

Graphical abstract

Keywords: Venom, Venom peptides, Pharmacological tools, Drug discovery, Therapeutics, Insecticides
Abstract
Venomous animals have evolved toxins that interfere with specific components of their victim’s core physiological systems, thereby causing biological dysfunction that aids in prey capture, defense against predators, or other roles such as intraspecific competition. Many animal lineages evolved venom systems independently, highlighting the success of this strategy. Over the course of evolution, toxins with exceptional specificity and high potency for their intended molecular targets have prevailed, making venoms an invaluable and almost inexhaustible source of bioactive molecules, some of which have found use as pharmacological tools, human therapeutics, and bioinsecticides. Current biomedically-focused research on venoms is directed towards their use in delineating the physiological role of toxin molecular targets such as ion channels and receptors, studying or treating human diseases, targeting vectors of human diseases, and treating microbial and parasitic infections. We provide examples of each of these areas of venom research, highlighting the potential that venom molecules hold for basic research and drug development.
1. Introduction
Venomous animals have taken advantage of the inherent biological complexity of their victims by developing toxins that target one or all of the nervous, musculoskeletal, or cardiovascular systems in order to facilitate prey capture or defense against predators. For example, many predatory venoms contain neurotoxins that disrupt neuromuscular transmission in order to rapidly paralyse envenomated prey [1]. Due to the inherent specificity and potency of most venom molecules, venoms have become an invaluable source of modulators to study a wide range of molecular targets, especially ligand-activated [2], [3], [4] and voltage-gated ion channels [5], [6], [7]. While research on many potential drug targets is hampered by the lack of selective modulators, our increasing knowledge of venom pharmacology is beginning to shed light on some of these poorly studied targets (e.g. [8]). Here we provide examples of how venom compounds have been used to study diverse molecular targets and aid our understanding of their role in human disease. We use knowledge gained from approved venom-derived drugs [9], [10] to illustrate how basic research can lead all the way from venom to a human therapeutic. Finally, we touch on other potential uses of toxins for human health, such as targeting vectors of human diseases, and development of antimicrobial and antiparasitic drugs (Fig. 1 ).
Fig. 1.
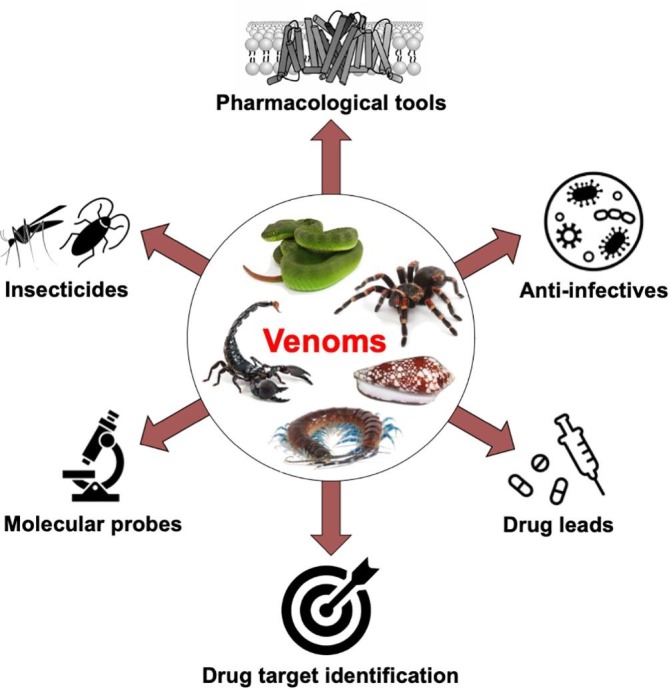
Fields of application in basic research and human health to which venom compounds have been applied. Venom compounds have the potential to be used as pharmacological tools, therapeutics, insecticides, or for targeting vectors of human disease or disease-inducing organisms.
2. The molecular toolkit of bioactive venom compounds
During the course of evolution, venom has evolved independently at least 100 times across eight different phyla [11]. It has been estimated that there are >220,000 venomous species on the planet, representing ~15% of extant animal biodiversity [12], spanning invertebrates such as annelids, arthropods, cnidarians, molluscs, nematodes, sea urchins, star fish, and vertebrates such as snakes, lizards, fish, shrews, and platypus [1], [12], [13], [14]. Chemically, venoms are complex mixtures of salts, small molecules, peptides, and proteins [15], [16], with an estimated 20 million compounds in spider venoms alone [17]. Most of these compounds are toxins, which are defined as substances that cause dose-dependent pathophysiological injury to a living organism, thereby reducing their viability [18]. The high metabolic cost of venom production [19] has ensured that over the course of evolution only the most potent toxins have prevailed. Venoms can therefore be considered natural libraries of highly optimised bioactive compounds [20], [21]. Given the vastly different phyletic origins of venomous species and the even greater diversity of their target organisms, it is expected that venom toxins modulate an extremely diverse range of molecular targets (Fig. 2 ).
Fig. 2.

The diversity in molecular structures of venom-derived toxins and their targets. The Protein Data Bank accession codes are indicated. (A) Snake toxin complexes: α-bungarotoxin bound to the α1 extracellular domain of the nicotinic acetylcholine receptor (2qc1) (nAChR) [65]; botrocetin bound to von Willebrand Factor (vWF) and platelet glycoprotein Ib (GPIb) (1u0n) [250]; cobra venom factor (CVF) bound to complement factor B (3hrz) [251]; captopril bound to angiotensin converting enzyme (ACE) (1uzf) [252]; triflin bound to small serum protein 2 (SSP2) (6imf) [253]; MitTx-α and –β bound to acid sensing ion channel 1 (ASIC1) (4ntw) [254]. (B) Cone snail toxin complexes: α-conotoxin ImI bound to acetylcholine binding protein (AChBP) (2byp) [255]; μ-conotoxin KIIIA bound to voltage-gated sodium channel (NaV)1.2/auxiliary β2 subunit (6j8e) [256]. (C) Spider toxin complexes: PcTx1 bound to ASIC1 (4fz0) [257]; Dc1a and tetrodotoxin (TTX) bound to NaVPaS (6a95) [228]; DkTx and resiniferatoxin (RTX) bound to transient receptor potential cation channel subfamily V member 1 (TRPV1) (5irx) [258]; ProTx-II bound to NaV1.7 voltage sensor domain II/NaVAB chimera (6n4i) [243]. (D) Gila monster toxin exendin-4 to bound glucagon-like peptide 1 receptor (GLP-1R) (3c5t) [259]. (E) Scorpion toxin complexes: charybdotoxin (CTX) bound to voltage-gated potassium channel (KV) 1.2–2.1 paddle chimera (4jta) [260]; α-mammal toxin AaH2 bound to human-cockroach hybrid NaV channel (6nt4) [246]. Structures are shown as a cartoon representation and from two angles (90° rotation). Toxin components are shown in purple and with a transparent surface representation where possible (note: TTX and RTX are shown as red sticks). Where applicable for the target structure, protein subunits or domains are colored differently. Structures are not to scale.
The dominant components of most venoms are small peptides. The high potency, selectivity and biological stability of many venom-derived peptides has made them particularly attractive from a drug discovery perspective. There are already two FDA-approved venom-derived peptides in clinical use — ziconotide, an analgesic drug from the venom of a marine cone snail [22], and exenatide, an anti-diabetic drug from a venomous lizard [23] — plus numerous others in clinical trials [24], [25] or preclinical development [9], [10]. The larger size of peptides compared to small molecules provides a larger binding surface with their molecular target, typically yielding higher potency and greater target specificity, thereby minimising unwanted side-effects. However, the larger size and polar nature of peptides translates into poor oral bioavailability. Other disadvantages of peptides can be poor membrane permeability and low metabolic stability [26], although these disadvantages can often be overcome by chemical modification [27], [28], [29]. In addition, progress in the chemical synthesis [30], [31], [32] and recombinant production [33], [34] of complex peptides has facilitated cost-effective venom-peptide manufacture.
Venomous animals have evolved peptide toxins with a huge diversity of molecular frameworks, most of them with tertiary structures stabilised by disulfide bonds [35]. The most well-known of these disulfide-rich frameworks is the ubiquitous inhibitor cystine knot (ICK) or knottin motif [36], [37], which is defined as an antiparallel β-sheet stabilised by a cystine knot [38]. A major advantage of ICK peptides for drug development is their resistance to proteases, high temperature, and harsh chemicals, which has been attributed to their knotted structure [39], [40], [41]. However, despite our extensive knowledge about a limited number of disulfide frameworks present in animal venoms, there are many families of disulfide-rich venom peptides that remain completely unexplored (e.g. [42], [43], [44], [45]). Further research on these novel disulfide-rich frameworks is therefore required to unleash any potential they might hold for drug development.
Recent advances in proteomics and transcriptomics have now made it possible to rapidly elucidate the venom proteome of even miniature venomous animals [46]. While these developments have greatly accelerated our understanding of the structure, function and evolution of venom proteins, they have led to a general neglect of non-proteinaceous venom molecules. A wide variety of small molecules have been found in venoms including salts, amino acids, hormones, nucleosides, neurotransmitters, and polyamines [47], [48], [49]. In addition to playing important roles in envenomation [47], some of these molecules are valuable pharmacological tools; for example, the polyamine toxins found in spider and wasp venoms are potent modulators of glutamate receptors and nicotinic acetylcholine receptors [50], [51].
3. Screening strategies — Finding the right assay
Early toxinological research focused on the study of venoms that adversely affect humans, with the aim of understanding their mechanism of action. The molecular targets of these venoms were typically delineated using low-throughput approaches such as in vivo and ex vivo tissue studies, manual patch-clamp recording, or biochemical approaches such as radioligand binding studies [52]. These bioassays can be highly informative because they provide information on integrated tissue or cellular responses that can be used to provide target information [52]. However, they require a priori knowledge about well-defined receptor populations and signalling pathways in target tissues, as well as the availability of selective agonists or antagonists.
In addition to providing fundamental insights into the molecular targets and mode of action of venoms and toxins, these traditional approaches yielded key insights into mammalian physiology, as exemplified below. However, these approaches neglected toxins that are of no consequence to human health, those that are present in low abundance in venom, and toxins that act on novel targets that are not represented in conventional bioassays. These neglected toxins may nonetheless offer fundamental insights into human disease, and some may even have therapeutic benefit. Accordingly, high-throughput screening approaches have been developed over the past decade to exploit the exceptional biodiversity of animal toxins for drug discovery. Since many venoms and toxins exert these biological effects through actions on cell membranes, receptors and ion channels, high-throughput techniques assessing changes in cellular signalling have proven particularly insightful. These include electrophysiological techniques [53], fluorescent imaging of Na+, Ca2+, K+ or Cl– flux [54], [55], enzyme-linked immunosorbent assays (ELISAs), as well as assays based on detection of bioluminescence, fluorescence polarisation, fluorescence-resonance energy transfer (FRET), bioluminescence resonance energy transfer (BRET), and scintillation proximity [56]. Invariably, these assays are designed to determine pharmacological activity at specific targets of interest, although each technique has limitations with regard to assay flexibility, throughput, amount of toxin required, and specificity (for a review see [52]). Thus, the choice of assay will depend on whether the intention is to identify the biological target of venom components that exert effects on biological processes either at the level of the cell or organism, or whether the screen is aimed at discovery of novel bioactivity at specific molecular targets or processes of interest.
4. Venom compounds as tools for understanding human physiology
4.1. Venom toxins provide key insights into the structure and function of acetylcholine receptors
It has been known since the early 1960s that the specific and virtually irreversible blocking action of α-bungarotoxin, a venom peptide from the banded krait Bungarus multicinctus, at the vertebrate neuromuscular junction is due to functional antagonism of the depolarising effects of acetylcholine [57]. However, the identity of the presumed target of α-bungarotoxin, the cholinergic receptor protein, was unclear until a single toxin-bound membrane protein was isolated from electric tissues of the marbled electric ray Torpedo marmorata [58] and the electric eel Electrophorus electricus [59]. It is now known that acetylcholine receptors (AChRs) can be classified into muscarinic (mAChR) and nicotinic (nAChR) subtypes, with the latter subtype being the target of α-bungarotoxin and numerous other venom peptides. mAChRs are metabotropic receptors whereas nAChRs are pentameric ligand-gated ion channels.
The profound impact of α-neurotoxins on various biological processes highlighted the importance of nAChRs in both physiology and pathology. Early medical applications included the surgical use of tubocurarine, a plant-derived toxin that potently inhibits muscle nAChRs and is famous for its use as a paralysing agent in poison arrows employed by South American Indians [60]. The use of tubocurarine as a muscle relaxant allowed patients undergoing surgery to be paralysed without using dangerously high doses of general anaesthetics [61]. More recently, nAChRs have gained attention as putative therapeutic targets for a range of human disorders. For example, a number of nAChR subtypes are thought to play key roles in chronic pain and inflammation [62], and several α-conotoxins with good selectivity for these subtypes are being developed as drug leads [63], [64].
The high affinity interaction of α-neurotoxins with nAChRs also provided key insights into the structure and mode of action of these receptors [65], and they remain important pharmacological tools due to their exquisite selectivity for specific AChR subtypes. For example, fluorescent derivatives of α-bungarotoxin are excellent probes for identifying α7 nAChRs [66], while subtype-selective α-conotoxins from the venom of marine cone snails can distinguish between nAChR subtypes containing common subunits [67], [68]. Since ablation of nAChR subunits using gene editing approaches would lead to complete loss of all nAChR isoforms containing the deleted subunit, α-neurotoxins remain invaluable tools for dissecting the physiological roles of specific nAChR subtypes and their contribution to human disease.
4.2. Uncovering new analgesic targets using animal toxins
In addition to being a valuable source of molecules that modulate known drug targets, venoms can also be used to discover new therapeutic targets. For example, The Centre of Excellence for New Target Discovery in Brazil, which is co-funded by GlaxoSmithKline, is focused on using venoms to identify and validate new drug targets.
One therapeutic area in which venom peptides have been successfully used to identify new drug targets is chronic pain [8], [69]. Although the primary role of venom for most animals that possess a venom system is predation, most venomous animals also use venom for defense. Some animals, such as bees, caterpillars, stonefish, and stingrays, use venom exclusively for this purpose. In contrast to the diverse mechanisms by which predatory venoms debilitate prey, defensive venoms are largely algogenic, that is they are designed to cause intense pain (as most people that have been stung by a bee or wasp can testify). Unlike death, pain induces a lasting memory that can be conveyed to conspecific predators. Since pronociceptive venom toxins have evolved to target vertebrate sensory neurons they have the potential to uncover new components of mammalian pain signalling pathways and to serve as pharmacological tools for studying these components [69], [70].
From a screen of venoms that activate mouse sensory neurons, Osteen et al. recently isolated two homologous peptides (Hm1a and Hm1b) from venom of the African tarantula Heteroscodra maculata that activated only a subset of sensory neurons [8]. The effect of these peptides was blocked by tetrodotoxin (TTX), a non-selective inhibitor of voltage-gated sodium (NaV) channels. This ultimately led to the discovery that these peptides are highly potent and selective agonists of NaV1.1 channels [8], which were not previously thought to be involved in pain signalling. NaV1.1 was shown to regulate the excitability of Aδ nerve fibres that transmit mechanical pain signals to the spinal cord, thereby highlighting this ion channel for the first time as a potential analgesic target [8]. Moreover, NaV1.1 was shown to be present in sensory neurons that innervate the gut, and its expression was upregulated in a mouse model of irritable bowel syndrome (IBS) [8]. This suggests that compounds that specifically inhibit NaV1.1 might be useful for treatment of IBS-related gut pain, and indeed NaV1.1 inhibitors were recently shown to reduce mechanical hypersensitivity in several models of chronic visceral pain [71].
