

Abstract
Investigation of intercellular interactions often requires discrete labeling of specific cell populations and precise protein localization. The zebrafish embryo is an excellent tool for examining such interactions with an in vivo model. Whole-mount immunohistochemical and immunofluorescence assays are frequently applied in zebrafish embryos to assess protein expression. However, it can be difficult to achieve accurate mapping of co-localized proteins in three-dimensional space. In addition, some studies may require the use of two antibodies that are not compatible with the same technique (e.g., antibody 1 is only suitable for immunohistochemistry and antibody 2 is only suitable for immunofluorescence). The purpose of the method described herein is to perform sequential immunofluorescence and/or immunohistochemistry on individual cryosections derived from early-stage zebrafish embryos. Here we describe the use of sequential rounds of immunofluorescence, imaging, immunohistochemistry, imaging for a single cryosection in order to achieve precise identification of protein expression at the single-cell level. This methodology is suitable for any study in early-stage zebrafish embryos that requires accurate identification of multiple protein targets in individual cells.
Keywords: Biology, Issue 147, Zebrafish, embryos, immunohistochemistry, immunofluorescence, colocalization, intercellular interaction
Introduction
The zebrafish is an extremely robust model organism that is currently used across a wide variety of disciplines in biomedical research. In particular, the rapid external development and translucency of zebrafish embryos provide an excellent tool for in vivo studies. Herein, we describe a method for sequential immunofluorescence (IF) and immunohistochemical (IHC) analyses of cryosectioned zebrafish embryos. This novel procedure uses sequential application of two antibodies on a single slide, enabling accurate identification of colocalized proteins at the cellular level while conserving tissue sections. This protocol is particularly useful for studies with the zebrafish model, since a relatively small number of antibodies have been validated for use in IF and/or IHC applications in zebrafish compared to mouse models.
The observation of intercellular interactions is an essential element in many studies, and may provide key insights into molecular mechanisms functioning at the cellular level that underlie phenotypes at the organismal level. Additionally, protein expression can provide information about cellular function, especially when examining expression of multiple proteins simultaneously within the cell (co-localization). Although whole-mount IHC and IF are commonly used techniques for analyzing protein expression in zebrafish embryos1,2,3,4,5, whole mount procedures can be problematic for achieving precise colocalization data. In our experience, it can be difficult to differentiate between layers of tissue and visualize protein expression at the single-cell level in whole-mount specimens. Imaging software programs may generally be unable to distinguish between surface staining versus deeper staining. Non-surface level protein expressions can be obscured by more brightly expressed surface level expression, leading to inaccuracies in quantification. Additionally, most traditional zebrafish clearing methods are quite toxic6 and thus less desirable for use.
Antibody-based techniques, such as IF and IHC, are often used to detect protein expression in sectioned material, simplifying the identification of discrete cell populations that express a particular protein within complex tissues. IHC is commonly used for colocalization, most frequently by using two different antibodies conjugated in different host species and visualized with different colored chromogens4,7,8,9,10. However, using multiple chromogens can lead to nonspecific background staining or incompatibility of colors11,12.
We developed a novel protocol for detection of multiple proteins by sequential IF and IHC on cryosectioned early-stage zebrafish embryos. Cryosectioning is particularly well-suited to delicate tissues such as zebrafish embryos, and cryosections are superior to paraffin-embedded sections for fluorescence-based assays13,14. We chose to optimize combined IF and IHC rather than dual-color IF or IHC, to circumvent the problem of antibody incompatibility for a single assay type. These problems are particularly relevant for research involving zebrafish due to the limited number of commercially available antibodies that are validated for use in zebrafish. In fact, a study of four large companies showed that commercially available antibodies for use in mouse was about 112,000 versus about 5,300 for use in zebrafish15. Finally, we chose to develop a protocol that could be performed on a single cryosection, which is essential when working with small or limited tissue samples such as are obtained from zebrafish embryos.
This protocol was designed to assess the proliferative behavior of donor cells in 48 h post-fertilization chimeric zebrafish embryos that were generated by blastula-to-blastula transplant as described by Carmany-Rampey and Moens16. Donor embryos were injected at the one-cell stage with a fluorescently labeled dextran conjugate prior to transplant of donor cells into recipient embryos. We used immunofluorescence for Ser 10 phosphorylated Histone H3 (pH3) to detect proliferating cells followed by immunohistochemistry for the labeled dextran to detect donor cells in chimeric zebrafish embryos. Sequential detection of pH3 and the labeled dextran within a single cryosection enabled us to identify and quantify individual cells that expressed both markers.
This sequential IF/IHC protocol for cryosectioned zebrafish will provide a useful tool for zebrafish researchers who desire a colocalization protocol for protein expression. The problems that this protocol was designed to address, such as small tissue specimens and limited antibody availability, are not unique to the zebrafish model. This method may therefore be of use to any researcher desiring to perform sequential IF/IHC.
