

Abstract
BACKGROUND & AIMS:
Intestinal microbes and their metabolites affect the development of colorectal cancer (CRC). Short-chain fatty acids are metabolites generated by intestinal microbes from dietary fiber. We investigated the mechanisms by which free fatty acid receptor 2 (FFAR2), a receptor for short-chain fatty acids that can affect the composition of the intestinal microbiome, contributes to the pathogenesis of CRC.
METHODS:
We performed studies with ApcMin/+ mice, ApcMin/+ Ffar2−/− mice, mice with conditional disruption of Ffar2 in dendritic cells (DCs) (Ffar2fl/flCD11c-Cre mice), ApcMin/+ Ffar2fl/flCD11c-Cre mice, and Ffar2fl/fl mice (controls); some mice were given dextran sodium sulfate to induce colitis, with or without a FFAR2 agonist or an antibody against interleukin 27 (IL27). Colon and tumor tissues were analyzed by histology, quantitative polymerase chain reaction, and 16S ribosomal RNA gene sequencing; lamina propria and mesenteric lymph node tissues were analyzed by RNA sequencing and flow cytometry. Intestinal permeability was measured after gavage with fluorescently labeled dextran. We collected data on colorectal tumors from The Cancer Genome Atlas.
RESULTS:
ApcMin/+ Ffar2−/− mice developed significantly more spontaneous colon tumors than ApcMin/+ mice and had increased gut permeability before tumor development, associated with reduced expression of E-cadherin. Colon tumors from ApcMin/+Ffar2−/− mice had a higher number of bacteria than tumors from ApcMin/+ mice, as well as higher frequencies of CD39+CD8+ T cells and exhausted or dying T cells. DCs from ApcMin/+Ffar2−/− mice had an altered state of activation, increased death, and higher production of IL27. Administration of an antibody against IL27 reduced the numbers of colon tumors in ApcMin/+ mice with colitis. Frequencies of CD39+CD8+ T cells and IL27+ DCs were increased in colon lamina propria from Ffar2fl/flCD11c-Cre mice with colitis compared with control mice or mice without colitis. ApcMin/+Ffar2fl/flCD11c-Cre mice developed even more tumors than ApcMin/+Ffar2fl/fl mice, and their tumors had even higher numbers of IL27+ DCs. ApcMin/+ mice with colitis given the FFAR2 agonist developed fewer colon tumors, with fewer IL27+ DCs, than mice not given the agonist. DCs incubated with the FFAR2 agonist no longer had gene expression patterns associated with activation or IL27 production.
CONCLUSIONS:
Loss of FFAR2 promotes colon tumorigenesis in mice by reducing gut barrier integrity, increasing tumor bacterial load, promoting exhaustion of CD8+ T cells, and overactivating DCs, leading to their death. Antibodies against IL27 and an FFAR2 agonist reduce tumorigenesis in mice and might be developed for the treatment of CRC.
Keywords: SCFA, Microbial Metabolite Receptor, Cytokine: Cytotoxicity
Graphical Abstract
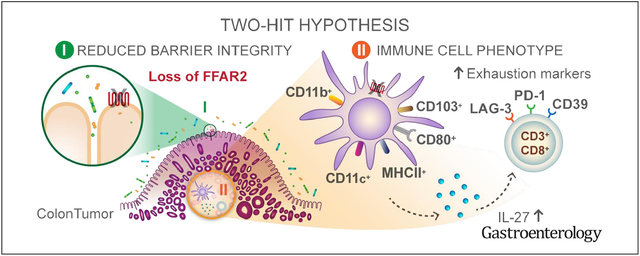
The gut microbiota furnishes an enormity of beneficial immune modulators for the host and holds tremendous opportunities for colorectal cancer (CRC). However, the close proximity of the microbiota to the gut epithelium presents homeostatic challenges that can contribute to a protumorigenic environment.1–3 Gut microbes can breach the epithelial barrier and trigger chronic, smoldering inflammation that contributes to tumorigenesis.4,5 How a breakdown in gut barrier integrity and inflammation affect the mucosal immune system during tumorigenesis is less well understood. In contrast to triggering inflammation and epithelial barrier breach, the gut microbiota can also reinforce barrier function and reduce gut inflammatory tone. Short-chain fatty acids (SCFAs), the most abundant microbial-derived metabolites in the colon, are produced by microbial fermentation of dietary fiber and have epithelial barrier–reinforcing and immune-modulating properties.6 High-fiber diets have been associated with a decreased risk for CRC,7,8 and a reduction in gut SCFA-producing bacteria is associated with CRC.9 SCFA serves as an energy source for colonocytes10 but can also signal through metabolite-sensing G-protein coupled receptors, including free fatty acid receptor 2 (FFAR) 2, FFAR3, GPR109, and Olfr78.11 Previous studies have shown that Ffar2−/− mice have increased tumor burden in mouse models of cancer,12–15 but how FFAR2 signaling contributes to tumorigenesis is not understood.