An unexpected consequence of the discovery of Hm1a and Hm1b was their application to Dravet syndrome (DS) epilepsy [72], [73], which is caused by heterozygous loss-of-function mutations in the gene encoding NaV1.1. DS is a pharmacoresistant epileptic encephalopathy characterised by childhood-onset polymorphic seizures, cognitive impairment, psychomotor regression, autistic traits, and increased risk of sudden death [72], [73]. In the central nervous system, NaV1.1 is found in the axon initial segments of GABAergic inhibitory interneurons, and consequently epileptic seizures in DS are thought to result from the reduced inhibitory activity of these neurons [72]. Remarkably, in a mouse model of DS, intracerebroventricular infusion of Hm1a restored the function of inhibitory interneurons and led to almost complete abolition of seizures within 3 days, which rescued the mice from premature death [72].
In a similar approach to that described above, a screen of snake venoms on somatosensory neurons revealed robust activation of trigeminal ganglia by Texas coral snake venom [74]. This activity could only be reproduced when two toxin components were combined to form the high affinity heteromeric MitTx complex. Subsequent patch-clamp studies revealed the target of MitTx to be acid-sensing ion channels (ASICs). ASICs are unusual homo- or heterotrimeric channels that are activated by protons and expressed throughout the nervous system [75], [76]. Although MitTx promotes activation of several ASIC subtypes, it is ~100-fold more potent at the splice-isoforms ASIC1a and ASIC1b compared with other subtypes. Texas coral snake envenomation causes intense pain in humans, and indeed MitTx elicited robust nocifensive behaviour when injected into the hindpaw of mice. This response was absent in pan-ASIC1 knockout mice, but present in an ASIC3 knockout, revealing the contribution of ASIC1 to peripheral pain sensation.
A subsequent study further delineated the specific roles of ASIC1a and ASIC1b in different pain pathways using mambalgin toxins isolated from black mamba venom [77]. Mambalgins were found to block ASICs expressed in the central (ASIC1a, ASIC1a/2a, and ASIC1a/2b) and peripheral (ASIC1b and ASIC1a/1b) nervous system. Intrathecal and intracerebroventricular injection of mambalgins in mice produced potent analgesia that was naloxone resistant and dependent on the presence of ASIC1a, suggesting an opioid independent mode of action proposed to be via central inhibition of ASIC1a/2a channels. Strikingly, peripheral intraplantar injection of mambalgins in mice produced analgesic effects in wild-type and ASIC1a knockout mice. This demonstrated for the first time that ASIC1b-containing channels in rodent nociceptors, as opposed to the splice isoform ASIC1a, are important for peripheral analgesia. More recent work using an ASIC1b knockout animal confirmed the role of this subtype in peripheral pain sensing [78]. In this model, peripheral injection of mambalgin had no effect on acid-induced hyperalgesia in ASIC1b knockout mice. Thus, the combined use of genetic techniques and venom toxins (MitTx and mambalgins) enabled dissection of the roles of ASIC1 splice isoforms in pain signalling.
4.3. Using toxins to elucidate the mechanisms underlying cold pain
In addition to venom peptides, non-peptidic marine toxins have proven to be invaluable tools for elucidating diverse pain pathways. Ciguatoxins (CTX) are temperature-stable polyethers produced by various species of Gambierdiscus, a benthic dinoflagellate [79]. The consumption of fish with high levels of ciguatoxins present in their flesh and viscera, a result of bioaccumulation, causes ciguatera fish poisoning [80]. These effects are due the action of CTX on ion channels including NaV channels [80]. The most potent congener P-CTX-1 has been used to pharmacologically probe neuropathic pain (Fig. 3 ). Ciguatera is characterised by both gastrointestinal and neurological symptoms including paresthesia, pruritus and cold allodynia, and it can even lead to death in severe cases [81]. The pathognomonic cold allodynia observed in ciguatera patients is also reported in a broad range of clinical indications including in chemotherapy patients, complex regional pain syndrome, and fibromyalgia [82], [83], [84]. Defined by sensitivity to innocuous cooling temperatures, cold allodynia is a poorly understood phenomenon. Subcutaneous injection of P-CTX-1 into the rodent hindpaw causes robust and concentration-dependent cold allodynia [85], and therefore it is uniquely positioned to elucidate the molecular basis of cold allodynia.
Fig. 3.

Toxins providing insight into the pathophysiological role of specific ion channels in sensory neurons. Subtype-selective NaV channel activators, including δ-TRTX-Hm1a and the scorpion toxins Cn2 and OD1, have highlighted modality-specific nociceptive roles for NaV1.1, NaV1.6 and NaV1.7, respectively, in sensory neurons. Activation of NaV1.7 by the scorpion toxin OD1 leads to spontaneous action potential firing in myelinated A-fibres and unmyelinated C-fibres, while activation of NaV1.1 leads to symptoms of mechanical allodynia and increased mechanical sensitivity. Cn2, a selective NaV1.6 activator, elicits pain behaviour and mechanical allodynia after local administration consistent with effects on myelinated A-fibres. Similarly, cold pain induced by ciguatoxin is elicited by selective activation of unmyelinated peripheral sensory neurons expressing the cold-sensitive TRPA1 and NaV1.8 channels, while α-dendrotoxin revealed a crucial role for KV1.1 in setting the activation threshold of cold-sensitive C-fibre neurons.
The large family of thermosensitive transient receptor potential (TRP) channels plays a critical role in temperature sensation [86]. Interestingly, TRPM8, the canonical cold sensing channel that is sensitive to menthol, is not associated with P-CTX-1-induced cold allodynia, as TRPM8 null mice administered P-CTX-1 display robust pain behaviours in response to innocuous cooling [85]. Rather, the combined findings from ex vivo skin-saphenous nerve recordings, in vivo studies and in vitro assays highlighted a role for TRPA1 in P-CTX-1-induced cold allodynia [85]. However, TRPA1 is not directly activated by P-CTX-1. Both TTX-sensitive and TTX-resistant NaV channels were responsible for the altered neuronal excitability, which drives a TRPA1-dependent change in cold sensing [85]. Although the precise role of different TRP channel isoforms in cold sensing remains contentious, the use of this marine toxin has proved valuable in delineating these complex mechanisms.
P-CTX-1 was also utilised to assess a range of analgesic treatments for cold allodynia. Using a suite of selective pharmacological agents including venom-derived peptides (such as analgesic conotoxins; recently reviewed in [87]), P-CTX-1-induced cold allodynia was found to be dependent on NaV1.6 and NaV1.8 [88]. Oxaliplatin-induced cold allodynia was completely blocked by μ-conotoxin GIIIA, a NaV1.6 inhibitor from cone snail venom [89], although intraplantar administration of Cn2, a NaV1.6-selective agonist from scorpion venom [90], elicited spontaneous pain and mechanical allodynia but did not enhance cold sensitivity. The mechanisms underlying neuronal cold sensitivity were further delineated using a class of presynaptic neurotoxins produced by mamba snakes (Dendroaspis sp.) (Fig. 3). Specifically, in trigeminal neurons, application of α-dendrotoxin-K, a blocker of Kv1.1 channels, elicited a reversible, concentration-dependent shift in the cold threshold [91].
4.4. Using scorpion toxins to study the function of peripheral sensory neurons
Subtype-selective NaV channel toxins have confirmed a crucial role for both NaV1.6 and NaV1.7 in pain signalling (Fig. 3). The NaV1.6 subtype is highly expressed in the central and peripheral nervous system, in particular at the nodes of Ranvier [92]. Due to the expression of NaV1.6 on motor neurons, NaV1.6 null mice are not viable beyond post-natal day 14 (P14) [93]. This greatly impeded the study of NaV1.6 in peripheral sensory neurons, where its role in excitability was less well defined. The subtype-selective scorpion toxin Cn2 was therefore used to probe the role of this channel in nociception. Cn2 causes NaV1.6 to open at hyperpolarised (more negative) potentials, and it enhances resurgent currents in large diameter sensory neurons in vitro and increases excitability of medium-diameter A fibres ex vivo [90], [94]. Therefore, it follows that in vivo nocifensive behaviours elicited by Cn2 administration are primarily mediated by medium-diameter rather than smaller sensory neurons. Critically, this demonstrates how toxins can be used to pharmacologically isolate a target of interest (in this case NaV1.6) in the presence of highly similar homologs of the target (NaV1.1, NaV1.7, NaV1.8 and NaV1.9).
The toxin OD1, isolated from venom of the Iranian yellow scorpion Odonthobuthus doriae, potently enhances the activity of NaV1.7 by inducing a hyperpolarising shift in the voltage-dependence of activation, inhibiting channel fast inactivation, and increasing peak current [95]. Injection of OD1 into the footpad of mice elicits pain behaviours including licking, lifting and flinching of the injected paw that can be reversed by local or systemic treatment with selective NaV1.7 inhibitors. Thus, the analgesic efficacy of NaV1.7 inhibitors can be rapidly profiled in vivo using OD1-induced pain behaviour, demonstrating that even toxins with no therapeutic potential can be effective tools for understanding the complexities of peripheral pain sensing [70] (Fig. 3).
4.5. Snake venom components act on almost every factor involved in hemostasis
Snakes are the most well-studied venomous animal, and hundreds of snake-venom proteins have been isolated over several decades. These venom components have improved our understanding of venom toxicity, significantly advanced our the knowledge of hemostatic disease mechanisms, and led to the development of diagnostic agents and life-saving drugs [96], [97], [98]. Due to the immense amount of research on snake venoms, we focus here on just a few examples where studies of snake venom have advanced our understanding of hemostasis and associated diseases.
4.5.1. Vasoconstrictor peptides from stiletto snakes are orthlogs of human endothelin
Studies into the lethal effects of envenomation by the burrowing asp Atractaspis engaddensis led to the discovery of a novel peptide named sarafotoxin (SFTX). SFTX is a potent vasoconstrictor of coronary blood vessels that can induce cardiac arrest [99], [100], [101]. Several SFTX isoforms were soon isolated from other stiletto snakes [102], [103], and it was subsequently discovered that the SFTXs are in fact orthologs of mammalian vasoconstrictor peptides known as endothelins (ETs) [104]. However, a major difference between these peptides is that a protease cleavage site involved in ET inactivation is not present in the SFTXs [105], [106], making them more stable in vivo. This allowed the SFTXs to be used as molecular tools to study vasoconstriction and expanded the available pharmacological toolbox beyond the ETs. The SFTXs and ETs have both been used to characterise ET receptors and downstream signalling pathways [107], [108].
4.5.2. The snake-venom protein botrocetin sheds light on von Willebrand factor function
von Willebrand factor (vWF) is important for platelet adhesion, and its deficiency in humans leads to a common blood coagulation disorder known as von Willebrand disease (vWD). Botrocetin is a heterodimeric protein found in venom of the South American pit viper Bothrops jararaca and it functions as a platelet-aggregating protein. It was initially named venom coagglutinin [109] as it leads to agglutination and aggregation of platelets by forming a complex with vWF and enhancing its affinity for the platelet receptor glycoprotein Ibα (GPIbα). This effect makes botrocetin an excellent diagnostic tool to assay for vWF binding to GPIbα in diseases such as vWD, or thrombopathies such as Bernard-Souler disease [110], [111]. Botrocetin has also been used as a tool to study the vWF-platelet interaction and its role in disease [112], [113]. Unlike other vWF binding proteins such as the antibiotic ristocetin, botrocetin-induced platelet agglutination activity is independent of the vWF multimer size. This is advantageous as it facilitates quantitation of vWF and allows assessment of vWD severity. The combination of botrocetin and ristocetin has allowed identification of missense mutations that cause Type B vWD [114].
4.6. Viper venom promotes nerve growth
A fortuitous observation using viper venom to study neurite outgrowth in healthy and tumorous cells led to the Nobel Prize in 1986. In studies of the regulation of cell growth, a fraction from mouse sarcoma extracts was shown to induce nerve cell growth in chick embryos [115]. This fraction was termed nerve growth factor (NGF) [116], and it was shown to be composed of both nucleic acid and protein. Snake venom from the cottonmouth viper Agkistrodon piscivorus was known to contain phosphodiesterase activity, and thus it was proposed that this venom could degrade the nucleic acid component (which was thought to be a contaminant) and reveal the active ingredient. Surprisingly, minute amounts of crude snake venom strongly promoted nerve growth, approximately 3000–6000 times more potently than the NGF-containing sarcoma fraction [117]. Comparison of the sarcoma extract and venom contents ultimately led to isolation and characterisation of the proteinaceous NGF. NGF has since been found in several snake venoms from the Viperidae, Crotalidae, and Elapidae families and it has been used widely to study cell growth regulation [118], [119]. The use of snake venom in this case not only led to a serendipitous finding with widespread importance to basic science, but NGF has also been important for elucidating the etiology of diseases such as developmental/growth deformations, tumours, and delayed wound healing.
4.7. Cobra venom elucidates complement system activity in innate immunity
Cobra venom factor (CVF) from the venom of cobra (Naja sp.) snakes has the intriguing property of disrupting activation of the complement component of the innate immune system. The complement system is comprised of a set of interacting proteins, and it can be activated in three different ways for it to play a role in pathogen clearance. The alternative pathway of activation involves spontaneous hydrolysis of complement factor C3 to C3b, which allows the plasma protein complement factors B and D to bind to C3b and transform it to C3-convertase on the external surface of an invading pathogen membrane. Subsequent recruitment of additional components of the complement pathway system leads to assembly of the membrane attack complex, which forms pores in the target cell membrane that kill or damage the pathogen [120], [121].
CVF is a structural and functional analogue of the human complement factor C3, and it acts in a similar manner to human factors C3b and C3c to activate and deplete the complement cascade [122], [123]. These properties of CVF have made it possible to independently study the role of complement in host defense, immune responses, and pathogenesis of disease. CVF was in fact pivotal to the discovery of complement factor B itself, which facilitated further characterisation of this complex pathway [120], [124], [125], [126], [127]. CVF is not only an immensely useful research tool, but it has recently been used as an immunosuppressant in tissue and organ transplants and cancer therapies [128], [129], and CVF derivatives are being designed to activate the complement system in disease states where it has been depleted [130], [131].
4.8. Spider toxins highlight the role of ASIC1a in stroke-induced brain damage
Stroke is the second leading cause of death worldwide [132], yet therapeutic options are limited to dissolution of blood clots with tissue plasminogen activator (tPA). Unfortunately, due to the risk of inducing an intracranial haemorrhage, tPA is used in <5% of stroke patients [133], and consequently there is an unmet need for compounds that can protect the brain from stroke-induced injury. Clinical trials of numerous agents, especially glutamate receptor antagonists, have failed to provide effective neuroprotection in stroke patients, which has spurred interest in better understanding the etiology of ischemia-induced brain injury [133]. Spider-venom peptides have been crucial for uncovering the key role of ASICs in stroke-induced brain damage, and validating these channels as a target for neuroprotective drugs [134], [135], [136], [137].
During ischemic conditions the brain switches from oxidative metabolism of glucose, its primary fuel, to anaerobic glycolysis, which results in lactic acidosis. The brain extracellular pH can fall from ~7.3 to as low as 6.0 in the ischemic core [134], [138], [139], which is sufficiently acidic to robustly activate proton-gated ASICs. The ASIC1a isoform is the dominant subtype in rodent and human brain [140], [141] and it primarily conducts sodium ions, although it is the only ASIC subtype that is also permeable to calcium (PNa/PCa ~ 15) [75]. It has been proposed that ASIC1a activation contributes to toxic intracellular calcium overload during stroke via both direct calcium permeability and downstream activation of voltage-gated calcium (CaV) channels and NMDA receptor-gated channels via ASIC-induced membrane depolarisation [75]. However, the primary mechanism by which ASIC1a activation leads to neuronal cell death appears to be recruitment and activation of RIPK1 [142], [143], a master regulator of necroptosis.
