

Abstract
Endothelial inflammation and mitochondrial dysfunction have been implicated in cardiovascular diseases, yet a unifying mechanism tying them together remains limited. Mitochondrial dysfunction is frequently associated with mitochondrial fission/fragmentation mediated by the GTPase dynamin-related protein 1 (Drp1). Nuclear factor (NF)-κB, a master regulator of inflammation, is implicated in endothelial dysfunction and resultant complications. Here, we explore a causal relationship between mitochondrial fission and NF-κB activation in endothelial inflammatory responses. In cultured endothelial cells, tumor necrosis factor-α or lipopolysaccharide induces mitochondrial fragmentation. Inhibition of Drp1 activity or expression suppresses mitochondrial fission, NF-κB activation, vascular cell adhesion molecule-1 induction, and leukocyte adhesion induced by these pro-inflammatory factors. Moreover, attenuations of inflammatory leukocyte adhesion were observed in Drp1 hetero-deficient mice as well as endothelial Drp1 silenced mice. Intriguingly, inhibition of the canonical NF-κB signaling suppresses endothelial mitochondrial fission. Mechanistically, NF-κB p65/RelA appears to mediate inflammatory mitochondrial fission in endothelial cells. In addition, the classical anti-inflammatory drug, salicylate, appears to maintain mitochondrial fission/fusion balance against tumor necrosis factor-α via inhibition of NF-κB. In conclusion, our results suggest a previously unknown mechanism whereby the canonical NF-κB cascade and a mitochondrial fission pathway interdependently regulate endothelial inflammation.
Keywords: Cell Signaling, Endothelium, Inflammation, Mitochondria, Nuclear Factor-κB, Basic Science Research
summary
We found that genetic reduction of Drp1 or pharmacological inhibition attenuated endothelial mitochondrial fission and inflammation in vitro as well as in mouse models. Inhibition of NF-kB also reduced endothelial mitochondrial fission in vitro and in mice with salicylate. Further investigation is needed to evaluate the translational potential of intervening in this pathway as a means to prevent inflammatory cardiovascular diseases.
Graphical Abstract
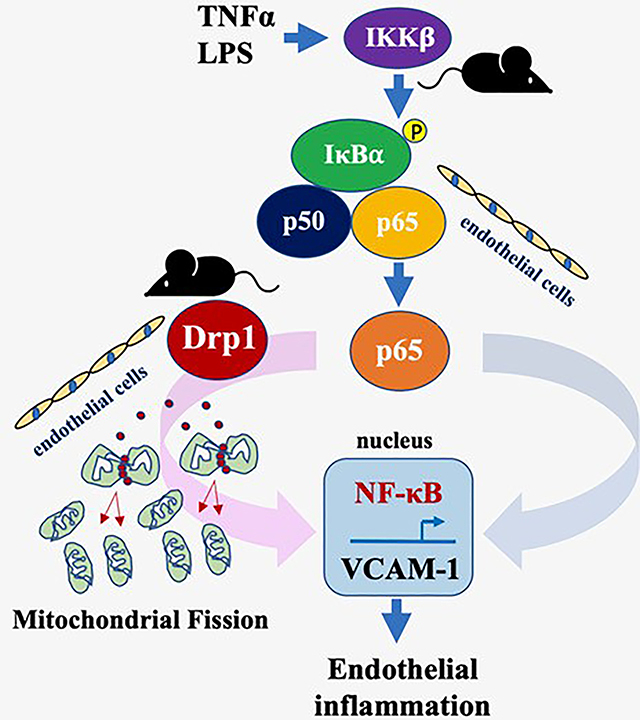
Introduction
Mitochondria are subcellular organelles that play a critical role in cellular metabolism in mammalian physiology. As part of their dynamic nature, mitochondria undergo continuous fission and fusion mainly mediated by dynamin-related protein 1 (Drp1) and mitofusins (Mfn1/2), respectively, in order to maintain mitochondrial quality and cellular homeostasis 1. Disruptions in the fission/fusion balance (primarily a shift toward fission) perturb cellular physiology and have been implicated in a variety of diseases including those seen in the cardiovascular system 2. Accordingly, various stressors such as high glucose, hypoxia and oxidative stress have been shown to induce Drp1-dependent mitochondrial fission in endothelial cells and mediate endothelial pathologies including impairment of endothelium-dependent relaxation, reduction in micro-vessels and defects in wound healing and angiogenesis 3–5. However, the mechanistic contribution of Drp1 as well as mitochondrial fission in endothelial inflammation has not been fully explored.
Endothelial dysfunction is an early independent risk factor for cardiovascular diseases such as hypertension and coronary artery disease. Endothelial inflammation and oxidative stress are the major characteristics associated with endothelial dysfunction 6. While the evolutionarily conserved nuclear factor-κB (NF-κB) pathway is indispensable to maintaining cellular homeostasis in response to transient stress 7, chronic NF-κB activation appears to mediate vascular inflammation and exaggerate endothelial dysfunction 8. Canonical signaling leading to NF-κB-dependent gene induction involves an upstream kinase, IκB kinase beta (IKKβ), and its substrate, IκBα. Phosphorylation and subsequent degradation of IκBα releases NF-κB to translocate to the nucleus where it induces genes involved in inflammation, immune responses, and tissue remodeling 7. Of importance to NF-κB-dependent cell adhesion molecules, vascular cell adhesion molecule-1 (VCAM-1) appears critical in vascular inflammation 9.