ETHICS STATEMENT:
All animal studies were approved by the Institutional Animal Care and Use Committee, North Carolina State University, Raleigh, North Carolina, USA.
Protocol
1. Embryo Preparation
-
Fix 48 h post-fertilization (hpf) chimeric zebrafish embryos generated by blastula-to-blastula transplant between AB wild-type zebrafish embryos in 4% paraformaldehyde (PFA) overnight with rocking at 4 °C. Perform two 5 min washes with rocking at room temperature using 500 μL of 1x phosphate buffered saline containing 0.1% of a non-ionic surfactant (1x PBSt; see the Table of Materials)
NOTE: Paraformaldehyde is toxic and a carcinogen and must be properly disposed of per institutional regulations. Work with paraformaldehyde should be performed in a chemical hood, and appropriate personal protective equipment (gloves, lab coat, and safety glasses) should always be used.
-
Dehydrate embryos by washing sequentially with 500 μL of 30%, 50%, and 70% methanol (MeOH) diluted with 1x PBSt for 10 min each at room temperature with rocking. Incubate embryos in 500 μL of 100% MeOH at −20 °C for at least 14 h.
NOTE: Methanol is toxic and must be properly disposed of per institutional regulations. Work with methanol should be performed in a chemical hood, and appropriate personal protective equipment (gloves, lab coat, and safety glasses) should always be used.
Rehydrate embryos by washing sequentially with 500 μL of 70%, 50%, and 30% MeOH diluted with 1x PBSt for 10 min each at room temperature with rocking. Perform two 5 min washes with 500 μL of 1x PBSt at room temperature with rocking.
Remove the 1x PBSt and incubate in 500 μL of 30% sucrose diluted with deionized water at room temperature with rocking until the embryos sink to the bottom of the tube (approximately 1 h). Replace sucrose with 500 μL of 15% fish gelatin/25% sucrose (15/25; see the Table of Materials) and incubate at room temperature with rocking overnight.
Replace approximately half of the volume of 15/25 with optimal cutting temperature medium (OCT medium) and incubate at room temperature with rocking until the embryos sink to the bottom of the tube (approximately 1 h).
Replace approximately half of the volume with OCT medium and incubate at room temperature with rocking for 1 h. Repeat this step one time.
During incubation steps with 15/25 and OCT medium, invert or flick the tube as needed to combine these reagents.
Materials
| Name | Company | Catalog Number | Comments |
|---|---|---|---|
| 15/25 | N/A | N/A | 15% fish gelatin/25% sucrose w/v, made in dH2O |
| Alexa 555 secondary antibody | Invitrogen | A21429 | used at 1:2,000 dilution in block buffer for IF |
| Antibody Diluent | Dako | S3022 | |
| Background Buster | Innovex | NB306 | |
| block buffer (IF) | N/A | N/A | 5% normal goat serum, 0.1 % Triton X-100 in 1x PBS |
| cellSens imaging software | Olympus | cellSens | imaging software used with compound light microscope |
| chimeric zebrafish embryos | N/A | N/A | AB wild-type chimera zebrafish embryos analyzed at 48 hpf |
| Compound light microscope | Olympus | BX51 | used with digital camera and cellSens imaging software for image capture after IHC |
| Confocal fluorescence microscope | Leica | DM 2500 | used with digital camera and Leica Application Suite Advanced Fluorescence imaging software for image capture after IF |
| Disposable plastic molds, 15 mm × 15 mm × 5 mm | Ted Pella | 27147–2 | |
| Digital camera, light microscope | Olympus | DP27 | |
| Digital camera, fluorescence microscope | Leica | TCS SPE | |
| Ethanol | Koptec | V1001 | |
| Fish gelatin | Sigma | G7041 | |
| Glass slides | Fisher | 12-550-15 | |
| Hydrogen peroxide, 30% | Fisher | H325–100 | |
| ImmPRESS HRP Polymer detection kit | Vector | MP-7401 | anti-rabbit secondary antibody, HRP polymer |
| Leica Application Suite Advanced Fluorescence imaging software | Leica | LAS AF 2.3.6 | imaging software used with confocal fluorescence microscope |
| Methanol | Fisher | A452-4 | |
| Modified Meyer's Hematoxylin | Thermo | 72804 | |
| Normal Goat Serum | MP Biomedical | 191356 | |
| OCT medium | Tissue-Tek/Fisher | 4583/4585 | |
| anti-Oregon Green antibody | Molecular Probes/Life Technologies | A889 | used at 1:7500 dilution for IHC |
| Oregon Green Dextran | Invitrogen | P7171 | used at 2.5% in 0.2 M KCl |
| Paraformaldehyde, 4% | Acros Organics | 416780030 | made in lab in 1x PBS |
| Permount | Fisher | SP15 | |
