

Abstract
To assess the role of core metabolism genes in bacterial virulence - independently of their effect on growth - we correlated the genome, the transcriptome and the pathogenicity in flies and mice of 30 fully sequenced Pseudomonas strains. Gene presence correlates robustly with pathogenicity differences among all Pseudomonas species, but not among the P. aeruginosa strains. However, gene expression differences are evident between highly and lowly pathogenic P. aeruginosa strains in multiple virulence factors and a few metabolism genes. Moreover, 16.5%, a noticeable fraction of the core metabolism genes of P. aeruginosa strain PA14 (compared to 8.5% of the non-metabolic genes tested), appear necessary for full virulence when mutated. Most of these virulence-defective core metabolism mutants are compromised in at least one key virulence mechanism independently of auxotrophy. A pathway level analysis of PA14 core metabolism, uncovers beta-oxidation and the biosynthesis of amino-acids, succinate, citramalate, and chorismate to be important for full virulence. Strikingly, the relative expression among P. aeruginosa strains of genes belonging in these metabolic pathways is indicative of their pathogenicity. Thus, P. aeruginosa strain-to-strain virulence variation, remains largely obscure at the genome level, but can be dissected at the pathway level via functional transcriptomics of core metabolism.
Subject terms: Microbiology, Bacteriology
Introduction
Pseudomonas species include Gram-negative, rod-shaped, facultative anaerobic and polarly flagellated bacteria able to utilize a variety of organic compounds and colonize a wide range of niches1. Some Pseudomonas species can be pathogenic to humans, animals and plants1–3. While able to inhabit diverse environments, including soil and water, P. aeruginosa strains are common opportunistic human pathogens. They can also infect multiple model hosts, including insects, plants and mice using a remarkable variety of virulence factors4–6. In humans, P. aeruginosa causes acute infections in injured, burned and immunocompromised patients and persistent respiratory infections in individuals with cystic fibrosis7–12.
Previous comparative analyses of P. aeruginosa strain-specific genes revealed that different isolates tested share >90% of their gene content irrespective of the source (soil, wound, urinary tract, lung, blood or eyes) or the type of infection they were found to cause3,13,14. The high virulence of bacterial pathogens is usually attributed to the presence of pathogenicity islands, which are clusters of virulence-related genes often acquired through horizontal gene transfer15,16. The core genome of the highly virulent P. aeruginosa strain PA14 shares ~90% of its genes (as identical homologues) with the prototypic reference strain PAO1, but PA14 contains extra genes in genomic islands related to survival in diverse environments17,18. Despite the striking genomic similarity, P. aeruginosa strain-to-strain variation in virulence is vast17,19,20 and cannot be simply explained by the presence of virulence-related genes residing on pathogenicity islands3. This comes in contrast with genome decay often described in comparative genomic studies of pathogen adaptation to a host-associated lifestyle6,21,22.
The existence of multiple P. aeruginosa virulence factors and their direct regulators may indicate constant strain evolution at the virulence effector/regulator level or at a hierarchically higher gene network level. P. aeruginosa strains can evolve, for example, in the lung of cystic fibrosis patients, and some strains, such as CF5, are only weakly pathogenic in models of acute infection13,19. Nevertheless, extensive efforts to link gene content with pathogenicity of P. aeruginosa do not suffice to explain virulence by the mere presence or absence of known virulence factors3,13,23. Accordingly, P. aeruginosa pathogenicity has been characterized context-dependent, because genes required for pathogenicity in one strain may not necessarily contribute to virulence in other strains. We hypothesized that, in addition to gene presence, the expression of virulence factors and their direct or indirect regulators may explain the differences in pathogenicity among Pseudomonas strains.
In the current study, we compared 30 fully sequenced Pseudomonas genomes using large-scale comparisons at the protein sequence level. As a first step we aimed to re-examine the differences in the presence and absence of genes in relation to differences in pathogenicity among Pseudomonas strains by expanding our search to non-P. aeruginosa strains. We thus established an extensive set of known virulence factors (VFs), the expression rather than the gene presence of which correlated more reliably with the highly versus the lowly virulent strains. In addition, core metabolism gene expression correlated with pathogenicity, but at the pathway rather than at the gene level. Estimating the percentage of the core metabolism genes participating in the virulence of P. aeruginosa strain PA14, we found core metabolism genes to affect virulence at least as often as non-metabolic genes. Despite the known interconnectivity between bacteria core metabolism and growth24, most of core metabolism gene mutants tested were found defective in virulence and compromised in known virulence mechanisms, without being essential for growth in culture or in the host.
Methods
Bacterial strains
The catalog of the core metabolic genes of P. aeruginosa strain PA14 was produced using the KEGG Database in 201425. The 553 metabolic gene mutants were picked from the 96-well plates of the publicly available PA14 Transposon Insertion Mutant Library, while the 95 non-metabolic gene mutants were randomly selected from sequential plates of the same library verifying that none of them is linked in the KEGG database with bacterial metabolism (http://ausubellab.mgh.harvard.edu/cgi-bin/pa14/home.cgi). The 30 wild type sequenced Pseudomonas strains used in this study are described in Supplementary Table 1.
Intranasal mouse lung infection assay
The intranasal infection achieves the spreading of the bacteria from the upper airways to the intestine and low airways, thus mimics the pathology seen in acute bacterial pneumonia26,27. PA14 wild type and mutant strains were grown in LB liquid cultures overnight. Cultures were then diluted 1:100 and were grown over day to OD600nm: 3.0 (~3 × 109 CFU/ml). One ml of each bacterial culture was centrifuged at 4610 rcf for 2 min and the supernatant was removed. The pellet was washed twice and finally diluted in sterile saline (0.9%). Mice were intranasally infected under very light anesthesia, as previously described28,29, by placing 10 μl of a bacterial suspension in each nostril (20 μl in total) to reach the desired infectious dose of 2 × 107 CFUs per mouse. Mortality counts were taken every day for 7 days.
Ethics statement
Animal protocols were approved by the Cyprus Veterinary Service inspectors under the license number CY/EXP/PR.L6/2018 for the Laboratory of Prof. Apidianakis at the University of Cyprus. The veterinary services act under the auspices of the Ministry of Agriculture in Cyprus and the project number is CY.EXP101. These national services abide by the National Law for Animal Welfare of 1994 and 2013 and the Law for Experiments with Animals of 2013 and 2017. All experiments were performed in accordance with these guidelines and regulations.
Fly pricking assay
Male Oregon R flies were pricked in the thoracic cuticular epithelium with a tungsten needle dipped in a bacteria suspension, as previously described30. The infection mix contained 980 μl ddH2O, 10 μl 1 M MgSO4 and 10 μl of bacteria OD600nm: 3. Vials were transferred at 25 °C and survival was measured every day. Two vials of 20 flies each were used for each mutant.
Fly wound colonization assay
20–25 Oregon R male flies were pump-injected dorsoventrally with 100 CFUs of a single bacterial strain grown at OD600nm: 3 and transferred into vials with fresh food at 25 °C, as previously described30. 24 hours later flies were ground in saline, plated on LB plates incubated for 16 hours at 37 °C before counting CFUs for each mutant strain in comparison to the wild type strain PA14.
Fly feeding assay
Female Oregon R flies 3–7 days old were starved for 5–6 hours and then were transferred in vials containing a cotton ball soaked with 5 ml of the following oral infection mix: 0.5 ml of bacteria OD600nm: 3, 1 ml 20% sucrose and 3.5 ml ddH2Ο. Vials were transferred at 25 °C and survival was measured every day. Three vials with 10 flies each were used for each mutant.
Fly survival
To determine the mortality rate of the infected flies, daily observation of the experimentations was necessary. Uninfected flies can live up to 60 days; however, infection using P. aeruginosa PA14 usually kills flies in less than 10 days. Fly infections were repeated in technical replicates two independent times for each bacterial strain for both assays (oral and wound infection). The fly survival was calculated based on the total number of flies surviving.
Fly intestinal colonization assay
Oregon R female flies 3–7 days old, previously starved for 5–6 hours, were fed with the above described oral infection mix for one day. Flies were next transferred into 50 ml Eppendorf tubes with 12 holes (1.2 mm in diameter) on the lid, as previously described31. Flies were able to reach food (Whatman filter paper disc soaked with 200 μl of 4% sucrose and 10% LB set on the lid and covered with parafilm) only through the holes of the lid. Flies were transferred in clean 50 ml Eppendorf tubes every day for 3 days to avoid contamination. On the third day, plates were cultured with a solution of ground flies to count CFUs and observe the colonization ability of each mutant strain, compared to the wild type strain.
Strain clustering based on pathogenicity
A survival graph was obtained for the flies infected by each bacterial strain and the survival curves were statistically analyzed using the Kaplan-Meier method32 using IBM SPSS. The resulting survival curves were compared pairwise in an all-against-all fashion, using a chi-square test (χ2). Applying the unweighted pair group method with the arithmetic mean (UPGMA) agglomerative clustering procedure33 (using the respective χ2-values as a pairwise distance) produces a tree-like representation for the strains analyzed. This “natural” hierarchical clustering was further used to separate Pseudomonas strains in three groups (corresponding to low, medium and high virulence) by cutting the tree in the second bifurcation from the root. Visual inspection of the Kaplan-Meier survival curves and the requirement that the well-characterized avirulent (i.e. CF5) or highly virulent strains (i.e. PA14) should be assigned as “low” and “high”, respectively, led to the designation of labels to the resulting clusters.
Phylogenetic analysis of Pseudomonas strains
We produced a reliable phylogeny of the 30 studied Pseudomonas species/strains which was correlated with the pathogenicity scale. We followed the approach described in Duan et al.34, but rpoB and rpoD homologs could not be identified in the genome of P. aeruginosa 2192. Therefore, a phylogenetic tree was constructed using the sequences of the universally present gyrase B (gyrB) and 16S rRNA genes for the 30 studied species. Escherichia coli K12 and Cellvibrio japonicus strain Ueda107 were used as outgroups. Sequences were retrieved from the Pseudomonas Genome Database35 and the NCBI nucleotide database36 for the outgroups, and were multiply aligned using ClustalO37 with default parameters. The resulting multiple sequence alignments were concatenated using the MEGA software package38 and the data matrix produced was further analyzed with MrBayes v3.239 (using the default parameters: lset nst = 6 rates = invgamma, mcmc ngen = 20000 samplefreq = 100 printfreq = 100 diagnfreq = 1000), which performs Bayesian inference of phylogeny. An essentially identical Pseudomonas phylogenetic tree was built with the complete gene matrix for the 29 species (i.e. excluding P. aeruginosa 2192) using the same procedure.