Among several cytokines implicated in endothelial inflammation, tumor necrosis factor-α (TNF-α) appears to be critical to induce endothelial dysfunction and expression of cell adhesion molecules and chemokines via NF-κB activation, thus contributing to leukocyte recruitment 10. While activation of the toll like receptors (TLRs) involved in the innate immune system are the first line of host defense mechanism in mammals, chronic activation of the endothelial TLRs, which are activated by microbial endotoxins including lipopolysaccharide (LPS) as well as damage-associated molecular pattern molecules, has also been implicated in NF-κB-driven vascular inflammation 11.
Given the association between vascular inflammation and perturbations in mitochondrial homeostasis in human and animal models of cardiovascular diseases 12, 13, we aimed to study the potential mechanisms that tie the mitochondrial fission signaling to endothelial inflammation such as those induced by TNF-α and LPS.
Methods
This article adheres to the American Heart Association Journals implementation of the Transparency and Openness Promotion Guidelines. The data that support the findings of this study are available from the corresponding author on reasonable request. Detailed methods and materials are available in the online-only Data Supplement and Table S1.
Results
TNF-α induces mitochondrial fission in endothelial cells via Drp1
In primary rat aortic endothelial cells (ECs), we observed a time-dependent (1 hour~6 hours) increase in mitochondrial fragmentation in response to TNF-α with maximum responses occurring at 3 hours (Figure 1A). Drp1 is a critical positive regulator of mitochondrial fission in mammalian cells 14. Genetic inhibition of Drp1 using adenoviruses encoding the GTPase deficient dominant-negative Drp1K38A mutant 15 (Figure 1B) or Drp1 small interfering RNA (siRNA), siDrp1, (Figure 1C) suppressed TNF-α-induced mitochondrial fission in cultured ECs. Pharmacologic inhibition of mitochondrial fission using mdivi1 16 also suppressed TNF-α-induced mitochondrial fragmentation (Figure 1D). Drp1 Ser616 phosphorylation is known to enhance Drp1 activity and mitochondrial fission. In contrast, Drp1 Ser637 phosphorylation is known to be inhibitory 17. A prior study reported that TNF-α increased both total and Ser616 phosphorylated Drp1 in H9C2 cells 18. TNF-α also down-regulated Mfn2 in 3T3 cells 19. In cultured ECs, TNF-α transiently down-regulated expression of IκBα. However, TNF-α did not alter Drp1 expression, Drp1 phosphorylation or Mfn2 expression in ECs (Figures S1), suggesting that the mechanism of Drp1 activation and fission induced by TNF-α may be cell type specific.
Figure 1. Mitochondrial fission inducer Drp1 mediates TNF-α)-induced mitochondrial fragmentation in endothelial cells.

A. Rat aortic ECs were stimulated with 10 ng/mL rat TNF-α for indicated times and mitochondrial fragmentation count (MFC) was measured. B. Rat aortic ECs transduced with adenovirus encoding dominant-negative Drp1K38A mutant or control GFP at 100 multiplicity of infection (moi) for 48 hours were stimulated with 10 ng/mL TNF-α for 3 hours and MFC was measured. C. Rat aortic ECs transduced with adenovirus encoding siDrp1 or control non-silencing RNA (siCon) at 100 moi for 48 hours were stimulated with 10 ng/mL TNF-α for 3 hours and MFC was measured. D. Rat aortic ECs pretreated with 50 μmol/L mitochondrial division inhibitor-1 (mdivi1) or vehicle (0.1% DMSO) for 1 hour were stimulated with 10 ng/mL TNF-α (TNF) for 3 hours and MFC was measured. con: basal control. The bars show the mean±SEM from 3~4 independent experiments as indicated. Scale bar is 15 μm and 4x zoomed pictures are included. **p<0.01, ***p<0.001, ****p<0.0001 (1-way ANOVA).
Drp1 and its mitochondrial receptor Mff mediate inflammatory NF-κB activation and VCAM-1 induction in endothelial cells
NF-κB is a master regulator of inflammation 7. ECs transduced with Drp1K38A showed significant inhibition in TNF-α-induced NF-κB-driven promoter activity (Figure 2A). Accordingly, we aimed to test the potential contribution of mitochondrial fission in the inflammatory VCAM-1 induction. TNF-α induced VCAM-1 induction in ECs. This VCAM-1 induction was attenuated by overexpression of Drp1K38A (Figure 2B) or silencing of Drp1 (Figure 2C). In contrast, silencing of Drp1 was unable to block TNF-α-induced p65 nuclear translocation in ECs (Figure 2D).
Figure 2. Drp1 mediates TNF-α-induced NF-κB activation and VCAM-1 induction in endothelial cells.