| 1x phosphate-buffered saline | made in lab | N/A | 50 mL 10x PBS, up to 500 mL volume with dH2O |
| 10x phoshpate-buffered saline | made in lab | N/A | use Cold Spring Harbor protocol |
| 1x PBSt | made in lab | N/A | 50 mL 10x PBS, 1 mL 10% Tween 20, up to 500 mL volume with dH2O |
| anti-Ser10 phosphorylated Histone H3 antibody | Santa Cruz | sc-8656-R | used at 1:500 dilution for IF |
| Scot’s Tap Water | Electron Microscopy Sciences | 26070-06 | have used other brands successfully in addition to EMS |
| Sucrose | Amresco | 335 | |
| Tween-20 | Fisher | BP337 | |
| TO-PRO3 | Invitrogen | T3605 | used at 1:1,000 in 1x PBS for IF |
| 1x Tris-buffered saline | made in lab | N/A | 50 mL 10x TBS, up to 500 mL volume with dH2O |
| 10x Tris-buffered saline | made in lab | N/A | 85 g NaCl, 61 g Tris base, up to 1 L volume with dH2O |
| 1x TBSt | made in lab | N/A | 50 mL 10x TBS, 1 mL 10% Tween 20, up to 500 mL volume with dH2O |
| Triton X-100 | Acros Organics | 327371000 | |
| Vectashield | Vector | H-1000 | non-hardening mounting media |
| Vector NovaRed substrate | Vector | SK-4800 | HRP substrate |
| Xylene | Fisher | X3P-1GAL |
2. Embryo Embedding and Preparation of Cryosections
Transfer embryos to a plastic mold (see the Table of Materials) with forceps, minimizing any transfer of the 15/25-OCT mixture. Fill the mold approximately half full with OCT medium and gently mix the embryos in OCT medium.
Prepare labeled plastic molds and transfer desired embryos (usually 1–3 embryos) into the empty, labeled plastic molds, minimizing carry over of OCT medium. Once the desired embryos are in the plastic mold, gently fill with OCT medium to the top of the mold.
Use forceps or 25 G needles to arrange embryos to the desired orientation, using a light stereomicroscope for visualization (Figure 1A,B).
Freeze the prepared molds on dry ice in an insulated container with a metal platform. Place an ice bucket or foam cooler gently over the metal platform to create a cold chamber (Figure 1C,D)
- Prepare 10–12 μm thick cryosections using a cryostat set at −20 °C.
Figure 1: Preparation of frozen OCT blocks.

(A) 50x microscopic view of embryos in OCT in a cryogenic mold prepared for freezing. Red arrow indicates head of zebrafish embryo position against the bottom surface of the mold; black arrow indicates tail pointing up towards the user. (B) Plastic molds with embryos and OCT placed on chilled metal platform. (C) Foam ice bucket placed over plastic molds to create freezing chamber. (D) OCT turns white from transparent once the block is fully frozen.
Figure 2: Cryosectioning of frozen OCT blocks.

(A), Application of fresh OCT onto cryostat disc and placement of frozen block on OCT, rotated 180° from the freezing orientation of the block. (B) Side view of block frozen to sectioning disc. (C) View of sectioning block with roll plate in position. (D) Pick-up of sections using a charged slide from the metal platform of the cryostat.
3. Immunofluorescence for pH3
Perform three 5 min washes of the slides in 1x PBS in an appropriate container, such as a Coplin jar. If tissues are not well adhered to the slide, perform washes by laying slides on a flat surface and gently pipetting 500 μL of 1x PBS onto the surface. Pour off 1x PBS between washes by gently tipping the slides.
Outline the sections with a barrier pen or wax pencil to keep liquid on the slides (Figure 3).
Lay the slides on a flat surface in a humid chamber and pipette 200 μL of block buffer per section onto the slides. Incubate in block buffer for 2 h at room temperature or overnight at 4 °C.
Prepare the primary antibody dilution (rabbit anti-pH3 antibody, 1:200; see Table of Materials) in block buffer and mix well by pipetting. Gently tip the slides to drain off the block buffer, return to the humid chamber, and pipette 200 μL of primary antibody solution per section onto the slides. For the secondary-only control, pipette 200 μL of block buffer onto the appropriate sections.
Incubate the slides at 4 °C overnight in a humid chamber filled with deionized water. Seal the edges of the humid chamber to help retain moisture.
Perform three 5 min washes of the slides in 1x PBS in an appropriate container, such as a Coplin jar. During wash steps, prepare the secondary antibody dilution (anti-rabbit fluorescent secondary antibody conjugate, 1:2,000; see Table of Materials) in block buffer and mix well by pipetting.