Sequence analysis and clustering
Protein sequences encoded in the Pseudomonas strains of interest were downloaded from the Pseudomonas Genome Database40 and were internally codified following the style of the COGENT database for consistency and easy manipulation from computational tools41. Sequences were filtered with CAST42 (using default parameters) and converted in database records for the NCBI BLAST suite of tools43,44 as previous studies have shown that filtering sequences in regions of extreme amino acid composition can eliminate most of the false-positive results in BLAST searches without sacrificing sensitivity45. In this study, a new, more effective version of the CAST algorithm was used46 offering optimized performance for pan-genome analyses. We used a local installation of BLASTP for performing all-against-all pairwise comparisons (e-value threshold: 10–6; Composition Based Statistics: off; masking mode: off; all other parameters left to their default values). BLAST results were used for sequence clustering using the MCL software47 employed with the default parameters as suggested by the authors for the delineation of Pseudomonas protein families.
Virulence factor (VF) selection
We collected annotated VFs encoded in the P. aeruginosa PAO1 genome based on the relevant section of the Pseudomonas Genome Database. Next, in order to obtain a comprehensive dataset of known VFs for most of the 30 studied species, an exhaustive literature search was manually realized in PubMed using keywords such as, “virulence factor”, “pathogenic capacity”, “secretion system”, in correlation with “Pseudomonas” species. Our list was further expanded by consulting the Virulence Factors Database (VFDB)48 as a complementary reference list. In this way, a list containing 254 virulence factors was created and VFs were then separated in five groups according to their function: type II secretion system (19 genes), type III secretion system (37 genes), type VI secretion system (18 genes), quorum sensing (44 genes) and other genes that did not enter any of these categories (136) (Supplementary Table 2).
Metabolic genes
We identified the proteins annotated to participate in distinct metabolic pathways and categories based on the KEGG database (Kyoto Encyclopedia of Genes and Genomes, www.kegg.jp 25. For the assessment of RNAseq transcriptomics data (see below) against metabolic pathways, we deliberately used an independent pathway definition provided by BioCyc49, in order to avoid any potential biases due to our initial selection of metabolic genes.
Phylogenetic profiles of Pseudomonas VFs and metabolic genes
We did not rely on external resources to infer homology among proteins encoded in the genomes of interest, but we used the bidirectional-best hit (BBH) as a more reliable approach. Briefly, parsing the all-against-all BLASTP results with custom-developed software, we identified cases where a protein protA from “Genome A” has protB as a best hit (according to the BLASTP score) in “Genome B”, which in turn has protA as its best hit in “Genome A”. This procedure rapidly identifies cases of 1–1 orthologs, with the drawback that some subtle cases (e.g. where recent gene duplication has occurred) may be missed50. Moreover, cases of wrongly predicted genes are expected to result in additional proteins that seem to be absent from some genomes of interest. In order to alleviate this drawback, all proteins of interest that appeared to be absent from particular Pseudomonas strains after the BBH analysis were further queried using translated BLAST searches (TBLASTN) against the respective target genomes and if reliable hits were found they were recorded as “present”. Phylogenetic profiles for Pseudomonas VFs and metabolic proteins were coded in Presence-Absence (PA) tables using a simple +1/−1 scheme (present/absent). PA tables were further visualised using the MeV software51 multiple array viewer feature and were subject to clustering using the k-means method in the R environment52. These clusters were then compared to the groups obtained by the pathogenicity ranking using the Adjusted Rand Index (ARI)53,54 as implemented in the mclust R package. The ARI is a measure for the chance grouping of elements between two clusterings accompanied by a measure of the statistical significance of the overlap of the groups compared.
Bacterial RNA isolation for RNA Seq
We extracted RNA from LB cultures at OD600nm 1 and 3 in biological replicates using the QIAzol lysis reagent (Qiagen). The quantity of the bacterial RNA samples was measured on a NanoDrop ND1000 system and their quality analyzed on the Agilent 2100 Bioanalyzer system with the Agilent RNA 6000 Nano kit protocol (Agilent Technologies), according to manufacturer’s instructions. Bacterial RNA samples with RNA Integrity Number (RIN) >7 were chosen for mRNA enrichment. The MICROBExpress Bacterial mRNA Enrichment Kit protocol (ThermoFisher Scientific) was performed on 5–10 μg of bacterial RNA, according to the manufacturer’s instructions. The quality and quantity of enriched mRNA were measured using the NanoDrop and the Bioanalyzer systems. After the assessment of the enriched bacterial mRNAs, libraries were prepared using the Ion Total RNA-Seq Kit v2 protocol and reagents (ThermoFisher Scientific) according to the manufacturer’s instructions.
The enriched mRNAs were fragmented, reverse transcribed and amplified after the addition of a specific (~10 bp) barcode to each sample. The quantity and quality of the prepared libraries were assessed on a Bioanalyzer using the DNA High Sensitivity Kit, according to the manufacturer’s instructions. All libraries were diluted to 1 nM and subsequently pooled together in pools of eight. 40 pM of pooled libraries were further processed for templating and enrichment, and loaded onto the Ion Proton PI V2 chips (ThermoFisher Scientific). The procedure was performed on the Ion Chef System using the Ion PI IC200 Chef kit, protocol and reagents (ThermoFisher Scientific), according to the manufacturer’s instructions. The sequencing of the loaded chips was performed using the Ion PI Sequencing 200 V3 kit on an Ion Proton system (ThermoFisher Scientific), according to the manufacturer’s instructions. Initial analysis, sample/barcode assignment, and genome mapping was performed on the Ion Proton server.
Short read mapping
The RNA-Seq FASTQ files obtained after Ion Proton sequencing (publicly accessible through the Gene Expression Omnibus database, GEO Series accession number GSE142464 and the following link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE142464) were mapped using tophat255 (https://ccb.jhu.edu/software/tophat/index.shtml), with default settings and using additional transcript annotation data for the Pseudomonas aeruginosa (UCBPP-PA14) genome (GCF_000014625.1_ASM1462v1_genomic.gff). Unmapped reads were submitted to a second round of mapping using Bowtie256 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) against the Pseudomonas aeruginosa (UCBPP-PA14) genome, with the very-sensitive switch turned on and merged with the initial mappings. Reads per gene were counted using featureCounts57 and those corresponding to rRNA genes were in silico depleted to avoid skewing the statistics of independent samples prior to processing with DESeq258 for the detection of differentially expressed genes.
Pathway enrichment analysis
Relevant gene lists were uploaded to BioCyc SmartTables. Pathway enrichment was performed using the respective BioCyc functionality implementing the Fischer exact test and the Parent-Child Intersection method that has been shown to be robust against false positives59. Multiple testing correction was performed with the Benjamini-Hochberg procedure. Adjusted p-values were considered significant when padj < 0.05.
Cluster analysis of patterns of differential gene expression
Cluster analysis and visualization of patterns of up-/down-regulation was performed with ClustViz60 on a matrix where up-/down-regulation of each gene was represented with +1/−1, respectively. Zero values were used to denote the absence of significant differential expression.
Minimal media assays
Bacteria that were grown in 3 ml LB O/N cultures, 500 μl were centrifuged at 8000 rpm for 2 min, the supernatant was removed, and the pellet was reconstituted and further diluted 1:100 in minimal media and incubated on a horizontal culture rotator at 37 °C. To prepare 50 ml of minimal medium solution we mixed 10 ml 5xM9, 100 μl 1 Μ MgSO4, 1 ml 20% glucose and 39 ml ddH2O. An alternative minimal medium solution was prepared by adding a final concentration of 5% ground and filtered fly extract to the above mix. For each mutant, two replicate cultures and three consecutive measurements at optical density OD600nm were taken.
Quantitative reverse transcription real-time PCR
RNA was extracted from log-phase bacteria grown in LB using the QIAzol Lysis Reagent. Briefly, 500 μl of bacteria OD600nm: 1.5 (~109) were spun down at 8000 rpm for 3 minutes. The pellet was dissolved by adding 500 μl of QIAzol and by pipetting up and down at 60 °C for 10 minutes. Next, 100 μl CHCl3 was added and tubes were inverted for 15 seconds. After a 5 minute incubation at room temperature, the supernatant (300 μl) was selected in a new tube, by a full-speed centrifugation at 4 °C for 15 minutes and then mixed with 300 μl iso-propanol by inverting the tubes. The tubes were then let on the bench for 5 minutes and, after a step of full-speed centrifugation at 4 °C for 10 minutes, the pellets were washed out with 500 μl of 70% EtOH. Finally, after centrifugation at 4 °C for 3 minutes and air drying, pellets were resuspended in 20–50 μl RNase free H2O by pipetting and stored at −80 °C. Bacterial RNA was reverse transcribed into complementary DNA (cDNA) using the PrimeScript RT reagent Kit (Perfect Real Time) (Takara: RR037A) according to the manufacturer’s instructions. Real-time PCR was performed, using a Bio-Rad CFX1000 thermal cycler and Bio-Rad CFX96 real-time imager with primer pairs listed below and iQ SYBR green supermix (Bio-Rad).
Primer sequences (5′-3′). “F” and “R” indicate the direction (forward or reverse) of each primer
rplU F: ATGGCGAAGACGTGAAAATC, R: GAACTTGATGATGCGGACCT
clpX F: GTTCGGTCTTATCCCCGAGT, R: AACAGCTTGGCGTACTGCTT
exsC F: CACCGTTTCGATCTGCATTTCG, R: CGAGAATCTGCGCATACAACTG
exoT F: GCCGAGATCAAGCAGATGAT, R: TTCGCCAGTCTCTCCTCTGT
pa1L F: GGTTGCACCCAATAATGTCC, R: CCAATATTGACGCTGAACGA
pilA F: CAGAGGCGACTGGTGAAATC, R: AGGGTAGAGTCAGCCGGAAT.
For each target gene two independent experiments were performed in biological triplicates. All samples were normalized to the expression of the control genes rplU and clpX via the Pfaffl method61.
Swarming motility
Swarming motility was assessed using Petri dishes each containing exactly 20 ml of medium with 5 g/l Bacto-agar (Difco), 8 g/l Nutrient Broth (Difco) and 5 g/l Dextrose. Bacterial cultures with the strains of interest were grown overnight in LB medium and 2 μl from each culture was added at the center of a swarming plate. The plates stayed open until the droplet was fully absorbed by the agar and then incubated for 24 hours at 37 °C. The diameter of the swarming zone on the plate was measured, and photos were taken.
Twitching motility
Twitching motility was assessed using Petri dishes containing 1.0% Bacto agar and 20 g/L LB broth. After the agar was solidified, the indicated strains were stabbed at the bottom of the plates with a sterile toothpick. The plates were incubated at 37 °C for 48 hours. The ability of the bacteria to adhere and form biofilms was examined by removing the agar, washing the unattached cells with water and staining the attached cells with crystal violet (1%). The stain solution was removed by carefully washing the plates with water. The diameter of the twitching zone on the plate was measured, and photos were taken.