A. Rat aortic ECs transduced with adenovirus encoding dominant-negative Drp1K38A mutant or control GFP at 100 moi for 48 hours were stimulated with 10 ng/mL TNF-α for 24 hours and NF-κB luciferase activity (fold) was measured. B. Rat aortic ECs transduced with adenovirus encoding dominant-negative Drp1K38A mutant or control GFP at 100 moi for 48 hours were stimulated with 10 ng/mL TNF-α (TNF) for 6 hours and VCAM-1 expression was measured. con: basal control. e: endogenous Drp1, m: mutant Drp1. For quantification, densitometry analysis was performed and normalized with corresponding GAPDH density. C. Rat aortic ECs transduced with adenovirus encoding siDrp1 or control non-silencing RNA (siCon) at 100 moi for 48 hours were stimulated with 10 ng/mL TNF-α for 6 hours and VCAM-1 expression was measured. The bars show the mean±SEM from 3~4 independent experiments as indicated. *p<0.05, ****p<0.0001 (1-way ANOVA). D. Rat aortic ECs transduced with adenovirus encoding siDrp1 or control non-silencing RNA (siCon) at 100 moi for 48 hours were stimulated with 10 ng/mL TNF-α for 20 min and p65 nuclear translocation was measured.
Drp1 mediates mitochondrial fission via its mitochondrial outer membrane receptors including mitochondria fission factor (Mff) 14. Over-expression of dominant negative Mff serine 155/172 alanine substitution mutant (MffS155/172A) 20 or silencing Mff expression suppressed TNF-α-induced mitochondrial fragmentation (Figures S2A and S2B). Dominant negative Mff attenuated TNF-α-induced NF-κB promoter activation (Figures S2C) and VCAM-1 induction (Figures S2D) in ECs. Silencing of Mff also attenuated TNF-α-induced VCAM-1 induction (Figures S2E).
TLR activation by LPS induces endothelial VCAM-1 via NF-κB activation 21. Accordingly, we have further tested if Drp1 and Mff are required for LPS responses in cultured ECs. Transduction of Drp1K38A or MffS155/172A attenuated LPS-induced mitochondrial fragmentation and VCAM-1 induction in ECs (Figures S3).
Drp1 inhibition attenuates monocyte adhesion and mitochondrial ROS production induced by TNF-α in endothelial cells
Endothelial NF-κB activation and VCAM-1 expression are causative factors in leukocyte adhesion and vascular inflammation 21, 22. In response to TNF-α, cultured ECs showed an increase in monocyte adhesion. ECs transduced with Drp1K38A (Figure 3A) or MffS155/172A (Figure 3B) were protected from TNF-α-induced monocyte adhesion. mdivi1 also attenuated TNF-α-induced monocyte adhesion in ECs (Figure 3C).
Figure 3. Drp1 mediates leucocyte endothelial adhesion in vitro.

A. aortic ECs transduced with adenovirus encoding dominant-negative Drp1K38A mutant or control GFP at 100 moi for 48 hours were stimulated with 10 ng/mL TNF-α (TNF) for 6 hours and THP-1 monocyte adhesion (fold) was measured. con: basal control. B. Rat aortic ECs transduced with adenovirus encoding dominant-negative MffS155/172A mutant or control GFP at 100 moi for 48 h were stimulated with 10 ng/mL TNF-α for 6 hours and THP-1 monocyte adhesion (fold) was measured. C. Rat aortic ECs pretreated with 50 μmol/L mdivi or vehicle (0.1% DMSO) for 1 hour were stimulated with 10 ng/mL TNF-α (TNF) for 6 hours and THP-1 monocyte adhesion (fold) was measured. Scale bar is 100 μm. The bars show the mean±SEM from 3~4 independent experiments as indicated. *p<0.05, ***p<0.005, ****p<0.001 (1-way ANOVA).
Since mitochondrial ROS production has been shown to enhance endothelial leukocyte adhesion 23 and ischemia/reperfusion-induced mitochondrial fission in human umbilical vein ECs 24, we have further studied the relationship between mitochondrial fission and oxidative stress. Genetic inhibition or silencing of Drp1 blocked TNF-α-induced mitochondrial oxidative stress as measured with a mitochondrial protein oxidization indicator, MitoTimer 25 (Figures S4A and S4B). However, scavenging of mitochondrial superoxide generation by superoxide dismutase 2 mimic mitoTempo did not alter TNF-α-induced VCAM-1 expression in ECs (Figure S4C), although it reduced mitochondrial oxidative stress (Figure S4D) and monocyte adhesion (Figure S4E). Taking these data together, the mitochondrial fission pathway via Drp1 appears to be critical for monocyte adhesion to ECs, which likely involves NF-κB-dependent VCAM-1 induction as well as mitochondrial oxidative stress.
Systemic as well as endothelial Drp1 silencing prevents leukocyte adhesion in mice
To determine in vivo significance of mitochondrial fission in regulation of vascular inflammation, heterozygous Drp1 mice (Drp1+/−) or control Drp1+/+ mice were infused with TNF-α. Subsequently, intravital microscopy was utilized to measure leukocyte adhesion in mouse mesenteric postcapillary venules. Compared with the control Drp1+/+ mice, TNF-α-induced leukocyte adhesion in mouse mesenteric postcapillary venules was attenuated in Drp1+/− mice (Figure 4A). Moreover, inducible silencing of endothelial Drp1 in the Drp1flox/flox VeCadTRE Cre mice attenuated TNF-α-induced leukocyte adhesion in mesenteric micro-circulation (Figure 4B and 4C). mdivi1 pretreatment also suppressed TNF-α-induced increases in leukocyte adhesion in C57BL/6 mice (Figure 4D). Taking these data together, Drp1 activation and subsequent mitochondrial fission appear to be prerequisites for NF-κB activation and subsequent endothelial inflammatory responses in vitro and in vivo.
