

Summary
Toll‐like receptors (TLRs) are evolutionarily conserved receptors essential for the host defence against pathogens. Both immune and non‐immune cells can express TLRs, although at different levels. Systemic sclerosis (SSc) is a chronic disease in which autoimmunity, dysregulated profibrotic mediator release and activation of fibroblasts lead to dysregulated collagen deposition and fibrosis. There is now increasing knowledge that the innate immune system and, in particular, TLRs take a part in SSc pathogenesis. The list of endogenous ligands that can stimulate TLRs in SSc is growing: these ligands represent specific danger‐associated molecular patterns (DAMPs), involved either in the initiation or the perpetuation of inflammation, and in the release of factors that sustain the fibrotic process or directly stimulate the cells that produce collagen and the endothelial cells. This review reports evidences concerning TLR signalling involvement in SSc. We report the new DAMPs, as well as the TLR‐linked pathways involved in disease, with emphasis on type I interferon signature in SSc, the role of plasmacytoid dendritic cells (pDCs) and platelets. The dissection of the contribution of all these pathways to disease, and their correlation with the disease status, as well as their values as prognostic tools, can help to plan timely intervention and design new drugs for more appropriate therapeutic strategies.
Keywords: fibrosis, immune cells, interferon signature, systemic sclerosis, Toll‐like receptor
TLRs are involved in the pathogenesis of Systemic Sclerosis and are activated by endogenous mediators (“danger signals”). This review is an updated report on the recognised endogenous mediators activating TLRs in Systemic Sclerosis, according to the most recent literature
Introduction
Systemic sclerosis (SSc) is an autoimmune and fibrotic disease with a high disease burden and mortality rate 1. Three most important hallmarks characterize SSc: autoimmunity, fibrosis and vasculopathy. Autoimmunity is an important component, as autoreactive T cells and autoantibodies play a central role in SSc pathogenesis 1, 2. Fibrosis is the most lethal feature responsible for organ failure 1, 2, 3. Microvascular constriction and endothelial damage, clinically expressed by Raynaud’s phenomenon (RP), are the first manifestation of SSc in 90–98% of cases and precede disease onset by years 4. According to the extension of the skin fibrosis, it is possible to define two major subsets of SSc: limited cutaneous SSc (lcSSc) and diffuse cutaneous SSc (dcSSc) 5. Currently, no effective drugs exist which can modify the disease course. Dysregulation of the innate immune system in genetically predisposed individuals plays a role in SSc, and aberrant Toll‐like receptors (TLRs) activation seems central to pathogenesis 6, 7. TLRs are a germline‐encoded group of pattern recognition receptors, which comprise 10 members (TLR‐1–TLR‐10) in humans and 12 members (TLR‐1–TLR‐9 and TLR‐11–TLR‐13) in mice 8. They are key for recognition of invading pathogens. As transmembrane receptors, they localize either at the cell surface or in the endosomal compartment. As they are involved in self‐ versus non‐self‐discrimination, TLRs have implications in various autoimmune and autoinflammatory conditions 9. Non‐immune cells, including fibroblasts, endothelial cells and platelets, also express TLRs and respond to a wide array of microbial molecules (pathogen‐associated molecular patterns, PAMPs). However, TLRs also respond to endogenous non‐microbial stimuli, referred to as danger‐associated molecular patterns (DAMPs) 10. Cellular stress and traumas induce DAMP release. The exact identification of the DAMPs involved in TLR stimulation in SSc is pivotal to the development of specific therapies 6.
DAMPs in autoimmunity
One of the first‐discovered DAMPs is the high‐mobility group protein 1 (HMGB1 11). DAMPs are generally inside the cells hidden to the immune system, but traumas or stress induce their release. HMGB1, the prototype of DAMPs, is normally expressed in the nuclei but if released can trigger, among other TLRs, TLR‐4. By binding to damaged DNA, HMGB1 can also favour TLR‐9 stimulation 12. Activated platelets in SSc blood release microparticles which contain HMGB1 13. HGMB1 also binds receptors for advanced glycation‐end‐product (RAGE), an immunoglobulin superfamily member 11.
In general, DAMPs are heterogeneous molecules: apart from HMGB1, other DAMPs comprise Ca2+, H2O2, reactive oxygen species (ROS), adenosine triphosphate (ATP), self‐nucleic acids, heat shock proteins (Hsps), S100 proteins (alarmins), fragments of the extracellular matrix (ECM), uric acid and heparin sulphate. Released intracellular mitochondria also represent DAMPs 14. Several DAMPs in SSc can contribute to disease pathogenesis 6.
Membrane TLRs and their DAMPs in SSc
In humans, TLR‐1, ‐2, ‐4, ‐5, ‐6 and ‐10 localize on the cell surface of immune and non‐immune cells. TLR‐4 recognizes bacterial‐derived lipopolysaccharides (LPS), a PAMP mainly expressed on the surface of Gram‐negative bacteria, as well as other factors acting as DAMPs; among these are Hsps, taxol, fibronectin extracellular matrix components (ECM), fatty acids, low‐density lipoprotein (LDL) and fibrinogen. TLR‐2 recognizes lipoproteins, peptidoglycans and lipopeptides, hyaluronic acid, Hsp 70 lipoarabinomannan from a variety of microorganisms, and even HMGB1 15, 16.
A role for TLR‐2 is possible in SSc pathogenesis 5, 6: TLR‐2 hyperexpressing SSc fibroblasts overproduce interleukin (IL)‐6, a critical molecule in fibrosis 17. Serum amyloid A, which is high in SSc 18, stimulates TLR‐2 in fibroblasts. A rare polymorphism of TLR‐2 was associated with diffuse SSc and with anti‐topoisomerase antibodies (ATA) and pulmonary arterial hypertension 19. TLR‐2 forms heterodimers with TLR‐1 or TLR‐6 (in a ligand‐specific manner) to recognize a variety of PAMPs, such as peptidoglycans, lipoteichoic acid, zymosan, and mannan. However, no information is available on the roles of TLR‐1 and TLR‐6 in SSc.
Bhattacharyya et al. demonstrated that the fibronectin extra domain A (EDA), an endogenous TLR‐4 binder, was elevated in the circulation and in the lesional skin biopsies of SSc patients, as well as in mice with experimentally induced cutaneous fibrosis 20. Disrupting TLR‐4‐signalling abrogated the deleterious effect of EDA, in that TLR‐4 stimulation induced collagen production and myofibroblast differentiation. In mice, the blockade of TRL‐4 mitigated experimentally induced fibrosis. Thus, the example of TLR‐4 and EDA is paradigmatic of how a DAMP, when out of control, activates multiple unwanted pathways leading to disease 20. An additional endogenous ligand for TLR‐4 studied by the same authors is tenascin C, which also mediates fibrosis in SSc. Tenascin C is not expressed normally, but is expressed transiently during wound healing and tissue remodelling. Tenascin C sustains fibrosis in a mouse model of SSc via TLR‐4 21. Accordingly, an enhanced TLR‐4‐responsive gene signature was present in SSc skin biopsies 22. Collectively, these studies point to the involvement of TLR‐4 signalling in fibrosis in SSc.