Results
Pathogenicity-based classification of Pseudomonas strains
To correlate the pathogenicity of 30 fully sequenced Pseudomonas strains (18 P. aeruginosa and 12 other Pseudomonas strains; Supplementary Table 1), we examined their virulence in two distinct types of acute infection: an oral and a wound Drosophila melanogaster infection assay (Fig. 1A,B)30,62. Kaplan-Meier survival analysis with a log-rank test and pairwise comparison over strata was used to analyze the results of fly survival after the infection. Even though it is common to group bacterial strains based on visual inspection of their survival curves, this procedure is subjective. To objectively partition the strains under study in terms of pathogenicity, the 30 bacterial strains were classified in three groups (low, medium and high), according their effect on fly survival (Fig. 1A,B and Supplementary Table 1), using hierarchical clustering (see Materials and Methods; Supplementary Tables 3-4). Based on published data, PA14 and CF5 were used for both infection type assays to define the “high” and “low” virulence clusters, respectively13,19.
Figure 1.

Comparative survival of all 30 Pseudomonas strains upon oral and wound infection in flies and lung infection in mice. (A,B) Three groups are distinguished, each representing species that are either high, medium or low virulent in fly oral (A) and wound (B) infection. Thin lines represent high virulent strains, thick lines represent medium virulent strains and dashed lines represent low virulent strains. Time is measured in hours. (C) Mouse survival (%) after intranasal mice infection with 3 lowly and 4 highly virulent P. aeruginosa strains. Twenty-microliter aliquots of a bacterial solution containing 2 × 107 bacteria were administered intranasally to each mouse. Infected mice were monitored for 5 days (n = 10).
Non-P. aeruginosa strains were grouped in the “low” and “medium” virulence clusters, with a notable exception of P. entomophila presence in the “high” virulence cluster. P. entomophila is a known entomopathogenic bacterium, able to infect and kill insects, including Drosophila63. P. aeruginosa strains B136-33, MTB-1, PA14, 213BR, 19BR, 9BR and 39016 were consistently virulent, while strains CF5 and C3719 consistently low in virulence regardless of the infection assay. Some P. aeruginosa strains were less consistently grouped between assays, such as PAO1, which was grouped with the “high” and “medium” virulence cluster in the wound and oral infection assays, respectively. To validate and more rigorously select high and low in acute virulence strains we used a model of acute intranasal mouse lung infection. We assessed the mortality rate of four of the highly (B136-33, MTB-1, PA14, 213BR) and the three most lowly (C3719, CF5, PACS2) virulent in Drosophila P. aeruginosa strains. We found that all but one (213BR) of the 7 tested strains retained their virulence potential in the acute mouse lung infection model (Fig. 1C).
Presence-Absence of single genes cannot explain Pseudomonas pathogenicity
We inferred a phylogenetic tree of all the 30 Pseudomonas strains based on the concatenated alignments of two core housekeeping genes, the 16 S rRNA and gyrB (Supplementary Fig. 1) in agreement with the trees constructed using four housekeeping genes or 1679 orthologs per Duan et al.34. Our results were also in agreement with previous observations that place Pseudomonas sp. UW4 within the P. fluorescens clade, even though phylogenetic analysis based on 16S rRNA sequences alone indicates a closer relationship to the P. putida clade.
Next, we created a database of 254 virulence factors (VFs) previously found in the Pseudomonas species included in our analysis (see Methods). We classified these VFs according to their functions in five groups: type II secretion system (T2SS; 18 genes), type III secretion system (T3SS; 38 genes), type VI secretion system (18 genes), quorum sensing (44 genes) and other (136) (presented sequentially in the Supplementary Table 2). For each category a BBH BLASTP search allowed us to detect the phylogenetic profiles of VFs across the Pseudomonas species. Five multiple array viewers (one for each functional category) were obtained indicating the presence-absence of VFs. A sixth multiple array viewer was obtained by merging all VFs together (Fig. 2A). All VFs found to be absent in a particular genome were manually re-examined with a TBLASTN analysis to exclude cases where the observed absence was the result of erroneous gene-finding or incomplete annotation. We found that most VFs are present in all P. aeruginosa strains, even in the less virulent strains, such as the stain CF5 (8th lane of Fig. 2A and Supplementary Table 2). On the other hand, most P. aeruginosa VFs were absent in non-P. aeruginosa strains (last 12 lanes of Fig. 2A and Supplementary Table 2). Based on the VF presence-absence profile highly and lowly virulent P. aeruginosa strains cluster together rather than clearly apart, and separately from the other Pseudomonas strains (Fig. 2B). Therefore, the presence-absence differences in VFs likely reflect the differences between P. aeruginosa and other Pseudomonas strains, but not the highly from the lowly virulent P. aeruginosa strains. Regarding the type II secretion system (T2SS), all P. aeruginosa strains have all genes except from the strain PA7, which is missing the toxA gene, as previously published17. However, non-P. aeruginosa strains are missing almost all T2SS factors. For example, P. putida KT2440 is known for lacking the toxA gene, while the Xcp family is replaced by the Gsp family in this strain64.
Figure 2.

Virulence factor Presence-Absence follows phylogeny rather than pathogenicity. (A) Part of a multiple array viewer based on the Presence-Absence (PA) of virulence factors (VFs). Present genes are shown in light grey, absent genes are shown in dark grey. The 18 vertical lanes on the left correspond to the 18 P. aeruginosa strains, while the next 12 lanes to the other Pseudomonas strains. (B) Clustering based on the presence/absence of virulence factors. The 2 lowly virulent in both fly infection assays P. aeruginosa strains are shown in dashed-line boxes; and the 7 high virulent in both assays P. aeruginosa strains in full-line boxes. (C) Phylogeny rather than pathogenicity of the 30 Pseudomonas spp. correlates best with Presence-Absence of virulence factors.
Regarding the type III secretion system (T3SS), four P. aeruginosa effector proteins have been functionally identified so far, ExoU, ExoS, ExoT and ExoY65. In agreement with previous studies66–71, we observed that most of the P. aeruginosa strains have either the exoS or the exoU gene, but not both, while the PA7 strain carries neither of them (Supplementary Table 2). While the PA7 strain lacks the entire T3SS17, recent studies reveal that the cytolytic activity of PA7-like strains is based on Exolysin (ExlA), a secreted 172 kDa pore-forming toxin, exported by a two-partner secretion (TPS) system72–74. The P. aeruginosa 39016 strain is missing six T3SS genes (exoS, exsE, exsC, pcr3, popD, pscE), the LESB58 is missing four genes (exoU, pcrH, exsE, pscP) and the CF5 strain, known as a lowly virulent P. aeruginosa strain is only missing three genes (exoU, exoT, pscQ). Two T3SS genes are also missing from the strains 2192 (exoU, exsC) and SCV20265 (exoU, pscE). As with the T2SS, T3SS is nearly entirely missing from all non-P. aeruginosa strains, and this is in agreement with previous data on P. putida64.
Regarding the type VI secretion system, the 39016 strain is missing three genes (hcpC, tagT, vgrG1) and the C3719 strain is missing only one gene (dotU). Interestingly, P. fluorescens has the T6SS gene hcp and P. protegens has the vgrG, hcp, lip, icmF, dotU, clpV, ppka and pppA genes. Moreover, the P. aeruginosa quorum sensing genes pzhA1, pzhA2, phzB2, rhlI and rhlR involved in phenazine biosynthesis are missing, not only from the non-P. aeruginosa strains, but also from some P. aeruginosa strains, primarily the low in virulence strains CF5 and C3719 (Supplementary Table 2).
We proceed by clustering Pseudomonas strains based on the presence-absence of specific VFs for the five VF functional classes and for all VFs. The resulting clusters were further analyzed using the Adjusted Rand Index (see Materials and Methods) to reveal hidden correlations to (a) overall Pseudomonas phylogeny, (b) pathogenicity ranking according to the wound infection assay, and (c) pathogenicity ranking according to the oral infection assay. We found that phylogeny, but also pathogenicity to wound infection, correlate with VF gene content (Fig. 2C), indicating that VF gene content differences are due to overall Pseudomonas phylogeny and do not reflect directly the pathogenicity of the 30 Pseudomonas species. Accordingly, the phylogeny of the 30 strains correlated well with: (i) their virulence classification in the wound infection (but not in the oral infection assay); (ii) VF gene content; and (iii) major metabolic pathway gene content. To eliminate the effect of extreme phylogenetic differences due to the inclusion of non-P. aeruginosa strains, the analysis was repeated with only the P. aeruginosa strains, in which case no correlation was evident for any comparison. Thus, we reconfirm earlier works claiming that the difference in virulence potential of P. aeruginosa cannot be fully attributed to the mere presence or absence of known virulence factors13, but also to other genomic differences yet to be determined.
Gene expression of selected VFs, but not of core metabolism genes, correlates with pathogenicity
To identify differentially expressed genes relating to pathogenicity we performed a transcriptome analysis of the 6 P. aeruginosa strains validated in mice as “high” (B136-33, MTB-1, PA14) or “low” (C3719, CF5, PACS2) in virulence (Fig. 1C). RNA was extracted from bacteria grown at mid-exponential and early stationary phase (optical density OD600nm: 1 and 3, respectively). We selected genes differentially regulated in the same direction (either over- or under-expressed) between all pairs of “high” vs. “low” in virulence strains. Nine genes were differentially expressed at both growth phases (8 of which were overexpressed in highly virulent strains) (Fig. 3A). At OD600nm: 1, 15 genes were up, and 6 genes were down in all 3 highly versus all 3 lowly pathogenic species (Fig. 3A). At OD600nm: 3, 52 were up and 2 genes were down in all 3 highly versus all 3 lowly pathogenic species (Fig. 3A). Overall, 20 VFs (most of them being quorum sensing and T3SS related), 10 metabolism genes, 5 transcriptional regulators and a number of hypothetical proteins were differentially expressed (Supplementary Table 5).
Figure 3.

Differential strain-to-strain expression and role in virulence of P. aeruginosa metabolic genes. (A,B) Genes differentially expressed between 3 lowly and 3 highly virulent P. aeruginosa strains: (A) VENN diagrams of the 66 differentially expressed genes identified by RNAseq transcriptomics analysis using http://bioinformatics.psb.ugent.be/webtools/Venn/. At OD600nm: 1, 15 genes were up (VENN diagram or the left) and 6 genes were down (VENN diagram on the right) in all 3 highly vs. all 3 lowly pathogenic species. At OD600nm: 3, 52 were up (VENN diagram or the left) and 2 genes were down (VENN diagram or the right) in all 3 highly vs. all 3 lowly pathogenic species. (B) Mutants for all 10 general metabolism differentially expressed genes were tested for virulence in the fly oral infection model, and 2 of them were found attenuated. (C–F) Screen results for 482 metabolic and 94 non-metabolic PA14 genes using two Drosophila infection assays (feeding and pricking): (C,D) Z-score analysis of the fly survival after infection of flies with 553 PA14 metabolic mutants (corresponding to 482 genes) using two independent assays: Feeding Assay (C) and Pricking Assay (D). The time of 50% fly death (LT50%) was assessed for each mutant and condition and a Z-score analysis was used to select those with a score >1 for any of the two assays, from which upon retest, 78 were found significantly attenuated in virulence per Kaplan-Meier survival analysis with a log-rank test. (E,F) Z-score analysis of the fly survival after infection of flies with 95 randomly selected non-metabolic PA14 mutants (corresponding to 94 genes) using two independent assays: Feeding Assay (E), Pricking Assay (F). The time of 50% fly death (LT50%) was assessed for each mutant and condition and a Z-score analysis was used to select those with a score >1 for any of the two assays, from which upon retest, 8 were found significantly attenuated in virulence per Kaplan-Meier survival analysis with a log-rank test.