Figure 4. Drp1 mediates leucocyte endothelial adhesion in vivo.

A. Leukocyte adhesion was evaluated by intravital microscopy of mesenteric micro-circulation in 8~10 week old male Drp1+/− and control Drp1+/+ mice treated with 20 ng/g TNF-α or saline control for 6 hours. B. Leukocyte adhesion was evaluated by intravital microscopy of mesenteric micro-circulation in 8~10 week old male Drp1flox/flox VeCadTRE Cre+/− (Drp1f/f Cre+/−) and control Drp1flox/flox VeCadTRE Cre−/− (Drp1f/f Cre−/−) mice treated with 20 ng/g TNF-α or saline control for 6 hours. C. Immunohistochemical confirmation of endothelial Drp1 silencing in mouse aorta. Scale bar is 50 μm. 4x zoomed pictures are included. D. Leukocyte adhesion (fold) was evaluated by intravital microscopy of mesenteric micro-circulation in 8~10 week old male C57BL/6 mice treated with 20 ng/g mouse TNF-α (TNF) or saline control (con) for 6 hours with or without pretreatment of 25 μg/g mdivi1 for 18 hours. The bars show the mean±SEM from 5~6 independent experiments as indicated. *p<0.05, **p<0.01, ***p<0.005, ****p<0.001 (1-way ANOVA).
Drp1 activity is necessary for proinflammatory proteome induction in endothelial cells
To further explore contribution of this pathway in overall pro-inflammatory re-programming in ECs, we took advantage of shotgun proteomics combined with gene ontology analysis. Rat aortic ECs transduced with Drp1K38A or control GFP adenovirus were stimulated with TNF-α and cell lysates were analyzed with a mass spectroscopy (Figure S5A and Table S2). In ECs expressing control GFP, TNF-α significantly up-regulated specific groups of functional proteins including those involved with aging, wound healing, and apoptotic mitochondria. Enrichment of mitochondrial proteins was also observed. Kyoto Encyclopedia of Genes and Genomes/KEGG pathway analysis further suggest a premature aging condition with proteotoxicity induced by TNF-α in ECs. Significant interaction is predicted among the enriched proteins in mitochondria and those implicated in aging and age associated diseases (Figure S5B). In sharp contrast, these specific alterations by TNF-α were not observed in ECs transduced with Drp1K38A, whereas proteins involved in collagen fibril organization and poly(A) RNA binding were reduced. These data suggest that the endothelial pro-inflammatory responses including those associated with inflamm-aging 26 and metabolic reprogramming are dictated by the status of mitochondrial fission and Drp1 activity.
Inter-dependent relationship exists between NF-κB activation and mitochondrial fission in endothelial cells
Our findings suggest a potential cross-communication between the canonical NF-κB cascade and mitochondrial fission machinery involving Drp1. Therefore, we further explored the regulatory role of the NF-κB cascade for endothelial mitochondrial fragmentation. We found that rat aortic ECs pretreated with Bay 11–7085, the inhibitor of IκBα phosphorylation and degradation, showed a decrease in TNF-α-induced mitochondrial fragmentation. Similar inhibitions in IκBα degradation and mitochondrial fragmentation were noted in ECs pre-treated with the IKKβ inhibitor TPCA-1 (Figure 5A, S6A and S6B). Adenoviruses encoding IκBα serine 32 and 36 alanine (IκBαS32/36A) 27 and IKKβ serine 177 and 181 alanine (IKKβS177/181A) 28 dominant-negative substitution mutants were used to verify the contribution of IκBα and IKKβ in mitochondrial fission regulation. IκBαS32/36A and IKKβS177/181A transduction in rat aortic ECs attenuated TNF-α-induced mitochondrial fragmentation (Figure 5B). In addition, transduction of the Rel homology domain of p65 29, a dominant negative inhibitor of NF-κB attenuated mitochondrial fragmentation induced by TNF-α in ECs (Figure S6C). These data suggest that the canonical NF-κB cascade is required for TNF-α-induced mitochondrial fission. It has been reported that the components of NF-κB are present at mitochondria 30, 31. Subcellular fractionation of rat aortic ECs demonstrated expression of IκBα and p65 in the mitochondria fraction. TNF-α induced mitochondrial IκBα degradation that was attenuated with TPCA-1 (Figure S6D).
Figure 5. NF-κB activation mediates mitochondrial fission in response to TNF-α in endothelial cells.

A. Rat aortic ECs pretreated with NF-κB inhibitors (5 μmol/L TPCA-1 or 10 μmol/L Bay11–7085,) for 30 min were stimulated with 10 ng/mL TNF-α for 3 hours and MFC was measured. Scale bar is 15 μm and 4x zoomed pictures are included. B. Rat aortic ECs transduced with adenovirus encoding IκBαS32/36A (IκBαSA/SA), IKKβS177/181A (IKKβSA/SA), or control GFP at 100 moi for 48 hours were stimulated with 10 ng/mL TNF-α for 3 hours and MFC was measured. The bars show the mean±SEM from 4 independent experiments as indicated. ***p<0.005, ****p<0.001 (1-way ANOVA).