With regard to other TLRs, one study addressed the expression of TLR‐5 and TLR‐10 23 in SSc fibroblasts and found up‐regulation of both TLRs. TLR‐5 possibly exerts a suppressive effect on collagen expression, perhaps as an attempt to regulate fibrosis. Indeed, TLR‐5 triggering by the PAMP flagellin could inhibit collagen deposition in in‐vitro cultured fibroblasts 23. However, it is probably unlikely that flagellin stimulate TLR‐5 in SSc. It is clear that TLR‐5 is primarily the receptor for the flagellin 24, but additional functional roles for TLR‐5 are likely to be revealed in SSc and other autoimmune diseases 25. Interestingly, a study showed that HMGB1 also acts as an agonist of TLR‐5 and induces signalling that activates myeloid differentiation primary response protein (MyD88) and nuclear factor kappa B (NF‐kB) in other settings 26. In the study mentioned, HMGB1–TLR‐5 interaction induces the release of proinflammatory factors, which results in a higher sensitivity to pain in animals 26. However, the putative endogenous ligands for TLR‐5 in SSc is unknown. As HGMB1 is up‐regulated in SSc during cell damage 13, the role of TLR‐5 may deserve further investigation. In rheumatoid arthritis (RA), monocytes expressed high levels of TLR‐5 and this expression correlated with disease activity and tumour necrosis factor (TNF)‐α production 27. The authors of the study suspected that endogenous ligands present in the RA‐affected synovia could be responsible for TLR‐5 triggering, although the TLR‐5‐specific DAMP in RA also remains elusive. Some other studies point towards other ‘suspects’, including members of the Hsps, a family of proteins produced in response to stressful conditions 28. For instance, a study that analysed tongue squamous carcinoma cell lines reported that Hsp27 is indeed a ligand for TLR‐5. Engagement of TLR‐5 by Hsp27 induced NF‐kB activation in cancer cells. Interestingly, using proteomic analysis and immunohistochemistry, a study showed that Hsp27 is highly expressed in SSc skin 29 Thus, it will be worth exploring whether HMGB1 and Hsp27 can act as DAMPs for TLR‐5 in SSc and the effect of these interactions. TLR‐10 can form dimers with TLR‐2, and may exert an anti‐inflammatory function. Human TLR‐10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells (pDCs), and activates gene transcription through MyD88 30. This TLR is an interesting candidate for further studies in SSc, as a recent paper has shown that double‐strand RNA (dsRNA) is a ligand for TLR‐10. The authors proposed that TLR‐10 can regulate the interferon (IFN)‐I pathway by sequestering dsRNA from TLR‐3 (see below) to prevent TLR‐3 signalling in response to dsRNA 31.
DNA/RNA‐sensing TLR‐7, ‐8 and ‐9 and their specific DAMPs in SSc
TLR‐7, ‐8 and ‐9 have a high sequence homology and share dependency on the MyD88 pathway 6, 7, 8 (Fig. 1). TLR‐9 recognizes dsDNA expressing unmethylated cytosine–phosphate–guanosine (CpG) motifs. TLR‐7 recognizes single‐strand RNA (ssRNA), whereas TLR‐8 recognizes RNA products generated by the lysosomal endoribonuclease RNase T2 32. The release of nucleic acids during cell death, apoptosis, necrosis or extracellular traps release (ETosis) is a common event during acute infection/inflammation/traumas 33, 34. Thus, three levels of protection exist: the first relies on the endosomal localization of these TLRs, in that self‐DNA and self‐RNA usually fail to enter into the cells, whereas DNA/RNA of endocellular bacteria or endocytosed extracellular bacteria can reach endosomal TLRs more easily. A second level of protection relies on self‐nucleic acid degradation by nucleases to avoid persistence in the extracellular milieu. Thirdly, pathogen‐derived DNA, unlike human DNA, is usually unmethylated and contains more CpG islands; TLR‐9 is more prone to recognize DNA with such characteristics. Of note, in some autoimmune diseases, including SSc, defects of methylation are present and more unmethylated self‐DNA may be available to engage TLR‐9 35.
Figure 1.
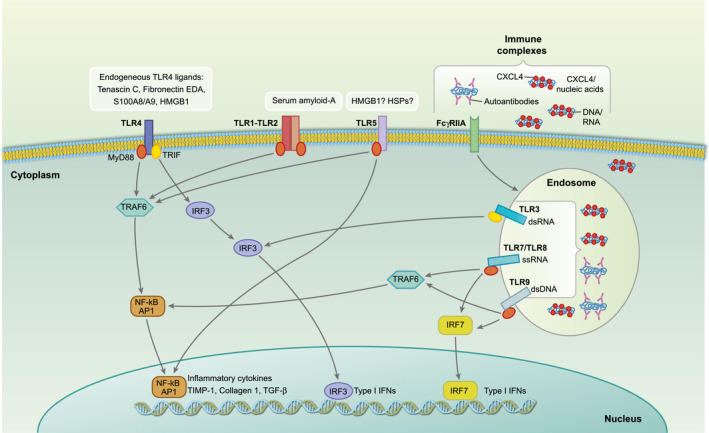
Endogenous Toll‐like receptor (TLR) ligands identified in systemic sclerosis (SSc). Binding of danger‐associated molecular patterns (DAMPs) (released by injured tissues) to TLR‐4 and TLR‐1 and ‐2 triggers the production of inflammatory cytokines [interleukin (IL)‐6, IL‐8, tumour necrosis factor (TNF)‐α, transforming growth factor (TGF)‐β], as well as factors involved in the extracellular matrix (ECM) deposition, such as the tissue inhibitor of metalloproteinases (TIMP‐1) and collagen 1. Whether TLR‐5 is triggered by endogenous ligand in SSc is unknown (possible candidates may be high‐mobility group protein 1 (HMGB1) or heat shock proteins (HSPs), according to studies in other settings (see text). SSc–immune complexes (SSc–IC), which include autoantibodies anti‐topoisomerase antibodies (ATA) and anti‐centromere antibodies (ACA), etc. as well as nanocristalline particles of platelet‐ and platelet dendritic cell (pDC)‐derived chemokine C‐X‐C ligand motif (CXCL4) bound to nucleic acids (DNA and RNA) can stimulate endosomal nucleic‐acid sensing TLRs TLR‐7, TLR‐8, TLR‐9 and TLR‐3, once internalized. SSc–IC also induces IL‐6, IL‐8, matrix metalloproteinase (MMP)‐2, monocyte chemoattractant protein 1 (MCP‐1), TGF‐β1 and pro‐collagen α1 by fibroblasts.