Although several small molecules and endogenous ligands of ASIC1a have been discovered to date, their lack of potency and selectivity limits their use as pharmacological tools for dissecting the role of ASICs in disease. The first potent and moderately selective ASIC1a ligand discovered is PcTx1, a 40-residue peptide from venom of the Trinidad chevron tarantula Psalmopoeus cambridgei [144]. PcTx1 inhibits homomeric ASIC1a (IC50 ~ 1 nM), ASIC1a/2b heteromers (IC50 ~ 3 nM), and ASIC1a/2a (35–85% inhibition at 50 nM) channels, and potentiates proton-evoked ASIC1b currents by over 3-fold control (EC50 ~ 25 nM) [144], [145], [146], [147], [148].
The key role of ASIC1a in stroke-induced brain damage was first highlighted in a seminal 2004 study which showed that genetic ablation of the ASIC1a-encoding gene reduced brain infarct size by ~60% in a mouse model of ischemic stroke [134]. However, since genetic ablation can give rise to compensatory upregulation of related channels with unknown consequences [149], [150], these researchers also used “PcTx1 venom” (i.e., crude P. cambridgei venom) as a pharmacological tool to validate the genetic studies. They showed that “PcTx1 venom” administered 30 min before and after stroke onset reduced infarct size by the same magnitude as genetic ablation of ASIC1a [134]. However, spider venoms are complex molecular cocktails that modulate an array of ion channels, and the venom of P. cambridgei is no exception. PcTx1 constitutes only ~0.4% of total P. cambridgei venom [136], and other components in this venom are known to target TRPV1 and voltage-gated potassium (KV) channels [151], [152]. This left open the question of whether the observed neuroprotective effects were induced by PcTx1, other components in the venom, or a combination of both. To address this question a more recent study used pure recombinant PcTx1 to confirm the involvement of ASIC1a in stroke [136]. Structure-activity relationship (SAR) studies of PcTx1 [153], [154] facilitated design of a weakly active PcTx1 mutant, which was not neuroprotective in the same rodent stroke model, thereby strengthening the evidence that ASIC1a inhibition reduces stroke-induced brain damage.
More recently, the 76-residue peptide Hi1a from venom of the Australian funnel-web spider Hadronyche infensa proved to be highly neuroprotective in a focal model of ischemic stroke, even when administered 8 h after the stroke onset [137]. In comparison to PcTx1, Hi1a is a slightly more potent inhibitor of ASIC1a (IC50 0.5 nM) and it is more selective, with >2000-fold higher potency for ASIC1a than homomeric ASIC1b, ASIC2a, and ASIC3 channels. Hi1a inhibition of ASIC1a is much less reversible than PcTx1 [137], and these venom peptides have a different mechanism of action [137], [155]. The different properties of PcTx1 and Hi1a broaden the pharmacological toolkit for studying ASIC1a and ASIC1a-related pathologies. Together these two venom peptides have provided striking pharmacological evidence that ASIC1a is involved in ischemic injuries of not just the brain, but also the heart [156] and kidney [157].
5. Venom compounds as leads for novel anti-infectives
The evolution of drug resistance across bacteria, fungi and parasites has created an unmet need for novel anti-infectives. The 2019 United Nations report on antimicrobial resistance estimated that >700,000 people die annually due to drug-resistant infections [158]. While most focus has been on human antibiotic resistance, control of parasitic nematodes in both livestock [159], [160] and companion animals [161], [162] is also seriously threatened by resistance. Despite the clear need for new anti-infectives, there is a diminishing pipeline of anti-infective drug candidates in both the human and veterinary spaces [163], [164], [165], which has created interest in venoms as a potential source of novel anti-infectives.
5.1. Antimicrobial toxins
Antimicrobial peptides (AMPs) are found in many animal venoms, and hundreds of examples have been reported with varying activity against fungi and both Gram-positive and Gram-negative bacteria [166], [167], [168], [169], [170], [171]. Venom-derived AMPs (vAMPs) are typically small, linear peptides that are rich in cationic residues (Lys, His and Arg), which facilitates their interaction with anionic membranes [166], [172]. They are often unstructured in aqueous solution but form ordered α-helices upon interaction with lipid membranes [172], [173].
Many vAMPs exhibit broad-spectrum cytolytic effects. This can be a double-edged sword for antimicrobial development as it often confers desirable broad-spectrum activity against a wide range of bacterial and fungal pathogens but also cytotoxicity against host cells. For example, the ponericin peptides isolated from the Amazonian stinging ant Neoponera goeldii have broad-spectrum activity against bacteria and fungi, but they are potently hemolytic [174]. Likewise, cupiennin 1a from venom of the wandering spider Cupiennius salei exhibits non-specific cytolytic activity against bacteria, insects, protozoan parasites, and human cells [175]. Mammalian cytotoxicity is typically assessed by screens against red blood cells or mammalian cell lines, but an often overlooked property of cytolytic vAMPs is that they can also cause pain. For example, venom-peptide Δ-myrtoxin-Mp1a from the Australian ant Myrmecia pilosula has submicromolar activity against bacterial pathogens but it causes mechanical allodynia when injected into mice [176]. Melittin, the primary component of honeybee venom, is potently antimicrobial but it also induces pain through direct and indirect activation of sensory neurons [177]. Hence, future research on vAMPs should place greater emphasis on screening across a broad range of cell lines and phenotypic readouts, particularly for cytolytic peptides.
A small number of vAMPs show some degree of inherent taxonomic selectivity. For example, bicarinalin from the ant Tetramorium bicarinatum is highly active against bacterial and fungal pathogens (minimal inhibitory concentration (MIC) of 0.7–25 µM, which is comparable to commercial anti-infectives), with at least 10-fold selectivity over human lymphocytes [178]. It also has similar potency to commercial antibiotics against clinical isolates of the Helicobacter pylori, a pathogen that causes stomach ulcers [179]. However, most vAMPs will require further engineering of their selectivity in order for them to be therapeutically useful [166], and several strategies have been employed successfully for this purpose. For example, shorter analogues of the scorpion-venom peptides hadrurin and vejovine were engineered with 100-fold improvement in selectivity for Escherichia coli over red blood cells [180]. Modification of the hydrophobicity and net charge of the wasp-venom peptide mastoparan through selective amino acid substitutions [181] and N-terminal acylation [182] increased its affinity for negatively-charged bacterial membranes and decreased its cytotoxicity towards mammalian cells.
Although most vAMPs have not progressed beyond basic in vitro studies, some have proven effective in in vivo infection models. ZY4 is a 17-residue cyclised derivative of cathelicidin-BF from venom of the snake Bungarus fasciatus with improved potency, selectivity and stability over the parent toxin [183]. In vitro, ZY4 kills multidrug-resistant isolates of the human pathogens Pseudomonas aeruginosa and Acinetobacter baumannii at lower concentrations than commercial antibiotics, and it reduces bacterial load and inflammation in the lungs of mice in a P. aeruginosa infection model, with no reported side-effects [183]. LyeTxI-b, an N-terminal acetylated derivative of LyeTxI from venom of the wolf spider Lycosa erythrognatha, has improved potency and selectivity against E. coli and also reduces both bacteraemia and inflammation in a mouse model of septic arthritis when administered intraarticularly [184].
Route of administration is a key issue that must be considered for therapeutic deployment of vAMPs; traditionally, peptides have poor oral bioavailability [185], and hence alternative methods of vAMP administration such as topical application should be considered. The effectiveness of this approach was demonstrated for Kn2-7, a derivative of the peptide BmKn2 from venom of the scorpion Mesobuthus martensii, which protected the skin of mice in a Staphylococcus aureus in vivo infection model [186]. Topical administration can also circumvent or alleviate off-target effects that are evident when the vAMP is present systemically. Finally, it might be possible to achieve additive or synergistic effects by combining vAMPs with existing antibiotics by utilising the pore-forming activity of the vAMP to enhance antibiotic delivery. For example, Garcia et al. found an additive effect when pathogen-specific antibiotics were combined with vAMPs isolated from the venom of arachnids [187].
5.2. Antiviral toxins
Viruses continue to pose an enormous threat to global health, as exemplified by the current global pandemic caused by SARS-CoV-2 [188]. Despite intensive intervention efforts, an estimated 770,000 people continue to die annually from human immunodeficiency virus acquired immunodeficiency syndrome (HIV-AIDS) [189]. Many serious viral diseases such as severe acute respiratory syndrome (SARS), Middle Eastern respiratory syndrome (MERS), dengue, zika, and Ebola lack vaccines or efficacious therapeutic treatments, highlighting the need for novel antivirals. Most reported venom toxins with antiviral activity are vAMPs with broad-spectrum activity; examples include melittin, snake venom PLA2 and L-amino oxidases, and mastoparan derivatives [190], [191]. Thus, a key challenge will be engineering these compounds to have selectivity for either the viral particles or the process by which they infect host cells. Mastoparan-7, derived from wasp-venom mastoparan, has broad-spectrum activity against lipid-enveloped viruses (including influenza and flaviviruses) through direct insertion and disruption of the envelope, but it remains highly cytotoxic to host cells in vitro [192]. Peptides with activity on specific viral protein targets rather than lipids may provide better selectivity. Lactarcin-1 from the central Asian spider Lachesana tarabaevi inhibits replication of dengue virus-2 (IC50 ~ 7 µM) by virtue of its ability to inhibit the viral protease NS2B-NS3, but it has low selectivity over mammalian cells [193]. The antibacterial activity of mucroporin, a vAMP from the scorpion Lychas mucronatus, was improved through histidine and arginine substitutions to create the analog mucroporin-M1 with increased net charge. Mucroporin-M1 has improved activity against both Gram-positive and Gram-negative bacteria [194], and these modifications also conferred micromolar antiviral activity in vitro (EC50 1–7 µM) against measles virus and pseudovirus models of the highly pathogenic SARS-CoV and avian influenza H5N1 viruses [195].
A key limitation of current research on venom-derived antiviral toxins is that few studies have progressed to in vivo testing. One example found that pre-treating vesicular stomatitis virus, a serious pathogen of cattle, with mastoparan-7 significantly improved clinical outcomes in a mouse challenge model through presumed inactivation of viral particles [192]. However, this needs to be confirmed in a more clinically relevant challenge model of viral infection followed by toxin administration to probe efficacy. Overall, further research is required to ascertain the potential of venom-derived peptides as viable antiviral drugs.
5.3. Antiparasitic toxins
Parasites remain one of the most serious threats to human and veterinary medicine, with current treatment options limited by drug resistance, side-effects and limited efficacy [196]. This has stimulated interest in venoms as a source of novel antiparasitic compounds (for a review see [197]). Most such studies have focused on protozoan parasites, which are responsible for a range of human diseases. The most problematic parasitic disease is malaria, which is caused by Plasmodium parasites; despite intensive intervention efforts, malaria kills an estimated 435,000 people each year and control is threatened by drug resistance [198]. A further 20 million people worldwide are infected by parasitic protozoans of the genera Leishmania and Trypanosoma, with approximately 3 million new infections per year [199].
Most antiparasitic compounds isolated from animal venoms are small cationic vAMPs, which again raises the issue of taxonomic selectivity. The vAMPs mastoparan [200] and melittin [199] inhibit development of Trypanosoma cruzi (the causative agent of Chagas disease), but selectivity over host cells is low. However, bicarinalin potently inhibits growth of Leishmania infantum (MIC 1.5 µM), the causative agent of infantile visceral leishmaniasis, with good selectivity over human lymphocytes [178]. Remarkably, crotamine, a peptide from venom of the South American rattlesnake Crotalus durissus terrificus, crosses infected red blood cell membranes to lyse the Plasmodium parasitophorous vacuole, thereby killing the parasite [201]. This may present an opportunity for combination therapy with commercial antimalarials, facilitating drug transport to the parasite. Further examples of venom components active against protozoan parasites include trimorphin, a PLA2 from venom of the North American rear-fanged snake Trimorphodon biscutatus lambda that inhibits growth of Leishmania major [202], and crovirin, a cysteine-rich secretory protein (CRISP) from the rattlesnake Crotalus viridis viridis that is active against protozoan parasites at concentrations that are not toxic to host cells [199]. The protozoan Toxoplasma gondii, which causes the key zoonotic disease toxoplasmosis, is susceptible to a venom peptide from the marine cone snail Conus californicus [203], while a C-type lectin and a PLA2 from B. pauloensis snake venom impair Toxoplasma adhesion and replication [204], [205].
The greatest burden of disability-adjusted life years in the neglected tropical diseases comes from helminth infections, including roundworms (Nematoda) and flatworms (Platyhelminthes) [189]. These worms cause important parasitic diseases in humans, including onchocerciasis, lymphatic filariasis, and schistosomiasis. About 200 million people worldwide suffer from schistosomiasis, and it is considered the second most socioeconomically devastating parasitic disease after malaria [206]. The burden of parasitic disease is also a key concern in animal health and production, with the worldwide cost of controlling livestock parasites amounting to tens of billions of dollars [207]. Despite this clear need for novel anthelmintics, it is an underexplored area of venom research with very few reports of anthelmintic venom toxins. Crotamine [208] and ε-latroinsectotoxin from venom of the black widow spider Latrodectus mactans tedecimgutattatus [209] both impair growth of the non-parasitic nematode Caenorhabditis elegans, but anthelmintic activity has not been reported against parasitic species. More recently, the spider-venom peptide Hi1a was shown to inhibit larval development of the barber’s pole worm Haemonchus contortus, a parasite of sheep production [210]. Gomesin, a peptide found in the venom [211] and hemocytes [212] of spiders, has broad-spectrum activity against bacteria, fungi, parasites and tumor cells [213], [214], and a cyclized version was recently shown to have improved stability and activity against Plasmodium in vitro [215].
To our knowledge, no venom-derived antiparasitics have yet been demonstrated to have in vivo efficacy. As for antibacterials, antifungals and antivirals, most venom-derived antiparasitics will likely require engineering to enhance their selectivity over host cells. An alternative strategy to developing venom compounds as anti-parasitic drugs is to instead impair Plasmodium development and transmission via transgenic mosquitoes engineered to express the anti-parasitic compound in the mosquito midgut, as recently demonstrated with scorpion-venom peptides [216].
As most research on venom compounds with antibacterial, antiparasitic, or antifungal activity has been performed in the last decade, we expect that more toxins with such activities will be discovered in the near future. Most work to date has focussed on human pathogens, but there is a clear need for novel veterinary anti-infectives which could be further explored in future. Due to the commonly found cytolytic nature of these peptides, a key challenge across all of these diseases will be engineering analogs with selectivity for the targeted pathogen.
6. Targeting vectors of human disease with venom-derived toxins
Arthropods, and in particular hematophagous (blood-sucking) species, are vectors for a wide-range of organisms that can cause human disease [217]. The most pernicious vectors are undoubtedly mosquitoes, which transmit protozoans (e.g. Plasmodium species that cause malaria), parasitic nematodes (e.g. Brugia species that cause filariasis), parasitic platyhelminths (e.g. trematodes that cause schistosomiases), and viruses (e.g. flaviviruses that cause yellow fever, West Nile fever, and dengue) [17], [217], [218]. Other important hematophagous disease vectors include triatomine bugs which transmit trypanosomes that cause Chagas disease, fleas which transmit the bacterium Yersinia pestis that causes bubonic plague, and ticks which transmit a wide variety of viral, bacterial and protozoan pathogens [17], [217]. Rather than directly targeting disease-causing organisms, venom toxins could be used instead to target the vectors of these diseases.
Insects are the dominant vectors of human diseases, and chemical insecticides are the primary means of controlling these insect pests [17], [218]. Unfortunately, all chemical insecticides target one of only a handful of molecular targets, and consequently their indiscriminate use has led to widespread resistance [17]. Thus, new insecticides with novel modes of action are much sought after. Spiders are professional insect killers, and they are the most speciose venomous animal, and consequently their venoms have been intensively studied as a potential source of novel bioinsecticides [17], [217], [218], [219], [220], [221]. As a result, many insecticidal toxins have been isolated from spider venoms, including peptides, proteins and polyamines, but they mostly lack the necessary taxonomic selectivity to be useful as insecticides.