Salicylate prevents endothelial mitochondrial fission in vitro and in vivo
To explore translational relevance of the NF-κB inhibition in endothelial mitochondrial fission and subsequent inflammatory responses, we examined the effect of a non-steroidal anti-inflammatory drug, sodium salicylate, which inhibits IKKβ 32. Sodium salicylate attenuated TNF-α-induced mitochondrial fragmentation, VCAM-1 induction and IκBα degradation in rat aortic ECs (Figure 6A – 6C). Sodium salicylate further prevented increases in leukocyte adhesion induced by TNF-α in mouse mesenteric micro-circulation (Figure 6D). Sodium salicylate or mdivi1 treatment also prevented reduction in mitochondrial aspect ratio in the TNF-α-treated mouse aorta (Figure 6E). Thus, these data uncover a new pharmaco-mechanism of a classical anti-inflammatory reagent, salicylate, to maintain mitochondrial fission fusion balance under an inflammatory stress.
Figure 6. Salicylate targets mitochondrial fission in endothelial cells.

A. Rat aortic ECs pretreated with 50 mM salicylate (salic) or vehicle (0.1% DMSO) for 30 min were stimulated with 10 ng/mL TNF-α (TNF) for 3 hours and MFC was measured. con: basal control. Scale bar is 15 μm and 4x zoomed pictures are included. B. Rat aortic ECs pretreated with 50 mM salicylate or vehicle (0.1% DMSO) for 30 min were stimulated with 10 ng/mL TNF-α for 6 hours and VCAM-1 expression was measured. C. Rat aortic ECs pretreated with 50 mM salicylate or vehicle (0.1% DMSO) for 30 min were stimulated with 10 ng/mL TNF-α for 20 min and IκBα degradation was measured with immunoblotting. D. Leukocyte adhesion was evaluated by intravital microscopy of mesenteric micro-circulation in mice treated with 20 ng/g TNF-α for 6hours with or without 200 μg/g sodium salicylate pretreatment for 7 hours. E. Endothelial mitochondrial aspect ratios were evaluated by electron microscopy in mouse aortas treated with 20 ng/g TNF-α for 6h with or without 200 μg/g sodium salicylate pretreatment for 7 hours or 25 μg/g mdivi1 for 18 hours. Scale bar is 50 nm. The bars show the mean±SEM from 3~5 independent experiments as indicated except E (41–57 mitochondria were analyzed from 15 pictures). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 (1-way ANOVA). F. Graphical representation of the overall findings. Left side description: In cultured endothelial cells, pharmacological and genetic inhibition of Drp1 activity attenuated TNF-α or LPS-induced NF-κB activation and subsequent inflammatory responses (VCAM-1 expression and THP-1 adhesion) in addition to mitochondrial fission inhibition. Presence of this cascade is also supported in mesenteric microcirculation with Drp1 knockout animals. Right side description: Intriguingly, inhibition of the canonical NF-κB cascade at multiple points (IKKβ, IκBα and p65) also attenuated mitochondrial fission in cultured ECs. Presence of this cascade is also supported in mesenteric microcirculation in mice with treatment of an IKKβ inhibitor, salicylate. Accordingly, our findings demonstrate an interdependent relationship between the canonical NF-κB cascade and the mitochondrial fission mechanism via Drp1 in mediating inflammatory responses in endothelial cells.
Discussion
Diseases associated with chronic vascular inflammation are typically marked by mitochondrial dysfunction and persistent NF-κB activation 33, 34. Mitochondrial regulation is critical for innate immune responses which involve mitochondrial antiviral signaling protein/MAVS to induce NF-κB activation 35. However, information regarding the potential cross-communication between the NF-κB cascade and mitochondrial morphological adaptation in inflammatory conditions has been not fully explored in the vascular system 36. Here we reconcile these two seemingly independent pathways by showing enhanced mitochondrial fission is not only a required element for the vascular inflammation (NF-κB promoter activation, VCAM-1 induction and leukocyte adhesion) but is also mediated by the the NF-κB component p65 (Figure 6F). However, several critical questions still remain unanswered in the potential cascade.
How does p65 feed into Drp1 and promote mitochondrial fission? In addition to Drp1 Ser616 phosphorylation or Ser637 de-phosphorylation, several post translational modifications such as sumoylation and nitrosylation are known to be involved in Drp1 activation 17. It is therefore likely that there is a signal cross-talk between the mitochondrial p65 and Drp1, which leads to the post translational modification(s) of Drp1 in mediating mitochondrial fission. In parallel to our findings, receptor-interacting serine/threonine-protein kinase 3/RIPK3 mediates Drp1-dependent fission downstream of the TNF-α receptor 37. In addition, NF-κB-inducing kinase activates Drp1 and induces mitochondrial fission in the absence of IKKs in mouse embryonic fibroblasts 38. In contrast, opposing regulatory relationships between NF-κB activity and mitochondrial fission were reported in cardiomyocytes 39. It is likely that the signaling mechanism to determine a regulatory relationship between Drp1 and NF-κB activation is dependent on the given cell type. Alternatively, it is possible that TNF-α or LPS alters expression of other fission/fusion machineries directly, or indirectly via other mitochondrial proteins in ECs. For examples, IKKα, a member of noncanonical NF-κB pathway, regulates mitochondrial fission via induction of optic atrophy 1 protein in fibroblasts 40. IκBα has been shown to regulate voltage-dependent anion channel 1 (VDAC1) at the outer mitochondrial membrane independently of NF-κB retention 31. Mitochondrial translocation of p65 via mortalin negatively regulates mitochondrial gene transcription and function 41, whereas positive regulation of mitochondrial respiration via p65 has also been reported 42. Further investigation is clearly needed to test some of these possibilities regulating Drp1 activation and mitochondrial fission in endothelial cells such as with assessments on protein-protein interactions and post translational modifications.