Kim et al.’s group demonstrated that SSc sera containing autoantibodies induced high levels of IFN‐α in healthy donor (HD) peripheral blood mononuclear cells (PBMCs) in a pDC‐ and RNA/DNA‐dependent manner 36. A paper by Eloranta et al. 37 also showed that sera from SSc patients, mixed with necrotic or apoptotic material, induced IFN‐α production in pDCs, apparently activated by immunoglobulin (Ig) immune complexes (ICs) formed by SSc autoantibodies. Thus, typical autoantibodies present in SSc, even several years before overt disease manifestation, are able to activate pDCs and the IFN‐α pathways. Stimulation depends on endosomal TLR receptors because bafilomycin, which prevents the acidification of the endosomes, inhibits IFN‐α production induced by IgG immune complexes. In our assays pDCs stimulated with SSc plasma produced IFN‐α, and this secretion was significantly inhibited by addition of an anti‐Fc receptor antibody, indicating that antibodies were at least partially responsible for IFN‐α production 38. Accordingly, Western blot analysis of IgG that was immune‐precipitated from SSc plasma revealed that IgG had DNA attached. Thus, typical SSc antibodies, and possibly other autoantibodies with unknown specificity, could also act as DAMPs because they bind nucleic acids. Of note, SSc autoantibodies also stimulated fibroblasts to secrete profibrotic mediators 39.
A study by Fang et al. reported a significant elevation of TLR‐9 expression in SSc dermis compared to control dermis and detection of a TLR‐9 signature in SSc skin 40. In‐vitro treatment of normal cutaneous fibroblasts with the TLR‐9 ligand unmethylated CpG induced a profibrotic profile, involving autocrine TGF‐β production. A recent paper detected a significant up‐regulation of TLR7 gene expression in PBMCs of a group of SSc patients compared to a non‐SSc group using the reverse transcription–quantitative polymerase chain reaction (RT–qPCR) technique 41. It is worth mention that several genes linked to autoimmunity are located on the X chromosome. Remarkably, among these genes are TLR‐8 and TLR‐7 41. Many autoimmune disorders, SSc included, are markedly sex‐biased and studies support a gene–dose effect of the X chromosome loci in systemic lupus erythematosus (SLE) predisposition and, perhaps, establishment 42. Souyris et al.’s group recently reported that B cells expressing TLR‐7 bi‐allelically (due to lack of X chromosome inactivation) were more responsive than monoallelic cells during TLR‐7‐driven B cell differentiation 43. It will be worthwhile to analyse this aspect in SSc, in that no studies have addressed this issue so far.
RNA sensing TLR‐3 and its DAMPs in SSc
Unlike the other endosomal nucleic acid sensors described above (TLR‐7, TLR‐8, TLR‐9), TLR‐3 is the only TLR to work in a MyD88‐independent manner. TLR‐3 associates with TIR‐domain‐containing adapter‐inducing IFN‐β (TRIF) (Fig. 1) and signals through interferon regulatory factor 3 (IRF‐3), a key factor involved in IFN‐β production and described to increase in SSc skin fibroblasts. The role of TLR‐3 is partly controversial. TLR‐3 activation (by polyinosinic:polycytidylic acid, Poly I:C)‐stimulated IFN‐I by fibroblasts but this, in turn, reduced fibroblast ability to produce ECM components. Conversely, Poly I:C stimulation promoted the expression of TGF‐β by the same cells, thus contributing to fibrosis 15, 44, 45. However, TLR‐3 function in SSc fibroblasts and endothelial cells is not only linked to the IFN‐I axis, but can be more complex. A paper by Farina et al. has shown that dsRNA induces endothelin 1 (EDN1) in endothelial cells and fibroblasts from SSc patients. EDN1 has a role in vascular complications in SSc, pulmonary hypertension and ulcers 46. Thus, dsRNA can be important in SSc to promote fibrosis, and not only for inducing IFN‐I.
The role of TLR and pDCs
The most recent literature has indicated the pDCs as an extremely important player in SSc. pDCs are the strongest producers of IFN‐I via TLR‐7/‐8/‐9 stimulation, and an IFN‐I signature is present in half of SSc patients 37, 47, 48, 49. A role for pDCs in SSc was already postulated by the fact that, as mentioned, SSc‐specific autoantibodies (ACA and ATA), were able to stimulate PBMCs of HD to produce IFN‐I. Targeting pDCs by anti‐BDCA2 antibodies blocked IFN‐α secretion 36, 37. Most recently, as well as IFN‐I release, pDCs have been shown to secrete, in SSc, CXCL4, a molecule originally identified as a chemokine but clearly exerting a plethora of different functions 50, 51, 52. A multi‐centre study indicated CXCL4 as an important SSc biomarker, which predicts a poor prognosis and correlates with lung fibrosis and pulmonary arterial hypertension 48.
Both van Bon et al. 48 and Ah Kioon et al. 51 observed that pDCs infiltrate the SSc‐involved skin, and both demonstrated that SSc pDCs release CXCL4. We also observed pDCs in SSc skin, where they appeared chronically activated, as they expressed Mx1 38, an IFN‐activated gene, as well as CXCL4. SSc pDCs over‐releasing CXCL4 produced much higher IFN‐α upon CpG (ODN2006, CpGb) challenge 48, 51. Ah Kioon et al. went on to show that SSc pDCs overproduce CXCL4 because they have an aberrant TLR‐8 expression compared to HD or SLE pDCs. Stimulation of TLR‐8 is the key event which mediates CXCL4 secretion by SSc pDCs. Both pDC‐derived CXCL4 and IFN‐α release are mediated by the phosphatidylinositol 3‐kinase d (PI3Kd), and the specific inhibition of this pathway can block the secretion of both CXCL4 and IFN‐α without affecting IL‐6 release. This is important, because it suggests that it could be possible to inhibit the chronic activation of pDCs in SSc by acting on this pathway 51. The physiological ligands that potentiate TLR‐8 signalling in SSc pDCs and the reason why TLR‐8 expression is up‐regulated in these cells remain elusive. In‐vivo depletion experiments further support a role for pDCs in SSc pathogenesis, as pDC depletion prevented disease in mouse models of scleroderma and could also revert fibrosis 51, 52. Thus, using depleting antibodies or targeting pDC function could be novel approaches to treat SSc patients. Although it was clearly shown that CXCL4 amplifies CpGb‐driven responses and responses elicited by artificial ligands for TLR‐7 and TLR‐8, the exact molecular mechanism of CXCL4’s contribution to the IFN‐I signature remains elusive.
CXCL4–DNA complexes as new DAMPs in SSc
The oligonucleotide CpGb and CXCL4 amplified pDC‐release of IFN‐α in vitro 48, 51. However, CpGb contains a phosphorothioate backbone, which makes the molecule resistant to the enzymatic degradation. Using natural DNA which, unlike CpGb, is sensitive to enzymatic degradation, we uncovered the underlined mechanistic link between CXCL4 and IFN‐α production by pDCs – CXCL4 forms nano‐crystalline complexes with DNA, and this enables otherwise non‐stimulatory natural DNA to induce immune amplification via TLR‐9‐activation. Notably, we demonstrated that CXCL4–DNA complexes are detectable and measurable in SSc plasma, and correlate with circulating IFN‐α 38. Crucial abilities relevant for CXCL4 to function as DAMP are (a) capacity to bind the DNA, (b) favour DNA internalization in immune cells and (c) protect DNA from degradation, as depicted in the cartoon in Fig. 1, which also reports the most relevant DAMPs in SSc. However, the emerging new paradigm is that the characteristics listed above are necessary but not sufficient to render a DNA/RNA‐binding molecule an efficient DAMP. It is crucial that the nucleic acid‐binding molecules organize the nucleic acid fragments into a molecular complex characterized by distance between the DNA/RNA ligands (internucleic acids spacing) optimal for the efficient triggering of the specific TLRs 53. We provide a scheme in Fig. 2 54. Thus, not all the DNA‐binding proteins possess the requirements to amplify TLR signalling. In this context, we may also hypothesize that some nucleic acid‐binding molecules could even interfere with TLR stimulation by sequestering released nucleic acids, thus working as decoy factors (and perhaps become tools for inhibitory strategies).This hypothesis may deserve investigation as a way to block the effects of CXCL4‐nucleic acids complexes in vivo.