An ideal insecticide should be inactive on humans and other vertebrates, as well as beneficial insects such as pollinators and natural predators of the targeted insect pest [221]. This narrow target-organism profile makes insecticide development particularly difficult. However, spider-venom peptides have been isolated with desirable taxonomic selectivity. For example, by carefully counter-screening insect toxins from the venom of Australian funnel-web spiders in vertebrate assays, a number of insect-selective neurotoxins were isolated [218]. One of these peptide toxins (GS-ω/κ-hexatoxin-Hv1a), which has complex polymodal pharmacology, has been developed commercially as a bioinsecticide by Vestaron Corporation; this peptide is active against a broad range of insect pests but is inactive against bees and vertebrates [221]. In order to target anopheline mosquitoes that transmit malaria, a gene encoding GS-ω/κ-hexatoxin-Hv1a was engineered into the entomopathogenic fungus Metarhizium pingshaense [222], and this weaponised entomopathogen proved highly successful in controlling malaria vectors in a field trial in Burkina Faso [223]. An advantage of this approach is that the range of susceptible species is determined by the entomopathogen rather than the venom peptide, thereby allowing deployment of insecticidal venom toxins that would otherwise lack the desired taxonomic selectivity [221], [224].
Some insecticidal venom peptides have exceptional taxonomic selectivity. For example, β-diguetoxin-Dc1a (Dc1a), a 56-residue knottin from venom of the desert bush spider Diguetia canities [225], and μ-theraphotoxin-Ae1a, a 39-residue knottin from venom of African tarantula Augacephalus ezendami [226], have potent insecticidal effects on German cockroaches (Blattella germanica), without significantly affecting American cockroaches (Periplaneta americana). Both knottins target insect NaV channels, and mutational analyses revealed that the susceptibility of insects to these toxins is determined by small sequence differences in their proposed binding site on the extracellular surface of the domain II voltage sensor (VSDII) [226], [227]. A high-resolution structure of Dc1a bound to a cockroach Nav channel was recently determined using cryoelectron microscopy [228], and it not only confirmed that VSDII is the binding site for Dc1a but revealed that the toxin’s inactivity against vertebrates [225] can be explained by sequence variations in the VSDII region of each of the human NaV channels. The Dc1a-NaV channel structure allows predictions to be made about which insects will be susceptible to Dc1a based on their NaV channel VSDII sequences, and it should allow use of structure-guided protein engineering to develop toxin analogs with improved potency and/or altered taxonomic selectivity.
7. Venom-derived drugs
In addition to highlighting how venom compounds can be used to elucidate disease mechanisms and uncover new drug targets, we want to provide examples of venom compounds that have progressed all the way from basic research to marketed drugs. There are currently six venom-derived drugs approved by the FDA, plus a snake-venom-derived serine protease (batroxobin; marketed as Defibrase®) that is approved for clinical use outside of the USA [9], [10]. Two further venom-derived drugs — dalazatide for treatment of autoimmune diseases [24] and tozuleristide for intraoperative imaging of brain tumours [25] — are currently undergoing clinical trials. The examples below demonstrate the potential for developing venom compounds into drugs [229] but, like any class of therapeutics, there are many challenges and the success rate is low [61].
The first venom-derived drug was captopril, an angiotensin-converting enzyme (ACE) inhibitor that is used to treat hypertension. The observation that patients envenomated by the lethal Brazilian viper Bothrops jararaca suffered from hypotension and severe intestinal contractions led to the discovery that the venom inhibits ACE [230], [231], [232]. Inhibition of ACE lowers blood pressure by preventing conversion of angiotensin I to the vasoconstrictive hormone angiotensin II, and by decreasing degradation of the vasodilatory peptide bradykinin. Researchers at Squibb isolated six ACE inhibitory peptides from B. jararaca venom, one of which (teprotide, 9 residues) was both potent and stable in vivo [230]. Detailed SAR studies of teprotide ultimately led to a smaller, orally active peptidomimetic known as captopril (D-2-methyl-3-mercaptopropanoyl-L-proline) [230], [232]. Captopril was approved by the FDA for treatment of hypertension in 1981, and by the mid 1990s it had become a blockbuster drug, reaching peak annual sales of more than USD $1.6 billion [233].
Another example of a reptile peptide that was developed into a human drug is exendin-4, a 39-residue peptide isolated from venom of the Gila monster Heloderma suspectum. In striking contrast to captopril, the marketed drug (exenatide) is identical to the peptide found in the lizard’s venom. Exenatide mirrors the effects of human hormone glucagon-like peptide-1 (GLP-1), an incretin that stimulates glucose-dependent insulin secretion, suppresses glucagon secretion, slows gastric emptying, and promotes satiety [234], but it is much more stable in vivo than human GLP-1 with a plasma half-life of 2.4 h [9]. Byetta®, a formulation of exenatide that requires twice daily subcutaneous injections, was approved by the FDA in 2005 for control of Type 2 diabetes, and it achieved worldwide sales of USD $800 million in fiscal year 2009 [9]. In order to improve patient acceptance and compliance, a new formulation was developed in which the peptide is embedded in biodegradable polymeric microspheres that extends the duration of exenatide release to such an extent that therapeutic levels can be maintained with weekly injections [235]. This new formulation (Bydureon®) was approved by the FDA in 2012, and annual sales have remained steady at USD $500–600 million despite the introduction of competing incretin mimetics.
Another drug that is an exact copy of a venom peptide is ziconotide, a synthetic version of ω-conotoxin MVIIA, a 25-residue knottin peptide found in venom of the marine cone snail Conus magus. Ziconotide is used to selectively inhibit CaV2.2 channels in the spinal cord, thereby preventing the propagation of pain signals from the spinal cord to processing centres in the brain [236]. It was approved by the FDA in 2004 for treating patients with chronic pain. Ziconotide is administered by intrathecal infusion, which limits its use to patients with intractable chronic pain that have failed on frontline analgesic drugs such as morphine. While ziconotide set a landmark as the first venom-derived drug with a neuronal target, its limited market acceptance due to its mode of administration and neuropsychiatric side-effects [237] has caused venom researchers to focus attention on analgesic targets in peripheral nociceptors rather than central neurons, as outlined in Section 4.2.
8. Conclusions and future directions
The integration of advanced venom proteomics with next-generation sequencing of venom-gland transcriptomes has made it possible to rapidly determine, at only moderate cost, the complete venom proteome of even miniature venomous animals [46]. As a result, novel venom peptides and proteins, often with unique three-dimensional folds, are being discovered at an unprecedented rate. The coupling of these omics workflows with high-throughput screening technologies [56], [238] is allowing the pharmacology of these compounds to be explored with unprecedented efficiency, often leading to the discovery of toxins with novel pharmacology [8], [74], [77], [239], [240]. All of these advances have combined to create tremendous interest in venoms as a potential source of drugs and bioinsecticides [12], [229].
While there have been some notable successes — there are seven venom-derived drugs in clinical use and several in Phase 2/3 clinical trials — the triumphs are scarcer than many predicted. No novel venom-derived drug has been approved by the FDA since 2005 [9], [10], and only one venom-derived compound has been commercialised as a bioinsecticide [221]. The reasons for this are manifold. Drug development is inherently difficult and the overall success rate is low [61]; venom-derived drug candidates are no different than any other class of therapeutic in this respect, although the overall approval rate for venom-derived drugs entering clinical trials is higher than other classes of therapeutics. Most venom researchers are not trained in the nuances of drug development, and this has led to over-hyped claims about the therapeutic potential of venom compounds, often in the absence of in vivo data. We encourage collaboration between venom researchers and those with experience in drug development so that, at a minimum, in vivo efficacy and toxicity can be assessed in validated disease models, using clinically viable routes of administration, before making claims about therapeutic potential. Renewed interest from pharmaceutical companies in venoms as source of drug leads and pharmacological tools [241], [242], [243], [244] will help to drive these collaborations.
Notwithstanding these caveats, there is little doubt that the field of venoms-based drug discovery has rapidly matured over the past decade and is now producing drug leads at an unprecedented rate. Advances in cryoelectron microscopy have facilitated determination of the three-dimensional structure of venom compounds bound to large membrane receptors [228], [243], [245], [246], [247], [248], and this should facilitate the rational design of drug candidates with improved potency and selectivity for their cognate molecular target. We predict that these advances this will lead to significant impact in some disease areas. For example, ion channels are now the third most common human drug target after kinases and G-protein-coupled receptors [249]; venoms are unquestionably the best natural source of potent ion channel modulators and venom-derived ion channel modulators have already attracted significant interest from pharmaceutical companies [24], [241], [242], [243], [244].
CRediT authorship contribution statement
Volker Herzig: Conceptualization, Supervision, Writing - review & editing, Funding acquisition. Ben Cristofori-Armstrong: Writing - review & editing, Funding acquisition. Mathilde R. Israel: Writing - review & editing, Funding acquisition. Samantha A. Nixon: Writing - review & editing, Funding acquisition. Irina Vetter: Conceptualization, Supervision, Writing - review & editing, Funding acquisition. Glenn F. King: Conceptualization, Supervision, Writing - review & editing, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We acknowledge funding from the Australian National Health & Medical Research Council (Principal Research Fellowship APP1136889, Program Grant APP1072113, and Development Grant APP1158521 to G.F.K., Career Development Fellowship APP1162503 to I.V., and C.J. Martin Fellowship APP1162427 to B.C-A.), the Australian Research Council (Future Fellowship FT190100482 to V.H.), Citizen’s United for Research in Epilepsy (Pediatric Epilepsies Award 353711 to G.F.K.), Westpac Bicentennial Foundation (Westpac Future Leaders Scholarship to S.A.N), and the Australian Government (Research Training Program Scholarships to M.R.I. and S.A.N).
References
- 1.Fry B.G., Roelants K., Champagne D.E., Scheib H., Tyndall J.D., King G.F. The toxicogenomic multiverse: convergent recruitment of proteins into animal venoms. Annu. Rev. Genomics Hum. Genet. 2009;10:483–511. doi: 10.1146/annurev.genom.9.081307.164356. [DOI] [PubMed] [Google Scholar]
- 2.Baron A., Diochot S., Salinas M., Deval E., Noel J., Lingueglia E. Venom toxins in the exploration of molecular, physiological and pathophysiological functions of acid-sensing ion channels. Toxicon. 2013;75:187–204. doi: 10.1016/j.toxicon.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Lebbe E.K., Peigneur S., Wijesekara I., Tytgat J. Conotoxins targeting nicotinic acetylcholine receptors: an overview. Mar. Drugs. 2014;12:2970–3004. doi: 10.3390/md12052970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dutertre S., Nicke A., Tsetlin V.I. Nicotinic acetylcholine receptor inhibitors derived from snake and snail venoms. Neuropharmacology. 2017;127:196–223. doi: 10.1016/j.neuropharm.2017.06.011. [DOI] [PubMed] [Google Scholar]
- 5.Olivera B.M., Miljanich G.P., Ramachandran J., Adams M.E. Calcium channel diversity and neurotransmitter release: the ω-conotoxins and ω-agatoxins. Annu. Rev. Biochem. 1994;63:823–867. doi: 10.1146/annurev.bi.63.070194.004135. [DOI] [PubMed] [Google Scholar]
- 6.King G.F., Escoubas P., Nicholson G.M. Peptide toxins that selectively target insect NaV and CaV channels. Channels. 2008;2:100–116. doi: 10.4161/chan.2.2.6022. [DOI] [PubMed] [Google Scholar]
- 7.Kalia J., Milescu M., Salvatierra J., Wagner J., Klint J.K., King G.F. From foe to friend: using animal toxins to investigate ion channel function. J. Mol. Biol. 2015;427:158–175. doi: 10.1016/j.jmb.2014.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osteen J.D., Herzig V., Gilchrist J., Emrick J.J., Zang C., Wang X. Selective spider toxins reveal a role for NaV1.1 channel in mechanical pain. Nature. 2016;534:494–499. doi: 10.1038/nature17976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King G.F. Venoms as a platform for human drugs: translating toxins into therapheutics. Expert. Opin. Drug Disc. 2011;11:1469–1484. doi: 10.1517/14712598.2011.621940. [DOI] [PubMed] [Google Scholar]
- 10.Pennington M.W., Czerwinski A., Norton R.S. Peptide therapeutics from venom: current status and potential. Bioorg. Med. Chem. 2018;26:2738–2758. doi: 10.1016/j.bmc.2017.09.029. [DOI] [PubMed] [Google Scholar]
- 11.Schendel V., Rash L.D., Jenner R.A., Undheim E.A.B. The diversity of venom: the importance of behavior and venom system morphology in understanding Its ecology and evolution. Toxins. 2019;11:666. doi: 10.3390/toxins11110666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holford M., Daly M., King G.F., Norton R.S. Venoms to the rescue. Science. 2018;361:842–844. doi: 10.1126/science.aau7761. [DOI] [PubMed] [Google Scholar]
- 13.Herzig V. Arthropod assassins: crawling biochemists with diverse toxin pharmacopeias. Toxicon. 2019;158:33–37. doi: 10.1016/j.toxicon.2018.11.312. [DOI] [PubMed] [Google Scholar]
- 14.Walker A.A., Robinson S.D., Yeates D.K., Jin J., Baumann K., Dobson J. Entomo-venomics: the evolution, biology and biochemistry of insect venoms. Toxicon. 2018;154:15–27. doi: 10.1016/j.toxicon.2018.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Rash L.D., Hodgson W.C. Pharmacology and biochemistry of spider venoms. Toxicon. 2002;40:225–254. doi: 10.1016/s0041-0101(01)00199-4. [DOI] [PubMed] [Google Scholar]
- 16.Kuhn-Nentwig L., Stoecklin R., Nentwig W. Venom composition and strategies in spiders: is everything possible? Adv. Insect Physiol. 2011:1–86. [Google Scholar]
- 17.King G.F., Hardy M.C. Spider-venom peptides: structure, pharmacology, and potential for control of insect pests. Annu. Rev. Entomol. 2013;58:475–496. doi: 10.1146/annurev-ento-120811-153650. [DOI] [PubMed] [Google Scholar]
- 18.Nelsen D.R., Nisani Z., Cooper A.M., Fox G.A., Gren E.C., Corbit A.G. Poisons, toxungens, and venoms: redefining and classifying toxic biological secretions and the organisms that employ them. Biol. Rev. Camb. Philos. Soc. 2014;89:450–465. doi: 10.1111/brv.12062. [DOI] [PubMed] [Google Scholar]
- 19.Morgenstern D., King G.F. The venom optimization hypothesis revisited. Toxicon. 2013;63:120–128. doi: 10.1016/j.toxicon.2012.11.022. [DOI] [PubMed] [Google Scholar]
- 20.Olivera B.M., Hillyard D.R., Marsh M., Yoshikami D. Combinatorial peptide libraries in drug design: lessons from venomous cone snails. Trends Biotechnol. 1995;13:422–426. doi: 10.1016/S0167-7799(00)88996-9. [DOI] [PubMed] [Google Scholar]
- 21.Sollod B.L., Wilson D., Zhaxybayeva O., Gogarten J.P., Drinkwater R., King G.F. Were arachnids the first to use combinatorial peptide libraries? Peptides. 2005;26:131–139. doi: 10.1016/j.peptides.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 22.Sanford M. Intrathecal ziconotide: a review of its use in patients with chronic pain refractory to other systemic or intrathecal analgesics. CNS Drugs. 2013;27:989–1002. doi: 10.1007/s40263-013-0107-5. [DOI] [PubMed] [Google Scholar]