Among the virulence genes overexpressed in highly pathogenic species are, exoU and spcU. This is simply because they do not exist in the lowly pathogenic species assessed. Furthermore, out of the 18 P. aeruginosa strains, 5 (PA14, B136-33, MTB-1, NCGM2.S1 and 39016) have the exoU gene, and all of them, except from 39016, have next to exoU the spcU gene, a chaperone required for efficient secretion of the ExoU cytotoxin75. Hypothetical proteins overexpressed in highly pathogenic species include: PA14_20600, a DUF3313 domain-containing protein (PFAM ID: PF11769), a domain defining a family of putative bacterial lipoproteins; PA14_04710, a 4Fe-4S ferredoxin that, like other ferredoxins, mediates electron transfer in a wide variety of metabolic reactions76; PA14_30690, a fimbrial protein playing a role in cell adhesion; and PA14_01120 that maps to the antitoxin Tsi6 of the structurally characterized Tse6::Tsi6 complex, a toxin-antitoxin system and is related to T6SS77. Quorum sensing genes, including, the autoinducer rhlI and its receptor rhlR, the rhamnolipid biosynthesis genes rhlA and rhlB, the proteases lasA and lasB, the pa1L and lecB lectins and the phenazine biosynthesis genes phzB1, phzC1, phzE1, phzM and phzS, were consistently overexpressed among the 3 highly vs. the 3 lowly virulent strains (Supplementary Table 5). Thus, contrary to the inability of VF presence-absence profiles to distinguish highly from lowly pathogenic P. aeruginosa strains, our data indicate that overexpression of particular VFs is indicative of virulence potential. However, VF control is very complex, and additional regulating factors need to be investigated to improve our understanding on their regulation.
Among the 10 metabolic genes, 8 were found upregulated in the highly virulent strains, but mutations in none of them exhibited defects in virulence in flies. Mutations, on the other hand, in the 2 downregulated in highly virulent strains metabolic genes exhibited clear defects in virulence (Fig. 3B). Of the latter, PA14_54640 (dspI) encodes for a putative enoyl-CoA hydratase involved in biofilm dispersion. Its mutation produces high biofilm formation, a condition that may promote chronicity to the expense of acute virulence78,79. The second downregulated in highly virulent strains metabolic gene PA14_72880 encodes for the putative short-chain dehydrogenase involved in fatty acid biosynthesis. Mutations of its orthologs in PAO1 reduce biofilm formation but also swarming motility and LasB protease activity80. The fact that a number of metabolic genes appear among the consistently differentially expressed genes (Fig. 3A) begs the question of the broader contribution of metabolism in P. aeruginosa virulence24.
Relative contribution of core metabolic and non-metabolic P. aeruginosa PA14 genes to virulence
To functionally assess the contribution of the core metabolism genes of P. aeruginosa to virulence, we took advantage of the PA14 unigene Transposon Insertion Mutant Library. The PA14 strain is highly pathogenic and well annotated. Its genome contains 6,537,648 nucleotides and 5,977 annotated genes3,19 794 of which were annotated as metabolic in the KEGG database in the early phase of this project in 2014 (961 genes were annotated as metabolic in October 2019). We investigated the virulence in flies of all 553 available and retrievable core metabolism gene mutants of the PA14 mutant library. These correspond to 482 core metabolism genes and 95 randomly selected mutants corresponding to 94 non-metabolic genes in the pricking (wound) and feeding (oral) infection fly assays. To avoid false negatives, we manually screened 3 replicates of 10 wild type orally infected and 2 replicates of 20 wild type wound infected flies with each of the 648 PA14 mutant strains. The time of 50% fly death (LT50%) was measured for each mutant in each assay and a Z-score analysis was used to select mutants allowing fly survival to infection by more than 1 standard deviation of the normalized mean (Fig. 3C-F). To avoid false positives, selected mutants were retested for virulence and the number of the attenuated mutants was finalized after performing Kaplan-Meier survival analysis with a log-rank test. Considering both assays, 16.2% (78/482) of the metabolic and 8.5% (8/94) of the non-metabolic P. aeruginosa genes, were found virulence-defective in flies (Fig. 4A). Even though the number of strains tested only marginally fails to statistically support that more metabolic than non-metabolic genes are implicated in virulence (Fisher exact test: p = 0.0583), the numbers clearly suggest that metabolism has a considerable contribution in P. aeruginosa virulence.
Figure 4.

P. aeruginosa core metabolism genes are abundantly represented among genes required specifically for full virulence in flies and mice. (A) Five hundred and fifty-three (553) P. aeruginosa metabolic Tn-mutants corresponding to 482 core metabolism genes and 95 randomly selected non-metabolic P. aeruginosa Tn-mutants corresponding to 94 non-metabolic genes were screened in the wound and oral infection Drosophila assays. Sixteen percent (16.2%) of the core metabolism and 8.5% of the non-metabolic P. aeruginosa genes were selected as required for full virulence. Examination of growth capacity of the selected mutants in minimal media and in the host revealed the percentage of core metabolism genes dispensable for growth, i.e. important specifically for full virulence, is at least as high as that of the non-metabolic genes. (B) Assignment of the 57 PA14 virulence-related metabolic genes in metabolic categories. The 57 core metabolism P. aeruginosa gene mutants that grow normally in minimal media and/or in the host belong in one or more of 7 general core metabolism categories. Each category is represented by at least 3 virulence-related genes, while nucleotide, carbohydrate and amino acid metabolism categories by 10 or more. (C) Survival kinetics of mice after intranasal inoculation with PA14 metabolic mutants. The graph shows the % mice survival after intranasal inoculation with 15 PA14 metabolic mutants and the wild type strain. All strains, but wild type PA14 (blue) and hom (light purple), overlap on the horizontal 100% survival line. Twenty-microliter of saline containing 2 × 107 bacteria was administered intranasally to each mouse. Infected mice were monitored for 6 days (n = 10–11).
Growth assessment of the virulence-defective PA14 metabolic mutants
The identified metabolism gene mutants might be impaired in growth (exhibit auxotrophy) rather than being directly involved in virulence factor production. Therefore, we sought to determine which of the 78 attenuated in virulence PA14 metabolic gene mutants are growth-essential in culture or necessary for host colonization during infection. We assessed growth in two types of minimal media (one without and one with fly extract as an extra nutrient), and the level of colonization during infection at the respective infection model (wound or oral) in which each mutant was found attenuated in virulence.
Using a standard minimal medium we initially identified 34 out of the 78 core metabolic mutants as prototrophs for being able to grow similarly to the wild type strain (Table 1). The remaining 44 core metabolic mutants did not grow at all in this medium, requiring additional nutrients for their growth. Nevertheless, if the needed nutrients are available in flies during infection, the lack of virulence should be due to a direct effect of the mutation in virulence rather than in growth. To assess this possibility, we inoculated minimal media supplemented with 5% fly extract with each of the 44 core metabolic mutants. This medium offers all the nutrients the bacterium can find in flies in the absence of the host defense against the bacteria. We found 12 of the 44 PA14 metabolic mutants to grow similarly to the wild type and considered them conditional prototrophs (Table 2 - Group A, B). All 8 virulence-defected non-metabolic mutant strains were also able to grow in this medium (Supplementary Fig. 2A,B) and we did not test them any further.
Table 1.
Thirty-four virulence-related prototrophs based on growth in glucose minimal media. From the 78 virulence-defective PA14 metabolic mutants, 34 can grow like the wild type in the M9 medium, indicating a connection of the corresponding genes with virulence.
| # | Metabolic PA14 mutants | In vitro growth | In vivo growth (colonization) | |
|---|---|---|---|---|
| Minimal medium (M9) | Intestinal | Wound | ||
| 1* | PA14_00120 (lipid A biosynthesis lauroyl acyltransferase) | + | − | − |
| 2 | PA14_04320 (ilvA1) | + | + | NT |
| 3 | PA14_07600 (folk) | + | + | NT |
| 4* | PA14_11250 (hypothetical protein) | + | − | − |
| 5 | PA14_11810 (aldehyde dehydrogenase) | + | + | NT |
| 6* | PA14_14680 (suhB) | + | − | − |
| 7 | PA14_17450 (surE) | + | + | NT |
| 8* | PA14_22050 (htrB) | + | − | − |
| 9 | PA14_20670 (glutamine synthetase) | + | + | NT |
| 10 | PA14_29860 (nuoM) | + | + | NT |
| 11 | PA14_29980 (nuoE) | + | + | NT |
| 12 | PA14_29990 (nuoD) | + | + | NT |
| 13 | PA14_31580 (acyl-CoA dehydrogenase) | + | + | NT |
| 14 | PA14_32690 (gtdA) | + | + | NT |
| 15 | PA14_38510 (hmgA) | + | + | NT |
| 16* | PA14_40980 (enoyl-CoA hydratase) | + | + | + |
| 17 | PA14_44070 (gltA) | + | + | NT |
| 18 | PA14_45010 (hyi) | + | + | NT |
| 19 | PA14_52050 (purN) | + | − | NT |
| 20 | PA14_52610 (hypothetical protein) | + | + | NT |
| 21 | PA14_52800 (acsA) | + | + | NT |
| 22 | PA14_65320 (miaA) | + | + | NT |
| 23 | PA14_66670 (ponA) | + | + | NT |
| 24 | PA14_68580 (pckA) | + | + | NT |
| 25 | PA14_70160 (bioA) | + | + | NT |
| 26 | PA14_71970 (wbpW) | + | + | NT |
| 27 | PA14_72850 (glutamine synthetase) | + | + | NT |
| 28 | PA14_00020 (dnaN) | + | NT | + |
| 29 | PA14_07170 (epd) | + | NT | + |
| 30 | PA14_08780 (rpoC) | + | NT | + |
| 31 | PA14_31700 (pgsA) | + | NT | + |
| 32 | PA14_32670 (hypothetical protein) | + | NT | + |
| 33 | PA14_41020 (adi) | + | NT | + |
| 34 | PA14_63990 (speA) | + | NT | + |
Feeding assay-selected: 1-27; Those selected in both assays are shown with asterisk (*); Pricking assay-selected only: 28-34; NT: Not Tested.
Table 2.
