In addition, LMAs from all groups had similar wall thickness and percent distensibility (Figures 3C and 3D, P>0.05), demonstrating wall thickness and distensibility were not changed during chronic hypertension, and neither captopril nor hydralazine treatment modified that.
Figure 3.
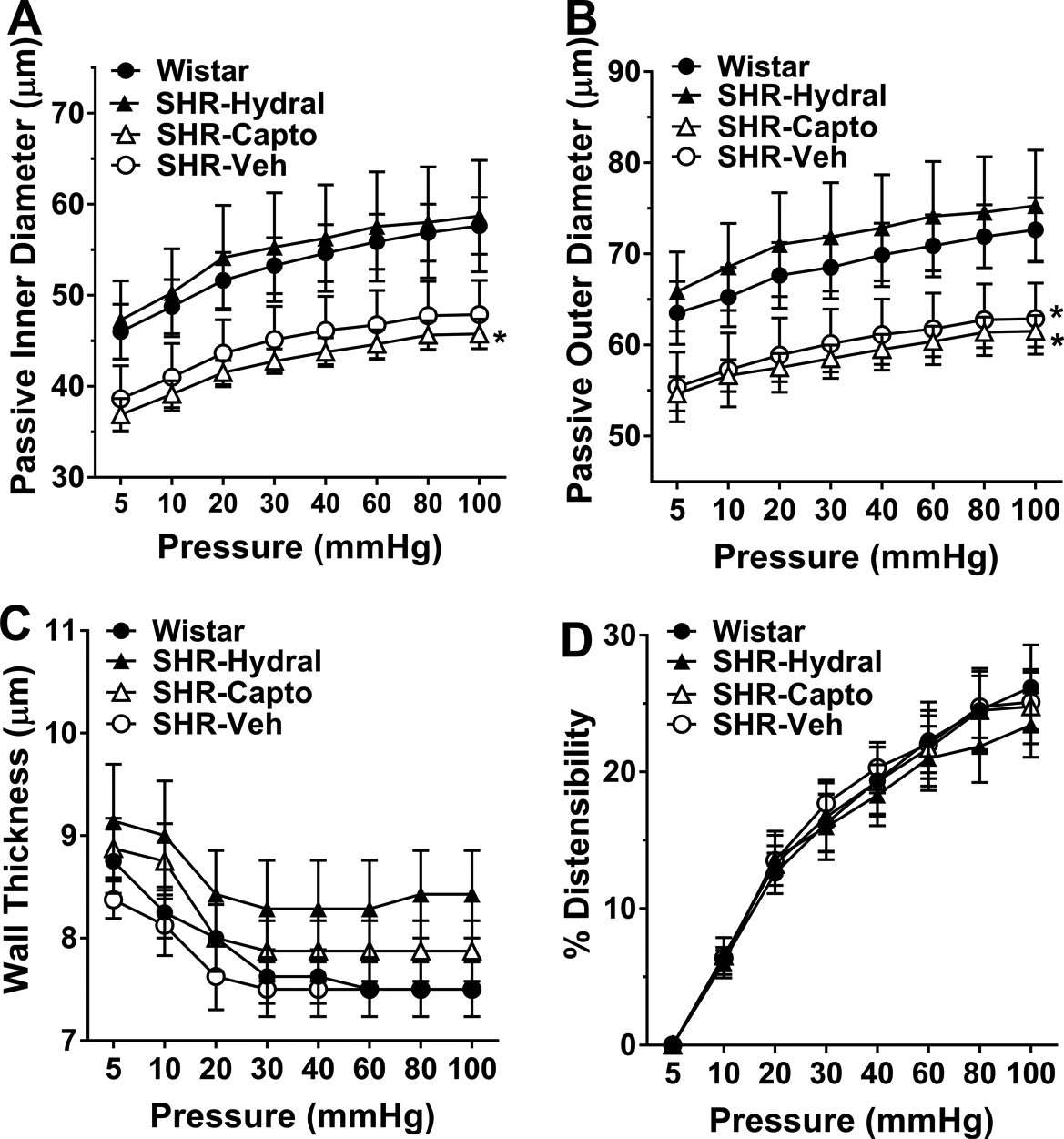
Passive structural measurements of LMAs from Wistar rats, SHRs treated with hydralazine, captopril or vehicle. A, Changes in passive inner diameter in response to increased intravascular pressure. There was a significant pressure effect [F(1, 213)=500.10, P<0.01], but no significance in pressure by group interaction or group effect [F(3, 213)=0.39, F(3, 27)=2.87; P>0.05]. *P<0.05 vs. Wistar and SHR-Hydral rats by pairwise group comparisons. B, Changes in passive outer diameter in response to increased intravascular pressure. There was a significant pressure effect [F(1, 213)=289.87, P<0.01], but no significance in pressure by group interaction or group effect [F(3, 213)=0.96, F(3, 27)=2.82; P>0.05]. *P<0.05 vs. SHR-Hydral rats by pairwise group comparisons. C, Changes in wall thickness in response to increased intravascular pressure. There was a significant pressure effect [F(1, 213)=115.50, P<0.01], but no significance in group by pressure interaction or group effect [F(3, 213)=1.68, F(3, 27)=0.90, P>0.05]. D, percent distensibility in response to increased intravascular pressure. There was a significant pressure effect [F(1, 213)=534.31, P<0.01], but no significance in pressure by group interaction or group effect [F(3, 213)=0.55, F(3, 27)=0.03; P>0.05]. Wistar: n=8; SHR-Hydral: n=7; SHR-Capto: n=8; SHR-Veh: n=8. Data were analyzed by mixed model repeated measures ANOVA.
Figure S2A and S2B show the passive inner and outer diameters of MCAs in response to increases in intravascular pressures. MCAs from Wistar rats had significantly larger inner (P<0.01) and outer (P<0.05) diameters than MCAs from all other SHR groups, suggesting MCAs from SHR-Veh were structurally smaller, and neither captopril nor hydralazine treatment reversed the inward remodeling. As shown in Figure S2C, MCAs from Wistar rats had smaller wall thickness compared to MCAs from SHR-Veh (P=0.06), SHR-Hydral (P<0.01) and SHR-Capto (P<0.01). Additionally, distensibility of MCAs was similar between groups (Figure S2D).