- 23.Triplitt C., Chiquette E. Exenatide: from the Gila monster to the pharmacy. J. Am. Pharm. Assoc. 2006;46:44–52. doi: 10.1331/154434506775268698. [DOI] [PubMed] [Google Scholar]
- 24.Tarcha E.J., Olsen C.M., Probst P., Peckham D., Munoz-Elias E.J., Kruger J.G. Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: a randomized phase 1b trial. PLoS ONE. 2017;12:e0180762. doi: 10.1371/journal.pone.0180762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patil C.G., Walker D.G., Miller D.M., Butte P., Morrison B., Kittle D.S. Phase 1 safety, pharmacokinetics, and fluorescence imaging study of tozuleristide (BLZ-100) in adults with newly diagnosed or recurrent gliomas. Neurosurgery. 2019;85:E641–E649. doi: 10.1093/neuros/nyz125. [DOI] [PubMed] [Google Scholar]
- 26.Craik D.J., Fairlie D.P., Liras S., Price D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013;81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 27.Fitches E., Audsley N., Gatehouse J.A., Edwards J.P. Fusion proteins containing neuropeptides as novel insect contol agents: snowdrop lectin delivers fused allatostatin to insect haemolymph following oral ingestion. Insect Biochem. Mol. Biol. 2002;32:1653–1661. doi: 10.1016/s0965-1748(02)00105-4. [DOI] [PubMed] [Google Scholar]
- 28.Clark R.J., Fischer H., Dempster L., Daly N.L., Rosengren K.J., Nevin S.T. Engineering stable peptide toxins by means of backbone cyclization: stabilization of the α-conotoxin MII. PNAS. 2005;102:13767–13772. doi: 10.1073/pnas.0504613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez M.C., Cudic M. Optimization of physicochemical and pharmacological properties of peptide drugs by glycosylation. Methods Mol. Biol. 2013;1081:107–136. doi: 10.1007/978-1-62703-652-8_8. [DOI] [PubMed] [Google Scholar]
- 30.Malins L.R., Payne R.J. Recent extensions to native chemical ligation for the chemical synthesis of peptides and proteins. Curr. Opin. Chem. Biol. 2014;22:70–78. doi: 10.1016/j.cbpa.2014.09.021. [DOI] [PubMed] [Google Scholar]
- 31.Werbitzky O., Giraud M. Manufacturing of venom- derived therapeutic peptides. In: King G.F., editor. Venoms to Drugs: Venoms as a Source for the Development of Human Therapeutics. Royal Society of Chemistry; London, UK: 2015. pp. 290–305. [Google Scholar]
- 32.Gentilucci L., Tosi P., Bauer A., De Marco R. Modern tools for the chemical ligation and synthesis of modified peptides and proteins. Future Med. Chem. 2016;8:2287–2304. doi: 10.4155/fmc-2016-0175. [DOI] [PubMed] [Google Scholar]
- 33.Klint J.K., Senff S., Saez N.J., Seshadri R., Lau H.Y., Bende N.S. Production of recombinant disulfide-rich venom peptides for structural and functional analysis via expression in the periplasm of E. coli. PLoS ONE. 2013;8:e63865. doi: 10.1371/journal.pone.0063865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turchetto J., Sequeira A.F., Ramond L., Peysson F., Bras J.L., Saez N.J. High-throughput expression of animal venom toxins in Escherichia coli to generate a large library of oxidized disulphide-reticulated peptides for drug discovery. Microb. Cell Fact. 2017;16:6. doi: 10.1186/s12934-016-0617-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavergne V., Alewood P.F., Mobli M., King G.F. The structural universe of disulfide-rich venom peptides. In: King G.F., editor. Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics London. Royal Society of Chemistry; UK: 2015. pp. 37–79. [Google Scholar]
- 36.Undheim E.A., Mobli M., King G.F. Toxin structures as evolutionary tools: using conserved 3D folds to study the evolution of rapidly evolving peptides. BioEssays. 2016;38:539–548. doi: 10.1002/bies.201500165. [DOI] [PubMed] [Google Scholar]
- 37.Postic G., Gracy J., Perin C., Chiche L., Gelly J.C. KNOTTIN: the database of inhibitor cystine knot scaffold after 10 years, toward a systematic structure modeling. Nucleic Acids Res. 2018;46:D454–D458. doi: 10.1093/nar/gkx1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pallaghy P.K., Nielsen K.J., Craik D.J., Norton R.S. A common structural motif incorporating a cystine knot and a triple-stranded β-sheet in toxic and inhibitory polypeptides. Protein Sci. 1994;3:1833–1839. doi: 10.1002/pro.5560031022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colgrave M.L., Craik D.J. Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: the importance of the cyclic cystine knot. Biochemistry. 2004;43:5965–5975. doi: 10.1021/bi049711q. [DOI] [PubMed] [Google Scholar]
- 40.Saez N.J., Senff S., Jensen J.E., Er S.Y., Herzig V., Rash L.D. Spider-venom peptides as therapeutics. Toxins. 2010;2:2851–2871. doi: 10.3390/toxins2122851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herzig V., King G.F. The cystine knot is responsible for the exceptional stability of the insecticidal spider toxin ω-hexatoxin-Hv1a. Toxins. 2015;7:4366–4380. doi: 10.3390/toxins7104366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gonzales D.T., Saloma C.P. A bioinformatics survey for conotoxin-like sequences in three turrid snail venom duct transcriptomes. Toxicon. 2014;92:66–74. doi: 10.1016/j.toxicon.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 43.Undheim E.A., Fry B.G., King G.F. Centipede venom: recent discoveries and current state of knowledge. Toxins. 2015;7:679–704. doi: 10.3390/toxins7030679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drukewitz S.H., Fuhrmann N., Undheim E.A.B., Blanke A., Giribaldi J., Mary R. A dipteran's novel sucker punch: evolution of arthropod atypical venom with a neurotoxic component in robber flies (Asilidae, Diptera) Toxins. 2018;10:29. doi: 10.3390/toxins10010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Touchard A., Ali S.R., Barassé V., Klopp C., Dejean A., Kini R.M. Venom peptide repertoire of the European myrmicine ant Manica rubida: identification of insecticidal toxins. J. Proteome Res. 2020;19:1800–1811. doi: 10.1021/acs.jproteome.0c00048. [DOI] [PubMed] [Google Scholar]
- 46.Kramer J., Pohl H., Predel R. Venom collection and analysis in the pseudoscorpion Chelifer cancroides (Pseudoscorpiones: Cheliferidae) Toxicon. 2019;162:15–23. doi: 10.1016/j.toxicon.2019.02.009. [DOI] [PubMed] [Google Scholar]
- 47.Inceoglu B., Lango J., Jing J., Chen L., Doymaz F., Pessah I.N. One scorpion, two venoms: prevenom of Parabuthus transvaalicus acts as an alternative type of venom with distinct mechanism of action. PNAS. 2003;100:922–927. doi: 10.1073/pnas.242735499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schroeder F.C., Taggi A.E., Gronquist M., Malik R.U., Grant J.B., Eisner T. NMR-spectroscopic screening of spider venom reveals sulfated nucleosides as major components for the brown recluse and related species. PNAS. 2008;105:14283–14287. doi: 10.1073/pnas.0806840105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walker A.A., Hernandez-Vargas M.J., Corzo G., Fry B.G., King G.F. Giant fish-killing water bug reveals ancient and dynamic venom evolution in Heteroptera. Cell. Mol. Life Sci. 2018;75:3215–3229. doi: 10.1007/s00018-018-2768-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mellor I.R., Usherwood P.N. Targeting ionotropic receptors with polyamine-containing toxins. Toxicon. 2004;43:493–508. doi: 10.1016/j.toxicon.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Strømgaard K., Jensen L.S., Vogensen S.B. Polyamine toxins: development of selective ligands for ionotropic receptors. Toxicon. 2005;45:249–254. doi: 10.1016/j.toxicon.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 52.Vetter I., Hodgson W.C., Adams D.J., McIntyre P. Venoms-based drug discovery: bioassays, electrophysiology, high-throughput screens and target identification. In: King G.F., editor. Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics. Royal Society of Chemistry; London, U.K.: 2015. pp. 97–128. [Google Scholar]
- 53.Dunlop J., Bowlby M., Peri R., Tawa G., LaRocque J., Soloveva V. Ion channel screening. Comb. Chem. High Throughput Screen. 2008;11:514–522. doi: 10.2174/138620708785204117. [DOI] [PubMed] [Google Scholar]
- 54.Tay B., Stewart T.A., Davis F.M., Deuis J.R., Vetter I. Development of a high-throughput fluorescent no-wash sodium influx assay. PLoS ONE. 2019;14:e0213751. doi: 10.1371/journal.pone.0213751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vetter I., Carter D., Bassett J., Deuis J.R., Tay B., Jami S. High-throughput fluorescence assays for ion channels and GPCRs. Adv. Exp. Med. Biol. 2020;1131:27–72. doi: 10.1007/978-3-030-12457-1_3. [DOI] [PubMed] [Google Scholar]
- 56.Prashanth J.R., Hasaballah N., Vetter I. Pharmacological screening technologies for venom peptide discovery. Neuropharmacology. 2017;127:4–19. doi: 10.1016/j.neuropharm.2017.03.038. [DOI] [PubMed] [Google Scholar]
- 57.Chang C.C., Lee C.Y. Isolation of neurotoxins from the venom of Bungarus multicinctus and their modes of neuromuscular blocking action. Arch. Int. Pharmacodyn. Ther. 1963;144:241–257. [PubMed] [Google Scholar]
- 58.Miledi R., Molinoff P., Potter L.T. Isolation of the cholinergic receptor protein of Torpedo electric tissue. Nature. 1971;229:554–557. doi: 10.1038/229554a0. [DOI] [PubMed] [Google Scholar]
- 59.Changeux J.P., Kasai M., Lee C.Y. Use of a snake venom toxin to characterize the cholinergic receptor protein. PNAS. 1970;67:1241–1247. doi: 10.1073/pnas.67.3.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown T.C. From arrow poison to neuromuscular blockers. Paediatr. Anaesth. 2013;23:865–867. doi: 10.1111/pan.12152. [DOI] [PubMed] [Google Scholar]
- 61.Harvey A.L. Toxins and drug discovery. Toxicon. 2014;92:193–200. doi: 10.1016/j.toxicon.2014.10.020. [DOI] [PubMed] [Google Scholar]
- 62.Hone A.J., McIntosh J.M. Nicotinic acetylcholine receptors in neuropathic and inflammatory pain. FEBS Lett. 2018;592:1045–1062. doi: 10.1002/1873-3468.12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hone A.J., Servent D., McIntosh J.M. α9-containing nicotinic acetylcholine receptors and the modulation of pain. Br. J. Pharmacol. 2018;175:1915–1927. doi: 10.1111/bph.13931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang H., Li X., Zhangsun D., Yu G., Su R., Luo S. The α9α10 nicotinic acetylcholine receptor antagonist αO-conotoxin GeXIVA[1,2] alleviates and reverses chemotherapy-induced neuropathic pain. Mar. Drugs. 2019;17:265. doi: 10.3390/md17050265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dellisanti C.D., Yao Y., Stroud J.C., Wang Z.Z., Chen L. Crystal structure of the extracellular domain of nAChR α1 bound to α-bungarotoxin at 1.94 Å resolution. Nat. Neurosci. 2007;10:953–962. doi: 10.1038/nn1942. [DOI] [PubMed] [Google Scholar]
- 66.Kim S.J., Kim J.E., Ko B.H., Moon I.S. Carbofuran induces apoptosis of rat cortical neurons and down-regulates surface ⍺7 subunit of acetylcholine receptors. Mol. Cells. 2004;17:242–247. [PubMed] [Google Scholar]
- 67.McIntosh J.M., Santos A.D., Olivera B.M. Conus peptides targeted to specific nicotinic acetylcholine receptor subtypes. Annu. Rev. Biochem. 1999;68:59–88. doi: 10.1146/annurev.biochem.68.1.59. [DOI] [PubMed] [Google Scholar]
- 68.Lewis R.J., Dutertre S., Vetter I., Christie M.J. Conus venom peptide pharmacology. Pharmacol. Rev. 2012;64:259–298. doi: 10.1124/pr.111.005322. [DOI] [PubMed] [Google Scholar]
- 69.Jami S., Erickson A., Brierley S.M., Vetter I. Pain-causing venom peptides: insights into sensory neuron pharmacology. Toxins. 2017;10:15. doi: 10.3390/toxins10010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bohlen C.J., Julius D. Receptor-targeting mechanisms of pain-causing toxins: How ow? Toxicon. 2012;60:254–264. doi: 10.1016/j.toxicon.2012.04.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salvatierra J., Castro J., Erickson A., Li Q., Braz J., Gilchrist J. Nav1.1 inhibition can reduce visceral hypersensitivity. JCI Insight. 2018;3:e121000. doi: 10.1172/jci.insight.121000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Richards K.L., Milligan C.J., Richardson R.J., Jancovski N., Grunnet M., Jacobson L.H. Selective NaV1.1 activation rescues Dravet syndrome mice from seizures and premature death. PNAS. 2018;115:E8077–E8085. doi: 10.1073/pnas.1804764115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chow Chun Yuen, Chin Yanni K.Y., Ma Linlin, Undheim Eivind A.B., Herzig Volker, King Glenn F. A selective NaV1.1 activator with potential for treatment of Dravet syndrome epilepsy. Biochem. Pharmacol. 2020:113991. doi: 10.1016/j.bcp.2020.113991. [DOI] [PubMed] [Google Scholar]
- 74.Bohlen C.J., Chesler A.T., Sharif-Naeini R., Medzihradszky K.F., Zhou S., King D. A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature. 2011;479:410–414. doi: 10.1038/nature10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gründer S., Chen X. Structure, function, and pharmacology of acid-sensing ion channels (ASICs): focus on ASIC1a. Int. J. Physiol. Pathophysiol. Pharmacol. 2010;2:73–94. [PMC free article] [PubMed] [Google Scholar]
- 76.Cristofori-Armstrong B., Rash L.D. Acid-sensing ion channel (ASIC) structure and function: insights from spider, snake and sea anemone venoms. Neuropharmacology. 2017;127:173–184. doi: 10.1016/j.neuropharm.2017.04.042. [DOI] [PubMed] [Google Scholar]
- 77.Diochot S., Baron A., Salinas M., Douguet D., Scarzello S., Dabert-Gay A.S. Black mamba venom peptides target acid-sensing ion channels to abolish pain. Nature. 2012;490:552–555. doi: 10.1038/nature11494. [DOI] [PubMed] [Google Scholar]
- 78.Chang C.T., Fong S.W., Lee C.H., Chuang Y.C., Lin S.H., Chen C.C. Involvement of acid-sensing ion channel 1b in the development of acid-induced chronic muscle pain. Front. Neurosci. 2019;13:1247. doi: 10.3389/fnins.2019.01247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Solino L., Costa P.R. Differential toxin profiles of ciguatoxins in marine organisms: chemistry, fate and global distribution. Toxicon. 2018;150:124–143. doi: 10.1016/j.toxicon.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 80.Lewis R.J. Ciguatera: Australian perspectives on a global problem. Toxicon. 2006;48:799–809. doi: 10.1016/j.toxicon.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 81.Bagnis R., Kuberski T., Laugier S. Clinical observations on 3,009 cases of ciguatera (fish poisoning) in the South Pacific. Am. J. Trop. Med. Hyg. 1979;28:1067–1073. doi: 10.4269/ajtmh.1979.28.1067. [DOI] [PubMed] [Google Scholar]
- 82.Berglund B., Harju E.L., Kosek E., Lindblom U. Quantitative and qualitative perceptual analysis of cold dysesthesia and hyperalgesia in fibromyalgia. Pain. 2002;96:177–187. doi: 10.1016/s0304-3959(01)00443-2. [DOI] [PubMed] [Google Scholar]
- 83.Windebank A.J., Grisold W. Chemotherapy-induced neuropathy. J. Peripher. Nerv. Syst. 2008;13:27–46. doi: 10.1111/j.1529-8027.2008.00156.x. [DOI] [PubMed] [Google Scholar]
- 84.Finch P.M., Knudsen L., Drummond P.D. Reduction of allodynia in patients with complex regional pain syndrome: a double-blind placebo-controlled trial of topical ketamine. Pain. 2009;146:18–25. doi: 10.1016/j.pain.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 85.Vetter I., Touska F., Hess A., Hinsbey R., Sattler S., Lampert A. Ciguatoxins activate specific cold pain pathways to elicit burning pain from cooling. EMBO J. 2012;31:3795–3808. doi: 10.1038/emboj.2012.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ramsey I.S., Delling M., Clapham D.E. An introduction to TRP channels. Annu. Rev. Physiol. 2006;68:619–647. doi: 10.1146/annurev.physiol.68.040204.100431. [DOI] [PubMed] [Google Scholar]
- 87.Munasinghe N.R., Christie M.J. Conotoxins that could provide analgesia through voltage gated sodium channel inhibition. Toxins. 2015;7:5386–53407. doi: 10.3390/toxins7124890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zimmermann K., Deuis J.R., Inserra M.C., Collins L.S., Namer B., Cabot P.J. Analgesic treatment of ciguatoxin-induced cold allodynia. Pain. 2013;154:1999–2006. doi: 10.1016/j.pain.2013.06.015. [DOI] [PubMed] [Google Scholar]
- 89.Cruz L.J., Kupryszewski G., LeCheminant G.W., Gray W.R., Olivera B.M., Rivier J. μ-conotoxin GIIIA, a peptide ligand for muscle sodium channels: chemical synthesis, radiolabeling, and receptor characterization. Biochemistry. 1989;28:3437–3442. doi: 10.1021/bi00434a043. [DOI] [PubMed] [Google Scholar]
- 90.Schiavon E., Sacco T., Cassulini R.R., Gurrola G., Tempia F., Possani L.D. Resurgent current and voltage sensor trapping enhanced activation by a β-scorpion toxin solely in Nav1.6 channel. Significance in mice Purkinje neurons. J. Biol. Chem. 2006;281:20326–20337. doi: 10.1074/jbc.M600565200. [DOI] [PubMed] [Google Scholar]