Lay the slides on a flat surface in a humid chamber and pipette 200 μL of secondary antibody solution per section onto the slides. Incubate the slides in secondary antibody solution(s) at room temperature, shielded from light, for 30 min.
Perform three 5 min washes of the slides in 1x PBS in an appropriate container, such as a Coplin jar, shielded from light. During wash steps, prepare a nuclear staining solution (see the Table of Materials).
Place the slides on a flat surface and pipette 200–500 μL (depending on the size of sample) of the nuclear staining solution onto each section. Incubate the slides in the nuclear staining solution for 10 min, shielded from light.
-
Drain off the nuclear staining solution and mount the slides with non-hardening fluorescent mounting media and a glass coverslip. Maintain the slides in the dark at 4 °C until imaging is performed.
NOTE: The best results are obtained when the slides are imaged on the same day or the next day.
Following IF, visualize and image the slides with a confocal fluorescence microscope and digital camera using 555 nm (red) and 645 nm (far-red) emission filters at 100x magnification (10x ocular magnification and 10x objective magnification). Keep the laser power consistent throughout imaging.
-
After imaging, place the slides in individual containers of 1x PBS and store flat at 4 °C overnight to prepare for coverslip removal. When placing the slides in 1x PBS, gently agitate the slides to assist in removal of the coverslip.
NOTE: Do not press down on the coverslip or attempt to remove the coverslip manually, as this can compromise quality of the sections. The coverslips must be removed horizontally with minimal forced movement.
Figure 3: View of prepared sections after placement on a slide.

Arrow indicates hydrophobic barrier, and dotted line delineates the zone containing sections within barrier perimeter.
4. Immunohistochemistry for Labeled Dextran
After the coverslips have been removed, gently transfer the slides into new 1x PBS in an appropriate container, such as a Coplin jar. Remove the 1x PBS and perform three 5 min incubations of the slides in 1x Tris-buffered saline with 0.1% of a non-ionic surfactant (1x TBSt) at room temperature.
-
Prepare 250 mL of 3% hydrogen peroxide (H2O2) diluted in deionized water in an appropriately sized container. Incubate the slides in 3% H2O2 solution at room temperature for 15 min.
NOTE: Hydrogen peroxide is corrosive and must be properly disposed of per institutional regulations. Appropriate personal protective equipment (gloves, lab coat, and safety glasses) should always be used.
Quickly rinse the slides with deionized water. Perform three 5 min washes of the slides in 1x TBSt. Carefully dry the area around the tissue sections and place the slides flat in a humid chamber. Outline the tissue sections with a barrier pen or wax pencil to keep liquid on the sections if needed.
Apply a ready-to-use 2.5% serum blocking solution provided with a secondary antibody labeling kit (see the Table of Materials) dropwise to each section. Incubate the slides in a humid chamber at room temperature for 20 min.
Prepare the primary antibody diluted in antibody diluent (rabbit anti-labeled dextran, 1:7,500; see the Table of Materials) and mix by gentle pipetting. Gently tip the slides to drain off the block buffer, dry the area around the sections, and place the slides flat in a humid chamber.
Do not wash before applying the primary antibody solution. Pipette 200 μL of primary antibody solution per section onto the slides and incubate in a humid chamber at 4 °C overnight. For the secondary-only control, pipette 200 μL of antibody diluent onto the appropriate sections.
Gently tip the slides to drain off the primary antibody solution and place in 1x TBSt in an appropriate container, such as a Coplin jar. Perform two 5 min washes of the slides in 1x TBSt. Dry the area around the sections and place flat in a humid chamber.
Apply a ready-to-use background-reducing blocking reagent (see the Table of Materials) dropwise to each section. Incubate the slides in a humid chamber at room temperature for 20 min.
Gently tip the slides to drain off the block buffer, carefully dry the area around the sections, and place the slides flat in a humid chamber. Do not wash before applying the secondary antibody solution.
Apply a ready-to-use secondary antibody solution (anti-rabbit horseradish peroxidase (HRP) polymer secondary antibody; see the Table of Materials) to each section dropwise and incubate in a humid chamber at room temperature for 30 min.
Gently tip the slides to drain off the secondary antibody solution and place in 1x TBSt in an appropriate container, such as a Coplin jar. Perform two 5 min washes of the slides in 1x TBSt.
Prepare the HRP chromogenic substrate immediately before use according to the manufacturer’s protocol (see the Table of Materials).
Dry the area around the sections, place the slides on a flat surface, and apply 200 μL of the HRP chromogenic substrate to each section. Incubate at room temperature for 3 min, starting a timer when the substrate has been applied to the first slide (Figure 4).
Drain off the substrate solution and rinse the slides briefly in a Coplin jar containing 1x PBS. Place the slides in deionized water in an appropriate container, such as a Coplin jar, and perform two 5 min washes in deionized water with gentle agitation.