FFAR2 is highly expressed on colonic epithelial cells16 and is expressed on immune cells, including myeloid cell populations17,18 and regulatory T cells.19 Ffar2−/− mice have impaired barrier integrity and increased microbial translocation to the mesenteric lymph node (MLN) at steady state20 and in a chemically induced colon cancer model.13,15 FFAR2 maintains epithelial barrier integrity in mice, in part, through regulation of antimicrobial peptides.21 Similar to epithelial cells, myeloid cells are important contributors to host defense and have both pro- and antitumorigenic effects. FFAR2 can regulate neutrophil chemotaxis and recruitment17,18,22,23 as well as macrophage cytokine expression.24 Dendritic cells (DCs) from FFAR2-deficient mice are unable to promote production of immunoglobulin A, which contributes to gut homeostasis.25 Although epithelial expression of FFAR2 regulates allergic responses via generation of tolerogenic CD103+ DCs in the gut,20 very little is known about how cell-intrinsic FFAR2 signaling regulates DC phenotype and function in the gut or tumor microenvironment. Additionally, whether there are FFAR2-dependent effects on the adaptive immune response independent of, or in conjunction with, bacterial translocation in CRC is not known.
Materials and Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled Wendy S. Garrett (wgarrett@hsph.harvard.edu), except for the FFAR2 agonist, for which Graeme Fraser (gfraser@epicstx.com) is the appropriate contact.
Experimental Model and Subject Details
Mice.
All mice are on the C57BL/6J background. Wild-type (WT), Ffar2−/−, Ffar2fl/fl, and Ffar2fl/fl CD11c-Cre male and female littermate mice were used between approximately 1.5 and 3 months. ApcMin/+ and ApcMin/+Ffar2−/− male and female mice were acquired from heterozygous breeders. For additional experimental details regarding mouse experiments, please see the Supplementary Materials. All experiments were approved and carried out in accordance with Harvard Medical School’s Standing Committee on Animals and the National Institutes of Health guidelines for animal use and care.
Fluorescein Isothiocyanate Dextran Feeding.
Mice were gavaged with 4 kDa fluorescein isothiocyanate (FITC) dextran (Sigma-Aldrich, St. Louis, MO; 46944–500MG-F) (10 mg/20 g mouse, 10 mg/100 μL in sterile phosphate-buffered saline [PBS] [Dulbecco’s calcium and magnesium free]). For additional details, see the Supplementary Materials.
Method Details
Epithelial Cell Isolation and Quantitative Polymerase Chain Reaction.
The colon was removed, opened longitudinally, and rinsed in PBS. The colon was incubated in PBS with 1 mmol/L dithiothreitol (Sigma-Aldrich, D0632–5G) on ice for 10 minutes. The epithelial layer was isolated using 2 rounds of 5 mmol/L EDTA (10 mL/colon) rotating at 37°C. Dislodged epithelial cell fractions were filtered (70-μm filter) and washed with PBS. Epithelial cells were resuspended in 1 mL Qiazol Lysis reagent (Qiagen, Hilden, Germany; 79306) and stored at −80°C. RNA was extracted following the manufacturer’s protocol. For additional details, please see the Supplementary Materials.
Lamina Propria, Mesenteric Lymph Nodes, and Colon Tumor Single-Cell Preparation.
The colon was removed, opened longitudinally, and rinsed in PBS. The epithelial layer was removed using 2 rounds (15 minutes each) of 3% fetal bovine serum (FBS) 5 mmol/L EDTA (10 mL/colon), rotating at 37°C (round 1 includes 1 mmol/L DTT). The lamina propria (LP) was washed in PBS and chopped into approximately 1-mm pieces. Tissue was digested in RPMI with glutamine (Sigma-Aldrich) with 10% FBS, 1% penicillin/ streptomycin (Corning, Corning, NJ), 0.5 mg/mL dispase (STEMCELL Technologies, Vancouver, British Columbia, Canada), 1 mg/mL collagenase D (Roche, Basel, Switzerland), 50 μg/mL DNAse (Roche) in 2 rounds of 30 minutes, rotating at 37°C. Isolated single cells were filtered (40-μm filter) and resuspended in 3% FBS 5 mmol/L EDTA, followed by washing with fluorescence-activated cell sorter (FACS) buffer (2% FBS 1 mmol/L EDTA). The MLNs were removed and crushed through a 40-μm filter and resuspended in FACS buffer. Tumors were removed and chopped into approximately 1-mm3 pieces in 1.5 mL digestion buffer (RPMI with glutamine, Sigma-Aldrich) with 10% FBS, 1% penicillin/streptomycin (Corning), 0.5 mg/mL dispase (STEMCELL Technologies), 0.5 mg/mL collagenase D (Roche), and 50 μg/mL DNAse (Roche) for 1 hour, rotating at 37°C. Cells were then passed through a 40-μm filter into 5 mL 3% FBS 5 mmol/L EDTA, followed by a wash with FACS buffer. All cells were counted on the hemocytometer followed by flow cytometry analysis.
Flow Cytometry.
Cells (colon LP, MLN, or tumor) were stained with LIVE/DEAD fixable yellow dead cell stain kit (Thermo Fisher Scientific, Waltham, MA; L34959) for 15 minutes at room temperature. Cells were washed with FACS buffer and then stained with FC block (BioLegend, San Diego, CA; 101310) for 10 minutes on ice, followed by mouse antibodies. For additional details, please see the Supplementary Materials.
Isolation of Primary Mouse Dendritic Cells and CD8D T Cells.