Figure 2.
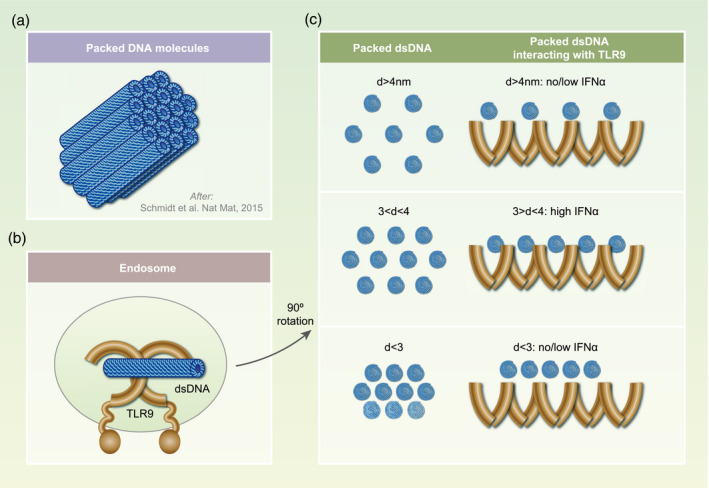
Toll‐like receptor (TLR)‐stimulation ability depends on the polycation–DNA complex structure, which influences the packaging of the DNA and the inter‐DNA spacing of contiguous DNA molecules. (a) Schematic representation of a structural type of DNA–polycation complex: cationic molecules bind and organize DNA chains (blue cylinders) into a columnar structure with a short‐ranged order. (b) Hypothetical structure of TLR‐9 and its interaction with dsDNA. (c) The optimal geometric spacing (inter‐DNA distance) between ordered dsDNA molecules bound to chemokine C‐X‐C ligand motif (CXCL4) [or other polycations, such as LL‐37 or human β‐defensin (HBD3)] is in the range between 3 and 4 nm (d). This amplitude almost matches the steric size of TLR‐9 and allows activate multiple TLR‐9 at the same time leading to the optimal interferon (IFN)‐α production. Outside this range (d smaller or larger than 3–4 nm), stimulation results in a modest or no IFN‐α production.
Given that self‐RNA can be a crucial DAMP for both TLR‐7/‐8 and TLR‐3, we anticipate that CXCL4 could also amplify RNA stimulation of these TLRs, as our preliminary studies indicate that CXCL4 can also condense human and bacterial RNA (Lande and Frasca, unpublished). Up to the present time, we have observed that pDCs also secrete IFN‐α when challenged with CXCL4–RNA complexes, but not when cultured with CXCL4 alone or RNA alone (Fig. 3a).
Figure 3.
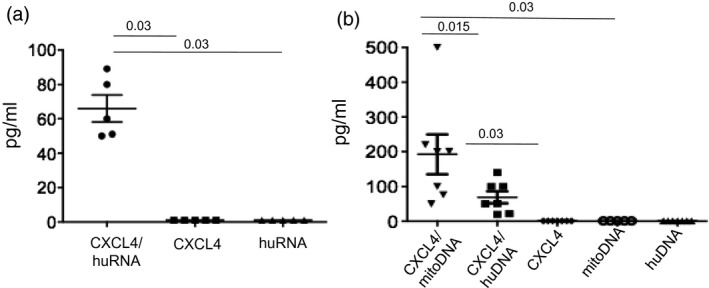
Chemokine C‐X‐C ligand 4 motif (CXCL4) in complex with human nucleic acids of various origin stimulates interferon (IFN)‐α release by platelet dendritic cells (pDCs). (a) Purified pDCs (175 × 103/ml) from five different healthy donor (HD) peripheral blood mononuclear cells (PBMCs) were treated with CXCL4 alone (1 μM), human RNA alone (huRNA, 20 μg/ml) or CXCL4 pre‐complexed with huRNA (at the same concentrations), overnight. The release of IFN‐α in the culture supernatants was tested by enzyme‐linked immunosorbent assay (ELISA), as described 29. (b) PDCs (175 × 103/ml) from seven different HD PBMCs were stimulated overnight with CXCL4 alone (1 μM), human DNA alone (huDNA, 10 μg/ml), mitochondrial DNA (mitoDNA, 10 μg/ml) or CXCL4 precomplexed with the two DNA types. IFN‐α in the culture supernatants was tested by ELISA. Horizontal bars are the means, vertical bars are standard errors of the mean, P‐values by Wilcoxon’s signed‐rank test.
The role of platelets in SSc
The role of platelets in SSc is established 44. In SSc there is a relatively high incidence of anti‐platelet antibodies, which mediate platelet activation 55. Platelets express at least functional TLR‐1/‐2/‐4 and TLR‐3/‐7/‐9 56. The triggering of TLR‐7 leads to cell surface exposure and release of CD40L (also known as CD154), which is a co‐stimulatory molecule located in platelet alpha granules 57. CD40L interacts with CD40 expressed by leucocytes (B cells, monocytes, DCs, neutrophils) and endothelial cells. The CD40–CD40L interactions are central in immunity as they mediate co‐stimulation, implement DC maturation and induce survival of B cells after antigen recognition by the B cell receptor 58, 59, 60. Interestingly, CD40L is commonly up‐regulated in SSc blood 61. Accumulating evidence points to a pathogenic role of the aberrantly activated platelets in the process of tissue damage and general inflammation in several autoimmune diseases 62, 63, 64. Platelets are probably involved in primary and/or secondary RP, as platelet activation markers are detectable during RP 65, 66. Platelets can also favour neutrophil‐extracellular trap release (NET) 13, the process that leads to release of huge amounts of DNA and autoantigens in the tissues, fuelling a harmful loop.
The important role of platelets in SSc is also worth mention with respect to CXCL4 up‐regulation: platelets are the major source of released CXCL4 upon infections or traumas 50. Indeed, platelets release huge amounts of CXCL4 after activation. Of interest, some studies uncovered that the lungs are sites of thrombopoiesis and reservoirs for platelets 67. Platelets regulate pulmonary vascular permeability of alveolar capillaries and have specialized activities in lung repair. Because CXCL4 elevation in SSc associates mainly with lung fibrosis and pulmonary arterial hypertension, it may be worthwhile to understand the exact CXCL4 expression in the SSc lung tissue. For instance, it can be of interest to clarify whether platelets or pDCs are the most important CXCL4 producers in the lung, and which ligand/receptor pairs are responsible for platelet activation and CXCL4 release, if occurring, in the lung.