-
Counterstain the slides by placing them in hematoxylin staining solution for up to 30 s.
NOTE: Hematoxylin is toxic and must be disposed of per institutional regulations. Appropriate personal protective equipment (gloves, lab coat, and safety glasses) should always be used.
Place the slides in deionized water in an appropriate container, such as a Coplin jar, and perform three 5 min washes in deionized water. Transfer the slides to a container containing Scott’s tap water (see the Table of Materials) and incubate for 1 min.
Place the slides in deionized water in an appropriate container, such as a Coplin jar, and perform three 5 min washes in deionized water.
-
Dehydrate the slides through a series of ethanol (EtOH) grades (diluted in deionized water) and xylene in a chemical hood. Perform one 2 min incubation in 70% EtOH, two 1 min incubations in 95% EtOH, two 1 min incubations in 100% EtOH, and three 1 min incubations in xylene. Remove the slides from xylene and place a coverslip while wet with a toluene-based mounting medium in a chemical hood.
NOTE: Xylene and toluene are toxic and flammable and must be properly disposed of per institutional regulations. Work with xylene and toluene should be performed in a chemical hood, and appropriate personal protective equipment (gloves, lab coat, and safety glasses) should always be used.
Following IHC, visualize and image the slides with a compound light microscope and digital camera at 100x magnification (10x ocular magnification and 10x objective magnification). (Figure 5).
Figure 4: Preparation for chromogenic substrate application during IHC.

The slide is placed on a flat surface covered with plastic wrap to allow uniform application of the chromogenic substrate while containing any spillage of hazardous material.
Figure 5: Representative IF and IHC labeling of cryosectioned 48 hpf chimeric zebrafish embryos generated by blastula-to-blastula transplant between AB wild-type zebrafish embryos.
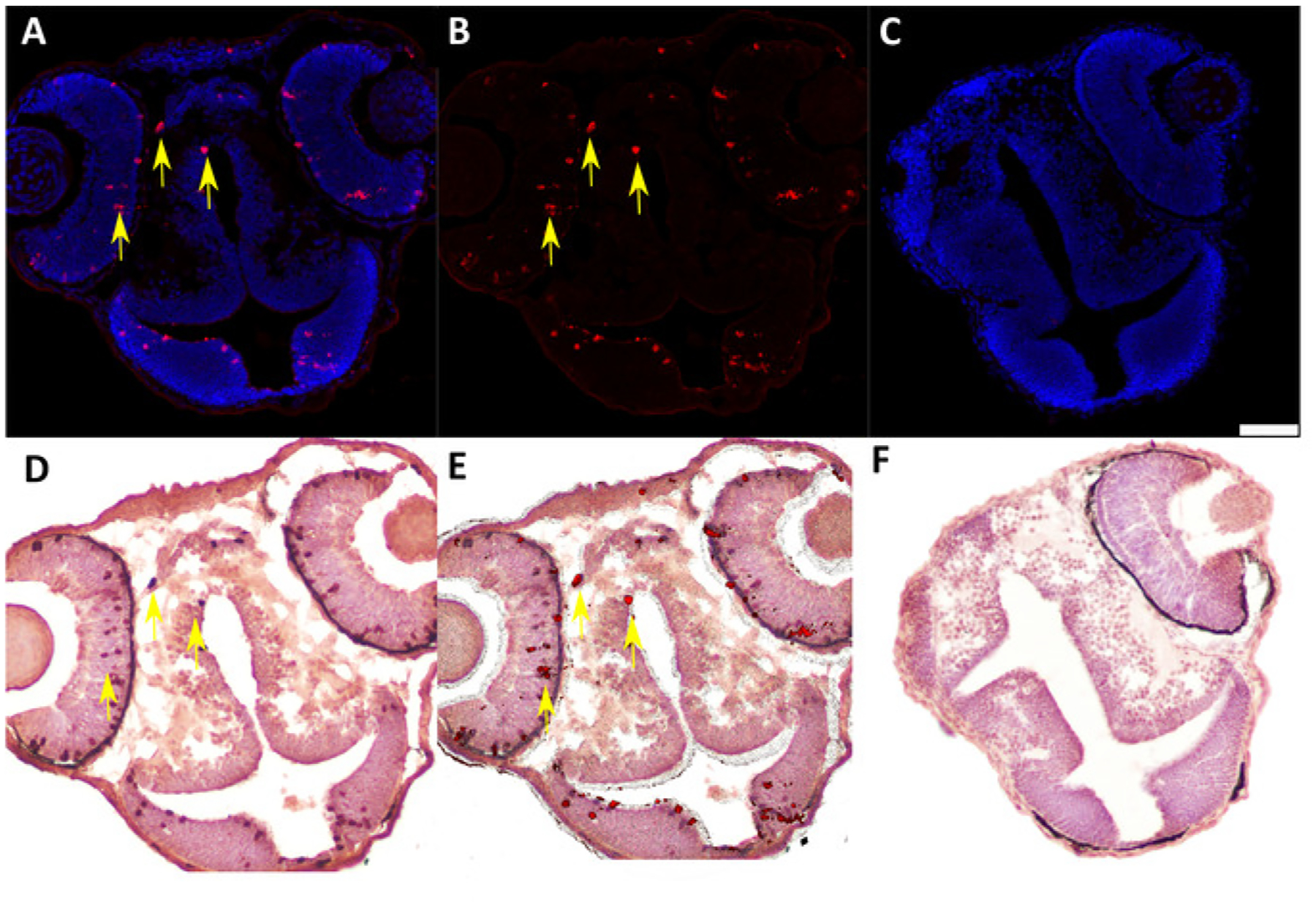
(A) IF to detect Ser 10 phosphorylated Histone H3 (anti-pH3, 1:200) expression in 48 hpf chimeric zebrafish embryo. Red = pH3-positive cells; blue = nuclei. Yellow arrows indicate examples of positive cells. (B) Subtraction of the blue nuclear stain in the digital image (ImageJ software) enhances visualization of pH3-positive cells for quantification and image overlay. (C) Negative control (secondary antibody only) for IF assay in cryosectioned 48 hpf chimeric zebrafish embryo. Scale bar represents 50 μm (applicable to all panels). (D) IHC to detect labeled dextran (anti-labeled dextran, 1:7,500) in chimeric 48 hpf zebrafish embryo generated by blastula-to-blastula transplant between AB wild-type zebrafish embryos. Donor embryos were injected with a fluorescently labeled dextran conjugate at the one-cell stage prior to use for blastula-to-blastula transplant. Red indicates cells labeled with a dextran conjugate; blue indicates nuclei. Yellow arrows indicate examples of positive cells. (E) Overlay of panel B (IF for pH3) and panel C (IHC for labeled dextran) showing colocalization of pH3 expression and dextran labeling in individual cells. Yellow arrows indicate examples of double-positive cells. (F) Negative control (secondary antibody only) for IHC assay in cryosectioned 48 hpf chimeric zebrafish embryo.
Representative Results