DCs were isolated from the MLN, colon LP, or tumors by using a CD11c+ MACS kit (Miltenyi Biotec, 130–108-338). CD11b+CD103+MHCII+CD11c+ DCs were sorted to > 95% purity using a FACSAria Ilu at Dana-Farber Cancer Institute Flow Cytometry Core. CD8+ T cells were isolated following the colon LP single cell isolation by sequential sorting first with the CD8+ MACS kit (Miltenyi Biotec, Bergisch Gladbach, Germany; 130–104-075), followed by the CD3+ MACS kit (Miltenyi Biotec, 130–095-130).
Histology and Colitis Scores.
For histology, standard protocols were followed for embedding and staining. Colons were blindly scored by JNG for monocyte infiltration, hyperplasia, injury, polymorphonuclear cell infiltration, and percent involvement. Cumulative histologic scores were quantified regarding the percent involvement by the disease process (<10%, 10%–25%, 30%–50%, and >50%) and presented as histologic colitis scores as follows: cumulative score percent involvement.
Tumor Quantification.
For ApcMin/+ mice and ApcMin/+ mice given dextran sulfate sodium (DSS), tumor counts were quantified by gross examination of colonic tumors.
16S Sequencing Analysis.
Seven tumor samples from ApcMin/+ mice and 8 tumor samples from ApcMin/+ Ffar2−/− mice were selected for 16S amplicon-based taxonomic profiling. The 16S ribosomal RNA (rRNA) gene sequencing protocol was adapted from the Earth Microbiome Project.26 For additional details, please see the Supplementary Materials.
RNA Sequencing Data Generation and Analysis.
MLNs (2 MLNs pooled/sample, N = 2) were removed from WT mice and were crushed through a 40-μm strainer. MLN DCs were sorted with a CD11c magnetically activated cell sorting (MACS) kit (Miltenyi Biotec, catalog no. 130–108-338). DCs from each sample (approximately 9 × 104 cells) were then split into 2 wells and resuspended in RPMI (Sigma-Aldrich) with 10% FBS and cultured in with either control (H2O) or with an agonist of FFAR2 (10 μmol/L) at 37°C for 6 hours. After incubation, RNA was immediately isolated using an RNeasy Micro kit (Qiagen, catalog number 74004). RNA quality verification, library preparation, and sequencing were performed at the Biopolymers Facility Next-Gen Sequencing Core Facility at Harvard Medical School. For additional details, please see the Supplementary Methods.
Quantification and Statistical Analysis
Statistical analysis was performed with Prism 7.0b (GraghPad Software, San Diego, CA; RRID:SCR_002798). Data are plotted as mean ± standard error of the mean (SEM), and statistical significance was determined by using the Mann–Whitney U test or unpaired t test. P < .05 was considered significant. For 16S rRNA gene amplicon surveys, operational taxonomic unit differential abundance testing between groups was evaluated by Mann–Whitney U tests, and none of these analyses resulted in P values that were statistically significant.
Data and Software Availability
All flow cytometry analysis was conducted using FlowJo 10.4.2 software (FlowJo, Ashland, OR).
All graphs and statistical analysis were generated using Prism 7 software (GraphPad Software, San Diego, CA).
The 16S rRNA gene amplicon survey data and RNA-sequencing data were submitted to the BioProject database, BioProject identification PRJNA550456. Data are available at http://www.ncbi.nlm.nih.gov/bioproject/550456.
Additional Resources
Human colon (COAD) and rectal (READ) adenocarcinoma expression data were obtained from The Cancer Genome Atlas (TCGA).
Results
FFAR2 Deficiency, Tumorigenesis, and Integrity of the Intestinal Barrier
We used ApcMin/+ mice to study FFAR2 signaling, intestinal barrier integrity, and the immune response against colorectal tumors. Although ApcMin/+ mice develop tumors in the small intestine and colon, we focused on colonic tumorigenesis, because human intestinal adenocarcinomas typically develop in the colon.5 As previously described,12,13 we observed that colonic tumor number increased in ApcMin/+Ffar2−/− mice compared with ApcMin/+ mice (Figure 1A). Prior studies have shown increased gut permeability in Ffar2−/− mice20 and colon tumor models.27,28 We verified that ApcMin/+Ffar2−/− mice also had increased gut permeability, using a serum FITC–dextran gut barrier leak assay (Figure 1B); this was previously observed in Ffar2−/− mice with and without colitis,15,20 This permeability defect occurred before the onset of macroscopic tumor development, supporting the idea that barrier compromise precedes tumor onset. To unravel factors contributing to barrier permeability in ApcMin/+Ffar2−/− mice, we measured E-cadherin (Cdh1) expression, which is important for barrier integrity29 and whose expression is increased by FFAR2 signaling.30 Cdh1 expression decreased in colonic epithelial cells from ApcMin/+Ffar2−/− mice (Figure 1C) before macroscopic tumor development and also decreased in colonic epithelial cells from Ffar2−/− mice (Supplementary Figure 1A).
Figure 1.