Increased Vasoreactivity of LMAs in Hypertensive Rats Is Ang II-dependent
To better understand the effects of Ang II and chronic hypertension on LMA vasoreactivity, we investigated the LMA lumen diameter change to vasoconstrictive and vasodilatory stimuli using pharmacological approaches. Figure 4A shows the vasoconstrictive response of LMAs to BK channel inhibitor paxilline. LMAs from Wistar rats constricted 21±2% to paxilline, demonstrating the presence of BK channels on LMAs and these channels are involved in opposing basal myogenic tone of LMAs.27 Additionally, LMAs from SHR-Veh showed significantly less vasoconstriction compared to Wistar rats, suggesting modified BK channel function in LMAs from hypertensive rats. Both hydralazine and captopril treatment increased the vasoconstrictive response which was similar to Wistar rats.
Figure 4.
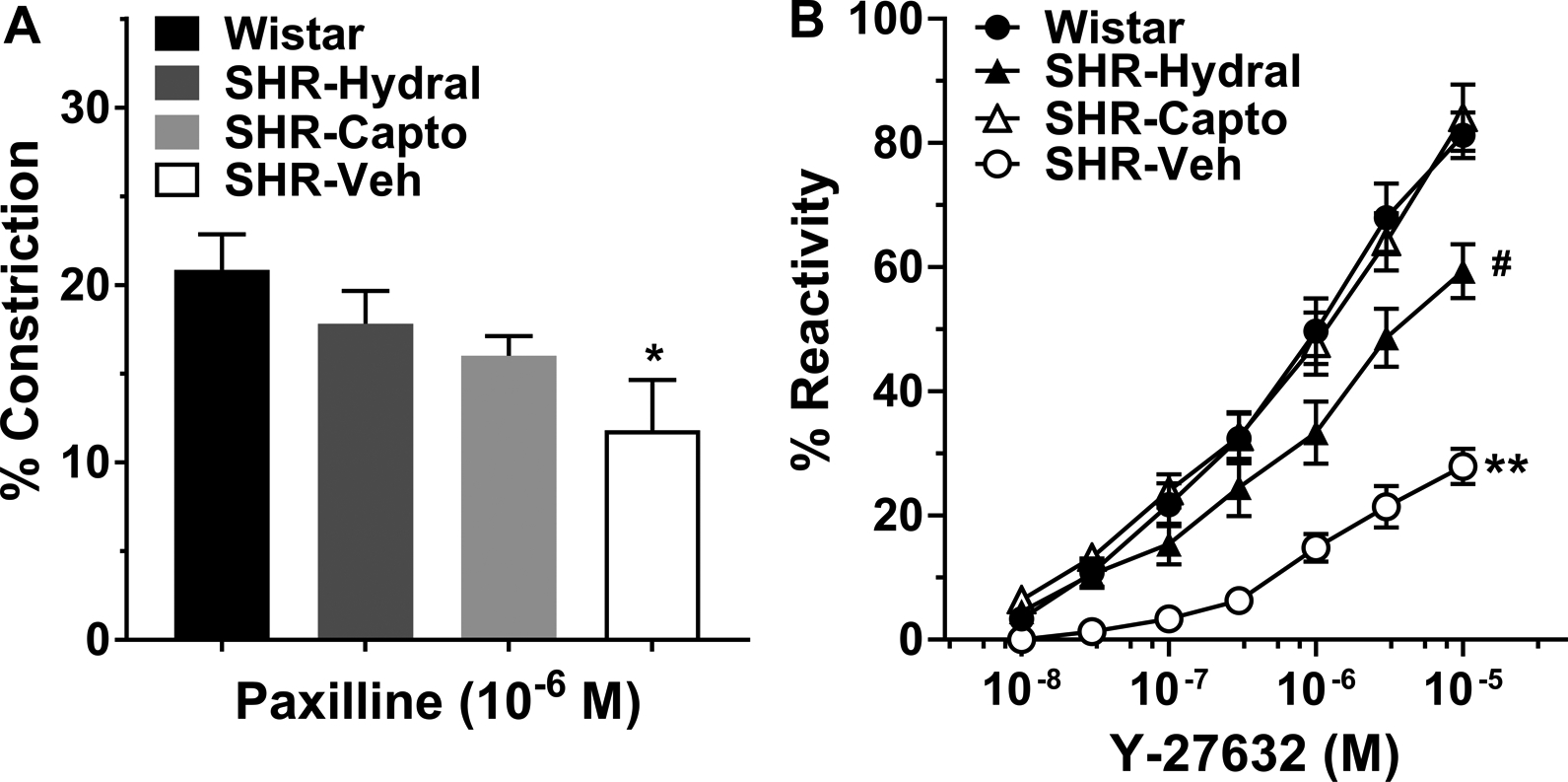
Reactivity of LMAs isolated from Wistar rats, and SHRs with or without treatment. A, Percent constriction to the BK channel inhibitor paxilline (10−6 M) in LMAs isolated from Wistar rats, and SHRs treated with hydralazine, captopril or vehicle. LMAs from SHR-Veh showed significantly less constrictive response compared to LMAs from Wistar rats. *P<0.05 vs. Wistar by one-way ANOVA with post-hoc Bonferroni test for multiple comparisons. B, Percent reactivity of LMAs to increasing concentrations of Y-27632 (10−8-10−5 M), a Rho-kinase inhibitor. The concentration by group interaction was not significant [F(3, 182)=0.74, P>0.05], however, there were significant concentration effect and group effect [F(1, 182)=217.11, F(3, 27)=22.27; P<0.01]. **P<0.01 vs. all groups, #P<0.05 vs. SHR-Capto and Wistar (P=0.056) by mixed model repeated measures ANOVA, square root transformation was used for reactivity outcomes. Wistar: n=8; SHR-Hydral: n=7; SHR-Capto: n=8; SHR-Veh: n=8.
The small G protein Rho and its target Rho-kinase have been shown to contribute to cerebral artery tone via the inhibition of myosin light chain phosphatase, and this effect is augmented in the cerebral circulation during hypertension.28 Thus, we studied if Rho-kinase regulated myogenic tone and if the function of Rho-kinase changes during hypertension. Figure 4B shows the percent reactivity of LMAs to increased concentration of Y-27632, a Rho-kinase inhibitor. LMAs from Wistar rats had 81±4% dilatory response at the maximum concentration (10−5 M); however, the dilatory response was significantly reduced in SHR-Veh (P<0.01). Captopril treatment completely restored the impaired dilatory response compared to Wistar rats. The mean dilatory response of LMAs from SHR-Hydral was larger than SHR-Veh (P<0.01), but less than LMAs from Wistar rats (P=0.057), indicating that hydralazine only partially restored the impaired dilatory response.