We developed this protocol in order to identify and analyze protein expression in individual donor cells in 48 h post-fertilization chimeric zebrafish embryos that were generated by blastula-to-blastula transplant between AB wild-type zebrafish embryos. Successful analysis of protein expression at the one-cell level required preparation of cryosections with appropriately oriented embryos (Figure 1 and Figure 2) and careful, sequential application of IF and IHC techniques to these cryosections (Figure 3 and Figure 4). Use of specific antibodies to simultaneously detect proliferating cells (anti-pH3, Figure 5A,B) and donor cells (anti-labeled dextran, Figure 5D) can be used to identify and quantify donor cells that are actively proliferating (Figure 5E). It is essential to generate high-quality images after both IF (Figure 2A,B) and IHC (Figure 2D) in order to accurately identify individual cells. An image analysis program should be used to perform image overlay (Figure 5E). Depending on the image analysis program used, it may be possible to perform automated counts of labeled cells should quantification be desired.
Discussion
We have presented a novel method for combined immunofluorescence and immunohistochemistry that represents an important step forward in performing colocalization experiments on cryosectioned zebrafish embryos. There is a critical lack of existing colocalization protocols that are optimized for use with small embryos and cryosectioned material17,18, both of which are otherwise commonly used in molecular studies14.
Existing colocalization protocols mainly focus on simultaneous visualization of two fluorophores17,18. While these protocols can work well, their usefulness is limited by the availability of antibodies that (i) can be used in zebrafish and (ii) are compatible with IF.
We feel that use of cryosectioning for zebrafish embryos provides a significant advantage in terms of preserved tissue morphology. As IHC is more commonly performed on paraffin sections than cryosections7,8, further exploration of cryosection-based IHC methods for detection of protein expression is warranted. The use of whole mount IF is a common method for visualizing expression levels in zebrafish embryos, and is more commonly described in the literature than IF using cryosections1,2,3,4,19. However, whole mount protocols have limitations, including the lack of accurate localization of proteins expressed in deep tissues and the potential for surface level expression to obscure protein expression in deeper tissues. We have consistently observed excellent maintenance of cellular morphology following cryopreservation, sectioning, and sequential IF and IHC. For applications that require precise localization of protein expression at the single-cell level, there is a significant advantage to section-based procedures as we have described in this protocol. Resin embedding provides an alternative option for performing IF or IHC assays on sections from early-stage zebrafish embryos20. Resin sections provide superior preservation of tissue morphology compared to cryosections, and relatively thinner tissue sections can be prepared. However, manufacturers generally do not recommend the use of resin sections for immuno-based assays, as some components of the resins cannot be removed from the sections and may mask antibody binding sites21.