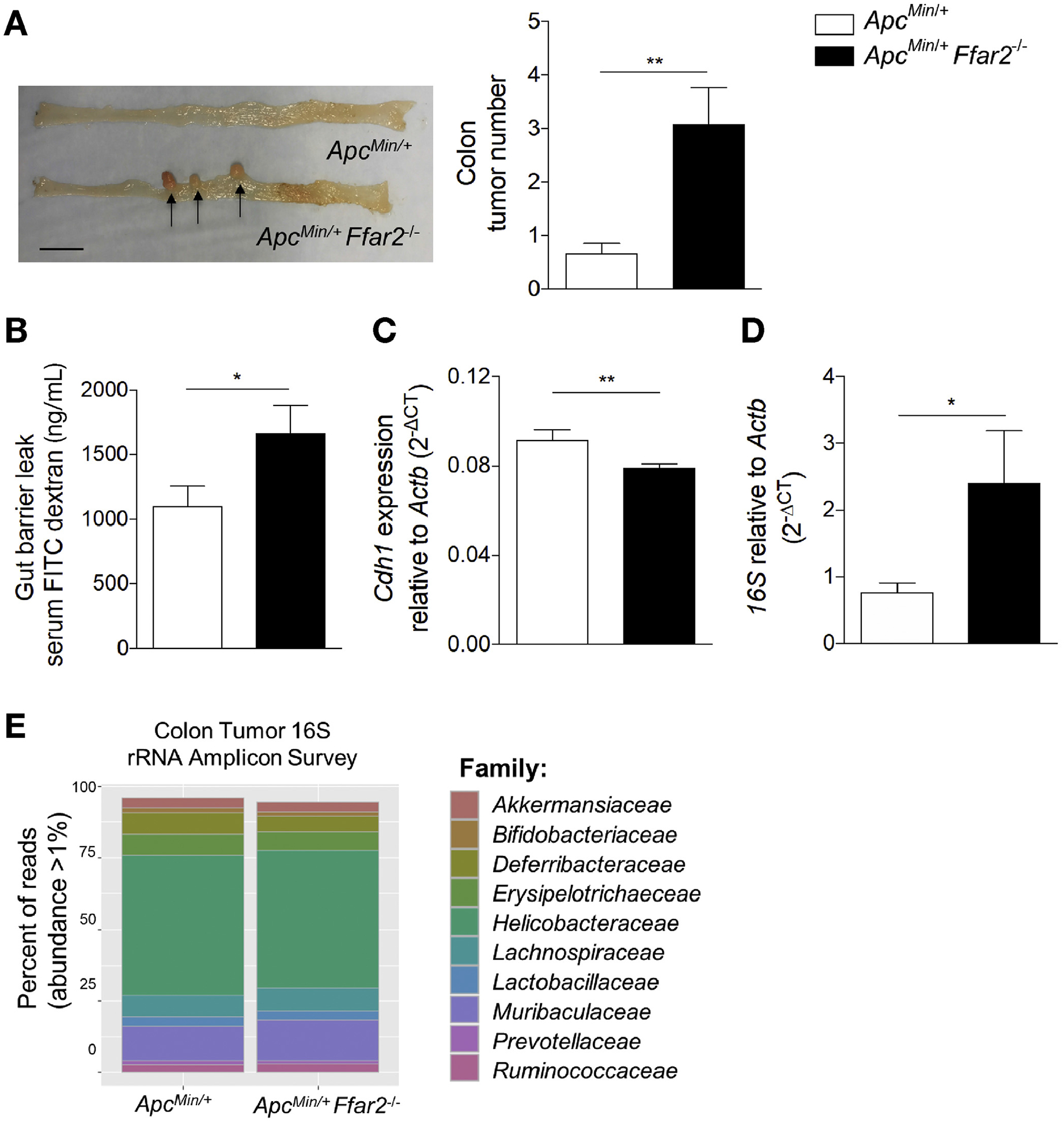
FFAR2 deficiency potentiates tumorigenesis and impairs gut barrier integrity. (A) Representative tumor image (left). Tumors indicated with arrows (scale bar, 1 cm). Colon tumor numbers from ApcMin/+ (N = 12) and ApcMin/+Ffar2−/− (N = 12) mice (right). (B) FITC-dextran serum concentration in ApcMin/+ (N = 8) and ApcMin/+Ffar2–/– (N = 8) mice before macroscopic tumor development. (C) Cdh1 expression in colon epithelial cells from ApcMin/+ (N = 7) and ApcMin/+Ffar2−/− (N = 8) pretumor mice. (D) Colon tumor 16S rRNA DNA levels from ApcMin/+ (N = 9) and ApcMin/+Ffar2−/− (N = 8) mice. (E) Family level analysis of 16S rRNA gene amplicon sequencing from ApcMin/+ (N = 7) and ApcMin/+Ffar2−/− (N = 8) colon tumors. Operational taxonomic unit differential abundance testing between groups was evaluated by Mann–Whitney U tests and were not significantly different. Data from A–D are from 3 independent experiments and are plotted as mean ± SEM. *P < .05, **P < .01, Mann-Whitney U test. CT, cycle threshold.