Next, we studied effect of hypertension and Ang II on LMA response to NO. Figure 5A shows that LMAs from all groups constricted to NO synthase inhibition with L-NAME that was not different between groups. Figure 5B shows the dilatory response to the NO donor SNP. LMAs from SHR-Veh had significantly impaired dilatory response compared to LMAs from Wistar rats (P<0.01), and this was restored by captopril, but not hydralazine.
Figure 5.
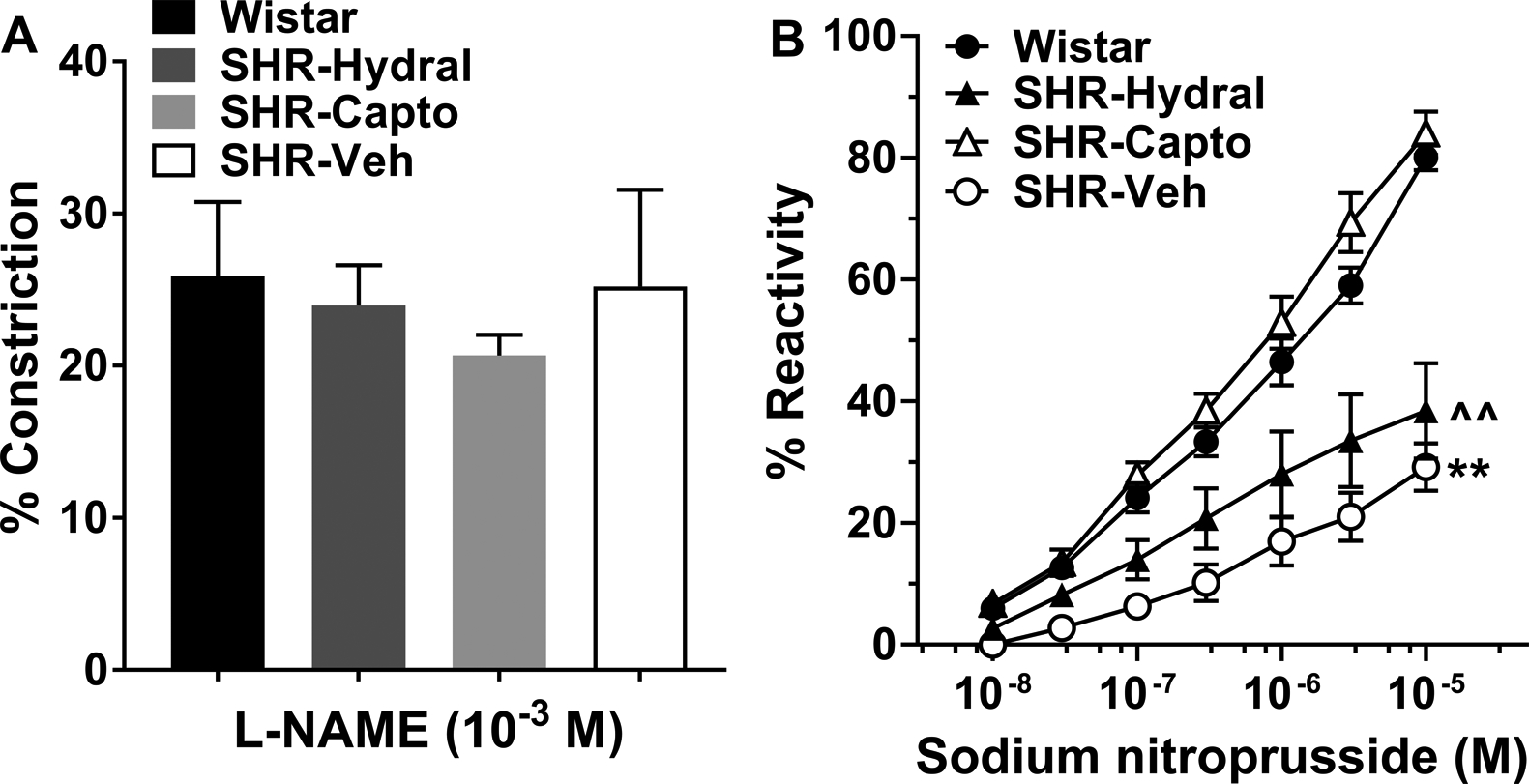
Reactivity of LMAs from Wistar rats and SHRs with or without treatment to NOS inhibition with L-NAME and dilation to SNP. A, Percent constriction to the NO synthase inhibitor L-NAME (10−3 M) in LMAs isolated from Wistar rats, SHRs treated with hydralazine, captopril or vehicle. LMAs from all groups had similar vasoconstriction to L-NAME, data were analyzed by Kruskal-Wallis and Dunn’s test for multiple comparisons. Wistar: n=7; SHR-Hydral: n=6; SHR-Capto: n=7; SHR-Veh: n=8. B, Percent reactivity to the NO donor SNP in LMAs isolated from Wistar rats, and SHRs treated with hydralazine, captopril or vehicle. The concentration by group interaction was not significant [F(3, 170)=1.69, P>0.05], however, there were significant concentration effect and group effect [F(1, 170)=161.05, F(3, 25)=18.32; P<0.01]. **P<0.01 vs. All; ^^P<0.01 vs. Wistar and SHR-Capto by mixed model repeated measures ANOVA. Wistar: n=7; SHR-Hydral: n=6; SHR-Capto: n=8; SHR-Veh: n=8.
AT1R Expression on LMAs
Although LMAs from SHRs responded to ACE inhibitors, it is not known if LMAs expressed AT1R in endothelial or smooth muscle cells. As shown in Figure 6A, positive AT1R staining was detected on LMAs, the co-expression of AT1R to endothelial and vascular smooth muscle cells were also observed in the images. In addition, isolated and perfused LMAs constricted significantly to increased concentration of Ang II (Figure 6B). With the presence of Ang II in the bath, an AT1R blocker candesartan (10−5 M) was added to the bath and caused 22±6% vasodilation. Together, these results indicate the expression of AT1R on both endothelial and smooth muscle cells of LMAs.