Forty-four virulence-defective mutants: 23 conditional prototrophs & 21 auxotrophs. These metabolic mutants can be grouped in metabolic pathways based on the KEGG database25. They are unable to grow in glucose minimal medium (M9), but 23 of them can grow “conditionally” like the wild type strain in M9 supplemented with 5% fly extract and/or in flies. The remaining 21 do not grow like the wild type in any condition tested.
| # | Major Pathways (KEGG database entry) | Metabolic PA14 mutants | In vitro growth | In vivo growth (colonization) | ||
|---|---|---|---|---|---|---|
| Glucose minimal medium (M9) with 5% Fly Extract | Intestinal | Wound | ||||
| 1 | Histidine metabolism (pau00340) | PA14_57770 (hisC1) | + | + | NT | Group (A) (# 6 mutants) Efficient growth in glucose minimal medium with 5% fly extract and in vivo |
| 2 | PA14_57780 (hisD) | + | + | NT | ||
| 3 | PA14_66950 (hisE) | + | + | NT | ||
| 4 | PA14_65250 (hisX) | + | + | NT | ||
| 5 | One carbon pool by folate (pau00670) | PA14_05590 (metF) | + | + | NT | |
| 6 | Pantothenate and CoA biosynthesis (pau00770) | PA14_62580 (panB) | + | + | NT | |
| 7 | PA14_62590 (panC) | + | − | NT | Group (B) (# 6 mutants) Efficient growth only in glucose minimal medium with 5% fly extract | |
| 8 | Purine metabolism (pau00230) | PA14_51240 (purC) | + | − | NT | |
| 9* | PA14_64220 (purD) | + | − | − | ||
| 10 | PA14_23920 (purF) | + | − | NT | ||
| 11* | PA14_64200 (purH) | + | − | NT | ||
| 12 | PA14_15740 (purL) | + | − | NT | ||
| 13* | Pyrimidine metabolism (pau00240) | PA14_05260 (pyrB) | − | + | − | Group (C) (# 11 mutants) Efficient growth only in vivo |
| 14* | PA14_05250 (pyrC) | − | − | + | ||
| 15* | Glycine, serine and threonine metabolism (pau00260) | PA14_16070 (hom) | − | + | + | |
| 16* | PA14_16090 (thrC) | − | − | + | ||
| 17 | Arginine biosynthesis (pau00220) | PA14_08480 (argC) | − | + | NT | |
| 18* | PA14_18740 (argG) | − | + | NT | ||
| 19 | PA14_18610 (argF) | − | NT | + | ||
| 20 | Glycolysis/Gluconeogenesis (pau00010) | PA14_66310 (aceF) | − | + | NT | |
| 21* | PA14_66290 (aceE) | − | + | NT | ||
| 22 | Cysteine and methionine metabolism (pau00270) | PA14_05620 (sahH) | − | − | + | |
| 23 | Lysine biosynthesis (pau00300) | PA14_69670 (lysA) | − | + | NT | |
| 24 | Phenylalanine, tyrosine and tryptophan biosynthesis (pau00400) | PA14_00450 (trpB) | Group (D) (# 21 mutants) Slow growth compared to the wild type PA14 in all the above assays | |||
| 25 | PA14_08360 (trpC) | |||||
| 26 | PA14_08350 (trpD) | |||||
| 27 | PA14_23850 (trpF) | |||||
| 28 | PA14_66600 (aroB) | |||||
| 29 | Valine, leucine and isoleucine biosynthesis (pau00290) | PA14_62130 (ilvC) | ||||
| 30 | PA14_04630 (ilvD) | |||||
| 31 | PA14_62150 (ilvH) | |||||
| 32 | PA14_62160 (ilvI) | |||||
| 33 | PA14_23790 (leuB) | |||||
| 34 | PA14_23750 (leuC) | |||||
| 35* | Pyrimidine metabolism (pau00240) | PA14_18710 (pyrC) | ||||
| 36* | PA14_24640 (pyrD) | |||||
| 37 | PA14_26890 (pyrF) | |||||
| 38* | PA14_62930 (carA) | |||||
| 39 | Pentose phosphate pathway (pau00030) | PA14_22910 (edd) | ||||
| 40 | PA14_23090 (edaA) | |||||
| 41 | Arginine biosynthesis (pau00220) | PA14_70280 (argB) | ||||
| 42 | PA14_69500 (argH) | |||||
| 43 | Glycine, serine and threonine metabolism (pau00260) | PA14_65560 (serB) | ||||
| 44 | Glycolysis/Gluconeogenesis (pau00010) | PA14_62830 (tpiA) | ||||
*: Found attenuated in both assays (pricking and feeding); NT: Not Tested.
Moreover, we sought to examine the 78 metabolic mutant strains during initial colonization, to identify auxotrophic strains able to colonize the flies, but also to study the colonization ability of the prototrophs. We adapted our wound and intestinal infection assays to assess colonization efficiency rather than fly survival. To assess intestinal colonization we infected flies for only one day with all flies being subsequently transferred to tubes with clean food, every day for 3 days. To assess wound colonization we injected the bacteria in the flies instead of pricking them with a tungsten needle to bypass the fly immune system that can easily eliminate attenuated in virulence bacteria at the wound site30. Calculating the number of retrievable bacteria per fly we found that 29 of the 34 prototrophs (Table 1) and 17 of the remaining 44 strains (Table 2 - Group A, C) were able to colonize the flies like the wild type. In summary, we found 34 prototrophs (Table 1), 23 conditional prototrophs able to grow either in the host or in minimal medium cultures supplemented with fly extract (Table 2, Group A-C) and 21 auxotrophs unable to grow in the host or in culture (Table 2, Group D). Accordingly, 11.8% (57/482) of the core metabolic and 8.5% (8/94) of the non-metabolic attenuated strains were categorized as virulence-related (Fig. 4A), suggesting that many and functionally disparate metabolism genes are connected to P. aeruginosa virulence.
Virulence-related metabolic genes of P. aeruginosa PA14 are part of core metabolism and are necessary for full virulence in mice
According to the KEGG database, the selected 57 virulence-related core metabolic genes belong to 7 metabolic categories and various metabolic gene pathways (Fig. 4B). Assessing the virulence in a mouse lung infection assay of mutants representing each core metabolic pathway as defined in the KEGG database we identified 15 mutants being virulence-defective in flies and mice. These mutants affect genes related to amino acid metabolism [PA14_57770 (hisC1), PA14_57780 (hisD), PA14_65250 (hisX), PA14_32690 (gtdA), PA14_16070 (hom)], nucleotide metabolism [PA14_17450 (surE), PA14_64220 (purD), PA14_52050 (purN)] and the metabolism of co-factors and vitamins [PA14_05590 (metF), PA14_07170 (epd), PA14_07600 (folKfolk)] (Fig. 4C and Table 3).
Table 3.
Pathways of the 15 PA14 metabolic genes corresponding to mutants found attenuated in mice lung infection. The names of the metabolic genes are listed in the first column. The second column shows the corresponding metabolic pathways based on the KEGG database25, while the last column shows the major central metabolic pathways in which those pathways belong.
| # | Genes/Names | Metabolic Pathways (KEGG database entry) | Major Central Metabolic Pathways |
|---|---|---|---|
| 1 | PA14_17450 (surE) | Purine (pau00230), Pyrimidine (pau00240) & Nicotinate and nicotinamide metabolism (pau00760) | Nucleotide metabolism Metabolism of cofactors and vitamins |
| 2 | PA14_64220 (purD) | Purine metabolism (pau00230) | |
| 3 | PA14_52050 (purN) | Purine metabolism (pau00230), One carbon pool by folate (pau00670) | |
| 4 | PA14_08780 (rpoC) | RNA polymerase (pau03020) | |
| 5 | PA14_57770 (hisC1) | Histidine metabolism (pau00340) & Phenylalanine, tyrosine and tryptophan biosynthesis (pau00400) | Amino acid metabolism |
| 6 | PA14_57780 (hisD) | Histidine metabolism (pau00340) | |
| 7 | PA14_65250 (hisX) | Histidine metabolism (pau00340) | |
| 8 | PA14_32690 (gtdA) | Tyrosine metabolism (pau00340) | |
| 9 | PA14_16070 (hom) | Glycine, serine and threonine metabolism (pau00260), Cysteine and methionine metabolism (pau00270) & Lysine biosynthesis (pau00300) | |
| 10 | PA14_05590 (metF) | One carbon pool by folate (pau00670) | Metabolism of cofactors and vitamins |
| 11 | PA14_07170 (epd) | Vitamin B6 metabolism (pau00750) | |
| 12 | PA14_07600 (folk) | Folate Biosynthesis (pau00790) | |
| 13 | PA14_68580 (pckA) | Glycolysis/Gluconeogenesis (pau00010), TCA cycle (pau00020), Pyruvate metabolism (pau00620) | Carbohydrate metabolism |
| 14 | PA14_65320 (miaA) | tRNA dimethylallyltransferase (pau01100) | Transfer RNA biogenesis |
| 15 | PA14_31700 (pgsA) | Glycerophospholipid metabolism (pau00564) | Lipid metabolism |
Virulence-related metabolic genes of P. aeruginosa PA14 are compromised in various aspects of virulence
To better understand the loss in virulence of the selected metabolic gene mutants, we studied them for the production of virulence factors associated with acute infection, namely, bacterial motility, and T3SS and quorum sensing gene expression. Swarming and twitching motility is very important for P. aeruginosa virulence81 and we tested both types of motility of 13 PA14 metabolic transposon (Tn) mutants found attenuated in both flies and mice. Mutants for epd, surE, gtdA, purN, folK, metF, hom and rpoC, were noticeably defective for swarming motility compared to the wild type strain (P < 0.001 for the first 7 and P < 0.01 for the last one), while pgsA and purD, exhibited increased swarming motility compared to the wild type (P < 0.05) (Fig. 5A). Regarding the twitching motility, the mutants surE, gtdA, miaA, rpoC, pgsA and purN exhibited a strong decrease in their ability to twitch compared to the wild type (P < 0.01) (Fig. 5B), but also the pckA, folK and epd mutants were significantly defective in twitching motility (P < 0.05) (Fig. 5B). Only hisD and purD Tn-mutants were not impaired in any of the two motilities.
Figure 5.
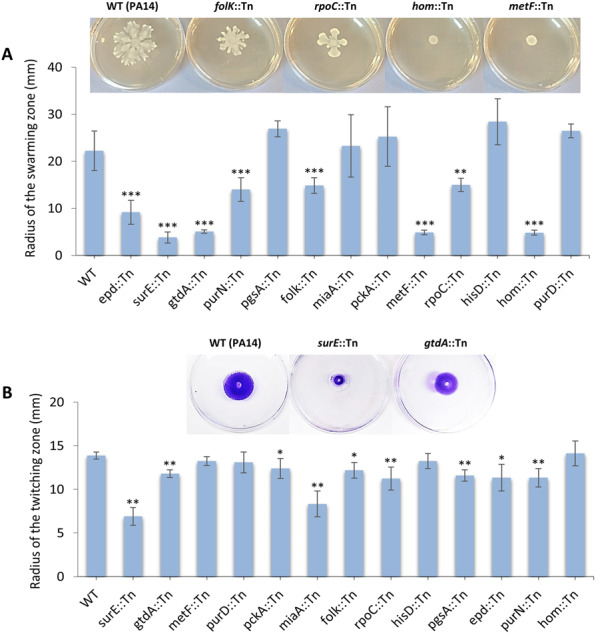
Swarming and twitching phenotypes of 13 PA14 metabolic gene mutants. (A) Swarming motility was measured by the length of the swarming zone from the center of the plate after inoculation of the cells at the center of the agar media and incubation at 37 °C for 24 h. The results are from 3 independent experiments. n = 3-13; **P < 0.01; ***P < 0.001 compared to WT by Mann-Whitney U test. Representative photos of Petri dishes exhibiting wild type PA14 and selected mutants with defective swarming motility, shown above the graph. (B) For the twitching motility the cells were stab-inoculated onto LB twitching plates (1% agar). The plates were incubated at 37 °C for 48 hours. The agar was removed, and the twitching zone was revealed by staining with crystal violet, measuring the maximum length of the twitching zone from the center of the plate. The results are from 3 independent experiments. n = 4-7; *P < 0.05; **P < 0.01 compared to WT by Mann-Whitney U test. Representative photos of Petri dishes exhibiting wild type PA14 and selected mutants with defective twitching motility, shown above the graph. In both (A) and (B) error bars depict standard deviation.