Interestingly, although anucleated, platelets contain mitochondrial DNA (mitoDNA), thus they can be a source of mitoDNA–CXCL4 complexes. We detected platelets in SSc skin, where they appeared to form aggregates and co‐localized with CXCL4 staining (Lande and Frasca, unpublished observations). Boudreau et al. demonstrated that, upon activation, platelets release mitochondria in the extracellular milieu 68. Of interest, we observed that mitoDNA, which is more similar to bacterial DNA when complexed with CXCL4, was able to activate pDCs even more effectively than huDNA (Fig. 3b).
Other possible DNA/RNA DAMPs in SSc
Two studies have demonstrated that the anti‐microbial peptide (AMP) cathelicidin LL‐37, a widely studied molecule amplifying TLR‐7/‐8/‐9 signalling 69, 70, is over‐expressed in SSc skin, especially in the dermis 71, 72. LL‐37 was the first anti‐microbial peptide shown to bind DNA and activate IFN‐α release in pDCs via TLR‐9 as a mechanism explaining the IFN‐α signature in psoriasis skin 69.
Later, other cationic proteins 73, 74, 75 and the AMP HBD2 and HBD3 (human β‐defensins) were shown to perform similar functions. HBD2 and HBD3 mRNA levels were higher in lesional skin of localized scleroderma patients compared to unaffected skin and skin from healthy volunteers 76. We have also analysed LL‐37, HBD2 and HBD3 expression in SSc skin by immunohistochemistry (Frasca, unpublished work). Occasionally we found HBD2 or HBD3 expression but, more often, we detected LL‐37 up‐regulation, especially in the dermis, and this coincided with IFN‐induced gene expression (Mx1) in accordance with Takahashi et al.’s 72 experiments (Frasca, unpublished). LL‐37 may be a putative TLR‐7/‐8 stimulator in SSc, as it stimulates TLR‐7/‐8 in myeloid DCs after forming complexes with human‐RNA, leading to TNF‐α and IL‐6 production 70.
TLRs and the IFN‐I signature in SSc
As mentioned previously, almost 50% of SSc patients exhibit an IFN‐I gene signature in blood and tissues. Moreover, a few polymorphisms of IFN‐regulatory genes can be associated with SSc 49. In some cases the type I interferons have been found to be beneficial in SSc animal models; for instance, in the bleomycin model, where IFN‐β attenuated the disease. However, many other papers indicate IFN‐I to be deleterious in SSc 49; a trial administering IFN‐α failed to cure SSc and was instead harmful to patients 77. In addition, IFN‐α treatment for other pathological conditions can induce SSc 78, 79. Although an IFN‐I signature can be detected both in early and long‐lasting SSc, it appears that this signature can be present even at very early SSc stages 49, 80.
We would like to emphasize here that, according to the literature, activation of the IFN‐I pathway as an early event in SSc is associated, together with high CXCL4, with more severe disease manifestation and poor prognosis 36, 37, 49, 80. This is in agreement with our observation that the presence in SSc plasma of CXCL4–DNA complexes correlated with the circulating IFN‐I signature, in particular in a group of patients with early active SSc 38. Thus, disrupting CXCL4–nucleic acid interaction or blocking excess of CXCL4 can be a possible intervention in early SSc. At later stages a correlation between plasma CXCL4–DNA complexes and circulating IFN‐α is also present 38, suggesting that CXCL4 contributes to the IFN‐I signature at any disease stages, thus long‐lasting patients can also benefit from anti‐IFN‐I treatment.
We estimate that treatment with drugs able to contrast DNA‐sensing TLR activation at very early stages, especially in those patients who immediately show a significant IFN‐I signature, could be of help. SSc patients could benefit from treatment with old and, therefore less expensive drugs such as chloroquine, already used for SLE (as a repositioning drug strategy). Moreover, chloroquine can also block TLR‐3 signalling 81.
In the context of the activation of the IFN‐I signature in SSc, a very recent paper demonstrated that interferon regulatory factor 7 (IRF7), a master regulator of the IFN‐I signature, is up‐regulated in SSc skin 82, and can represent a link between the prominent IFN‐I signature and fibrosis. The authors demonstrated that IRF7 associated with Smad3 in SSc fibroblasts, and that absence of IRF7 in the bleomycin mouse model attenuated fibrosis of the dermis.
Pathogens as activators of TLRs in SSc
The interplay between pathogenic viruses or bacteria and the immune system may contribute to autoimmune diseases 83, 84. Indications that herpesvirus infections play a role in SLE or multiple sclerosis are present in the literature 85, 86. One mechanism by which pathogens favour autoimmunity is the phenomenon of ‘molecular mimicry’ between self‐ and pathogen‐derived molecules, which can confound the immune system self–non‐self‐discrimination 87. As a second mechanism, the inability to clear the pathogens favours persistent infections and therefore continuous stimulation of innate immune cells via TLRs. Some infectious agents have been proposed as possible SSc triggering factors, such as parvovirus B19, cytomegalovirus, Epstein–Barr virus (EBV) and retroviruses 88.
Latent infections by EBV, which replicates in primary human monocytes, could trigger SSc. Induction of EBV viral lytic genes induced TLR‐8 expression in both HD and SSc monocytes infected with EBV 89. EBV can infect fibroblasts and endothelial cells of SSc skin, and this can lead to an aberrant TLR stimulation in these cells. Such stimulation could induce well‐known markers of fibrosis, including TGF‐β and EDN1, and conversion of fibroblasts into myofibroblasts 90.
Cytomegalovirus (HCMV) infections are also candidates for SSc induction, in that SSc can manifest shortly after an acute episode of infection with HCMV 91. Products of HCMV are involved in the induction of a fibrotic programme in human dermal fibroblasts and cause vasculopathy similar to that observed in SSc. However, a clear activation of TLRs in immune and non‐immune cells by HCMV is lacking 92. Conversely, B19 virus infection of monocytes from patients with SSc was found to induce TNF‐α, more frequently than in HD monocyte cultures 92. B19 virus‐induced production of TNF‐α positively correlated with the amount of viral DNA detected at the end of incubation time, suggesting a role for viral DNA in cytokine production, probably through TLR‐9 activation 93.
Conclusions
SSc is a disease in which innate immune cells play a role and probably support adaptive immune cell licensing and activation, favouring autoimmunity establishment. TLR engagement may activate immune cells. However, several studies report that non‐immune cells, namely fibroblasts and endothelial cells (cell types important in SSc pathogenesis) can express basal low levels of the same TLRs. Such expression can increase under inflammatory conditions. Therefore, these cells can also experience an aberrant TLR‐driven stimulation 94, 95, 96, 97, 98. Of note, endothelial cell dysfunctions and dysregulation of wound repair are typical of SSc. Several DAMPs have been identified in SSc which can trigger various TLRs, among which TLR‐4, TLR‐7, TLR‐8 and TLR‐9 ligands are probably the most important, although TLR‐3‐signalling can also play a role in SSc pathology. The IFN‐I activation pathway also plays a crucial role in SSc. The IFN‐I signature at onset or in early stages, as mentioned above, forecasts a poor prognosis, and early IFN‐I block can be of help. Newly discovered molecules, such as CXCL4, can represent a link between pDCs, the IFN‐I axis and the fibrotic process. We need more in‐vivo and in‐vitro studies to confirm the importance of the new players. New therapeutic agents should be tested in parallel with old treatments (for instance, some of those currently used in SLE) for the capacity to block the IFN‐I pathways, CXCL4 and pDCs.