Figure 6.
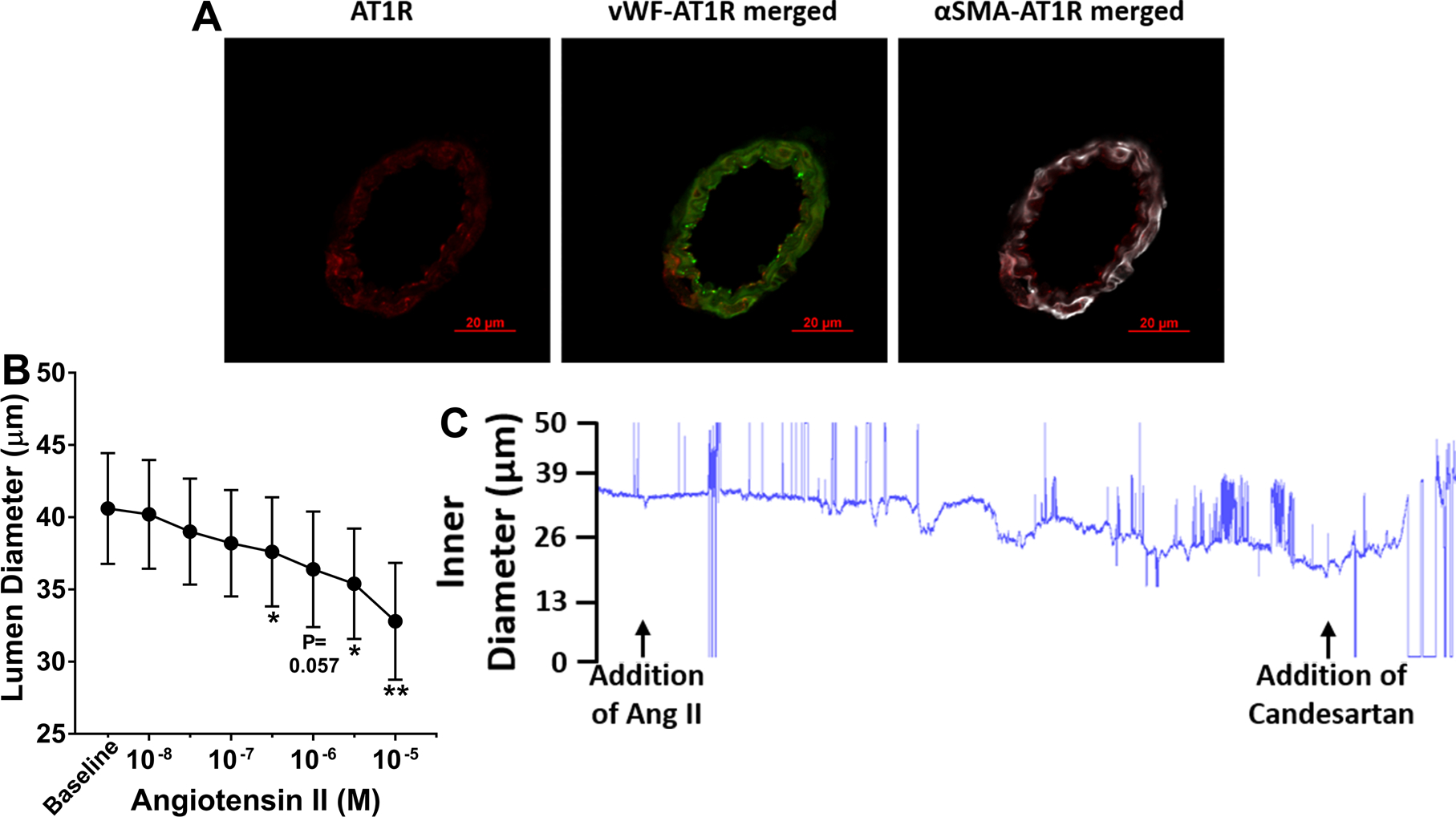
Expression of AT1R and vasoactivity of LMAs to Ang II and captopril. A, Representative immunohistochemical images of cross section of LMAs stained with vWF (green), αSMA (white) and AT1R (Red). AT1R: Ang II type 1 receptor; vWF: von Willebrand factor, an endothelial marker; αSMA: α-smooth muscle actin, a vascular smooth muscle marker. AT1R presented red, vWF was conjugated to ALEXA FLUOR 488 and presented green, αSMA was conjugated to Cy3 and presented white. Scale bar: 20 μm. B, Change in lumen diameter in response to increased concentration of Ang II in LMAs isolated from Wistar rats (n=5). Ang II caused concentration-dependent vasoconstriction. *P<0.05, **P<0.01 vs. Baseline by repeated measures ANOVA with post-hoc Bonferroni correction for multiple comparisons.
Discussion
In the present study, we found that LMAs from hypertensive rats had smaller lumens that was due to a combination of increased myogenic tone and inward remodeling. Though captopril and hydralazine lowered both systolic and diastolic BP in hypertensive rats that was similar to normotensive rats, only captopril treatment normalized the increased myogenic tone of LMAs. Interestingly, captopril did not normalize inward remodeling, but the effect was selective for functional changes. Maximum vasodilatory response of LMAs to Rho-kinase inhibitor was impaired during chronic hypertension that was completely restored by captopril, and only partially by hydralazine treatment. In addition, LMAs from all groups constricted similarly to the NOS inhibition. However, the maximum vasodilation to SNP was impaired in hypertensive rats in comparison to normotensive rats that was reversed only by captopril treatment, not hydralazine. In addition, we showed that AT1R was expressed on both endothelial and vascular smooth muscle cells, suggesting the underlying mechanisms by which ACE inhibition regulated LMA vasoreactivity and structural remodeling was likely via AT1R.