- 91.Madrid R., de la Pena E., Donovan-Rodriguez T., Belmonte C., Viana F. Variable threshold of trigeminal cold-thermosensitive neurons is determined by a balance between TRPM8 and Kv1 potassium channels. J. Neurosci. 2009;29:3120–3131. doi: 10.1523/JNEUROSCI.4778-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Caldwell J.H., Schaller K.L., Lasher R.S., Peles E., Levinson S.R. Sodium channel NaV1.6 is localized at nodes of ranvier, dendrites, and synapses. PNAS. 2000;97:5616–5620. doi: 10.1073/pnas.090034797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kearney J.A., Buchner D.A., De Haan G., Adamska M., Levin S.I., Furay A.R. Molecular and pathological effects of a modifier gene on deficiency of the sodium channel Scn8a (NaV1.6) Hum. Mol. Genet. 2002;11:2765–2775. doi: 10.1093/hmg/11.22.2765. [DOI] [PubMed] [Google Scholar]
- 94.Israel M.R., Tanaka B.S., Castro J., Thongyoo P., Robinson S.D., Zhao P. NaV1.6 regulates excitability of mechanosensitive sensory neurons. J. Physiol. 2019;597:3751–3768. doi: 10.1113/JP278148. [DOI] [PubMed] [Google Scholar]
- 95.Deuis J.R., Wingerd J.S., Winter Z., Durek T., Dekan Z., Sousa S.R. Analgesic effects of GpTx-1, PF-04856264 and CNV1014802 in a mouse model of NaV1.7-mediated pain. Toxins. 2016;8:78. doi: 10.3390/toxins8030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kini R.M. Toxins in thrombosis and haemostasis: potential beyond imagination. J. Thromb. Haemost. 2011;9(Suppl 1):195–208. doi: 10.1111/j.1538-7836.2011.04279.x. [DOI] [PubMed] [Google Scholar]
- 97.Koh C.Y., Kini R.M. From snake venom toxins to therapeutics-cardiovascular examples. Toxicon. 2012;59:497–506. doi: 10.1016/j.toxicon.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 98.McCleary R.J., Kini R.M. Non-enzymatic proteins from snake venoms: a gold mine of pharmacological tools and drug leads. Toxicon. 2013;62:56–74. doi: 10.1016/j.toxicon.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 99.Kochva E., Viljoen C.C., Botes D.P. A new type of toxin in the venom of snakes of the genus Atractaspis (Atractaspidinae) Toxicon. 1982;20:581–592. doi: 10.1016/0041-0101(82)90052-6. [DOI] [PubMed] [Google Scholar]
- 100.Weiser E., Wollberg Z., Kochva E., Lee S.Y. Cardiotoxic effects of the venom of the burrowing asp, Atractaspis engaddensis (Atractaspididae, Ophidia) Toxicon. 1984;22:767–774. doi: 10.1016/0041-0101(84)90159-4. [DOI] [PubMed] [Google Scholar]
- 101.Lee S.Y., Lee C.Y., Chen Y.M., Kochva E. Coronary vasospasm as the primary cause of death due to the venom of the burrowing asp, Atractaspis engaddensis. Toxicon. 1986;24:285–291. doi: 10.1016/0041-0101(86)90153-4. [DOI] [PubMed] [Google Scholar]
- 102.Becker A., Dowdle E.B., Hechler U., Kauser K., Donner P., Schleuning W.D. Bibrotoxin, a novel member of the endothelin/sarafotoxin peptide family, from the venom of the burrowing asp Atractaspis bibroni. FEBS Lett. 1993;315:100–103. doi: 10.1016/0014-5793(93)81142-m. [DOI] [PubMed] [Google Scholar]
- 103.Takasaki C., Tamiya N., Bdolah A., Wollberg Z., Kochva E. Sarafotoxins S6: several isotoxins from Atractaspis engaddensis (burrowing asp) venom that affect the heart. Toxicon. 1988;26:543–548. doi: 10.1016/0041-0101(88)90234-6. [DOI] [PubMed] [Google Scholar]
- 104.Yanagisawa M., Kurihara H., Kimura S., Tomobe Y., Kobayashi M., Mitsui Y. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 105.Skolovsky M., Galron R., Kloog Y., Bdolah A., Indig F.E., Blumberg S. Endothelins are more sensitive than sarafotoxins to neutral endopeptidase: possible physiological significance. PNAS. 1990;87:4702–4706. doi: 10.1073/pnas.87.12.4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vijayaraghavan J., Scicli A.G., Carretero O.A., Slaughter C., Moomaw C., Hersh L.B. The hydrolysis of endothelins by neutral endopeptidase 24.11 (enkephalinase) J. Biol. Chem. 1990;265:14150–14155. [PubMed] [Google Scholar]
- 107.Kloog Y., Bousso-Mittler D., Bdolah A., Sokolovsky M. Three apparent receptor subtypes for the endothelin/sarafotoxin family. FEBS Lett. 1989;253:199–202. doi: 10.1016/0014-5793(89)80958-5. [DOI] [PubMed] [Google Scholar]
- 108.Ducancel F. Endothelin-like peptides. Cell. Mol. Life Sci. 2005;62:2828–2839. doi: 10.1007/s00018-005-5286-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Read M.S., Shermer R.W., Brinkhous K.M. Venom coagglutinin: an activator of platelet aggregation dependent on von Willebrand factor. PNAS. 1978;75:4514–4518. doi: 10.1073/pnas.75.9.4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Matsui T., Hamako J. Structure and function of snake venom toxins interacting with human von Willebrand factor. Toxicon. 2005;45:1075–1087. doi: 10.1016/j.toxicon.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 111.Read M.S., Smith S.V., Lamb M.A., Brinkhous K.M. Role of botrocetin in platelet agglutination: formation of an activated complex of botrocetin and von Willebrand factor. Blood. 1989;74:1031–1035. [PubMed] [Google Scholar]
- 112.Marsh N., Williams V. Practical applications of snake venom toxins in haemostasis. Toxicon. 2005;45:1171–1181. doi: 10.1016/j.toxicon.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 113.Clemetson K.J., Lu Q., Clemetson J.M. Snake C-type lectin-like proteins and platelet receptors. Pathophysiol. Haemost. Thromb. 2005;34:150–155. doi: 10.1159/000092414. [DOI] [PubMed] [Google Scholar]
- 114.Rabinowitz I., Tuley E.A., Mancuso D.J., Randi A.M., Firkin B.G., Howard M.A. von Willebrand disease type B: a missense mutation selectively abolishes ristocetin-induced von Willebrand factor binding to platelet glycoprotein Ib. PNAS. 1992;89:9846–9849. doi: 10.1073/pnas.89.20.9846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Levi-Montalcini R., Meyer H., Hamburger V. In vitro experiments on the effects of mouse sarcomas 180 and 37 on the spinal and sympathetic ganglia of the chick embryo. Cancer Res. 1954;14:49–57. [PubMed] [Google Scholar]
- 116.Kostiza T., Meier J. Nerve growth factors from snake venoms: chemical properties, mode of action and biological significance. Toxicon. 1996;34:787–806. doi: 10.1016/0041-0101(96)00023-2. [DOI] [PubMed] [Google Scholar]
- 117.Cohen S., Levi-Montalcini R. A nerve growth-stimulating factor isolated from snake venom. PNAS. 1956;42:571–574. doi: 10.1073/pnas.42.9.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Angeletti R.H. Nerve growth factor from cobra venom. PNAS. 1970;65:668–674. doi: 10.1073/pnas.65.3.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Trummal K., Tõnismägi K., Paalme V., Järvekülg L., Siigur J., Siigur E. Molecular diversity of snake venom nerve growth factors. Toxicon. 2011;58:363–368. doi: 10.1016/j.toxicon.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 120.Müller-Eberhard H.J., Nilsson U.R., Dalmasso A.P., Polley M.J., Calcott M.A. A molecular concept of immune cytolysis. Arch. Pathol. 1966;82:205–217. [PubMed] [Google Scholar]
- 121.Vogt W., Dieminger L., Lynen R., Schmidt G. Alternative pathway for the activation of complement in human serum. Formation and composition of the complex with cobra venom factor that cleaves the third component of complement. Hoppe Seylers Z Physiol. Chem. 1974;355:171–183. doi: 10.1515/bchm2.1974.355.1.171. [DOI] [PubMed] [Google Scholar]
- 122.Eggertsen G., Lind P., Sjöquist J. Molecular characterization of the complement activating protein in the venom of the Indian cobra (Naja n. siamensis) Mol. Immunol. 1981;18:125–133. doi: 10.1016/0161-5890(81)90078-x. [DOI] [PubMed] [Google Scholar]
- 123.Vogel C.W., Smith C.A., Müller-Eberhard H.J. Cobra venom factor: structural homology with the third component of human complement. J. Immunol. 1984;133:3235–3241. [PubMed] [Google Scholar]
- 124.Götze O., Müller-Eberhard H.J. The C3-activator system: an alternate pathway of complement activation. J. Exp. Med. 1971;134:90s–108s. [PubMed] [Google Scholar]
- 125.Müller-Eberhard H.J., Fjellström K.E. Isolation of the anticomplementary protein from cobra venom and its mode of action on C3. J. Immunol. 1971;107:1666–1672. [PubMed] [Google Scholar]
- 126.Hunsicker L.G., Ruddy S., Austen K.F. Alternate complement pathway: factors involved in cobra venom factor (CoVF) activation of the third component of complement (C3) J. Immunol. 1973;110:128–138. [PubMed] [Google Scholar]
- 127.Alper C.A., Goodkofsky I., Lepow I.H. The relationship of glycine-rich -glycoprotein to factor B in the properdin system and to the cobra factor-binding protein of huan serum. J. Exp. Med. 1973;137:424–437. doi: 10.1084/jem.137.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Suhr B.D., Black S.M., Guzman-Paz M., Matas A.J., Dalmasso A.P. Inhibition of the membrane attack complex of complement for induction of accommodation in the hamster-to-rat heart transplant model. Xenotransplantation. 2007;14:572–579. doi: 10.1111/j.1399-3089.2007.00422.x. [DOI] [PubMed] [Google Scholar]
- 129.Vonk F.J., Jackson K., Doley R., Madaras F., Mirtschin P.J., Vidal N. Snake venom: from fieldwork to the clinic. BioEssays. 2011;33:269–279. doi: 10.1002/bies.201000117. [DOI] [PubMed] [Google Scholar]
- 130.Fritzinger D.C., Hew B.E., Thorne M., Pangburn M.K., Janssen B.J., Gros P. Functional characterization of human C3/cobra venom factor hybrid proteins for therapeutic complement depletion. Dev. Comp. Immunol. 2009;33:105–116. doi: 10.1016/j.dci.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 131.Vogel C.W., Fritzinger D.C. Cobra venom factor: structure, function, and humanization for therapeutic complement depletion. Toxicon. 2010;56:1198–1222. doi: 10.1016/j.toxicon.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 132.Woodruff T.M., Thundyil J., Tang S.C., Sobey C.G., Taylor S.M., Arumugam T.V. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 2011;6:11. doi: 10.1186/1750-1326-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Besancon E., Guo S., Lok J., Tymianski M., Lo E.H. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol. Sci. 2008;29:268–275. doi: 10.1016/j.tips.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 134.Xiong Z.G., Zhu X.M., Chu X.P., Minami M., Hey J., Wei W.L. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 135.Pignataro G., Simon R.P., Xiong Z.G. Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain. 2007;130:151–158. doi: 10.1093/brain/awl325. [DOI] [PubMed] [Google Scholar]
- 136.McCarthy C.A., Rash L.D., Chassagnon I.R., King G.F., Widdop R.E. PcTx1 affords neuroprotection in a conscious model of stroke in hypertensive rats via selective inhibition of ASIC1a. Neuropharmacology. 2015;99:650–657. doi: 10.1016/j.neuropharm.2015.08.040. [DOI] [PubMed] [Google Scholar]
- 137.Chassagnon I.R., McCarthy C.A., Chin Y.K., Pineda S.S., Keramidas A., Mobli M. Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a. PNAS. 2017;114:3750–3755. doi: 10.1073/pnas.1614728114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rehncrona S. Brain acidosis. Ann. Emerg. Med. 1985;14:770–776. doi: 10.1016/s0196-0644(85)80055-x. [DOI] [PubMed] [Google Scholar]
- 139.O'Bryant Z., Vann K.T., Xiong Z.G. Translational strategies for neuroprotection in ischemic stroke — focusing on acid-sensing ion channel 1a. Transl. Stroke Res. 2014;5:59–68. doi: 10.1007/s12975-013-0319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hoagland E.N., Sherwood T.W., Lee K.G., Walker C.J., Askwith C.C. Identification of a calcium permeable human acid-sensing ion channel 1 transcript variant. J. Biol. Chem. 2010;285:41852–41862. doi: 10.1074/jbc.M110.171330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li M., Inoue K., Branigan D., Kratzer E., Hansen J.C., Chen J.W. Acid-sensing ion channels in acidosis-induced injury of human brain neurons. J. Cereb. Blood Flow Metab. 2010;30:1247–1260. doi: 10.1038/jcbfm.2010.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang Y.Z., Wang J.J., Huang Y., Liu F., Zeng W.Z., Li Y. Tissue acidosis induces neuronal necroptosis via ASIC1a channel independent of its ionic conduction. Elife. 2015;4:e05682. doi: 10.7554/eLife.05682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang J.J., Liu F., Yang F., Wang Y.Z., Qi X., Li Y. Disruption of auto-inhibition underlies conformational signaling of ASIC1a to induce neuronal necroptosis. Nat. Commun. 2020;11:475. doi: 10.1038/s41467-019-13873-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Escoubas P., De Weille J.R., Lecoq A., Diochot S., Waldmann R., Champigny G. Isolation of a tarantula toxin specific for a class of proton-gated Na+ channels. J. Biol. Chem. 2000;275:25116–25121. doi: 10.1074/jbc.M003643200. [DOI] [PubMed] [Google Scholar]
- 145.Chen X., Kalbacher H., Grunder S. Interaction of acid-sensing ion channel (ASIC) 1 with the tarantula toxin psalmotoxin 1 is state dependent. J. Gen. Physiol. 2006;127:267–276. doi: 10.1085/jgp.200509409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sherwood T.W., Lee K.G., Gormley M.G., Askwith C.C. Heteromeric acid-sensing ion channels (ASICs) composed of ASIC2b and ASIC1a display novel channel properties and contribute to acidosis-induced neuronal death. J. Neurosci. 2011;31:9723–9734. doi: 10.1523/JNEUROSCI.1665-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Joeres N., Augustinowski K., Neuhof A., Assmann M., Grunder S. Functional and pharmacological characterization of two different ASIC1a/2a heteromers reveals their sensitivity to the spider toxin PcTx1. Sci. Rep. 2016;6:27647. doi: 10.1038/srep27647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cristofori-Armstrong B., Saez N.J., Chassagnon I.R., King G.F., Rash L.D. The modulation of acid-sensing ion channel 1 by PcTx1 is pH-, subtype- and species-dependent: importance of interactions at the channel subunit interface and potential for engineering selective analogues. Biochem. Pharmacol. 2019;163:381–390. doi: 10.1016/j.bcp.2019.03.004. [DOI] [PubMed] [Google Scholar]
- 149.El-Brolosy M.A., Kontarakis Z., Rossi A., Kuenne C., Gunther S., Fukuda N. Genetic compensation triggered by mutant mRNA degradation. Nature. 2019;568:193–197. doi: 10.1038/s41586-019-1064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ma Z., Zhu P., Shi H., Guo L., Zhang Q., Chen Y. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature. 2019;568:259–263. doi: 10.1038/s41586-019-1057-y. [DOI] [PubMed] [Google Scholar]
- 151.Choi S.J., Parent R., Guillaume C., Deregnaucourt C., Delarbre C., Ojcius D.M. Isolation and characterization of Psalmopeotoxin I and II: two novel antimalarial peptides from the venom of the tarantula Psalmopoeus cambridgei. FEBS Lett. 2004;572:109–117. doi: 10.1016/j.febslet.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 152.Siemens J., Zhou S., Piskorowski R., Nikai T., Lumpkin E.A., Basbaum A.I. Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature. 2006;444:208–212. doi: 10.1038/nature05285. [DOI] [PubMed] [Google Scholar]
- 153.Saez N.J., Mobli M., Bieri M., Chassagnon I.R., Malde A.K., Gamsjaeger R. A dynamic pharmacophore drives the interaction between Psalmotoxin-1 and the putative drug target acid-sensing ion channel 1a. Mol. Pharmacol. 2011;80:796–808. doi: 10.1124/mol.111.072207. [DOI] [PubMed] [Google Scholar]
- 154.Saez N.J., Deplazes E., Cristofori-Armstrong B., Chassagnon I.R., Lin X., Mobli M. Molecular dynamics and functional studies define a hot spot of crystal contacts essential for PcTx1 inhibition of acid-sensing ion channel 1a. Br. J. Pharmacol. 2015;172:4985–4995. doi: 10.1111/bph.13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chen X., Kalbacher H., Grunder S. The tarantula toxin psalmotoxin 1 inhibits acid-sensing ion channel (ASIC) 1a by increasing its apparent H+ affinity. J. Gen. Physiol. 2005;126:71–79. doi: 10.1085/jgp.200509303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Redd M.A., Scheuer S.E., Saez N.J., Gao L., Hicks M., Villanueva J.E. Enhanced donor heart preservation by therapeutic inhibition of acid sensing ion channel 1a. bioRxiv. 2020 doi: 10.1101/869826. [DOI] [Google Scholar]
- 157.Song N., Lu Z., Zhang J., Shi Y., Ning Y., Chen J. Acid-sensing ion channel 1a is involved in ischaemia/reperfusion induced kidney injury by increasing renal epithelia cell apoptosis. J. Cell Mol. Med. 2019;23:3429–3440. doi: 10.1111/jcmm.14238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Interagency Coordination Group on Antimicrobial Resistance, No time to wait: securing the future from drug-resistant infections. Report to the Secretary-General of the United Nations (2019) Available at www.who.int/antimicrobial-resistance/interagency-coordination-group/final-report/en/.