Gram-negative bacterial pathogens have evolved multiple protein secretion systems that facilitate the infection of eukaryotic hosts. P. aeruginosa deploys its type III secretion system (T3SS) to enhance its pathogenicity by injecting cytotoxic effector proteins into the host cells82. T3SS expression is regulated transcriptionally and post-transcriptionally in response to host cell contact and environmental Ca2+ levels83. We examined the expression of the T3SS regulatory gene exsC and the effector protein exoT, in the 13 Tn-mutants, under Ca2+ limiting conditions (LB supplemented with 5 mM EGTA). We observed reduced expression of the exoT in 10 of the 13 selected mutants and overexpression in 1 of them, compared to the wild type (Fig. 6A). Similarly, the expression of exsC is reduced in 5 of these mutants, with only purD remaining unaffected in the expression of any of the two genes (Fig. 6B). Nevertheless, purD as well as the miaA, rpoC, surE, purN and gtdA mutants were compromised in the expression of the type IV pilus biogenesis gene pilA (Fig. 6D), while the pa1L gene, which is controlled by quorum sensing is significantly affected by miaA, rpoC and tentatively by purD (Fig. 6C). miaA, is a tRNA isopentenyl transferase, a tRNA modification enzyme important for translation efficiency, while rpoC is the DNA-directed RNA polymerase beta subunit that has an important role in transcription. Considering that total disruption of rpoC might be lethal for P. aeruginosa84, the transposon mutant available for rpoC in the library we used might be a hypomorph with reduced gene expression or protein function due to the transposon insertion being at the end of the gene. Assuming off-target effects of transposon mutagenesis are absent from most of the mutants, our results suggest that loss of metabolic gene function impacts acute virulence by compromising one or more key mechanisms of P. aeruginosa virulence.
Figure 6.

Expression of various virulence factors of 13 PA14 metabolic gene mutants. (A,B) Expression of T3SS genes in PA14 metabolic mutants. (C,D) Expression of pa1L and type IV pilus gene pilA in PA14 metabolic mutants. Bacteria were grown to an OD600nm of 1 in LB with 5 mM EGTA before RNA extraction and cDNA synthesis. *P < 0.05; **P < 0.01 compared to WT by Mann-Whitney U test (n = 3–7). In all plots (A–D) the average fold change ± standard deviation is shown.
Core metabolism pathways containing necessary for full virulence and virulence-related differentially expressed genes
To pinpoint metabolic gene expression patterns important for P. aeruginosa virulence, we assessed the differential expression of the 78 core metabolism genes required for the full virulence of P. aeruginosa strain PA14. Comparisons were performed for the 3 selected as high vs. the 3 selected as low in virulence P. aeruginosa strains for each of the bacterial growth phases. 45 of the 78 functionally important metabolic genes did not exhibit differential expression among any of the 18 comparisons performed. Of the remaining 33 genes, 32 exhibited a significant differential expression in up to 8 of the total 18 pairwise combinations (Supplementary Fig. 3). Thus, no consistent differential gene expression is noted for any but one of the 78 core metabolism genes required for full virulence.
However, using the BioCyc Pathway/Genome Database Collection we identified 107 differentially expressed metabolic pathways containing genes differentially expressed between at least one highly (PA14, MTB-1, B136-33) vs. all 3 lowly (C3719, CF5, PACS2) virulent strains and pathways containing one or more of the 78 genes necessary for full virulence. Overlapping these pathways, we find 8 pathways containing genes upregulated in all 3 highly virulent strains vs. all 3 lowly in virulence strains and at least one gene required for full virulence when mutated (Fig. 7A; Supplementary Table 6). These common pathways are annotated as beta-oxidation, xylenol degradation, Gln biosynthesis, branched chain amino acid biosynthesis, succinate biosynthesis, citramalate biosynthesis, and chorismate biosynthesis (Fig. 7B); and their functions include: (i) the 4-hydroxyl-phenylacetate degradation and succinate production, (ii) the glutamine biosynthesis from glutamic acid, (iii) the shikimate and chorismate biosynthesis from D-erythrose 4-phosphate, (iv) the branched-chain amino acid biosynthesis of leucine, (v) the 2,5- and 3,5-xylenol degradation to citramalate, and (vi) the beta-oxidation of fatty acids. Of note, genes required for full virulence when mutated and those upregulated in each of the highly virulent strains are not usually the same, but they frequently belong in the same pathway (Supplementary Fig. 4). Therefore, differences in virulence among P. aeruginosa strains maybe partly due to the differential expression of genes belonging in these core metabolism pathways.
Figure 7.

(A) Overlap among 4 metabolic gene pathway groups. Three expression-based metabolic pathway groups arise from differentially expressed genes comparing each of the 3 high (B136–33, MTB-1 or PA14) with all 3 low (CF5, C3719 and PCS2) in virulence strains, while the 4th functionality-based metabolic pathway group arises from the 78 genes functionally important for full virulence in flies. Out of 137 metabolic pathways assessed for overlap based on the BioCyc Database Collection (https://biocyc.org), 21 were common to the 3 expression-based metabolic pathway groups, while 8 of them were also functionally important for virulence. We circle the numbers of the 8 functionality-based plus 13 more, totaling 21 common expression-based metabolic pathways. (B) Common metabolic pathways implicated in P. aeruginosa virulence. Highlighted are the common pathways shown in (A) within the Cellular Overview of P. Aeruginosa PA14 metabolism from BioCyc.
Discussion
Aspects of bacterial metabolism have been linked to virulence, including the T3SS85, the regulation of exotoxin A expression86,87 and the adaptation to the host nutritional environment88. Carbon source availability, for example, affects virulence genes presumably as an adjustment to the host micro-environment89. P. aeruginosa gene AaaA is an arginine-specific autotransporter providing a fitness advantage in chronic wound infections where the sole source of nitrogen is peptides with an aminoterminal arginine90. In addition, P. aeruginosa CbrA is a sensor kinase involved in carbon and nitrogen utilization, which affects swarming, biofilm formation, cytotoxicity, and antibiotic resistance91. Computational screening further elucidates interconnectivity between P. aeruginosa virulence factor synthesis and growth with a metabolic network as a denominator24. Such studies support the notion that core metabolism genes are growth-essential and unsuitable drug targets due to their high potential for resistance development24. Our functional transcriptomics approach though suggests the existence of a widespread growth-independent effect of core metabolism gene expression in virulence.
We defined as virulence genes those necessary to the full virulence of P. aeruginosa independently of their effect on bacterial growth or host colonization ability. Thus, we included as growth-independent, mutants able to grow similarly to the wild type strain in minimal media (M9) or M9 supplemented with fly extract. We also included attenuated in virulence mutants able to colonize the host as efficiently as the wild type isogenic strain PA14. We found that 11.8% of the growth-independent core metabolism genes and 8.5% of the non-metabolic genes are directly linked to virulence. This is in agreement with previous studies linking primary with secondary virulence-related metabolites for being synthesized using the same precursor molecules92. For example, chorismate is the last common precursor for the biosynthesis of the primary aromatic metabolites phenylalanine, tyrosine and tryptophan as well as of the secondary aromatic metabolites phenazine-1-carboxylic acid and pyocyanin, which are known virulence factors92. P. aeruginosa utilizes the siderophore pyochelin during iron limitation, which is synthesized by the conversion of chorismate to isochorismate by the isochorismate synthase PchA and of isochorismate to salicylate by the isochorismate-pyruvate lyase PchB93. In addition, the primary metabolite anthranilate is a precursor for the P. aeruginosa quinolone signal (PQS), which is a secondary metabolite having a crucial role in quorum sensing94. Of note, the other major siderophore produced by P. aeruginosa, pyoverdine, is synthesized from L-ornithine-derived hydroxamates and L-lysine biosynthesis intermediate products95.
In this study, histidine metabolism mutants were considered conditional prototrophs for being able to grow efficiently in minimal media supplemented with fly extract and to colonize the flies as efficiently as the wild type P. aeruginosa strain. The virulence attenuation of these mutants is more likely due to defects in virulence rather than in growth. Similarly, most of the purine metabolism mutants were considered conditional prototrophs for being able to grow in minimal media that contained fly extract, but these mutants were unable to colonize flies to the same extent as the wild type strain. This suggests that although there are enough nutrients to sustain growth in the host tissues, bacteria are unable to acquire them through their virulence factors. Interestingly, we also identified conditional prototrophs that although unable to grow efficiently in culture in any of the two types of minimal media, they could colonize flies normally, thus the corresponding genes may help to damage the host, irrespective of their contribution to growth.
To validate our approach, we assessed VF production in a subset of the metabolic gene mutants that were attenuated in virulence not only in flies but also in a mouse lung infection model. We found that all of them exhibited defects in at least one aspect of virulence and most of them are involved in metabolic steps absent from the human metabolism network. Thus, many metabolic genes could be targets for anti-infective therapies against acute P. aeruginosa virulence. It is nevertheless questionable whether prioritization in pharmacological targeting should be given to virulence-related metabolic genes also affecting growth or those that do not affect growth.
Moreover, we found differential expression of many known virulence factors and some metabolism genes through classical transcriptome analysis of highly vs. lowly pathogenic P. aeruginosa strains. Combinatorial functional transcriptomics analysis at the pathway level was much more informative, revealing metabolic pathways containing differentially expressed genes between all 3 highly virulent vs. all 3 lowly virulent strains and genes required for full virulence: (i) Aromatic compound degradation/biosynthesis, including the 4-hydroxyl-phenylacetate degradation for succinate production (Succinate biosynthesis), the 2,5- and 3,5-xylenol degradation to citramalate (Xylenol degradation) and shikimate and chorismate biosynthesis from D-erythrose 4-phosphate (Chorismate biosynthesis), (ii) Amino acid biosynthesis, including branched chain amino acid biosynthesis and the glutamine biosynthesis from glutamic acid (Gln biosynthesis), and (iii) Fatty acid and lipid degradation (beta-oxidation). Thus, P. aeruginosa virulence can be analyzed at the transcriptome and functional level using common core metabolism modules that control and indicate the virulence of disparate P. aeruginosa strains.