Disclosures
The authors declare no conflicts of interest
Author contributions
L. F. organized and wrote the manuscript and prepared the figures with the help of R. L. Both L. F. and R. L. planned and performed the experiments reported in the review.
Acknowledgements
We thank Raffaella Palazzo and Immacolata Pietraforte for technical assistance and Anna Mennella for critical reading of the manuscript.
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Innate immunity in systemic sclerosis. Clinical and Experimental Immunology 2020, 201: 12‐13.
Novel insights into dendritic cells in the pathogenesis of systemic sclerosis. Clinical and Experimental Immunology 2020, 201: 25‐33.
The role of innate immune cells in systemic sclerosis in the context of autologous hematopoietic stem cell transplantation. Clinical and Experimental Immunology 2020, 201: 34‐39.
References
- 1. Denton CP, Khanna D. Systemic sclerosis. Lancet 2017; 390:1685–99. [DOI] [PubMed] [Google Scholar]
- 2. Zanatta E, Codullo V, Avouac J, Allanore Y. Systemic sclerosis: recent insight in clinical management. Joint Bone Spine 2019. doi: 10.1016/j.jbspin.2019.09.015. [DOI] [PubMed] [Google Scholar]
- 3. O’Reilly S, Hugle T, van Laar JM. T cells in systemic sclerosis: a reappraisal. Rheumatology (Oxf) 2012; 51:1540–9. [DOI] [PubMed] [Google Scholar]
- 4. Abraham DJ, Krieg T, Distler J, Distler O. Overview of pathogenesis of systemic sclerosis. Rheumatology (Oxf) 2009; 48(Suppl 3):S3–7. [DOI] [PubMed] [Google Scholar]
- 5. Cutolo M, Soldano S, Smith V. Pathophysiology of systemic sclerosis: current understanding and new insights. Expert Rev Clin Immunol 2019; 15:753–64. [DOI] [PubMed] [Google Scholar]
- 6. O’Reilly S. Toll‐like receptors in systemic sclerosis: an emerging target. Immunol Lett 2018; 195:2–8. [DOI] [PubMed] [Google Scholar]
- 7. Laurent P, Sisirak V, Lazaro E et al Innate immunity in systemic sclerosis fibrosis: recent advances. Front Immunol 2018; 9:1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beutler BA. TLRs and innate immunity. Blood 2009; 113:1399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Theofilopoulos AN, Gonzalez‐Quintial R, Lawson BR et al Sensors of the innate immune system: their link to rheumatic diseases. Nat Rev Rheumatol 2010; 6:146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gong T, Liu L, Jiang W, Zhou R. DAMP‐sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol 2019; 20:95–112. [DOI] [PubMed] [Google Scholar]
- 11. Paudel YN, Angelopoulou E, Piperi C, Balasubramaniam V, Othman I, Shaikh MF. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: updates on receptor signalling. Eur J Pharmacol 2019; 858:172487. [DOI] [PubMed] [Google Scholar]
- 12. Tian J, Avalos AM, Mao SY et al Toll‐like receptor 9‐dependent activation by DNA‐containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 2007; 8:487–96. [DOI] [PubMed] [Google Scholar]
- 13. Maugeri N, Capobianco A, Rovere‐Querini P et al Platelet microparticles sustain autophagy‐associated activation of neutrophils in systemic sclerosis. Sci Transl Med 2018; 10:eaao3089. [DOI] [PubMed] [Google Scholar]
- 14. Zhang Q, Raoof M, Chen Y et al Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464:104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takeda K, Kaisho T, Akira S. Toll‐like receptors. Annu Rev Immunol 2003; 21:335–76. [DOI] [PubMed] [Google Scholar]
- 16. Park JS, Svetkauskaite D, He Q et al Involvement of toll like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 2004; 279:7370–7. [DOI] [PubMed] [Google Scholar]
- 17. O’Reilly S, Cant R, Ciechomska M et al Serum amyloid A induces interleukin‐6 in dermal fibroblasts via Toll‐like receptor 2, interleukin‐1 receptor‐associated kinase 4 and nuclear factor‐κB. Immunology 2014; 143:331–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brandwein SR, Medsger TA Jr, Skinner M, Sipe JD, Rodnan GP, Cohen AS. Serum amyloid A protein concentration in progressive systemic sclerosis (scleroderma). Ann Rheum Dis 1984; 43:586–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Broen JC, Bossini‐Castillo L, van Bon L et al A rare polymorphism in the gene for Toll‐like receptor 2 is associated with systemic sclerosis phenotype and increases the production of inflammatory mediators. Arthritis Rheum 2011; 64:264–71. [DOI] [PubMed] [Google Scholar]
- 20. Bhattacharyya S, Tamaki Z, Wang W et al Fibronectin EDA promotes chronic cutaneous fibrosis through Toll‐like receptor signaling. Sci Transl Med 2014; 6:232ra50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bhattacharyya S, Wang W, Morales‐Nebreda L et al Tenascin‐C drives persistence of organ fibrosis. Nat Commun 2016; 7:11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bhattacharyya S, Wang W, Qin W et al TLR4‐dependent fibroblast activation drives persistent organ fibrosis in skin and lung. JCI Insight 2018; 3:e98850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sakoguchi A, Nakayama W, Jinnin M et al The expression profile of the toll‐like receptor family in scleroderma dermal fibroblasts. Clin Exp Rheumatol 2014; 32(Suppl 86):S4–9. [PubMed] [Google Scholar]
- 24. Hayashi F, Smith KD, Ozinsky A et al The innate immune response to bacterial flagellin is mediated by Toll‐like receptor 5. Nature 2001; 410:1099–103. [DOI] [PubMed] [Google Scholar]
- 25. Yang J, Yan H. TLR5: beyond the recognition of flagellin. Cell Mol Immunol 2017; 14:1017–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Das N, Dewan V, Grace PM et al HMGB1 activates proinflammatory signaling via TLR5 leading to allodynia. Cell Rep 2016; 17:1128–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Charmberlain ND, Vila OM, Volin MV et al TLR5, a novel and unidentified inflammatory mediator in rheumatoid arthritis that correlates with disease activity score and joint TNF‐α levels. J Immunol 2012; 189:475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dubrez L, Causse S, Borges Bonan N, Dumétier B, Garrido C. Heat‐shock proteins: chaperoning DNA repair. Oncogene 2020; 39:516–29. [DOI] [PubMed] [Google Scholar]
- 29. Aden N, Shiwen X, Aden D et al Proteomic analysis of scleroderma lesional skin reveals activated wound healing phenotype of epidermal cell layer. Rheumatology 2008; 47:1754–60. [DOI] [PubMed] [Google Scholar]
- 30. Hasan U, Chaffois C, Gaillard C et al Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol 2005; 174:2942–50. [DOI] [PubMed] [Google Scholar]
- 31. Lee SM, Yip TF, Yan S et al Recognition of double‐stranded RNA and regulation of interferon pathway by toll‐like receptor 10. Front Immunol 2018; 9:516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Greulich W, Wagner M, Gaidt MM et al TLR8 is a sensor of RNase T2 degradation products. Cell 2019; 179:1264–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peltzer N, Walczak H. Cell death and inflammation – a vital but dangerous liaison. Trends Immunol 2019; 40:387–402. [DOI] [PubMed] [Google Scholar]