While the whole of the protocol is essential for accurate and successful analysis of expression patterns, a few specific steps are critical for experimental success. The first critical step is the handling of embryos during embedding in OCT13,14. It can be challenging to orient each embryo in the same transverse plane so that sections are cut from approximately the same area for all embryos. We have found that using small gauge needles to perform small, deliberate movements through the viscous OCT medium for embryo orientation is superior to using forceps, which are too large and displace too much OCT medium. A second critical step occurs during sectioning of frozen blocks, as it can be difficult to obtain high-quality sections that contain the desired tissue(s) without significant training and experience. We thus recommend use of test samples until the technique is mastered. A third critical step is coverslip removal, which has the potential to disturb or strip off tissue samples. We have found that a combination of gentle agitation to loosen the coverslip and slow removal of the slide from the PBS container is the most effective approach for removing coverslips. A gentle and steady hand is best; researchers may find that some trial and error is necessary to learn how to remove coverslips effectively.
Additional important steps in the protocol include antibody optimization (primary and secondary antibodies for both IF and IHC assays) and exposure of slides to the chromogenic substrate and counterstain. Thorough research regarding primary antibody specificity and target concentration is essential; generally, it is best to collect sufficient non-precious samples for at least two separate experiments for IF and IHC that will be performed to optimize primary antibody concentrations before beginning the IF/IHC combination. Including a known positive and negative control for each primary antibody is essential in each experiment to ensure that both IF and IHC assays are performing appropriately. Adjustments to secondary antibody concentration may also be necessary. Optimization of exposure of tissue sections to the chromogenic substrate and counterstain for the IHC assay is required, since manufacturer guidelines often describe a wide range of possible exposure times. Not all chromogens may be compatible with counterstains and endogenous tissue pigmentation, requiring careful planning with regard to chromogen selection.
There are numerous potential modifications that can be applied to the described protocol, and we have successfully performed this protocol with other combinations of antibodies that could not both be used for IF. As alluded to above, chromogenic substances have distinct compatibility with specific counterstains. We have found that these components are easily modifiable in the protocol. Additionally, the parameters for antibody incubation are quite flexible, and can be increased or decreased depending on desired staining intensity. The embedding process can also be modified. While we have focused on transverse sections, embryos can easily be oriented in any direction to address particular tissues of interest. Finally, there are multiple possible pause points in this protocol. Dehydrated embryos can be stored in 100% MeOH at −20 °C for a few months; frozen blocks can be stored at −80 °C for up to three months; and prepared slides can be stored at −80 °C for up to nine months in a slide box.
Due to the relative complexity of this combined IF/IHC protocol, there are multiple possible sources of variation or error that may require troubleshooting, ranging from tissue quality to researcher experience to sample handling. Most of the critical steps described above are considered critical because they either require optimization at the individual user level or they require particular dexterity, skill, and experience. However, we have found that nonspecific background staining and low signal intensity are the most significant issues requiring optimization. As with any IF or IHC protocol, addressing these issues can be time consuming and labor intensive, and can be compounded with this method since the two techniques are combined. For this reason, we highly recommend optimizing IF and IHC separately before proceeding with the combined protocol.
While we predict that this method will be useful for a broad range of experiments, there are potential limitations. Successful performance of this procedure is dependent on maintaining intact tissue samples through two sequential experiments that include numerous wash steps. For this reason, any problems that arise during sectioning or tissue handling, including use of older slides that may have degraded in quality, will significantly limit researchers’ ability to perform this protocol successfully. Although slides can potentially be stored at −80 °C for up to nine months, tissue adherence to slides declines over time and we had the best success with slides that are used within a month of preparation or ideally, the next day. A second limitation is the small footprint of zebrafish embryos. While we have found this experiment to be useful in visualizing proteins that are relatively abundantly expressed in early-stage embryos, proteins that are expressed inconsistently or at low levels may be very difficult to capture in a section. Finally, since the protocol uses 10–12 μm cryosections, it is best suited for evaluation of protein expression in larger structures, such as nuclei, rather than smaller sub-cellular components.
In summary, our combined IF/IHC protocol will be advantageous for a wide variety of studies in early-stage zebrafish embryos, and represents an important innovation in precise protein expression analysis in such specimens. Our method, shown successfully in Figure 5, will allow researchers to preserve the delicate morphology of early-stage zebrafish embryos in cryosections while working with the somewhat limited range of currently available primary antibodies that are validated for use in IF or IHC in this species. This protocol will likely be useful for studies in other fish and amphibian species (e.g., Medaka and Xenopus spp.), which are hampered by similar limitations to antibody availability, and may be applicable for traditional mammalian models as well.