While the impact of specific core metabolism genes is not discernible at the genomic level, interesting patterns emerge. By classifying the 30 fully sequenced Pseudomonas species experimentally in two different Drosophila infection assays (wound and oral infection) as low, medium or high in virulence, we found that the phylogeny of the 30 strains correlates well with their virulence classification in the wound infection, but not in the oral infection assay. This is probably because the wound infection is more acute and guided by highly efficient virulence-related genes, as opposed to the intestinal environment in which Pseudomonas is less virulent. The phylogeny of the 30 strains also correlates well with virulence factor gene content, which is expected since P. aeruginosa species have their own repertoire and group together in the phylogenetic tree and separately from the non-P. aeruginosa strains. Similarly, the phylogeny of the 30 strains also correlates with major metabolic pathway gene content. Thus, the existing correlations between pathogenicity of the 30 Pseudomonas strains and gene content cannot be attributed specifically to VFs or metabolic genes. The gene content of the 30 Pseudomonas spp. regarding five metabolic pathways (carbon, glycolysis, pyrimidine, propanoate metabolism and valine, leucine, isoleucine degradation) explains their virulence classification upon the wound infection. Nevertheless, when the analysis is repeated with only the P. aeruginosa strains, no correlation is evident for any comparison. Thus, the existing correlation between the pathogenicity of the 30 Pseudomonas strains and genomic content in metabolic genes cannot be clearly attributed to specific genes, but rather to broader interspecies differences. Given the difference in expression of known virulence factors and core metabolism pathways in distinguishing highly from lowly virulent strains, it is likely that gene promoter differences in addition to the presence of VFs and core metabolism genes may account for the total strain-to-strain variation in virulence potential.
Supplementary information
Acknowledgements
Bacterial RNA sequencing was performed at the BSRC Al. Fleming Genomics Facility. We thank Dr. Vaggelis Harokopos for the NGS experiments and Dr. Martin Reczko for initial bioinformatic analyses. We also thank Stelios Theofilou and Eleni Ioannidou for helping with the Drosophila infection screens and the UCY library for access to BioCyc.
Author contributions
S.P., K.G., V.J.P. and Y.A. wrote the paper. S.P., V.J.P. and Y.A. edited the paper. S.P. and T.C. performed experiments and contributed to the project management. K.G., S.T. and V.J.P. performed computational analysis. V.J.P. and Y.A. supervised the project, provided funding and conceptualized the work.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files), expect from the RNA-Seq FASTQ files, which are publicly accessible through the Gene Expression Omnibus database, GEO Series accession number GSE142464 and the following link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE142464
Materials and protocols generated during the current study are available from the corresponding authors on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Kaliopi Georgiades, Theodoulakis Christofi and Stella Tamana.
Contributor Information
Vasilis J. Promponas, Email: vprobon@ucy.ac.cy
Yiorgos Apidianakis, Email: apidiana@ucy.ac.cy.
Supplementary information
is available for this paper at 10.1038/s41598-020-66194-4.
References
- 1.Anzai Y, Kim H, Park JY, Wakabayashi H, Oyaizu H. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int. J. Syst. Evol. Microbiol. 2000;50(Pt 4):1563–1589. doi: 10.1099/00207713-50-4-1563. [DOI] [PubMed] [Google Scholar]
- 2.Yamamoto S, et al. Phylogeny of the genus Pseudomonas: intrageneric structure reconstructed from the nucleotide sequences of gyrB and rpoD genes. Microbiology. 2000;146(Pt 10):2385–2394. doi: 10.1099/00221287-146-10-2385. [DOI] [PubMed] [Google Scholar]
- 3.Wolfgang MC, et al. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA. 2003;100:8484–8489. doi: 10.1073/pnas.0832438100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stover CK, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 5.Mahajan-Miklos S, Rahme LG, Ausubel FM. Elucidating the molecular mechanisms of bacterial virulence using non-mammalian hosts. Mol. Microbiol. 2000;37:981–988. doi: 10.1046/j.1365-2958.2000.02056.x. [DOI] [PubMed] [Google Scholar]
- 6.Mathee K, et al. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. USA. 2008;105:3100–3105. doi: 10.1073/pnas.0711982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doring G. Serine proteinase inhibitor therapy in alpha(1)-antitrypsin inhibitor deficiency and cystic fibrosis. Pediatr. Pulmonol. 1999;28:363–375. doi: 10.1002/(sici)1099-0496(199911)28:5<363::aid-ppul9>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 8.Winstanley C, O’Brien S, Brockhurst MA. Pseudomonas aeruginosa Evolutionary Adaptation and Diversification in Cystic Fibrosis Chronic Lung Infections. Trends Microbiol. 2016;24:327–337. doi: 10.1016/j.tim.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moradali MF, Ghods S, Rehm BH. Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation. Survival, and Persistence. Front. Cell. Infect. Microbiol. 2017;7:39. doi: 10.3389/fcimb.2017.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hill PJ, et al. Modifications of Pseudomonas aeruginosa cell envelope in the cystic fibrosis airway alters interactions with immune cells. Sci. Rep. 2017;7:4761-017–05253-9. doi: 10.1038/s41598-017-05253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parkins, M. D., Somayaji, R. & Waters, V. J. Epidemiology, Biology, and Impact of Clonal Pseudomonas aeruginosa Infections in Cystic Fibrosis. Clin. Microbiol. Rev. 31, 10.1128/CMR.00019-18. Print 2018 Oct (2018). [DOI] [PMC free article] [PubMed]
- 12.La Rosa, R., Johansen, H. K. & Molin, S. Adapting to the Airways: Metabolic Requirements of Pseudomonas aeruginosa during the Infection of Cystic Fibrosis Patients. Metabolites9, 10.3390/metabo9100234 (2019). [DOI] [PMC free article] [PubMed]
- 13.Lee, D. G. et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 7, R90-2006-7-10-r90. Epub 2006 Oct 12 (2006). [DOI] [PMC free article] [PubMed]
- 14.Ernst RK, et al. Genome mosaicism is conserved but not unique in Pseudomonas aeruginosa isolates from the airways of young children with cystic fibrosis. Environ. Microbiol. 2003;5:1341–1349. doi: 10.1111/j.1462-2920.2003.00518.x. [DOI] [PubMed] [Google Scholar]
- 15.Hacker J, Kaper JB. Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 2000;54:641–679. doi: 10.1146/annurev.micro.54.1.641. [DOI] [PubMed] [Google Scholar]
- 16.He J, et al. The broad host range pathogen Pseudomonas aeruginosa strain PA14 carries two pathogenicity islands harboring plant and animal virulence genes. Proc. Natl. Acad. Sci. USA. 2004;101:2530–2535. doi: 10.1073/pnas.0304622101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roy PH, et al. Complete genome sequence of the multiresistant taxonomic outlier Pseudomonas aeruginosa PA7. PLoS One. 2010;5:e8842. doi: 10.1371/journal.pone.0008842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balasubramanian D, Mathee K. Comparative transcriptome analyses of Pseudomonas aeruginosa. Hum. Genomics. 2009;3:349–361. doi: 10.1186/1479-7364-3-4-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Apidianakis Y, et al. Profiling early infection responses: Pseudomonas aeruginosa eludes host defenses by suppressing antimicrobial peptide gene expression. Proc. Natl. Acad. Sci. USA. 2005;102:2573–2578. doi: 10.1073/pnas.0409588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winstanley C, et al. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool Epidemic Strain of Pseudomonas aeruginosa. Genome Res. 2009;19:12–23. doi: 10.1101/gr.086082.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moran NA. Microbial minimalism: genome reduction in bacterial pathogens. Cell. 2002;108:583–586. doi: 10.1016/s0092-8674(02)00665-7. [DOI] [PubMed] [Google Scholar]
- 22.Georgiades K, Raoult D. Defining pathogenic bacterial species in the genomic era. Front. Microbiol. 2011;1:151. doi: 10.3389/fmicb.2010.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vasquez-Rifo A, Veksler-Lublinsky I, Cheng Z, Ausubel FM, Ambros V. The Pseudomonas aeruginosa accessory genome elements influence virulence towards Caenorhabditis elegans. Genome Biol. 2019;20:270-019–1890-1. doi: 10.1186/s13059-019-1890-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartell JA, et al. Reconstruction of the metabolic network of Pseudomonas aeruginosa to interrogate virulence factor synthesis. Nat. Commun. 2017;8:14631. doi: 10.1038/ncomms14631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cannon CL, et al. In vitro and murine efficacy and toxicity studies of nebulized SCC1, a methylated caffeine-silver(I) complex, for treatment of pulmonary infections. Antimicrob. Agents Chemother. 2009;53:3285–3293. doi: 10.1128/AAC.00314-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munder A, et al. Acute intratracheal Pseudomonas aeruginosa infection in cystic fibrosis mice is age-independent. Respir. Res. 2011;12:148-9921–12-148. doi: 10.1186/1465-9921-12-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teichgraber V, et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat. Med. 2008;14:382–391. doi: 10.1038/nm1748. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, et al. Early production of IL-17 protects against acute pulmonary Pseudomonas aeruginosa infection in mice. FEMS Immunol. Med. Microbiol. 2011;61:179–188. doi: 10.1111/j.1574-695X.2010.00764.x. [DOI] [PubMed] [Google Scholar]
- 30.Apidianakis Y, Rahme LG. Drosophila melanogaster as a model host for studying Pseudomonas aeruginosa infection. Nat. Protoc. 2009;4:1285–1294. doi: 10.1038/nprot.2009.124. [DOI] [PubMed] [Google Scholar]
- 31.Kapsetaki SE, et al. The bacterial metabolite 2-aminoacetophenone promotes association of pathogenic bacteria with flies. Nat. Commun. 2014;5:4401. doi: 10.1038/ncomms5401. [DOI] [PubMed] [Google Scholar]
- 32.Kaplan EL, Meier P. Nonparametric Estimation from Incomplete Observations. Journal of the American Statistical Association. 1958;53:457–481. [Google Scholar]
- 33.Sokal RR, Michener CD. A statistical method for evaluating systematic relationships. University of Kansas Science Bulletin. 1958;38:1409–1438. [Google Scholar]
- 34.Duan, J., Jiang, W., Cheng, Z., Heikkila, J. J. & Glick, B. R. The complete genome sequence of the plant growth-promoting bacterium Pseudomonas sp. UW4. PLoS One 8, e58640 (2013). [DOI] [PMC free article] [PubMed]
- 35.Winsor GL, et al. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 2016;44:D646–53. doi: 10.1093/nar/gkv1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benson DA, et al. GenBank. Nucleic Acids Res. 2018;46:D41–D47. doi: 10.1093/nar/gkx1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sievers F, Higgins DG. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014;1079:105–116. doi: 10.1007/978-1-62703-646-7_6. [DOI] [PubMed] [Google Scholar]
- 38.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 40.Winsor GL, et al. Pseudomonas Genome Database: facilitating user-friendly, comprehensive comparisons of microbial genomes. Nucleic Acids Res. 2009;37:D483–8. doi: 10.1093/nar/gkn861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janssen P, et al. COmplete GENome Tracking (COGENT): a flexible data environment for computational genomics. Bioinformatics. 2003;19:1451–1452. doi: 10.1093/bioinformatics/btg161. [DOI] [PubMed] [Google Scholar]
- 42.Promponas, V. J. et al. CAST: an iterative algorithm for the complexity analysis of sequence tracts. Bioinformatics16, 915–922 (2000). [DOI] [PubMed]
- 43.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 44.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kreil DP, Ouzounis CA. Comparison of sequence masking algorithms and the detection of biased protein sequence regions. Bioinformatics. 2003;19:1672–1681. doi: 10.1093/bioinformatics/btg212. [DOI] [PubMed] [Google Scholar]
- 46.Kirmitzoglou I, Promponas VJ. LCR-eXXXplorer: a web platform to search, visualize and share data for low complexity regions in protein sequences. Bioinformatics. 2015;31:2208–2210. doi: 10.1093/bioinformatics/btv115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen L, et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33:D325–8. doi: 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karp PD, et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief Bioinform. 2019;20:1085–1093. doi: 10.1093/bib/bbx085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dalquen DA, Dessimoz C. Bidirectional best hits miss many orthologs in duplication-rich clades such as plants and animals. Genome Biol. Evol. 2013;5:1800–1806. doi: 10.1093/gbe/evt132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saeed AI, et al. TM4: a free, open-source system for microarray data management and analysis. BioTechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 52.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, https://www.R-project.org/ (2008).