- 34. Daniel C, Leppkes M, Munoz LE, Schley G, Schett G, Herrmann M. Extracellular DNA traps in inflammation, injury and healing. Nat Rev Nephrol 2019; 15:559–75. [DOI] [PubMed] [Google Scholar]
- 35. Hewagama A, Richardson B. The genetics and epigenetics of autoimmune diseases. J Autoimmun 2009; 33:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim D, Peck A, Santer D et al Induction of interferon‐alpha by scleroderma sera containing autoantibodies to topoisomerase I: association of higher interferon‐alpha activity with lung fibrosis. Arthritis Rheum 2008; 58:2163–73. [DOI] [PubMed] [Google Scholar]
- 37. Eloranta ML, Franck‐Larsson K, Lovgren T et al Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis 2010; 69:1396–402. [DOI] [PubMed] [Google Scholar]
- 38. Lande R, Lee EY, Palazzo R et al CXCL4 assembles DNA into liquid crystalline complexes to amplify TLR9‐mediated interferon‐alpha production in systemic sclerosis. Nat Commun 2019; 10:1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Raschi E, Chighizola CB, Cesana L et al Immune complexes containing scleroderma‐specific autoantibodies induce a profibrotic and proinflammatory phenotype in skin fibroblasts. Arthritis Res Ther 2018; 20:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fang F, Marangoni RG, Zhou X et al Toll‐like receptor 9 signaling is augmented in systemic sclerosis and elicits transforming growth factor beta‐dependent fibroblast activation. Arthritis Rheumatol 2016; 68:1989–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vreca M, Zekovic A, Damjanov N et al Expression of TLR7, TLR9, JAK2, and STAT3 genes in peripheral blood mononuclear cells from patients with systemic sclerosis. J Appl Genet 2018; 59:59–66. [DOI] [PubMed] [Google Scholar]
- 42. Souyris M, Mejia JE, Chaumeil J, Guery JC. Female predisposition to TLR7‐driven autoimmunity: gene dosage and the escape from X chromosome inactivation. Semin Immunopathol 2018; 41:153–64. [DOI] [PubMed] [Google Scholar]
- 43. Souyris M, Cenac C, Azar P et al TLR7 escapes X chromosome inactivation in immune cells. Sci Immunol 2018; 3:eaap8855. [DOI] [PubMed] [Google Scholar]
- 44. Farina GA, York MR, Di Marzio M et al Poly(I:C) drives type I IFN‐ and TGFbeta‐mediated inflammation and dermal fibrosis simulating altered gene expression in systemic sclerosis. J Invest Dermatol 2010; 130:2583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fang F, Ooka K, Sun X et al A synthetic TLR3 ligand mitigates profibrotic fibroblast responses by inducing autocrine IFN signaling. J Immunol 2013; 191:2956–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Farina G, York M, Collins C, Lafyatis R. dsRNA activation of endothelin‐1 and markers of vascular activation in endothelial cells and fibroblasts. Ann Rheum Dis 2011; 70:544–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Assassi S, Mayes MD, Arnett FC et al Systemic sclerosis and lupus: points in an interferon‐mediated continuum. Arthritis Rheum 2010; 62:589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Bon L, Affandi AJ, Broen J et al Proteome‐wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med 2014; 370:433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Skaug B, Assassi S. Type I interferon dysregulation in systemic sclerosis. Cytokine 2019; 23:154635. [DOI] [PubMed] [Google Scholar]
- 50. Vandercappellen J, Van Damme J, Struyf S. The role of the CXC chemokines platelet factor‐4 (CXCL4/PF‐4) and its variant (CXCL4L1/PF‐4var) in inflammation, angiogenesis and cancer. Cytokine Growth Factor Rev 2011; 22:1–18. [DOI] [PubMed] [Google Scholar]
- 51. Ah Kioon MD, Tripodo C, Fernandez D et al Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med 2018; 10:eaam8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kafaja S, Valera I, Divekar AA et al pDCs in lung and skin fibrosis in a bleomycin‐induced model and patients with systemic sclerosis. JCI Insight 2018; 3:98380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schmidt NW, Jin F, Lande R et al Liquid‐crystalline ordering of antimicrobial peptide‐DNA complexes controls TLR9 activation. Nat Mater 2015; 14:696–700. [DOI] [PubMed] [Google Scholar]
- 54. Lee EY, Lee MW, Wong GCL. Modulation of toll‐like receptor signaling by antimicrobial peptides. Semin Cell Dev Biol 2019; 88:173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Scherlinger M, Guillotin V, Truchetet ME et al Systemic lupus erythematosus and systemic sclerosis: all roads lead to platelets. Autoimmun Rev 2018; 17:625–35. [DOI] [PubMed] [Google Scholar]
- 56. Czirjak L, Molnar I, Csipo I, Szabolcs M, Mihaly A, Szegedi G. Anti‐platelet antibodies against gpIIb/IIIa in systemic sclerosis. Clin Exp Rheumatol 1994; 12:527–9. [PubMed] [Google Scholar]
- 57. Koupenova M, Vitseva O, MacKay CR et al Platelet‐TLR7 mediates host survival and platelet count during viral infection in the absence of platelet‐dependent thrombosis. Blood 2014; 124:791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ma DY, Clark EA. The role of CD40 and CD40L in dendritic cells. Semin Immunol 2009; 21:265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 2009; 229:152–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gordon J. CD40 and its ligand: central player in B lymphocyte survival, growth and differentiation. Blood Rev 1995; 9:53–6. [DOI] [PubMed] [Google Scholar]
- 61. Doran JP, Veale DJ. Biomarkers in systemic sclerosis. Rheumatology 2008; 47:36–8. [DOI] [PubMed] [Google Scholar]
- 62. Cognasse F, Nguyen KA, Damien P et al The inflammatory role of platelets via their TLRs and Siglec receptors. Front Immunol 2015; 6:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lievenss D, Zernecke A, Seijkens T et al Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood 2010; 116:4317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ntelis K, Bogdanos D, Dimitroulas T, Sakkas L, Daoussis D. Platelets in systemic sclerosis: the missing link connecting vasculopathy, autoimmunity, and fibrosis? Curr Rheumatol Rep 2019; 21:15. [DOI] [PubMed] [Google Scholar]
- 65. Pamuk GE, Turgut B, Pamuk ON et al Increased circulating platelet–leucocyte complexes in patients with primary Raynaud’s phenomenon and Raynaud’s phenomenon secondary to systemic sclerosis: a comparative study. Blood Coagul Fibrinolysis 2007; 18:297–302. [DOI] [PubMed] [Google Scholar]
- 66. Shemirani AH, Nagy B Jr, Takats AT et al Increased mean platelet volume in primary Raynaud’s phenomenon. Platelets 2011; 23:312–6. [DOI] [PubMed] [Google Scholar]