One of the most important considerations when treating acute ischemic stroke patients is to identify the extent of salvageable tissue, i.e. the penumbra. The penumbral tissue receives limited blood supply after onset of stroke to maintain neuronal integrity and thus is only salvageable within a limited time window. McCabe et al. showed that within one hour of stroke, hypertensive rats had significantly more ischemic damage and a smaller penumbra compared to Wistar rats.29 Letourneur et al. also reported that hypertensive rats had significantly smaller penumbra compared to normotensive rats after ischemia which progressed quickly to infarction,30 suggesting that hypertension shortens the duration of ischemic penumbra and thus the time window of therapeutic treatment. Collateral enhancing strategies therefore have been studied to restore blood flow and maintain penumbra during acute ischemia, particularly in patients who are not eligible for endovascular therapies or are outside the therapeutic window for intravenous thrombolysis. Several approaches have been used to increase collateral blood flow during or after ischemia, including induced hypertension, lying-flat head position (0°), partial aortic obstruction diverting splanchnic blood flow to the brain, remote ischemic perconditioning, and sphenopalatine ganglion stimulation.31,32 The efficacy of those approaches may be due to the passive response and larger lumens of LMAs from normotensive animals that provides less vascular resistance and hence increased collateral flow. However, the effect may not be the same in patients with chronic hypertension in which LMAs are vasoconstricted that increases with increased intravascular pressure. Our results show that ACE inhibition could reverse the increased myogenic tone of LMAs and this effect was independent of BP, suggesting that hypertensive patients prescribed with ACE inhibitors or AT1R blockers may respond better to collateral enhancing therapies than those taking other antihypertensive agents. Similarly, inhaled NO and Rho-kinase inhibitor Y-27632 have been shown to increase collateral blood flow during ischemia, reduce infarct size and improve neurologic outcome in mouse models.33,34 However, our results demonstrate the impaired dilatory responses of LMAs to SNP and Y-27632 were completely reversed by ACE inhibition treatment, again indicating that hypertensive patients with ACE inhibitors and AT1R blockers may benefit more from those therapies.
Though the cause of hypertension varies, it is well-established that chronic hypertension is associated with cerebral vascular wall hypertrophy and reduced vasodilatory responses.35 Chronic hypertension has a number of effects on the cerebral circulation, including increased stiffness of the large cerebral pial vessels, making them more resistant to passive dilation.36 Consistent with these findings, our study showed that MCAs from SHR-Veh had significantly smaller passive inner and outer diameters, and increased wall thickness compared to Wistar normotensive rats. Additionally, neither hydralazine nor captopril treatment changed the structural remodeling. In contrast, LMAs from SHR-Veh showed smaller passive inner and outer diameters but wall thickness remained similar compared to LMAs from Wistar rats. Interestingly, hydralazine not captopril treatment reversed the structural remodeling. LMAs have unique hemodynamics. Under normal physiological conditions, LMAs are relatively passive compared to similar-sized pial arterioles that do not anastomose and LMAs dilate in response to increased intravascular pressure.8 However, in the current study we showed that LMAs from hypertensive rats developed considerable myogenic tone and constricted significantly to increased pressure, an effect that could limit retrograde blood flow during an occlusion. Consistent with our findings in vitro, studies have reported poor collateral recruitment and flow during permanent MCAO in stroke-prone SHR, that was associated with less penumbra and significantly more ischemic damage compared to normotensive rats.29,37 Our lab also showed that selectively increasing pial collateral flow using pharmacological agents can decrease infarction in SHRs.23
During chronic hypertension, Ang II has emerged as a critical factor in the detrimental cerebrovascular effects, including remodeling, shifting the CBF autoregulatory curve, promoting vascular inflammation and oxidative stress, and inhibiting endothelium-dependent relaxation.38–42 Thus, interventions targeting the renin-angiotensin system are predicted to have substantial influence on cerebral circulation and BP and reverse the deleterious cerebrovascular effects induced by Ang II. The ACE inhibitor captopril was the first drug shown to maintain CBF while reducing both the lower and upper limits of autoregulation of hypertensive rats,43 and increasing lumen diameter of large arteries.44 More interestingly, captopril preserved collateral blood supply in the lower limb of hypertensive patients via the lack of Ang II-induced vasoconstriction or reducing breakdown of bradykinin.45 In addition, ACE inhibitors have been shown to increase regional CBF in the frontoparietal regions46 and normalize CBF reserve via decreasing thickness of medial layer of carotid artery, MCAs and larger pial arteries.47 These results indicate that ACE inhibition provides beneficial effects on the cerebral circulation that are particularly important during ischemic conditions. Our current study adds to the knowledge of the effects of Ang II on LMA function and reactivity during chronic hypertension.
The beneficial effects of antihypertensive medications on collateral flow were recently confirmed by Fujita et al. who reported a significantly positive association between chronic hypertension and poor collateral status in patients with acute ischemic stroke.48 The proportion of patients with poor collateral status increased in a stepwise manner in patients without chronic hypertension having the best collateral flow, hypertensive patients without antihypertensive medications having the worst, and hypertensive patients treated with antihypertensive medications were in between. Though the antihypertensive agents also included calcium channel blockers and β blockers that are Ang II-independent, 60% of patients had ACE inhibitors or AT1R blockers and rank as the most commonly prescribed antihypertensive agents. Similar to animal studies, this clinical study demonstrated that chronic hypertension has a detrimental effect on pial collateral flow in patients with large-vessel occlusion. This is consistent with our current findings of increased vasoconstriction and impaired dilatory capacity of LMAs during chronic hypertension. For the preexisting LMAs to be fully recruited and function as collateral vessels during ischemia, the improvement of vasodilatory responses involving both endothelial and vascular smooth muscle cells are critical. Thus, the goal of daily BP control may have gone beyond what has been addressed so far, which is not only to reduce the increased risks of cardiovascular and cerebrovascular morbidity and mortality that are associated with elevated BP, but also to prepare the brain with healthy pial collaterals.
One interesting finding in the current study was that LMA vasodilatory response to Y-27632 was significantly impaired from SHRs compared to normotensive Wistar rats. In contrast, Chrissobolis et al. showed that Y-27632 caused significantly greater cerebral vasodilatory response in basilar artery from hypertensive rats compared to normotensive rats.28 Enhanced vasoconstrictive responses can occur via multiple mechanisms, including Ca2+ sensitization, increased phosphorylation of myosin light chain kinase by protein kinase C,49 decreased myosin phosphatase activity and increased Rho-associated kinase activity.50 Raina et al. showed that Y-27632 caused increased Ca2+ sensitization; however, had little effect on intracellular Ca2+ or myosin light chain kinase activation in mouse mesenteric arteries.51 Thus, decreased LMA vasodilation to Rho kinase inhibition might be relevant to modified Ca2+ sensitization during chronic hypertension. Additionally, LMAs are unique pial vessels and the regulation of Rho kinase during chronic hypertension might be distinct from other cerebral vessels. Our study also has several limitations. First, we did not study the presence or expression of AT2R on LMAs that could have been involved in vascular remodeling. AT2R is responsible for mediating Ang II-induced vasodilation in cerebral pial arterioles52 and cerebral parenchymal vessels.53 However, due to the lack of evidence that AT2R expression is affected by captopril treatment in SHRs,54 we did not study AT2R expression on LMAs. Second, the AT1R antibody used lacks verification of specificity and was not tested in AT1R knockout tissue. Thus, we confirmed AT1R presence using isolated LMAs and showed Ang II treatment induced vasoconstriction.