Altered barrier permeability can facilitate bacteria closely associating with the gut epithelium and infiltrating tumoral tissue.31,32 We confirmed the bacterial dependence of tumor development in ApcMin/+ mice by generating germ-free ApcMin/+ mice in which we observed no colonic tumors at 6 months of age compared with age-matched conventional specific pathogen–free mice (Supplementary Figure 1B), supporting the contribution of the gut microbiota to tumorigenesis. We next examined the bacterial 16S rRNA gene amplicon levels in ApcMin/+Ffar2−/− tumors and observed higher levels by qPCR compared to ApcMin/+ tumors (Figure 1D), suggesting that there is an overall increase in abundance of tumor-associated bacteria. Given that loss of Ffar2 can alter the composition of the fecal microbiota,13,20 we assessed whether taxonomic differences between ApcMin/+ and ApcMin/+Ffar2−/− colon tumors could affect tumorigenesis. We conducted 16S rRNA gene amplicon sequencing analysis on colon tumors from ApcMin/+ and ApcMin/+Ffar2−/− mice. Although we observed a high abundance of the family Helicobacteraceae in both ApcMin/+ and ApcMin/+Ffar2−/− tumors (Figure 1E), taxon relative abundance from the phylum to the genus level was not significantly different between ApcMin/+ and ApcMin/+Ffar2−/− colon tumors (Supplementary Figure 1D and E). Regardless of the absence of host FFAR2, we observed no difference in fatty acid biosynthesis (Supplementary Figure 1F) or fatty acid metabolism (Supplementary Figure 1G) with inferred functional analysis using PICRUSt (https://huttenhower.sph.harvard.edu/galaxy). These data indicate that loss of FFAR2 is associated with gut barrier integrity breakdown, reduced colon epithelial cell expression of Cdh1, and increased abundance of tumor-associated bacteria, all of which could contribute to tumorigenesis.
FFAR2 Deficiency and Exhaustion of CD8+ T Cells in Tumor
Given the findings of altered barrier function and tumoral bacterial load, we proposed that chronic inflammation associated with increased tumoral bacteria may affect CD8+ T-cell phenotype and function in the tumor microenvironment. We focused on CD8+ T cells, because they are cytotoxic lymphocytes essential for generating a productive antitumor immune response.33 Also, increased expression of coinhibitory molecules such as CD39, PD-1, and LAG-3 and, subsequently, decreased CD8+ T-cell function are associated with chronic infection34 and decreased antitumor immune responses.35,36 We evaluated CD8+ T-cell exhaustion status in tumors from ApcMin/+ and ApcMin/+Ffar2−/− mice and found an increase in CD39+ (Figure 2A), PD1+ (Figure 2B), and LAG-3+ (Figure 2C) CD8+ T-cell frequencies in ApcMin/+ Ffar2−/− tumors (gating strategy in Supplementary Figure 2A). Intratumoral CD8+ T-cell frequency decreased (Figure 2D), whereas CD8+ T-cell death increased in ApcMin/+Ffar2−/− tumors (Figure 2E), a phenotype associated with T-cell exhaustion. To confirm CD8+ T-cell functional exhaustion, we examined interferon gamma (IFNG)+ CD39+CD8+ T-cell frequency and observed an approximately 50% decrease in ApcMin/+Ffar2−/− tumors (Figure 2F). To determine if CD8+ T-cell exhaustion in ApcMin/+Ffar2−/− tumors could be attributed to a requirement for Ffar2 expression in CD8+ T cells, we assessed Ffar2 expression in sorted CD3+CD8+ T cells from colon lamina propria of WT mice and found no evidence of expression (Supplementary Figure 2B). These data are consistent with prior findings of no FFAR2 expression in CD8+ T cells.17 Our findings show that CD8+ T-cell exhaustion and increased cell death in Ffar2−/− tumors are independent of CD8+ T-cell Ffar2 expression.