How do Drp1 (and/or mitochondrial fission) and p65 intersect and coordinate NF-κB activation in ECs? As p65 still translocates to the nucleus with Drp1 inhibition, it is likely that Drp1 may positively regulate NF-κB activation such as via an enhancer system 43 independent of p65 as illustrated in Fig 6F. Increase in mitochondrial energy production in response to mitochondrial fission may also fulfill the need for enhanced transcription independently of p65. Drp1 is abundant in cytosol and also functions independently from mitochondria 14. However, our confirmation that inhibition of a mitochondrial Drp1 receptor Mff attenuates NF-κB activation and VCAM-1 induction support the role of mitochondrial Drp1 and fission in regulation of inflammation. Thus, our data extend a new mitochondrial branch of the NF-κB regulation via Drp1/Mff-dependent mitochondrial fission which dictates the canonical NF-κB signaling to transmit vascular inflammation. Our concept of the vital role of Drp1 in promotion of stress-induced inflammatory responses is supported by the literature demonstrating the requirement of Drp1 in T cell proliferation and immune responses 44, 45.
Is mitochondrial fission necessarily detrimental to EC according to our data? In general, mitochondrial fission fusion machineries are both important to maintain mitochondrial health and quality control 1. In ECs, Drp1 appears indispensable for angiogenesis 5. NF-κB mediates critical defense mechanisms in the immune systems including those interacting with endothelium 7. Thus, mitochondrial fission is not necessarily detrimental to EC but may provide us with an alternative way to potentially control undesired inflammatory response in certain chronic conditions. It is well accepted that mitochondria orchestrate cell reprogramming from a metabolic perspective. Recent studies demonstrated that Drp1-dependent mitochondrial fission may be critical in such cell reprograming 46. Available literature 47 as well as our proteomic data indicate that the metabolic re-programming of endothelial cells is also vital to the development of vascular pathology. In addition, our data suggest that activation of Drp1 and/or mitochondrial fission contributes to mitochondrial superoxide production, which is most likely due to enhanced mitochondrial oxygen consumption and ATP production. The enhanced mitochondrial superoxide production via Drp-1 activation has also been reported in other cell systems 48. However, these data cannot exclude the possibility that Drp1 and mitochondrial fission also affect oxidative stress in mitochondria via alteration on mitochondrial antioxidant defense mechanisms such as Sirt3 expression/activity 49, glutathione levels 50 as well as changes in mitochondrial NAD+ levels 51.
What is the clinical significance of the salicylate data? Salicylate is one of the most frequently utilized anti-inflammatory reagents known to suppress inflammatory responses such as those with NF-κB activation 32. Salicylate is also known to inhibit mitochondrial ATP production and activate AMP-activated protein kinase/AMPK 52. Our findings indicate that salicylate may be effective to maintain mitochondrial fission fusion balance under inflammatory condition. Thus, salicylate may also be beneficial in other pathological conditions, where disruptions of the fission fusion balance have been observed.
Perspectives
The present study has demonstrated a previously unknown mechanism whereby the NF-κB cascade and a mitochondrial fission pathway operated by Drp1 interdependently regulate endothelial inflammation. Therefore, endothelial Drp1 appears to be a unique therapeutic target against inflammatory cardiovascular diseases.
Supplementary Material
Novelty and Significance.
What is new?
A GTPase, dynamin-related protein-1 (Drp1) and mitochondrial fission are important players in endothelial NF-kB activation and inflammation.
What is relevant?
Inhibiting Drp1 and mitochondrial fission prevented endothelial inflammatory responses in endothelial culture and mouse models which introduces a potential therapeutic target, Drp1 or mitochondrial dynamics in general, as a possible approach.
Acknowledgements
This study was supported by National Institute of Health grants, HL128324 (S.E. and V.R.), HL133248 (S.E.), DK111042 (R.S. and S.E.), NS109382 (S.E.), GM123266 (H.S.), F31HL127971 (S.J.F.), F31HL146081 (M.J.B.) and F30HL146006 (H.A.C.), American Heart Association grants, 16GRNT30130013 (V.R.), 16GRNT30410007 (S.E.), 19PRE34430038 (M.J.B.) and 19PRE34430037 (H.A.C), and Japan Grant-in-Aid for Scientific Research, 18KK0437 (M.M.).
Footnotes
Conflict of Interest
None.
Contributor Information
Steven J. Forrester, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, U.S.A.
Kyle J. Preston, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, U.S.A.
Hannah A. Cooper, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, U.S.A.
Michael J. Boyer, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, U.S.A.
Kathleen M. Escoto, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, U.S.A.
Anthony J. Poltronetti, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, U.S.A.
Katherine J. Elliott, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, U.S.A.
Ryohei Kuroda, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, U.S.A..