In summary, LMAs from hypertensive animals had considerable myogenic tone and impaired vasodilatory responses that were Ang II-dependent. Chronic ACE inhibition completely reversed the increased vasoconstriction and impaired vasodilation, suggesting the restored and improved collateral circulation in the presence of occlusion. These results suggest that chronic ACE inhibition may be beneficial to collateral flow when stroke occurs in the setting of chronic hypertension.
Perspectives
Our results show that the increased vasoconstriction and impaired vasodilation of LMAs are Ang II-dependent and impaired LMA function during chronic hypertension can be ameliorated with ACE inhibitor treatment. Thus ACE inhibitors or AT1R blockers should be preferable to other Ang II-independent antihypertensive agents, such as hydralazine or calcium channel blockers to improve cerebrovascular health. It was recently reported that stroke patients with good pial collaterals were associated with reduced ischemic core growth but not improved neurologic outcome, and that pial collateral status did not impact the treatment effect of endovascular thrombectomy.55 This suggests that LMA function may be impacted by ischemia and reperfusion over time. Therefore, further studies are required to understand LMA function and structure changes after ischemia and reperfusion, as well as after reperfusion therapies.
Supplementary Material
Novelty and Significance.
What Is New?
This study demonstrates that increased vasoconstriction and impaired vasodilation of pial collaterals during chronic hypertension are Ang II-dependent.
What Is Relevant?
Our results suggest that ACE inhibition during chronic hypertension restores hyperconstriction and vascular dysfunction of pial collaterals that could improve stroke outcome by increasing collateral perfusion. This study contributes to the ongoing efforts to understand collateral function with the intent to improve outcome from acute ischemic stroke.
Acknowledgements
We thank the Microscopy Imaging Center, Larner College of Medicine, University of Vermont for help with immunohistochemical staining of vessel images. We also want to thank biostatistician Dr. Mike DeSarno from Biomedical Statistics Research Core at University of Vermont, for his tremendous help and support on statistical analyses.
Sources of Funding
This work was generously supported by National Institutes of Health, National Institute of Neurologic Disorders and Stroke grant R01 NS093289, and National Center for Research Resources grant # 1S10OD025030-01, the Cardiovascular Research Institute of Vermont, the Totman Medical Research Trust, and American Heart Association grant # 19PRE34430175/Zhaojin Li/2019-2020.
Footnotes
Disclosures
None.
Summary
This study explains the underlying mechanisms of hyperconstriction and vascular dysfunction of LMAs during hypertension. Additionally, lowering BP alone without inhibiting Ang II may not provide same protective effects of pial collaterals.
References
- 1.Benjamin EJ, Muntner P, Bittencourt MS. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139(10):e56–e528. doi: 10.1161/CIR.0000000000000659 [DOI] [PubMed] [Google Scholar]
- 2.Pan J, Li X, Peng Y. Remote ischemic conditioning for acute ischemic stroke: dawn in the darkness. Reviews in the Neurosciences. 2016;27(5):501–510. doi: 10.1515/revneuro-2015-0043 [DOI] [PubMed] [Google Scholar]
- 3.Kaufmann AM, Firlik AD, Fukui MB, Wechsler LR, Jungries CA, Yonas H. Ischemic core and penumbra in human stroke. Stroke. 1999;30(1):93–99. doi: 10.1161/01.STR.30.1.93 [DOI] [PubMed] [Google Scholar]
- 4.Astrup J, Siesjö BK, Symon L. Thresholds in cerebral ischemia-the ischemic penumbra. Stroke. 1981;12(6):723–725. doi: 10.1161/01.STR.12.6.723 [DOI] [PubMed] [Google Scholar]
- 5.Brozici M, Van Der Zwan A, Hillen B. Anatomy and functionality of leptomeningeal anastomoses: a review. Stroke. 2003;34(11):2750–2762. doi: 10.1161/01.STR.0000095791.85737.65 [DOI] [PubMed] [Google Scholar]
- 6.Liebeskind DS, Jahan R, Nogueira RG, Zaidat OO, Saver JL. Impact of collaterals on successful revascularization in Solitaire FR with the intention for thrombectomy. Stroke. 2014;45(7):2036–2040. doi: 10.1161/STROKEAHA.114.004781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beard DJ, McLeod DD, Logan CL, et al. Intracranial pressure elevation reduces flow through collateral vessels and the penetrating arterioles they supply. A possible explanation for ‘collateral failure’and infarct expansion after ischemic stroke. Journal of Cerebral Blood Flow & Metabolism. 2015;35(5):861–872. doi: 10.1038/jcbfm.2015.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan S-L, Sweet JG, Bishop N, Cipolla MJ. Pial collateral reactivity during hypertension and aging: understanding the function of collaterals for stroke therapy. Stroke. 2016;47(6):1618–1625. doi: 10.1161/STROKEAHA.116.013392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang H, Chalothorn D, Faber JE. Collateral Vessels Have Unique Endothelial and Smooth Muscle Cell Phenotypes. International journal of molecular sciences. 2019;20(15):3608. doi: 10.3390/ijms20153608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’donnell MJ, Xavier D, Liu L, et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. The Lancet. 2010;376(9735):112–123. doi: 10.1016/S0140-6736(10)60834-3 [DOI] [PubMed] [Google Scholar]
- 11.Kjeldsen SE. Hypertension and cardiovascular risk: general aspects. Pharmacological Research. 2018;129:95–99. doi: 10.1016/j.phrs.2017.11.003 [DOI] [PubMed] [Google Scholar]