- 159.Geurden T., Chartier C., Fanke J., di Regalbono A.F., Traversa D., von Samson-Himmelstjerna G. Anthelmintic resistance to ivermectin and moxidectin in gastrointestinal nematodes of cattle in Europe. Int. J. Parasitol. Drugs Drug. Resist. 2015;5:163–171. doi: 10.1016/j.ijpddr.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kotze A.C., Prichard R.K. Anthelmintic resistance in Haemonchus contortus: history, mechanisms and diagnosis. Adv. Parasitol. 2016;93:397–428. doi: 10.1016/bs.apar.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 161.Wolstenholme A.J., Evans C.C., Jimenez P.D., Moorhead A.R. The emergence of macrocyclic lactone resistance in the canine heartworm Dirofilaria immitis. Parasitology. 2015;142:1249–1259. doi: 10.1017/S003118201500061X. [DOI] [PubMed] [Google Scholar]
- 162.Jimenez Castro P.D., Howell S.B., Schaefer J.J., Avramenko R.W., Gilleard J.S., Kaplan R.M. Multiple drug resistance in the canine hookworm Ancylostoma caninum: an emerging threat? Parasit Vectors. 2019;12:576. doi: 10.1186/s13071-019-3828-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Panic G., Duthaler U., Speich B., Keiser J. Repurposing drugs for the treatment and control of helminth infections. Int. J. Parasitol. Drugs Drug. Resist. 2014;4:185–200. doi: 10.1016/j.ijpddr.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Blaskovich M.A., Butler M.S., Cooper M.A. Polishing the tarnished silver bullet: the quest for new antibiotics. Essays Biochem. 2017;61:103–114. doi: 10.1042/EBC20160077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Woods D.J., Lauret C., Geary T. Anthelmintic discovery and development in the animal health industry. Expert Opin. Drug Discov. 2007;2:S25–S33. doi: 10.1517/17460441.2.S1.S25. [DOI] [PubMed] [Google Scholar]
- 166.Kuhn-Nentwig L. Antimicrobial and cytolytic peptides of venomous arthropods. Cell. Mol. Life Sci. 2003;60:2651–2668. doi: 10.1007/s00018-003-3106-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Harrison P.L., Abdel-Rahman M.A., Miller K., Strong P.N. Antimicrobial peptides from scorpion venoms. Toxicon. 2014;88:115–137. doi: 10.1016/j.toxicon.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fratini F., Cilia G., Turchi B., Felicioli A. Insects, arachnids and centipedes venom: a powerful weapon against bacteria. A literature review. Toxicon. 2017;130:91–103. doi: 10.1016/j.toxicon.2017.02.020. [DOI] [PubMed] [Google Scholar]
- 169.Samy R.P., Stiles B.G., Franco O.L., Sethi G., Lim L.H.K. Animal venoms as antimicrobial agents. Biochem. Pharmacol. 2017;134:127–138. doi: 10.1016/j.bcp.2017.03.005. [DOI] [PubMed] [Google Scholar]
- 170.Primon-Barros M., Jose Macedo A. Animal venom peptides: potential for new antimicrobial agents. Curr. Top. Med. Chem. 2017;17:1119–1156. doi: 10.2174/1568026616666160930151242. [DOI] [PubMed] [Google Scholar]
- 171.Perez-Peinado C., Defaus S., Andreu D. Hitchhiking with Nature: snake venom peptides to fight cancer and superbugs. Toxins. 2020;12:255. doi: 10.3390/toxins12040255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Schlamadinger D.E., Gable J.E., Kim J.E. Toxins and antimicrobial peptides: interactions with membranes. Proc. SPIE Int. Soc. Opt. Eng. 2009;7397:73970J. doi: 10.1117/12.827439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dubovskii P.V., Vassilevski A.A., Kozlov S.A., Feofanov A.V., Grishin E.V., Efremov R.G. Latarcins: versatile spider venom peptides. Cell. Mol. Life Sci. 2015;72:4501–4522. doi: 10.1007/s00018-015-2016-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Orivel J., Redeker V., Le Caer J.P., Krier F., Revol-Junelles A.M., Longeon A. Ponericins, new antibacterial and insecticidal peptides from the venom of the ant Pachycondyla goeldii. J. Biol. Chem. 2001;276:17823–17829. doi: 10.1074/jbc.M100216200. [DOI] [PubMed] [Google Scholar]
- 175.Kuhn-Nentwig L., Willems J., Seebeck T., Shalaby T., Kaiser M., Nentwig W. Cupiennin 1a exhibits a remarkably broad, non-stereospecific cytolytic activity on bacteria, protozoan parasites, insects, and human cancer cells. Amino Acids. 2011;40:69–76. doi: 10.1007/s00726-009-0471-0. [DOI] [PubMed] [Google Scholar]
- 176.Dekan Z., Headey S.J., Scanlon M., Baldo B.A., Lee T.H., Aguilar M.I. △-myrtoxin-Mp1a is a helical heterodimer from the venom of the jack jumper ant that has antimicrobial, membrane-disrupting, and nociceptive activities. Angew. Chem. Int. Ed. Engl. 2017;56:8495–8499. doi: 10.1002/anie.201703360. [DOI] [PubMed] [Google Scholar]
- 177.Chen J., Guan S.M., Sun W., Fu H. Melittin, the major pain-producing substance of bee venom. Neurosci. Bull. 2016;32:265–272. doi: 10.1007/s12264-016-0024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tene N., Bonnafe E., Berger F., Rifflet A., Guilhaudis L., Segalas-Milazzo I. Biochemical and biophysical combined study of bicarinalin, an ant venom antimicrobial peptide. Peptides. 2016;79:103–113. doi: 10.1016/j.peptides.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 179.Guzman J., Tene N., Touchard A., Castillo D., Belkhelfa H., Haddioui-Hbabi L. Anti-Helicobacter pylori properties of the ant-venom peptide bicarinalin. Toxins. 2017;10:21. doi: 10.3390/toxins10010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sánchez-Vásquez L., Silva-Sanchez J., Jimenez-Vargas J.M., Rodriguez-Romero A., Munoz-Garay C., Rodriguez M.C. Enhanced antimicrobial activity of novel synthetic peptides derived from vejovine and hadrurin. Biochim. Biophys. Acta, Mol. Cell. Biol. Lipids. 1830;2013:3427–3436. doi: 10.1016/j.bbagen.2013.01.028. [DOI] [PubMed] [Google Scholar]
- 181.Irazazabal L.N., Porto W.F., Ribeiro S.M., Casale S., Humblot V., Ladram A. Selective amino acid substitution reduces cytotoxicity of the antimicrobial peptide mastoparan. Biochim. Biophys. Acta, Mol. Cell. Biol. Lipids. 1858;2016:2699–2708. doi: 10.1016/j.bbamem.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 182.Etzerodt T., Henriksen J.R., Rasmussen P., Clausen M.H., Andresen T.L. Selective acylation enhances membrane charge sensitivity of the antimicrobial peptide mastoparan-x. Biophys. J . 2011;100:399–409. doi: 10.1016/j.bpj.2010.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mwangi J., Yin Y., Wang G., Yang M., Li Y., Zhang Z. The antimicrobial peptide ZY4 combats multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii infection. PNAS. 2019;116:26516–26522. doi: 10.1073/pnas.1909585117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Reis P.V.M., Boff D., Verly R.M., Melo-Braga M.N., Cortes M.E., Santos D.M. LyeTxI-b, a synthetic peptide derived from Lycosa erythrognatha spider venom, shows potent antibiotic activity in vitro and in vivo. Front. Microbiol. 2018;9:667. doi: 10.3389/fmicb.2018.00667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Otvos L., Jr., Wade J.D. Current challenges in peptide-based drug discovery. Front. Chem. 2014;2:62. doi: 10.3389/fchem.2014.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cao L., Dai C., Li Z., Fan Z., Song Y., Wu Y. Antibacterial activity and mechanism of a scorpion venom peptide derivative in vitro and in vivo. PLoS ONE. 2012;7:e40135. doi: 10.1371/journal.pone.0040135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Garcia F., Villegas E., Espino-Solis G.P., Rodriguez A., Paniagua-Solis J.F., Sandoval-Lopez G. Antimicrobial peptides from arachnid venoms and their microbicidal activity in the presence of commercial antibiotics. J. Antibiot. 2013;66:3–10. doi: 10.1038/ja.2012.87. [DOI] [PubMed] [Google Scholar]
- 188.Wu D., Wu T., Liu Q., Yang Z. The SARS-CoV-2 outbreak: what we know. Int. J. Infect. Dis. 2020;94:44–48. doi: 10.1016/j.ijid.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.GBD Disease and Injury Incidence Prevalence Collaborators Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1789–1858. doi: 10.1016/S0140-6736(18)32279-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.da Mata E.C., Mourao C.B., Rangel M., Schwartz E.F. Antiviral activity of animal venom peptides and related compounds. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017;23:3. doi: 10.1186/s40409-016-0089-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Vilas Boas L.C.P., Campos M.L., Berlanda R.L.A., de Carvalho Neves N, Franco O.L. Antiviral peptides as promising therapeutic drugs. Cell. Mol. Life Sci. 2019;76:3525–3542. doi: 10.1007/s00018-019-03138-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sample C.J., Hudak K.E., Barefoot B.E., Koci M.D., Wanyonyi M.S., Abraham S. A mastoparan-derived peptide has broad-spectrum antiviral activity against enveloped viruses. Peptides. 2013;48:96–105. doi: 10.1016/j.peptides.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Rothan H.A., Bahrani H., Rahman N.A., Yusof R. Identification of natural antimicrobial agents to treat dengue infection: in vitro analysis of latarcin peptide activity against dengue virus. BMC Microbiol. 2014;14:140. doi: 10.1186/1471-2180-14-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Dai C., Ma Y., Zhao Z., Zhao R., Wang Q., Wu Y. Mucroporin, the first cationic host defense peptide from the venom of Lychas mucronatus. Antimicrob. Agents Chemother. 2008;52:3967–3972. doi: 10.1128/AAC.00542-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Li Q., Zhao Z., Zhou D., Chen Y., Hong W., Cao L. Virucidal activity of a scorpion venom peptide variant mucroporin-M1 against measles, SARS-CoV and influenza H5N1 viruses. Peptides. 2011;32:1518–1525. doi: 10.1016/j.peptides.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Maser P., Wittlin S., Rottmann M., Wenzler T., Kaiser M., Brun R. Antiparasitic agents: new drugs on the horizon. Curr. Opin. Pharmacol. 2012;12:562–566. doi: 10.1016/j.coph.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 197.Adade C.M., Souto-Padrón T. Venoms as sources of novel anti-parasitic agents. In: Gopalakrishnakone P., editor. Toxins and Drug Discovery. Springer; Dordrecht: 2015. [Google Scholar]
- 198.Phillips M.A., Burrows J.N., Manyando C., van Huijsduijnen R.H., Van Voorhis W.C., Wells T.N.C. Malaria. Nat. Rev. Dis. Primers. 2017;3:17050. doi: 10.1038/nrdp.2017.50. [DOI] [PubMed] [Google Scholar]
- 199.Adade C.M., Oliveira I.R., Pais J.A., Souto-Padron T. Melittin peptide kills Trypanosoma cruzi parasites by inducing different cell death pathways. Toxicon. 2013;69:227–239. doi: 10.1016/j.toxicon.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 200.Vinhote J.F.C., Lima D.B., Menezes R., Mello C.P., de Souza B.M., Havt A. Trypanocidal activity of mastoparan from Polybia paulista wasp venom by interaction with TcGAPDH. Toxicon. 2017;137:168–172. doi: 10.1016/j.toxicon.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 201.El Chamy Maluf S, Dal Mas C., Oliveira E.B., Melo P.M., Carmona A.K., Gazarini M.L. Inhibition of malaria parasite Plasmodium falciparum development by crotamine, a cell penetrating peptide from the snake venom. Peptides. 2016;78:11–16. doi: 10.1016/j.peptides.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 202.Peichoto M.E., Tavares F.L., Dekrey G., Mackessy S.P. A comparative study of the effects of venoms from five rear-fanged snake species on the growth of Leishmania major: identification of a protein with inhibitory activity against the parasite. Toxicon. 2011;58:28–34. doi: 10.1016/j.toxicon.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 203.De Leon-Nava M.A., Romero-Nunez E., Luna-Nophal A., Bernaldez-Sarabia J., Sanchez-Campos L.N., Licea-Navarro A.F. In vitro effect of the synthetic cal14.1a conotoxin, derived from Conus californicus, on the human parasite Toxoplasma gondii. Mar. Drugs. 2016;14:66. doi: 10.3390/md14040066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Borges I.P., Castanheira L.E., Barbosa B.F., de Souza D.L., da Silva R.J., Mineo J.R. Anti-parasitic effect on Toxoplasma gondii induced by BnSP-7, a Lys49-phospholipase A homologue from Bothrops pauloensis venom. Toxicon. 2016;119:84–91. doi: 10.1016/j.toxicon.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 205.Castanheira L. Naves de Souza DL, Silva RJ, Barbosa B, Mineo JR, Tudini KA, et al., Insights into anti-parasitism induced by a C-type lectin from Bothrops pauloensis venom on Toxoplasma gondii. Int. J. Biol. Macromol. 2015;74:568–574. doi: 10.1016/j.ijbiomac.2014.11.035. [DOI] [PubMed] [Google Scholar]
- 206.WHO . World Health Organization; 2016. Water-related Diseases. [Google Scholar]
- 207.Roeber F., Jex A.R., Gasser R.B. Impact of gastrointestinal parasitic nematodes of sheep, and the role of advanced molecular tools for exploring epidemiology and drug resistance — an Australian perspective. Parasit. Vectors. 2013;6:153. doi: 10.1186/1756-3305-6-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Dal Mas C., Moreira J.T., Pinto S., Monte G.G., Nering M.B., Oliveira E.B. Anthelmintic effects of a cationic toxin from a South American rattlesnake venom. Toxicon. 2016;116:49–55. doi: 10.1016/j.toxicon.2015.11.021. [DOI] [PubMed] [Google Scholar]