Masashi Miyao, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, U.S.A.; Department of Forensic Medicine, Kyoto University Graduate School of Medicine, Kyoto, Japan
Hiromi Sesaki, Department of Cell Biology, Johns Hopkins School of Medicine, Baltimore, MD, U.S.A..
Tomoko Akiyama, Advanced Medical Research Center, Yokohama City University, Yokohama, Japan.
Yayoi Kimura, Advanced Medical Research Center, Yokohama City University, Yokohama, Japan.
Victor Rizzo, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, U.S.A..
Rosario Scalia, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, 3500 N. Broad Street, Philadelphia, PA19140.
Satoru Eguchi, Cardiovascular Research Center, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, U.S.A..
References
- 1.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–87. [DOI] [PubMed] [Google Scholar]
- 2.Hall AR, Burke N, Dongworth RK and Hausenloy DJ. Mitochondrial fusion and fission proteins: novel therapeutic targets for combating cardiovascular disease. Br J Pharmacol. 2014;171:1890–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess MA, Levit A, Kim B, Hartman ML, Joseph L, Shirihai OS and Vita JA. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124:444–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diebold I, Hennigs JK, Miyagawa K, Li CG, Nickel NP, Kaschwich M, Cao A, Wang L, Reddy S, Chen PI, Nakahira K, Alcazar MA, Hopper RK, Ji L, Feldman BJ and Rabinovitch M. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 2015;21:596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim YM, Youn SW, Sudhahar V, Das A, Chandhri R, Cuervo Grajal H, Kweon J, Leanhart S, He L, Toth PT, Kitajewski J, Rehman J, Yoon Y, Cho J, Fukai T and Ushio-Fukai M. Redox Regulation of Mitochondrial Fission Protein Drp1 by Protein Disulfide Isomerase Limits Endothelial Senescence. Cell Rep. 2018;23:3565–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daiber A, Steven S, Weber A, Shuvaev VV, Muzykantov VR, Laher I, Li H, Lamas S and Munzel T. Targeting vascular (endothelial) dysfunction. Br J Pharmacol. 2017;174:1591–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayden MS and Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gimbrone MA Jr. and Garcia-Cardena G Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res. 2016;118:620–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inflammation Libby P. and cardiovascular disease mechanisms. Am J Clin Nutr. 2006;83:456S–460S. [DOI] [PubMed] [Google Scholar]
- 10.Pober JS and Sessa WC. Inflammation and the blood microvascular system. Cold Spring Harb Perspect Biol. 2014;7:a016345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salvador B, Arranz A, Francisco S, Cordoba L, Punzon C, Llamas MA and Fresno M. Modulation of endothelial function by Toll like receptors. Pharmacol Res. 2016;108:46–56. [DOI] [PubMed] [Google Scholar]
- 12.Dikalov SI and Ungvari Z. Role of mitochondrial oxidative stress in hypertension. Am J Physiol Heart Circ Physiol. 2013;305:H1417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freigang S, Ampenberger F, Weiss A, Kanneganti TD, Iwakura Y, Hersberger M and Kopf M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1alpha and sterile vascular inflammation in atherosclerosis. Nat Immunol. 2013;14:1045–53. [DOI] [PubMed] [Google Scholar]
- 14.Sesaki H, Adachi Y, Kageyama Y, Itoh K and Iijima M. In vivo functions of Drp1: lessons learned from yeast genetics and mouse knockouts. Biochim Biophys Acta. 2014;1842:1179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smirnova E, Shurland DL, Ryazantsev SN and van der Bliek AM. A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol. 1998;143:351–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR and Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otera H, Ishihara N and Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013;1833:1256–68. [DOI] [PubMed] [Google Scholar]
- 18.Shen YL, Shi YZ, Chen GG, Wang LL, Zheng MZ, Jin HF and Chen YY. TNF-alpha induces Drp1-mediated mitochondrial fragmentation during inflammatory cardiomyocyte injury. Int J Mol Med. 2018;41:2317–2327. [DOI] [PubMed] [Google Scholar]
- 19.Bach D, Naon D, Pich S, Soriano FX, Vega N, Rieusset J, Laville M, Guillet C, Boirie Y, Wallberg-Henriksson H, Manco M, Calvani M, Castagneto M, Palacin M, Mingrone G, Zierath JR, Vidal H and Zorzano A. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin-6. Diabetes. 2005;54:2685–93. [DOI] [PubMed] [Google Scholar]
- 20.Toyama EQ, Herzig S, Courchet J, Lewis TL Jr., Loson OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC and Shaw RJ. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milstone DS, Ilyama M, Chen M, O’Donnell P, Davis VM, Plutzky J, Brown JD, Haldar SM, Siu A, Lau AC, Zhu SN, Basheer MF, Collins T, Jongstra-Bilen J and Cybulsky MI. Differential role of an NF-kappaB transcriptional response element in endothelial versus intimal cell VCAM-1 expression. Circ Res. 2015;117:166–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D and Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- 23.Zinovkin RA, Romaschenko VP, Galkin II, Zakharova VV, Pletjushkina OY, Chernyak BV and Popova EN. Role of mitochondrial reactive oxygen species in age-related inflammatory activation of endothelium. Aging (Albany NY). 2014;6:661–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giedt RJ, Yang C, Zweier JL, Matzavinos A and Alevriadou BR. Mitochondrial fission in endothelial cells after simulated ischemia/reperfusion: role of nitric oxide and reactive oxygen species. Free Radic Biol Med. 2012;52:348–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laker RC, Xu P, Ryall KA, Sujkowski A, Kenwood BM, Chain KH, Zhang M, Royal MA, Hoehn KL, Driscoll M, Adler PN, Wessells RJ, Saucerman JJ and Yan Z. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J Biol Chem. 2014;289:12005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrucci L and Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S and Baeuerle PA. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 1995;14:2876–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A and Rao A. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278:860–6. [DOI] [PubMed] [Google Scholar]