- 12.Folkow B The pathophysiology of hypertension. Drugs. 1993;46(2):3–7. doi: 10.2165/00003495-199300462-00003 [DOI] [PubMed] [Google Scholar]
- 13.Veerasingham SJ, Raizada MK. Brain renin–angiotensin system dysfunction in hypertension: recent advances and perspectives. British journal of pharmacology. 2003;139(2):191–202. doi: 10.1038/sj.bjp.0705262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sowers JR. Hypertension, angiotensin II, and oxidative stress. Mass Medical Soc; 2002. doi: 10.1056/NEJMe020054 [DOI] [PubMed] [Google Scholar]
- 15.Dzau VJ. Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension. 2001;37(4):1047–1052. doi: 10.1161/01.HYP.37.4.1047 [DOI] [PubMed] [Google Scholar]
- 16.Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension. 2001;38(3):581–587. doi: 10.1161/hy09t1.096249 [DOI] [PubMed] [Google Scholar]
- 17.Itoh T, Kajikuri J, Tada T, Suzuki Y, Mabuchi Y. Angiotensin II-induced modulation of endothelium-dependent relaxation in rabbit mesenteric resistance arteries. The Journal of physiology. 2003;548(3):893–906. doi: 10.1113/jphysiol.2002.034116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Didion SP, Faraci FM. Angiotensin II produces superoxide-mediated impairment of endothelial function in cerebral arterioles. Stroke. 2003;34(8):2038–2042. doi: 10.1161/01.STR.0000081225.46324.AA [DOI] [PubMed] [Google Scholar]
- 19.Nishimura Y, Ito T, Saavedra JM. Angiotensin II AT1 blockade normalizes cerebrovascular autoregulation and reduces cerebral ischemia in spontaneously hypertensive rats. Stroke. 2000;31(10):2478–2486. doi: 10.1161/01.str.31.10.2478 [DOI] [PubMed] [Google Scholar]
- 20.Ito T, Nishimura Y, Saavedra J. Pre-treatment with candesartan protects from cerebral ischaemia. Journal of the Renin-Angiotensin-Aldosterone System. 2001;2(3):174–179. doi: 10.3317/jraas.2001.024 [DOI] [PubMed] [Google Scholar]
- 21.Forder JP, Munzenmaier DH, Greene AS. Angiogenic protection from focal ischemia with angiotensin II type 1 receptor blockade in the rat. American Journal of Physiology-Heart and Circulatory Physiology. 2005;288(4):H1989–H1996. doi: 10.1152/ajpheart.00839.2004 [DOI] [PubMed] [Google Scholar]
- 22.Kumai Y, Ooboshi H, Ago T, et al. Protective effects of angiotensin II type 1 receptor blocker on cerebral circulation independent of blood pressure. Experimental neurology. 2008;210(2):441–448. doi: 10.1016/j.expneurol.2007.11.028 [DOI] [PubMed] [Google Scholar]
- 23.Cipolla MJ, Linfante I, Abuchowski A, Jubin R, Chan S-L. Pharmacologically increasing collateral perfusion during acute stroke using a carboxyhemoglobin gas transfer agent (Sanguinate™) in spontaneously hypertensive rats. Journal of Cerebral Blood Flow & Metabolism. 2018;38(5):755–766. doi: 10.1177/0271678X17705567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Z, Tremble SM, Cipolla MJ. Implications for understanding ischemic stroke as a sexually dimorphic disease: the role of pial collateral circulations. American Journal of Physiology-Heart and Circulatory Physiology. 2018;315(6):H1703–H1712. doi: 10.1152/ajpheart.00402.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daugherty A, Rateri DL, Hong L, Balakrishnan A. Measuring blood pressure in mice using volume pressure recording, a tail-cuff method. Journal of visualized experiments: JoVE. 2009. doi: 10.3791/1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cocks T, Kemp B, Pruneau D, Angus J. Comparison of contractile responses to 5-hydroxytryptamine and sumatriptan in human isolated coronary artery: synergy with the thromboxane A2-receptor agonist, U46619. British journal of pharmacology. 1993;110(1):360–368. doi: 10.1111/j.1476-5381.1993.tb13818.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256(5056):532–535. doi: 10.1126/science.1373909 [DOI] [PubMed] [Google Scholar]
- 28.Chrissobolis S, Sobey CG. Evidence that Rho-kinase activity contributes to cerebral vascular tone in vivo and is enhanced during chronic hypertension: comparison with protein kinase C. Circulation research. 2001;88(8):774–779. doi: 10.1161/hh0801.090441 [DOI] [PubMed] [Google Scholar]
- 29.McCabe C, Gallagher L, Gsell W, Graham D, Dominiczak AF, Macrae IM. Differences in the evolution of the ischemic penumbra in stroke-prone spontaneously hypertensive and Wistar-Kyoto rats. Stroke. 2009;40(12):3864–3868. doi: 10.1161/STROKEAHA.109.559021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Letourneur A, Roussel S, Toutain J, Bernaudin M, Touzani O. Impact of genetic and renovascular chronic arterial hypertension on the acute spatiotemporal evolution of the ischemic penumbra: a sequential study with MRI in the rat. Journal of Cerebral Blood Flow & Metabolism. 2011;31(2):504–513. doi: 10.1038/jcbfm.2010.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma J, Ma Y, Dong B, Bandet MV, Shuaib A, Winship IR. Prevention of the collapse of pial collaterals by remote ischemic perconditioning during acute ischemic stroke. Journal of Cerebral Blood Flow & Metabolism. 2017;37(8):3001–3014. doi: 10.1177/0271678X16680636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bang OY, Goyal M, Liebeskind DS. Collateral circulation in ischemic stroke: assessment tools and therapeutic strategies. Stroke. 2015;46(11):3302–3309. doi: 10.1161/STROKEAHA.115.010508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terpolilli NA, Kim S-W, Thal SC, et al. Inhalation of nitric oxide prevents ischemic brain damage in experimental stroke by selective dilatation of collateral arterioles. Circulation research. 2012;110(5):727–738. doi: 10.1161/CIRCRESAHA.111.253419 [DOI] [PubMed] [Google Scholar]