- 53.Hubert L, Arabie P. Comparing Partitions. Journal of Classification. 1985;2:193–218. [Google Scholar]
- 54.Steinley D. Properties of the Hubert-Arabie adjusted Rand index. Psychol. Methods. 2004;9:386–396. doi: 10.1037/1082-989X.9.3.386. [DOI] [PubMed] [Google Scholar]
- 55.Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36-2013-14-4-r36 (2013). [DOI] [PMC free article] [PubMed]
- 56.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 58.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550-014–0550-8. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grossmann S, Bauer S, Robinson PN, Vingron M. Improved detection of overrepresentation of Gene-Ontology annotations with parent child analysis. Bioinformatics. 2007;23:3024–3031. doi: 10.1093/bioinformatics/btm440. [DOI] [PubMed] [Google Scholar]
- 60.Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015;43:W566–70. doi: 10.1093/nar/gkv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pfaffl MW. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Research. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Apidianakis Y, Pitsouli C, Perrimon N, Rahme L. Synergy between bacterial infection and genetic predisposition in intestinal dysplasia. Proc. Natl. Acad. Sci. USA. 2009;106:20883–20888. doi: 10.1073/pnas.0911797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vodovar N, et al. Complete genome sequence of the entomopathogenic and metabolically versatile soil bacterium Pseudomonas entomophila. Nat. Biotechnol. 2006;24:673–679. doi: 10.1038/nbt1212. [DOI] [PubMed] [Google Scholar]
- 64.Nelson KE, et al. Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ. Microbiol. 2002;4:799–808. doi: 10.1046/j.1462-2920.2002.00366.x. [DOI] [PubMed] [Google Scholar]
- 65.Yahr TL, Vallis AJ, Hancock MK, Barbieri JT, Frank DW. ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc. Natl. Acad. Sci. USA. 1998;95:13899–13904. doi: 10.1073/pnas.95.23.13899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feltman H, et al. Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology. 2001;147:2659–2669. doi: 10.1099/00221287-147-10-2659. [DOI] [PubMed] [Google Scholar]
- 67.Lomholt JA, Poulsen K, Kilian M. Epidemic population structure of Pseudomonas aeruginosa: evidence for a clone that is pathogenic to the eye and that has a distinct combination of virulence factors. Infect. Immun. 2001;69:6284–6295. doi: 10.1128/IAI.69.10.6284-6295.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garey KW, Vo QP, Larocco MT, Gentry LO, Tam VH. Prevalence of type III secretion protein exoenzymes and antimicrobial susceptibility patterns from bloodstream isolates of patients with Pseudomonas aeruginosa bacteremia. J. Chemother. 2008;20:714–720. doi: 10.1179/joc.2008.20.6.714. [DOI] [PubMed] [Google Scholar]
- 69.Pirnay JP, et al. Pseudomonas aeruginosa population structure revisited. PLoS One. 2009;4:e7740. doi: 10.1371/journal.pone.0007740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bradbury RS, Roddam LF, Merritt A, Reid DW, Champion AC. Virulence gene distribution in clinical, nosocomial and environmental isolates of Pseudomonas aeruginosa. J. Med. Microbiol. 2010;59:881–890. doi: 10.1099/jmm.0.018283-0. [DOI] [PubMed] [Google Scholar]
- 71.Ozer EA, Nnah E, Didelot X, Whitaker RJ, Hauser AR. The Population Structure of Pseudomonas aeruginosa Is Characterized by Genetic Isolation of exoU+ and exoS+ Lineages. Genome Biol. Evol. 2019;11:1780–1796. doi: 10.1093/gbe/evz119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Basso P, et al. Multiple Pseudomonas species secrete exolysin-like toxins and provoke Caspase-1-dependent macrophage death. Environ. Microbiol. 2017;19:4045–4064. doi: 10.1111/1462-2920.13841. [DOI] [PubMed] [Google Scholar]
- 73.Basso, P. et al. Pseudomonas aeruginosa Pore-Forming Exolysin and Type IV Pili Cooperate To Induce Host Cell Lysis. mBio8, 10.1128/mBio.02250-16 (2017). [DOI] [PMC free article] [PubMed]
- 74.Trouillon, J. et al. Species-specific recruitment of transcription factors dictates toxin expression. Nucleic Acids Res. (2020). [DOI] [PMC free article] [PubMed]
- 75.Finck-Barbancon V, et al. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol. Microbiol. 1997;25:547–557. doi: 10.1046/j.1365-2958.1997.4891851.x. [DOI] [PubMed] [Google Scholar]
- 76.Giastas P, et al. The structure of the 2[4Fe-4S] ferredoxin from Pseudomonas aeruginosa at 1.32-A resolution: comparison with other high-resolution structures of ferredoxins and contributing structural features to reduction potential values. J. Biol. Inorg. Chem. 2006;11:445–458. doi: 10.1007/s00775-006-0094-9. [DOI] [PubMed] [Google Scholar]
- 77.Whitney JC, et al. An interbacterial NAD(P)(+) glycohydrolase toxin requires elongation factor Tu for delivery to target cells. Cell. 2015;163:607–619. doi: 10.1016/j.cell.2015.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Amari DT, Marques CN, Davies DG. The putative enoyl-coenzyme A hydratase DspI is required for production of the Pseudomonas aeruginosa biofilm dispersion autoinducer cis-2-decenoic acid. J. Bacteriol. 2013;195:4600–4610. doi: 10.1128/JB.00707-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Feinbaum RL, et al. Genome-wide identification of Pseudomonas aeruginosa virulence-related genes using a Caenorhabditis elegans infection model. PLoS Pathog. 2012;8:e1002813. doi: 10.1371/journal.ppat.1002813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo QQ, et al. Characterization of 3-Oxacyl-Acyl Carrier Protein Reductase Homolog Genes in Pseudomonas aeruginosa PAO1. Front. Microbiol. 2019;10:1028. doi: 10.3389/fmicb.2019.01028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yeung AT, et al. Swarming of Pseudomonas aeruginosa is controlled by a broad spectrum of transcriptional regulators, including MetR. J. Bacteriol. 2009;191:5592–5602. doi: 10.1128/JB.00157-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hauser AR. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 2009;7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dasgupta N, Ashare A, Hunninghake GW, Yahr TL. Transcriptional induction of the Pseudomonas aeruginosa type III secretion system by low Ca2+ and host cell contact proceeds through two distinct signaling pathways. Infect. Immun. 2006;74:3334–3341. doi: 10.1128/IAI.00090-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brinkman CL, et al. Characterization of the effects of an rpoC mutation that confers resistance to the Fst peptide toxin-antitoxin system toxin. J. Bacteriol. 2013;195:156–166. doi: 10.1128/JB.01597-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wilharm G, Heider C. Interrelationship between type three secretion system and metabolism in pathogenic bacteria. Front. Cell. Infect. Microbiol. 2014;4:150. doi: 10.3389/fcimb.2014.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Daddaoua A, et al. Genes for carbon metabolism and the ToxA virulence factor in Pseudomonas aeruginosa are regulated through molecular interactions of PtxR and PtxS. PLoS One. 2012;7:e39390. doi: 10.1371/journal.pone.0039390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Udaondo Z, Ramos JL, Segura A, Krell T, Daddaoua A. Regulation of carbohydrate degradation pathways in Pseudomonas involves a versatile set of transcriptional regulators. Microb. Biotechnol. 2018;11:442–454. doi: 10.1111/1751-7915.13263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rohmer L, Hocquet D, Miller SI. Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol. 2011;19:341–348. doi: 10.1016/j.tim.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Poncet S, et al. Correlations between carbon metabolism and virulence in bacteria. Contrib. Microbiol. 2009;16:88–102. doi: 10.1159/000219374. [DOI] [PubMed] [Google Scholar]
- 90.Luckett JC, et al. A novel virulence strategy for Pseudomonas aeruginosa mediated by an autotransporter with arginine-specific aminopeptidase activity. PLoS Pathog. 2012;8:e1002854. doi: 10.1371/journal.ppat.1002854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yeung AT, Bains M, Hancock RE. The sensor kinase CbrA is a global regulator that modulates metabolism, virulence, and antibiotic resistance in Pseudomonas aeruginosa. J. Bacteriol. 2011;193:918–931. doi: 10.1128/JB.00911-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sterritt, O. W. et al. Structural and functional characterisation of the entry point to pyocyanin biosynthesis in Pseudomonas aeruginosa defines a new 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase subclass. Biosci. Rep. 38, 10.1042/BSR20181605. Print 2018 Oct 31 (2018). [DOI] [PMC free article] [PubMed]
- 93.Serino L, et al. Structural genes for salicylate biosynthesis from chorismate in Pseudomonas aeruginosa. Mol. Gen. Genet. 1995;249:217–228. doi: 10.1007/BF00290369. [DOI] [PubMed] [Google Scholar]
- 94.Calfee MW, Coleman JP, Pesci EC. Interference with Pseudomonas quinolone signal synthesis inhibits virulence factor expression by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA. 2001;98:11633–11637. doi: 10.1073/pnas.201328498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ge L, Seah SY. Heterologous expression, purification, and characterization of an l-ornithine N(5)-hydroxylase involved in pyoverdine siderophore biosynthesis in Pseudomonas aeruginosa. J. Bacteriol. 2006;188:7205–7210. doi: 10.1128/JB.00949-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article (and its Supplementary Information files), expect from the RNA-Seq FASTQ files, which are publicly accessible through the Gene Expression Omnibus database, GEO Series accession number GSE142464 and the following link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE142464
Materials and protocols generated during the current study are available from the corresponding authors on reasonable request.