- 29.Liu Q, Chen Y, Auger-Messier M and Molkentin JD. Interaction between NFkappaB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ Res. 2012;110:1077–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cogswell PC, Kashatus DF, Keifer JA, Guttridge DC, Reuther JY, Bristow C, Roy S, Nicholson DW and Baldwin AS Jr. NF-kappa B and I kappa B alpha are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-kappa B. J Biol Chem. 2003;278:2963–8. [DOI] [PubMed] [Google Scholar]
- 31.Pazarentzos E, Mahul-Mellier AL, Datler C, Chaisaklert W, Hwang MS, Kroon J, Qize D, Osborne F, Al-Rubaish A, Al-Ali A, Mazarakis ND, Aboagye EO and Grimm S. IkappaBetaalpha inhibits apoptosis at the outer mitochondrial membrane independently of NF-kappaB retention. EMBO J. 2014;33:2814–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin MJ, Yamamoto Y and Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77–80. [DOI] [PubMed] [Google Scholar]
- 33.Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW 2nd, Kitsis RN, Otsu K, Ping P, Rizzuto R, Sack MN, Wallace D, Youle RJ, American Heart Association Council on Basic Cardiovascular Sciences CoCC, Council on Functional G and Translational B. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ Res. 2016;118:1960–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van der Heiden K, Cuhlmann S, Luong le A, Zakkar M and Evans PC. Role of nuclear factor kappaB in cardiovascular health and disease. Clin Sci (Lond). 2010;118:593–605. [DOI] [PubMed] [Google Scholar]
- 35.West AP, Shadel GS and Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koshiba T, Yasukawa K, Yanagi Y and Kawabata S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci Signal. 2011;4:ra7. [DOI] [PubMed] [Google Scholar]
- 37.Wang Z, Jiang H, Chen S, Du F and Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–43. [DOI] [PubMed] [Google Scholar]
- 38.Jung JU, Ravi S, Lee DW, McFadden K, Kamradt ML, Toussaint LG and Sitcheran R. NIK/MAP3K14 Regulates Mitochondrial Dynamics and Trafficking to Promote Cell Invasion. Curr Biol. 2016;26:3288–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nan J, Hu H, Sun Y, Zhu L, Wang Y, Zhong Z, Zhao J, Zhang N, Wang Y, Wang Y, Ye J, Zhang L, Hu X, Zhu W and Wang J. TNFR2 Stimulation Promotes Mitochondrial Fusion via Stat3- and NF-kB-Dependent Activation of OPA1 Expression. Circ Res. 2017;121:392–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laforge M, Rodrigues V, Silvestre R, Gautier C, Weil R, Corti O and Estaquier J. NF-kappaB pathway controls mitochondrial dynamics. Cell Death Differ. 2016;23:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson RF, Witzel II and Perkins ND. p53-dependent regulation of mitochondrial energy production by the RelA subunit of NF-kappaB. Cancer Res. 2011;71:5588–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L, Moretti M, De Smaele E, Beg AA, Tergaonkar V, Chandel NS and Franzoso G. NF-kappaB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol. 2011;13:1272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM, Bair S, Newton G, Lichtman A, Kung A, Yang T, Wang H, Luscinskas FW, Croce K, Bradner JE and Plutzky J. NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56:219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simula L, Pacella I, Colamatteo A, Procaccini C, Cancila V, Bordi M, Tregnago C, Corrado M, Pigazzi M, Barnaba V, Tripodo C, Matarese G, Piconese S and Campello S. Drp1 Controls Effective T Cell Immune-Surveillance by Regulating T Cell Migration, Proliferation, and cMyc-Dependent Metabolic Reprogramming. Cell Rep. 2018;25:3059–3073 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang YJ, Bang BR, Han KH, Hong L, Shim EJ, Ma J, Lerner RA and Otsuka M. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat Commun. 2015;6:8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prieto J, Leon M, Ponsoda X, Sendra R, Bort R, Ferrer-Lorente R, Raya A, Lopez-Garcia C and Torres J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat Commun. 2016;7:11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eelen G, de Zeeuw P, Simons M and Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116:1231–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qi X, Qvit N, Su YC and Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. 2013;126:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qiu X, Brown K, Hirschey MD, Verdin E and Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–7. [DOI] [PubMed] [Google Scholar]
- 50.Mari M, Morales A, Colell A, Garcia-Ruiz C, Kaplowitz N and Fernandez-Checa JC. Mitochondrial glutathione: features, regulation and role in disease. Biochim Biophys Acta. 2013;1830:3317–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao W, Wang RS, Handy DE and Loscalzo J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid Redox Signal. 2018;28:251–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rena G and Sakamoto K. Salicylic acid: old and new implications for the treatment of type 2 diabetes? Diabetol Int. 2014;5:212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.