Figure 2.
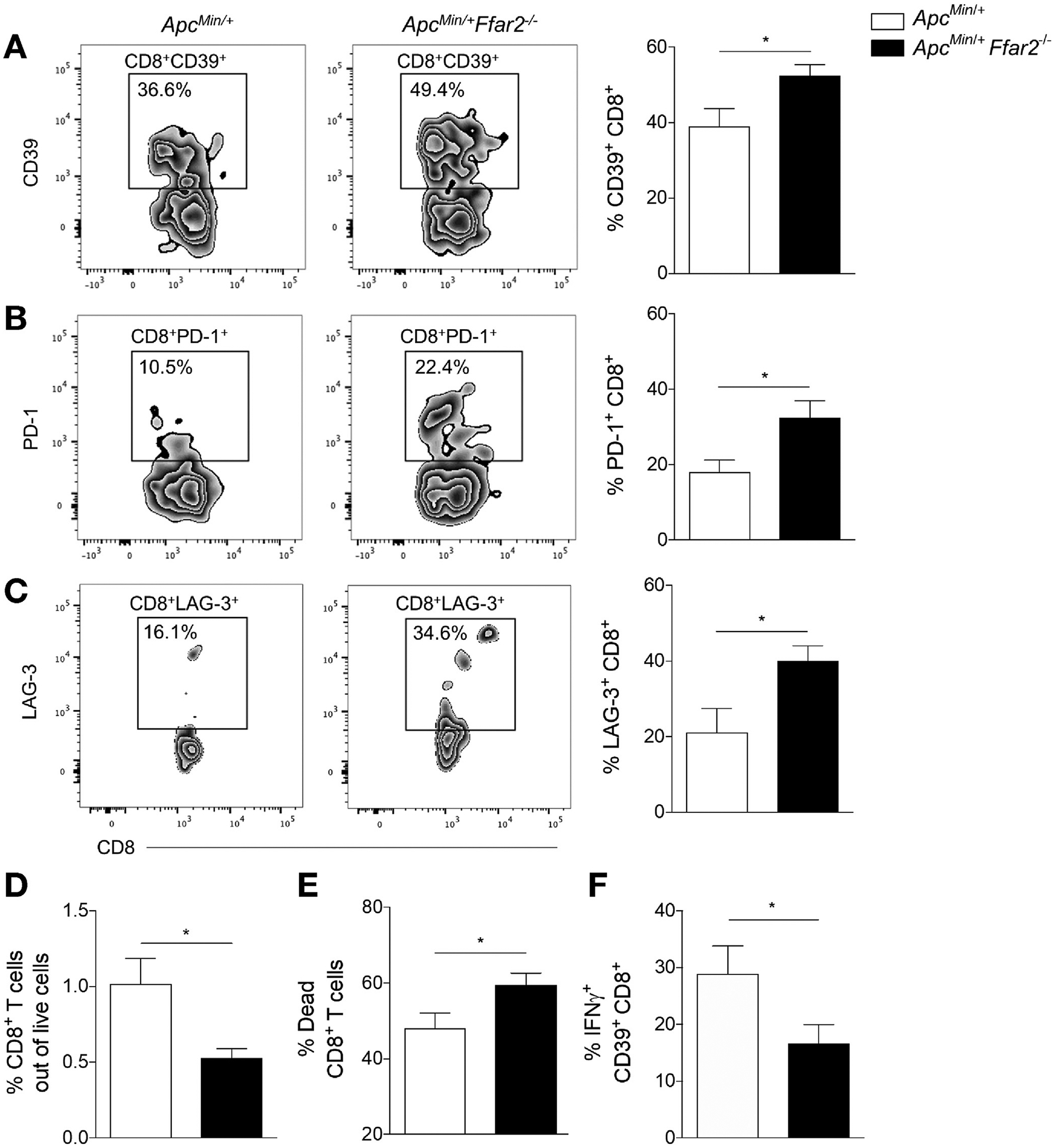
FFAR2 deficiency increases exhausted CD8+ T cells in colon tumors. (A) Flow cytometric analysis of tumor CD39+CD8+ cell frequency in ApcMin/+ (N = 10) and ApcMin/+Ffar2−/− (N = 13) mice. (B) Tumor PD-1+CD8+ T-cell frequency in ApcMin/+ (N = 10) and ApcMin/+Ffar2−/− (N = 13) mice. (C) Tumor LAG-3+CD8+ T-cell frequency in ApcMin/+ (N = 4) and ApcMin/+Ffar2−/− (N = 7) mice. (D) Tumor live CD3+CD8+ T-cell frequency in ApcMin/+ (N = 8) and ApcMin/+Ffar2−/− (N = 8) mice. (E) Frequency of dead CD3+CD8+ T cells in tumors from ApcMin/+ (N = 8) and ApcMin/+ IFNG+ Ffar2−/− (N = 8) mice. (F) Tumor Data from A–F are from Min/+Ffar2−/− (N = 12) mice. cell frequency out of CD39+CD8+ cells in ApcMin/+ (N = 7) and Apc ≥3 independent experiments and are plotted as mean ± SEM. *P < .05, unpaired t test.
Alterations in FFAR2-Deficient Dendritic Cells in Tumors
The lack of FFAR2 expression in colonic CD8+ T cells prompted us to explore additional cell types that may be responsible for altering CD8+ T-cell phenotype and function during tumorigenesis. Previous studies showed that many types of myeloid cells, including neutrophils, eosinophils,17 and DCs,20 can express FFAR2. Because DCs are key components in both antibacterial responses and the acquisition and presentation of tumor antigens to T cells, we focused on the role of FFAR2 in tumoral DCs. We sorted DCs from ApcMin/+ and ApcMin/+Ffar2−/− tumors and found that those from ApcMin/+ tumors expressed detectible levels of FFAR2 (Figure 3A), indicating that FFAR2 deficiency in DCs could potentially alter their function. In human CRC, reduced intratumoral DC frequency is correlated with worse prognosis.37,38 We found that DC frequency was reduced in ApcMin/+Ffar2−/− tumors (Figure 3B and for gating strategy, see Supplementary Figure 3A) compared with ApcMin/+ tumors and that this reduced frequency correlated with increased intratumoral DC death (Figure 3C). Next, we looked at levels of CD80, a costimulatory molecule up-regulated during DC activation, and observed increased CD80hi DC frequency in ApcMin/+Ffar2−/− tumors (Figure 3D). These observations suggest that tumor Ffar2−/− DCs have an altered activation state similar to DCs overactivated with lipopolysaccharide that can exhibit increased cell death.39 Increased DC death was likely due to activation status and not loss of FFAR2, because cell death was not increased in total CD45+ cells from the same ApcMin/+ Ffar2−/− colon tumors (Supplementary Figure 3B). In addition to cell death, it is possible that there are differences in the chemokines responsible for DC recruitment in ApcMin/+ Ffar2−/− tumors. However, expression of chemokines related to DC recruitment was not different between ApcMin/+ and ApcMin/+Ffar2−/− colon tumors (Supplementary Figure 3C). Although data on how the tumor microenvironment regulates tumor-infiltrating DC activation can be conflicting,40 our data (Figures 1 and 2) support the idea that increased levels of tumoral bacteria may contribute to increased activation and cell death in FFAR2-deficient DCs.