- 209.Mee C.J., Tomlinson S.R., Perestenko P.V., De Pomerai D., Duce I.R., Usherwood P.N. Latrophilin is required for toxicity of black widow spider venom in Caenorhabditis elegans. Biochem. J. 2004;378:185–191. doi: 10.1042/BJ20031213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Nixon S.A., Saez N.J., Herzig V., King G.F., Kotze A.C. The antitrypanosomal diarylamidines, diminazene and pentamidine, show anthelmintic activity against Haemonchus contortus in vitro. Vet. Parasitol. 2019;270:40–46. doi: 10.1016/j.vetpar.2019.05.008. [DOI] [PubMed] [Google Scholar]
- 211.S.S. Pineda, Y.K.Y. Chin, E.A.B. Undheim, S. Senff, M. Mobli, C. Dauly, et al., Structural venomics reveals evolution of a complex venom by duplication and diversification of an ancient peptide-encoding gene, Proc. Natl. Acad. Sci. USA (2020) in press. [DOI] [PMC free article] [PubMed]
- 212.Silva P.I., Jr., Daffre S., Bulet P. Isolation and characterization of gomesin, an 18-residue cysteine-rich defense peptide from the spider Acanthoscurria gomesiana hemocytes with sequence similarities to horseshoe crab antimicrobial peptides of the tachyplesin family. J. Biol. Chem. 2000;275:33464–33470. doi: 10.1074/jbc.M001491200. [DOI] [PubMed] [Google Scholar]
- 213.Tanner J.D., Deplazes E., Mancera R.L. The biological and biophysical properties of the spider peptide gomesin. Molecules. 2018;23:1733. doi: 10.3390/molecules23071733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ikonomopoulou M.P., Fernandez-Rojo M.A., Pineda S.S., Cabezas-Sainz P., Winnen B., Morales R.A.V. Gomesin inhibits melanoma growth by manipulating key signaling cascades that control cell death and proliferation. Sci. Rep. 2018;8:11519. doi: 10.1038/s41598-018-29826-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Chan L.Y., Zhang V.M., Huang Y.H., Waters N.C., Bansal P.S., Craik D.J. Cyclization of the antimicrobial peptide gomesin with native chemical ligation: influences on stability and bioactivity. ChemBioChem. 2013;14:617–624. doi: 10.1002/cbic.201300034. [DOI] [PubMed] [Google Scholar]
- 216.Possani L.D., Corona M., Zurita M., RodrÌguez M.H. From noxiustoxin to scorpine and possible transgenic mosquitoes resistant to malaria. Arch. Med. Res. 2002;33:398–404. doi: 10.1016/s0188-4409(02)00370-3. [DOI] [PubMed] [Google Scholar]
- 217.Windley M.J., Herzig V., Dziemborowicz S.A., Hardy M.C., King G.F., Nicholson G.M. Spider-venom peptides as bioinsecticides. Toxins. 2012;4:191–227. doi: 10.3390/toxins4030191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tedford H.W., Sollod B.L., Maggio F., King G.F. Australian funnel-web spiders: master insecticide chemists. Toxicon. 2004;43:601–618. doi: 10.1016/j.toxicon.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 219.Smith J.J., Herzig V., King G.F., Alewood P.F. The insecticidal potential of venom peptides. Cell. Mol. Life Sci. 2013;70:3665–3693. doi: 10.1007/s00018-013-1315-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Saez N.J., Herzig V. Versatile spider venom peptides and their medical and agricultural applications. Toxicon. 2019;158:109–126. doi: 10.1016/j.toxicon.2018.11.298. [DOI] [PubMed] [Google Scholar]
- 221.King G.F. Tying pest insects in knots: the deployment of spider-venom-derived knottins as bioinsecticides. Pest Manag. Sci. 2019;75:2437–2445. doi: 10.1002/ps.5452. [DOI] [PubMed] [Google Scholar]
- 222.Bilgo E., Lovett B., Fang W., Bende N., King G.F., Diabate A. Improved efficacy of an arthropod toxin expressing fungus against insecticide-resistant malaria-vector mosquitoes. Sci. Rep. 2017;7:3433. doi: 10.1038/s41598-017-03399-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lovett B., Bilgo E., Millogo S.A., Ouattarra A.K., Sare I., Gnambani E.J. Transgenic Metarhizium rapidly kills mosquitoes in a malaria-endemic region of Burkina Faso. Science. 2019;364:894–897. doi: 10.1126/science.aaw8737. [DOI] [PubMed] [Google Scholar]
- 224.Fang W., Lu H.L., King G.F., St Leger R.J. Construction of a hypervirulent and specific mycoinsecticide for locust control. Sci. Rep. 2014;4:7345. doi: 10.1038/srep07345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Krapcho K.J., Kral R.M., Jr., Vanwagenen B.C., Eppler K.G., Morgan T.K. Characterization and cloning of insecticidal peptides from the primitive weaving spider Diguetia canities. Insect Biochem. Mol. Biol. 1995;25:991–1000. doi: 10.1016/0965-1748(95)00029-u. [DOI] [PubMed] [Google Scholar]
- 226.Herzig V., Ikonomopoulou M., Smith J.J., Dziemborowicz S., Gilchrist J., Kuhn-Nentwig L. Molecular basis of the remarkable species selectivity of an insecticidal sodium channel toxin from the African spider Augacephalus ezendami. Sci. Rep. 2016;6:29538. doi: 10.1038/srep29538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Bende N.S., Dziemborowicz S., Mobli M., Herzig V., Gilchrist J., Wagner J. A distinct sodium channel voltage-sensor locus determines insect selectivity of the spider toxin Dc1a. Nat. Commun. 2014;5:4350. doi: 10.1038/ncomms5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Shen H.Z., Li Z.Q., Jiang Y., Pan X.J., Wu J.P., Cristofori-Armstrong B. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science. 2018;362:eaau2596. doi: 10.1126/science.aau2596. [DOI] [PubMed] [Google Scholar]
- 229.King G.F. Royal Society of Chemistry; UK: 2015. Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics London. [Google Scholar]
- 230.Cushman D.W., Ondetti M.A. History of the design of captopril and related inhibitors of angiotensin converting enzyme. Hypertension. 1991;17:589–592. doi: 10.1161/01.hyp.17.4.589. [DOI] [PubMed] [Google Scholar]
- 231.Opie L.H., Kowolik H. The discovery of captopril: from large animals to small molecules. Cardiovasc. Res. 1995;30:18–25. [PubMed] [Google Scholar]
- 232.Cushman D.W., Ondetti M.A. Design of angiotensin converting enzyme inhibitors. Nat. Med. 1999;5:1110–11103. doi: 10.1038/13423. [DOI] [PubMed] [Google Scholar]
- 233.Li J.J. Oxford University Press; New York: 2014. Blockbuster Drugs: The Rise and Decline of the Pharmaceutical Industry. [Google Scholar]
- 234.Barnett A. Exenatide. Expert Opin. Pharmacother. 2007;8:2593–2608. doi: 10.1517/14656566.8.15.2593. [DOI] [PubMed] [Google Scholar]
- 235.Malone J., Trautmann M., Wilhelm K., Taylor K., Kendall D.M. Exenatide once weekly for the treatment of type 2 diabetes. Expert Opin. Invest. Drugs. 2009;18:359–367. doi: 10.1517/13543780902766802. [DOI] [PubMed] [Google Scholar]
- 236.Miljanich G.P. Ziconotide: neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004;11:3029–3040. doi: 10.2174/0929867043363884. [DOI] [PubMed] [Google Scholar]
- 237.Backryd E. Do the potential benefits outweigh the risks? An update on the use of ziconotide in clinical practice. Eur. J. Pain. 2018;22:1193–1202. doi: 10.1002/ejp.1229. [DOI] [PubMed] [Google Scholar]
- 238.Vetter I., Davis J.L., Rash L.D., Anangi R., Mobli M., Alewood P.F. Venomics: a new paradigm for natural products-based drug discovery. Amino Acids. 2011;40:15–28. doi: 10.1007/s00726-010-0516-4. [DOI] [PubMed] [Google Scholar]
- 239.Sun P., Wu F., Wen M., Yang X., Wang C., Li Y. A distinct three-helix centipede toxin SSD609 inhibits Iks channels by interacting with the KCNE1 auxiliary subunit. Sci. Rep. 2015;5:13399. doi: 10.1038/srep13399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ma L., Chin Y.K.Y., Dekan Z., Herzig V., Chow C.Y., Heighway J. Novel venom-derived inhibitors of the human EAG channel, a putative antiepileptic drug target. Biochem. Pharmacol. 2018;158:60–72. doi: 10.1016/j.bcp.2018.08.038. [DOI] [PubMed] [Google Scholar]
- 241.Revell J.D., Lund P.E., Linley J.E., Metcalfe J., Burmeister N., Sridharan S. Potency optimization of Huwentoxin-IV on hNaV1. 7: a neurotoxin TTX-S sodium-channel antagonist from the venom of the Chinese bird-eating spider Selenocosmia huwena. Peptides. 2013;44:40–46. doi: 10.1016/j.peptides.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 242.Murray J.K., Ligutti J., Liu D., Zou A., Poppe L., Li H. Engineering potent and selective analogues of GpTx-1, a tarantula venom peptide antagonist of the NaV1.7 sodium channel. J. Med. Chem. 2015;58:2299–2314. doi: 10.1021/jm501765v. [DOI] [PubMed] [Google Scholar]
- 243.Xu H., Li T., Rohou A., Arthur C.P., Tzakoniati F., Wong E. Structural basis of NaV1.7 inhibition by a gating-modifier spider toxin. Cell. 2019;176:702–715. doi: 10.1016/j.cell.2018.12.018. [DOI] [PubMed] [Google Scholar]
- 244.Neff R.A., Flinspach M., Gibbs A., Shih A.Y., Minassian N.A., Liu Y. Comprehensive engineering of the tarantula venom peptide huwentoxin-IV to inhibit the human voltage-gated sodium channel hNaV1.7. J. Biol. Chem. 2020;295:1315–1327. doi: 10.1074/jbc.RA119.011318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Twomey E.C., Yelshanskaya M.V., Vassilevski A.A., Sobolevsky A.I. Mechanisms of channel block in calcium-permeable AMPA receptors. Neuron. 2018;99:956–968. doi: 10.1016/j.neuron.2018.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Clairfeuille T., Cloake A., Infield D.T., Llongueras J.P., Arthur C.P., Li Z.R. Structural basis of ⍺-scorpion toxin action on NaV channels. Science. 2019;363:eaav8573. doi: 10.1126/science.aav8573. [DOI] [PubMed] [Google Scholar]
- 247.Shen H., Liu D., Wu K., Lei J., Yan N. Structures of human NaV1.7 channel in complex with auxiliary subunits and animal toxins. Science. 2019;363:1303–1308. doi: 10.1126/science.aaw2493. [DOI] [PubMed] [Google Scholar]
- 248.Pan X., Li Z., Huang X., Huang G., Gao S., Shen H. Molecular basis for pore blockade of human Na+ channel NaV1.2 by the μ-conotoxin KIIIA. Science. 2019;363:1309–1313. doi: 10.1126/science.aaw2999. [DOI] [PubMed] [Google Scholar]
- 249.Santos R., Ursu O., Gaulton A., Bento A.P., Donadi R.S., Bologa C.G. A comprehensive map of molecular drug targets. Nat. Rev. Drug. Discov. 2017;16:19–34. doi: 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Fukuda K., Doggett T., Laurenzi I.J., Liddington R.C., Diacovo T.G. The snake venom protein botrocetin acts as a biological brace to promote dysfunctional platelet aggregation. Nat. Struct. Mol. Biol. 2005;12:152–159. doi: 10.1038/nsmb892. [DOI] [PubMed] [Google Scholar]
- 251.Janssen B.J., Gomes L., Koning R.I., Svergun D.I., Koster A.J., Fritzinger D.C. Insights into complement convertase formation based on the structure of the factor B-cobra venom factor complex. EMBO J. 2009;28:2469–2478. doi: 10.1038/emboj.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Natesh R., Schwager S.L., Evans H.R., Sturrock E.D., Acharya K.R. Structural details on the binding of antihypertensive drugs captopril and enalaprilat to human testicular angiotensin I-converting enzyme. Biochemistry. 2004;43:8718–8724. doi: 10.1021/bi049480n. [DOI] [PubMed] [Google Scholar]
- 253.Shioi N., Tadokoro T., Shioi S., Okabe Y., Matsubara H., Kita S. Crystal structure of the complex between venom toxin and serum inhibitor from Viperidae snake. J. Biol. Chem. 2019;294:1250–1256. doi: 10.1074/jbc.RA118.006840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Baconguis I., Bohlen C.J., Goehring A., Julius D., Gouaux E. X-ray structure of acid-sensing ion channel 1-snake toxin complex reveals open state of a Na+-selective channel. Cell. 2014;156:717–729. doi: 10.1016/j.cell.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Hansen S.B., Sulzenbacher G., Huxford T., Marchot P., Taylor P., Bourne Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005;24:3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Pan X., Li Z., Huang X., Huang G., Gao S., Shen H. Molecular basis for pore blockade of human Na+ channel Nav1.2 by the μ-conotoxin KIIIA. Science. 2019;363:1309–1313. doi: 10.1126/science.aaw2999. [DOI] [PubMed] [Google Scholar]
- 257.Baconguis I., Gouaux E. Structural plasticity and dynamic selectivity of acid-sensing ion channel-spider toxin complexes. Nature. 2012;489:400–405. doi: 10.1038/nature11375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Gao Y., Cao E., Julius D., Cheng Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature. 2016;534:347–351. doi: 10.1038/nature17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Runge S., Thogersen H., Madsen K., Lau J., Rudolph R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J. Biol. Chem. 2008;283:11340–11347. doi: 10.1074/jbc.M708740200. [DOI] [PubMed] [Google Scholar]
- 260.Banerjee A., Lee A., Campbell E., Mackinnon R. Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K+ channel. Elife. 2013;2:e00594. doi: 10.7554/eLife.00594. [DOI] [PMC free article] [PubMed] [Google Scholar]