- 67. Lefrancais E, Ortiz‐Munoz G, Caudrillier A et al The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017; 544:105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Boudreau LH, Duchez AC, Cloutier N et al Platelets release mitochondria serving as substrate for bactericidal group IIA‐secreted phospholipase A2 to promote inflammation. Blood 2014; 124:2173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lande R, Gregorio J, Facchinetti V et al Plasmacytoid dendritic cells sense self‐DNA coupled with antimicrobial peptide. Nature 2007; 449:564–9. [DOI] [PubMed] [Google Scholar]
- 70. Ganguly D, Chamilos G, Lande R et al Self‐RNA‐antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med 2009; 206:1983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim HJ, Cho DH, Lee KJ et al LL‐37 suppresses sodium nitroprusside‐induced apoptosis of systemic sclerosis dermal fibroblasts. Exp Dermatol 2011; 20:843–5. [DOI] [PubMed] [Google Scholar]
- 72. Takahashi T, Asano Y, Nakamura K et al A potential contribution of antimicrobial peptide LL‐37 to tissue fibrosis and vasculopathy in systemic sclerosis. Br J Dermatol 2016; 175:1195–203. [DOI] [PubMed] [Google Scholar]
- 73. Tewary P, de la Rosa G, Sharma N et al β‐defensin 2 and 3 promote the uptake of self or CpG DNA, enhance IFN‐α production by human plasmacytoid dendritic cells, and promote inflammation. J Immunol 2013; 191:865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lande R, Chamilos G, Ganguly D et al Cationic antimicrobial peptides in psoriatic skin cooperate to break innate tolerance to self‐DNA. Eur J Immunol 2015; 45:203–13. [DOI] [PubMed] [Google Scholar]
- 75. Meller S, Di Domizio J, Voo KS et al TH17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol 2015; 16:970–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kreuter A, Hyun J, Skrygan M et al Ultraviolet Al‐induced downregulation of human beta‐defensins and interleukin‐6 and interleukin‐8 correlates with clinical improvement in localized scleroderma. Br J Dermatol 2006; 155:600–7. [DOI] [PubMed] [Google Scholar]
- 77. Black CM, Silman AJ, Herrick AI et al Interferon‐alpha does not improve outcome at one year in patients with diffuse cutaneous scleroderma: results of a randomized, double‐blind, placebo‐controlled trial. Arthritis Rheum 1999; 42:299–305. [DOI] [PubMed] [Google Scholar]
- 78. Beretta L, Caronni M, Vanoli M, Scorza R. Systemic sclerosis after interferon‐alfa therapy for myeloproliferative disorders. Br J Dermatol 2002; 147:385–6. [DOI] [PubMed] [Google Scholar]
- 79. Powell A, Myles ML, Yacyshyn E. The development of systemic sclerosis in a female patient with multiple sclerosis following beta interferon treatment. Clin Rheumatol 2008; 27:1467–8. [DOI] [PubMed] [Google Scholar]
- 80. Brkic Z, van Bon L, Cossu M et al The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann Rheum Dis 2016; 75:1567–73. [DOI] [PubMed] [Google Scholar]
- 81. Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol 2011; 186:4794–804. [DOI] [PubMed] [Google Scholar]
- 82. Wu M, Skaug B, Bi X et al Interferon regulatory factor 7 (IRF7) represents a link between inflammation and fibrosis in the pathogenesis of systemic sclerosis. Ann Rheum Dis 2019; 78:1583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vogel A, Manns MP, Strassburg CP. Autoimmunity and viruses. Clin Liver Dis 2002; 6:739–53. [DOI] [PubMed] [Google Scholar]
- 84. Smatti MK, Cyprian FS, Nasrallah GK, Al Thani AA, Almishal RO, Yassine HM. Viruses and autoimmunity: a review on the potential interaction and molecular mechanisms. Viruses 2019; 11:E762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Owens GP, Gilden D, Burgoon MP, Yu X, Bennett JL. Viruses and multiple sclerosis. Neuroscientist 2012; 17:659–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li ZX, Zeng S, Wu HX, Zhou Y. The risk of systemic lupus erythematosus associated with Epstein–Barr virus infection: a systematic review and meta‐analysis. Clin Exp Med 2018; 19:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rashid T, Ebringer A. Autoimmunity in rheumatic diseases is induced by microbial infections via crossreactivity or molecular mimicry. Autoimmune Dis 2012; 21:539282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Randone SB, Guiducci S, Cerinic MM. Systemic sclerosis and infections. Autoimmun Rev 2008; 8:36–40. [DOI] [PubMed] [Google Scholar]
- 89. Farina A, Peruzzi G, Lacconi V et al Epstein–Barr virus lytic infection promotes activation of Toll‐like receptor 8 innate immune response in systemic sclerosis monocytes. Arthritis Res Ther 2017; 19:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Farina A, Cirone M, York M et al Epstein–Barr virus infection induces aberrant TLR activation pathway and fibroblast–myofibroblast conversion in scleroderma. J Invest Dermatol 2014; 134:954–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Janahi EMA, Das S, Bhattacharya SN et al Cytomegalovirus aggravates the autoimmune phenomenon in systemic autoimmune diseases. Microb Pathog 2018; 120:132–9. [DOI] [PubMed] [Google Scholar]
- 92. Efthymiou G, Dardiotis E, Liaskos C et al A comprehensive analysis of antigen‐specific antibody responses against human cytomegalovirus in patients with systemic sclerosis. Clin Immunol 2019; 207:87–96. [DOI] [PubMed] [Google Scholar]
- 93. Zakrzewska K, Arvia R, Torcia MG et al Effects of parvovirus B19 in vitro infection on monocytes from patients with systemic sclerosis: enhanced inflammatory pathways by caspase‐1 activation and cytokine production. J Invest Dermatol 2019; 139:2125–33.e1. [DOI] [PubMed] [Google Scholar]
- 94. Stawski L, Marden G, Trojanowska M. The activation of human dermal microvascular cells by Poly(I:C), lipopolysaccharide, imiquimod, and ODN2395 is mediated by the Fli1/FOXO3A pathway. J Immunol 2018; 200:248–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Fitzner N, Clauberg S, Essmann F, Liebmann J, Kolb‐Bachofen V. Human skin endothelial cells can express all 10 TLR genes and respond to respective ligands. Clin Vacc Immunol 2008; 15:138–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wang JF, Hori K, Ding J et al Toll‐like receptors expressed by dermal fibroblasts contribute to hypertrophic scarring. J Cell Physiol 2011; 226:1265–73. [DOI] [PubMed] [Google Scholar]
- 97. Yao C, Oh JH, Lee DH, Bae JS, Jin CL, Park CH, Chung JH. Toll‐like receptor family members in skin fibroblasts are functional and have a higher expression compared to skin keratinocytes. Int J Mol Med 2015; 35:1443–50. [DOI] [PubMed] [Google Scholar]
- 98. Fukui A, Ohta K, Nishi H et al Interleukin‐8 and CXCL10 expression in oral keratinocytes and fibroblasts via Toll‐like receptors. Microbiol Immunol 2013; 57:198–206. [DOI] [PubMed] [Google Scholar]