Figure 3.
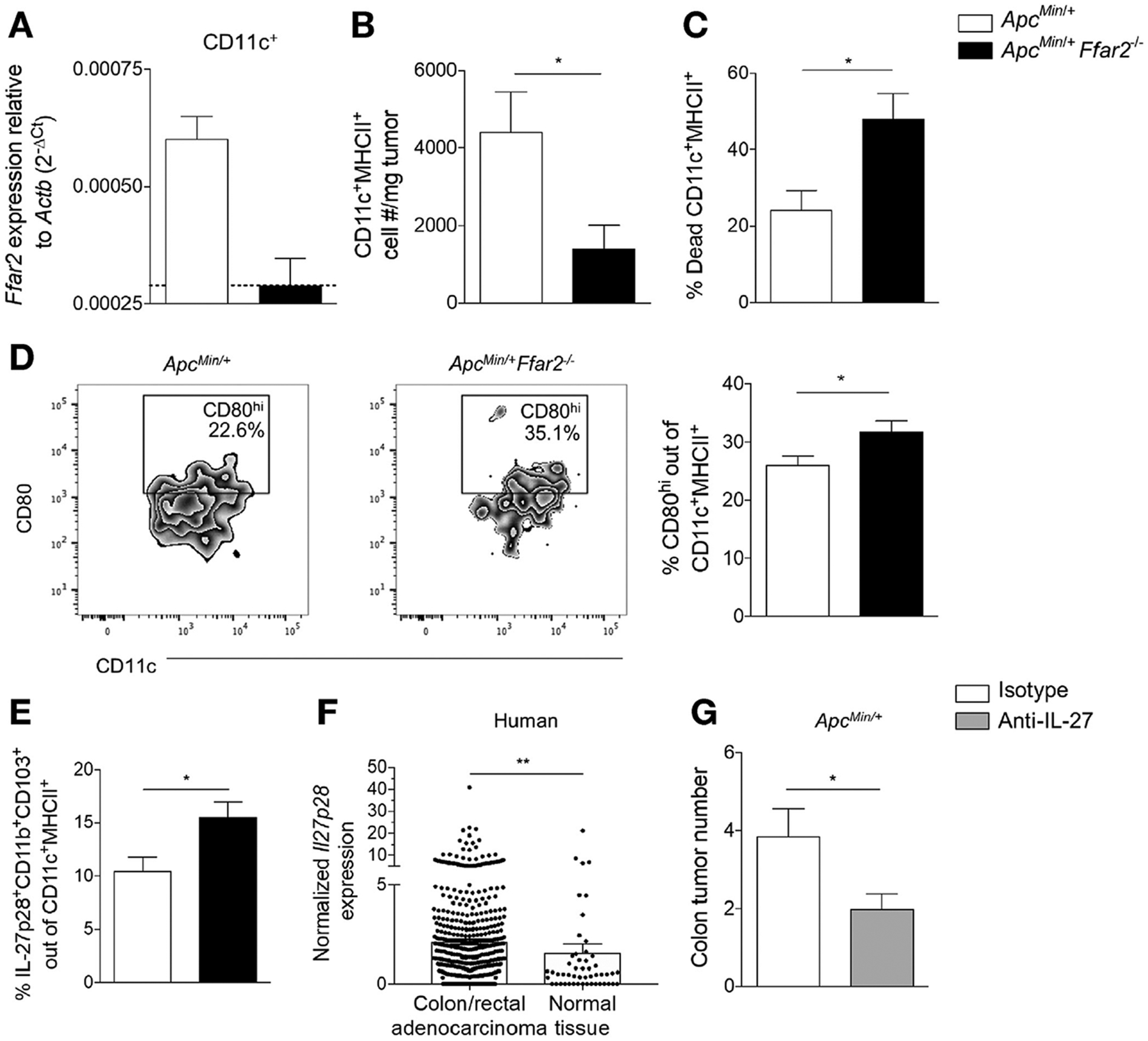
Roles of FFAR2 in DCs and effects of anti-IL27 on colonic tumorigenesis. (A) Expression of FFAR2 in sorted tumor DCs from ApcMin/+ (N = 3) and ApcMin/+Ffar2−/− (N = 3) mice. Dotted line indicates the limit of detection for Ffar2 primers. (B) CD11c+MHCII+ cell number/milligram of tumor from ApcMin/+ (N = 5) vs ApcMin/+Ffar2−/− (N 7) mice. (C) Dead colon tumor CD11c+MHCII+ cell frequency from ApcMin/+ (N = 7) vs ApcMin/+Ffar2−/− (N = 12) mice. (D) Tumor CD80hiCD11c+MHCII+ cell frequency from ApcMin/+ (N = 7) vs ApcMin/+Ffar2−/− (N = 11) mice. (E) Tumor IL27+ DC frequency from ApcMin/+ (N = 6) and ApcMin/+Ffar2−/−(N = 7) mice. (F) Normalized IL27p28 expression in human colon and rectal adenocarcinoma tissue (N = 622) vs normal tissue (N = 51). (G) Tumor numbers from ApcMin/+ mice with DSS-induced colits injected with isotype (N = 8) or anti-IL27 (N = 9) antibody. Data from A are from 2 independent experiments. Data from B–E and G are from ≥3 independent experiments. Data from F are from the TCGA COAD and READ data sets. Data from A–E and G are plotted as mean ± SEM. *P < .05, **P < .01, Mann–Whitney U test.