- 34.Rikitake Y, Kim H-H, Huang Z, et al. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke. 2005;36(10):2251–2257. doi: 10.1161/01.STR.0000181077.84981.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iadecola C, Davisson RL. Hypertension and cerebrovascular dysfunction. Cell metabolism. 2008;7(6):476–484. doi: 10.1016/j.cmet.2008.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Izzard AS, Horton S, Heerkens EH, Shaw L, Heagerty AM. Middle cerebral artery structure and distensibility during developing and established phases of hypertension in the spontaneously hypertensive rat. Journal of hypertension. 2006;24(5):875–880. doi: 10.1097/01.hjh.0000222757.54111.06 [DOI] [PubMed] [Google Scholar]
- 37.Biose IJ, Dewar D, Macrae IM, McCabe C. Impact of stroke co-morbidities on cortical collateral flow following ischaemic stroke. Journal of Cerebral Blood Flow & Metabolism. 2019:0271678X19858532. doi: 10.1177/0271678X19858532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang M, Mao Y, Ramirez S, Tuma R, Chabrashvili T. Angiotensin II induced cerebral microvascular inflammation and increased blood–brain barrier permeability via oxidative stress. Neuroscience. 2010;171(3):852–858. doi: 10.1016/j.neuroscience.2010.09.029 [DOI] [PubMed] [Google Scholar]
- 39.Ando H, Zhou J, Macova M, Imboden H, Saavedra JM. Angiotensin II AT1 receptor blockade reverses pathological hypertrophy and inflammation in brain microvessels of spontaneously hypertensive rats. Stroke. 2004;35(7):1726–1731. doi: 10.1161/01.STR.0000129788.26346.18 [DOI] [PubMed] [Google Scholar]
- 40.Saavedra JM, Nishimura Y. Angiotensin and cerebral blood flow. Cellular and molecular neurobiology. 1999;19(5):553–573. doi: 10.1023/a:1006995016403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Didion SP, Sigmund CD, Faraci FM. Impaired endothelial function in transgenic mice expressing both human renin and human angiotensinogen. Stroke. 2000;31(3):760–764. doi: 10.1161/01.STR.31.3.760 [DOI] [PubMed] [Google Scholar]
- 42.Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through nox-2–derived radicals. Arteriosclerosis, thrombosis, and vascular biology. 2006;26(4):826–832. doi: 10.1161/01.ATV.0000205849.22807.6e [DOI] [PubMed] [Google Scholar]
- 43.Barry DI, Jarden JO, Paulson OB, Graham DI, Strandgaard S. Cerebrovascular aspects of converting-enzyme inhibition I: Effects of intravenous captopril in spontaneously hypertensive and normotensive rats. Journal of hypertension. 1984;2(6):589–597. doi: 10.1097/00004872-198412000-00003 [DOI] [PubMed] [Google Scholar]
- 44.Simon AC, Levenson J, Bouthier J, Safar M. Captopril-induced changes in large arteries in essential hypertension. The American journal of medicine. 1984;76(5):71–75. doi: 10.1016/0002-9343(84)90888-X [DOI] [PubMed] [Google Scholar]
- 45.Roberts D, Mcloughlin G, Tsao Y, Breckenridge A. Placebo-controlled comparison of captopril, atenolol, labetalol, and pindolol in hypertension complicated by intermittent claudication. The Lancet. 1987;330(8560):650–653. doi: 10.1016/s0140-6736(87)92441-x [DOI] [PubMed] [Google Scholar]
- 46.Kobayashi S, Yamaguchi S, Okada K, Suyama N, Bokura K, Murao M. The effect of enalapril maleate on cerebral blood flow in chronic cerebral infarction. Angiology. 1992;43(5):378–388. doi: 10.1177/000331979204300502 [DOI] [PubMed] [Google Scholar]
- 47.Clozel J-P, Kuhn H, Hefti F. Effects of cilazapril on the cerebral circulation in spontaneously hypertensive rats. Hypertension. 1989;14(6):645–651. doi: 10.1161/01.hyp.14.6.645 [DOI] [PubMed] [Google Scholar]
- 48.Fujita K, Tanaka K, Yamagami H, et al. Detrimental Effect of Chronic Hypertension on Leptomeningeal Collateral Flow in Acute Ischemic Stroke. Stroke. 2019:STROKEAHA. 119.025142. doi: 10.1161/STROKEAHA.119.025142 [DOI] [PubMed] [Google Scholar]
- 49.Venema RC, Raynor R, Noland T Jr, Kuo J. Role of protein kinase C in the phosphorylation of cardiac myosin light chain 2. Biochemical Journal. 1993;294(2):401–406. doi: 10.1042/bj2940401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimura K, Ito M, Amano M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 1996;273(5272):245–248. doi: 10.1126/science.273.5272.245 [DOI] [PubMed] [Google Scholar]
- 51.Raina H, Zacharia J, Li M, Wier W. Activation by Ca2+/calmodulin of an exogenous myosin light chain kinase in mouse arteries. The Journal of physiology. 2009;587(11):2599–2612. doi: 10.1113/jphysiol.2008.165258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vincent J-M, Kwan YW, Lung Chan S, Perrin-Sarrado C, Atkinson J, Chillon J-M. Constrictor and dilator effects of angiotensin II on cerebral arterioles. Stroke. 2005;36(12):2691–2695. doi: 10.1161/01.STR.0000190002.79052.bf [DOI] [PubMed] [Google Scholar]
- 53.Takao M, Kobari M, Tanahashi N, et al. Dilatation of cerebral parenchymal vessels mediated by angiotensin type 1 receptor in cats. Neuroscience letters. 2002;318(2):108–112. doi: 10.1016/S0304-3940(01)02493-4 [DOI] [PubMed] [Google Scholar]
- 54.Brouwers S, Smolders I, Massie A, Dupont AG. Angiotensin II Type 2 Receptor–Mediated and Nitric Oxide–Dependent Renal Vasodilator Response to Compound 21 Unmasked by Angiotensin-Converting Enzyme Inhibition in Spontaneously Hypertensive Rats In Vivo. Hypertension. 2013;62(5):920–926. doi: 10.1161/HYPERTENSIONAHA.112.00762 [DOI] [PubMed] [Google Scholar]
- 55.de Havenon A, Mlynash M, Kim-Tenser MA, et al. Results from DEFUSE 3: good collaterals are associated with reduced ischemic core growth but not neurologic outcome. Stroke. 2019;50(3):632–638. doi: 10.1161/STROKEAHA.118.023407 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.