Acknowledgments
This work was supported by NIH grant 5K01OD021419-02 and NC State University College of Veterinary Medicine.
Footnotes
Video Link
The video component of this article can be found at https://www.jove.com/video/59344/
Disclosures
The authors have nothing to disclose.
References
- 1.Whole-mount IHC protocol for zebrafish embryos [Internet]. Irvine: GeneTex; Available from: http://www.genetex.com/uploaddata/Protocol/Document/IHC-WM.pdf. (2013). [Google Scholar]
- 2.Inoue D, Wittbrodt J One for all--a highly efficient and versatile method for fluorescent immunostaining in fish embryos. PLoS One. 6 (5), (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kogata N, Howard BA A whole-mount immunofluorescence protocol for three-dimensional imaging of the embryonic mammary primordium. Journal of Mammary Gland Biology and Neoplasia. 18 (2), 227–231 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Santos D, Monteiro S, Luzio A General Whole-Mount Immunohistochemistry of Zebrafish (Danio rerio) Embryos and Larvae Protocol. Methods in Molecular Biology. 1797, 365–371 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Shive HR, West RR, Embree LJ, Sexton JM, Hickstein DD Expression of KRASG12V in Zebrafish Gills Induces Hyperplasia and CXCL8-Associated Inflammation. Zebrafish. 12 (3), 221–229 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karlsson J, von Hofsten J, Olsson PE Generating transparent zebrafish: a refined method to improve detection of gene expression during embryonic development. Marine Biotechnology (NY). 3 (6), 522–527 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Macdonald R Zebrafish Immunohistochemistry In: Guille M, editor. Molecular Methods in Developmental Biology. 127th ed., p. 77–88. Humana Press; New York: (1999). [DOI] [PubMed] [Google Scholar]
- 8.Santos C, Pinto ML Immunohistochemical Assessment as a Tool for Investigating Developmental Toxicity in Zebrafish (Danio rerio). Methods in Molecular Biology. 1797, 461–476 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Shive HR, West RR, Embree LJ, Azuma M, Sood R, Liu P, et al. brca2 in zebrafish ovarian development, spermatogenesis, and tumorigenesis. Proceedings of the National Academy of Sciences. 107 (45), 19350–19355 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shive HR, West RR, Embree LJ, Golden CD, Hickstein DD BRCA2 and TP53 collaborate in tumorigenesis in zebrafish. PLoS One. 9 (1), (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Loos CM Multiple Immunoenzyme Staining: Methods and Visualizations for the Observation With Spectral Imaging. Journal of Histochemistry and Cytochemistry. 56 (4), 313–328 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van der Loos CM Immunoenzyme Multiple Staining Methods. Bios Scientific Publishers; New York: (1999). [Google Scholar]
- 13.OCT Embedding For Cryostat Sectioning Of Embryos or Larvae [Internet]. The Zebrafish Book, 5th edition University of Oregon Press; Available from: https://wiki.zfin.org/display/prot/OCT+Embedding+For+Cryostat+Sectioning+Of+Embryos+Or+Larvae. (2007). [Google Scholar]
- 14.Fischer AH, Jacobson KA, Rose J, Zeller R Preparation of Cells and Tissues for Fluorescence Microscopy In: Spector and Goldman, editors. Basic Methods in Microscopy. Cold Spring Harbor Laboratory Press; New York: (2006). [Google Scholar]
- 15.Torres E The Lack of Commercial Antibodies for Model Organisms (and How You Can Deal with It) [Internet]. BenchSci Blog. https://blog.benchsci.com/the-lack-of-commercial-antibodies-for-model-organisms-and-how-you-can-deal-with-it (2018). [Google Scholar]
- 16.Carmany-Rampey A, Moens CB Modern mosaic analysis in the zebrafish. Methods. 39, 228–238 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Da’as SI, Balci TB, Berman JN Mast cell development and function in the zebrafish. Methods in Molecular Biology. 1220, 29–57 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Quan FB et al. Comparative distribution and in vitro activities of the urotensin II-related peptides URP1 and URP2 in zebrafish: evidence for their colocalization in spinal cerebrospinal fluid-contacting neurons. PLoS One. 10 (3), (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westerfield M The zebrafish book A guide for the laboratory use of zebrafish (Danio rerio). 4th ed University of Oregon Press; Eugene: (2000). [Google Scholar]
- 20.Sullivan-Brown J, Bisher ME, Burdine RD Embedding, serial sectioning and staining of zebrafish embryos using JB-4 resin. Nature Protocols. 6 (1), 46–55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.JB-4 Embedding Kit. [Internet]. Polysciences, Inc. http://www.polysciences.com/default/catalog-products/life-sciences/histology-microscopy/plastic-embedding-media-kits/jb-4-sup-r-sup-embedding-kits-reagents/jb-4supsup-embedding-kit/ (2018). [Google Scholar]