Given these provocative DC findings, we next determined if there were differences in cytokine production in DCs with vs without FFAR2. Interleukin (IL) 12 family cytokines, including IL12, IL35, IL23, and IL27, mediate antitumor immunity.41 There were no differences in levels of IL12B+ and p70+ cells by flow cytometry in peripheral (Supplementary Figure 4A) or tumor DCs (Supplementary Figure 4C) in the absence of Ffar2 mRNA or Il12b mRNA (based on quantitative reverse-transcription polymerase chain reaction [PCR]), which encodes the receptor subunit shared by IL12 and IL23, in sorted DCs (Supplementary Figure 4B). This prompted us to focus on IL27.
IL27 increases levels of CD39 and PD1 in cultured CD8+ T cells,42 and tumor-infiltrating CD8+ T cells from IL27 receptor-knockout mice have decreased expression of multiple coinhibitory receptors,43 indicating that IL27 signaling in T cells may contribute to their exhaustion. DCs regulate tumor CD8+ T cells within the tumor microenvironment,44 so we proposed that IL27 might be altered in Ffar2−/− DCs in ApcMin/+ colon tumors and contribute to the CD8+ T-cell exhaustion we observed (Figure 2). The population frequency of IL27-producing DCs increased in ApcMin/+Ffar2−/− tumors compared with ApcMin/+ tumors (Figure 3E). We also observed increased IL27 in DCs from the MLNs of Ffar2−/− mice (Supplementary Figure 4D) and increased expression of IL27 in sorted MLN Ffar2−/− DCs (Supplementary Figure 4E); loss of FFAR2 might affect production of IL27 by DCs.
To assess whether our findings hold relevance for patients with CRC, we examined levels of Il27 mRNA in the TCGA colorectal data sets COAD and READ (https://cancergenome.nih.gov/). There was increased expression of IL27 mRNA in CRC tissue compared with normal tissue (Figure 3F). These results indicate that human colorectal tumors overexpress IL27. Because of the increased IL27+ DC frequency we observed in ApcMin/+Ffar2−/− tumors and the increase in IL27 mRNA in human colorectal tumors, we evaluated the effect of IL27 neutralization on tumorigenesis in mice. In ApcMin/+ mice treated with DSS colitis given the antibody against IL27, we found that treatment with an anti-IL27 antibody reduced colon tumor numbers (Figure 3G). These data suggest an important role for IL27+ DCs in modulating the antitumor immune response.
Disruption of FFAR2 in Dendritic Cells of Mice With Intestinal Inflammation
Because we observed alterations in DC activation state and phenotype in ApcMin/+Ffar2−/− mice, we investigated whether the effects of FFAR2 were cell intrinsic to DCs by generating mice bearing a CD11c-mediated conditional deletion of FFAR2. We evaluated Ffar2fl/fl vs Ffar2fl/flCD11c-Cre mice to determine if changes in IL27+ DCs occurred within the colon LP, which could contribute to colon tumorigenesis in ApcMin/+ mice. We verified FFAR2 deficiency in Ffar2fl/flCD11c-Cre mice (Supplementary Figure 5A) and then we gave Ffar2fl/fl and Ffar2fl/flCD11c-Cre mice 1.5% DSS, to induce a mild gut barrier integrity breach. We observed minimal weight loss, as is expected in mice given low-dose DSS (Figure 4A), and no difference in histology-based colitis score (Figure 4B) in Ffar2fl/flCD11c-Cre vs Ffar2fl/fl mice. Gut barrier permeability, as measured by serum FITC–dextran levels, of Ffar2fl/flCD11c-Cre mice was similar to that of control mice and mice given DSS (Figure 4C), indicating that loss of FFAR2 from CD11c+ cells does not alter barrier integrity. CD39+CD8+ T-cell frequency increased in the colon LP of Ffar2fl/flCD11c-Cre mice after administration of DSS but not, as expected, in mice given water alone (Figure 4D). We examined the activation status and found that Ffar2fl/flCD11c-Cre mice had an increased frequency of CD80hi DCs in the colon LP (Supplementary Figure 5B). This was similar to what we observed in colon tumors from ApcMin/+Ffar2−/− (Figure 3D). IL27+ DC frequency in the colon LP increased in Ffar2fl/flCD11c-Cre mice given DSS but was not altered in mice given only water (Figure 4E). Because our results show that CD8+ T cells do not express Ffar2 (Supplementary Figure 2B), we attribute increased CD39+CD8+ T cells to a loss of Ffar2 in DCs. These data show that barrier integrity disruption is required to increase IL27+ DCs and CD39+CD8+ T cells in Ffar2fl/flCD11c-Cre mice and that DC activation is enhanced by barrier integrity disruption.
Figure 4.
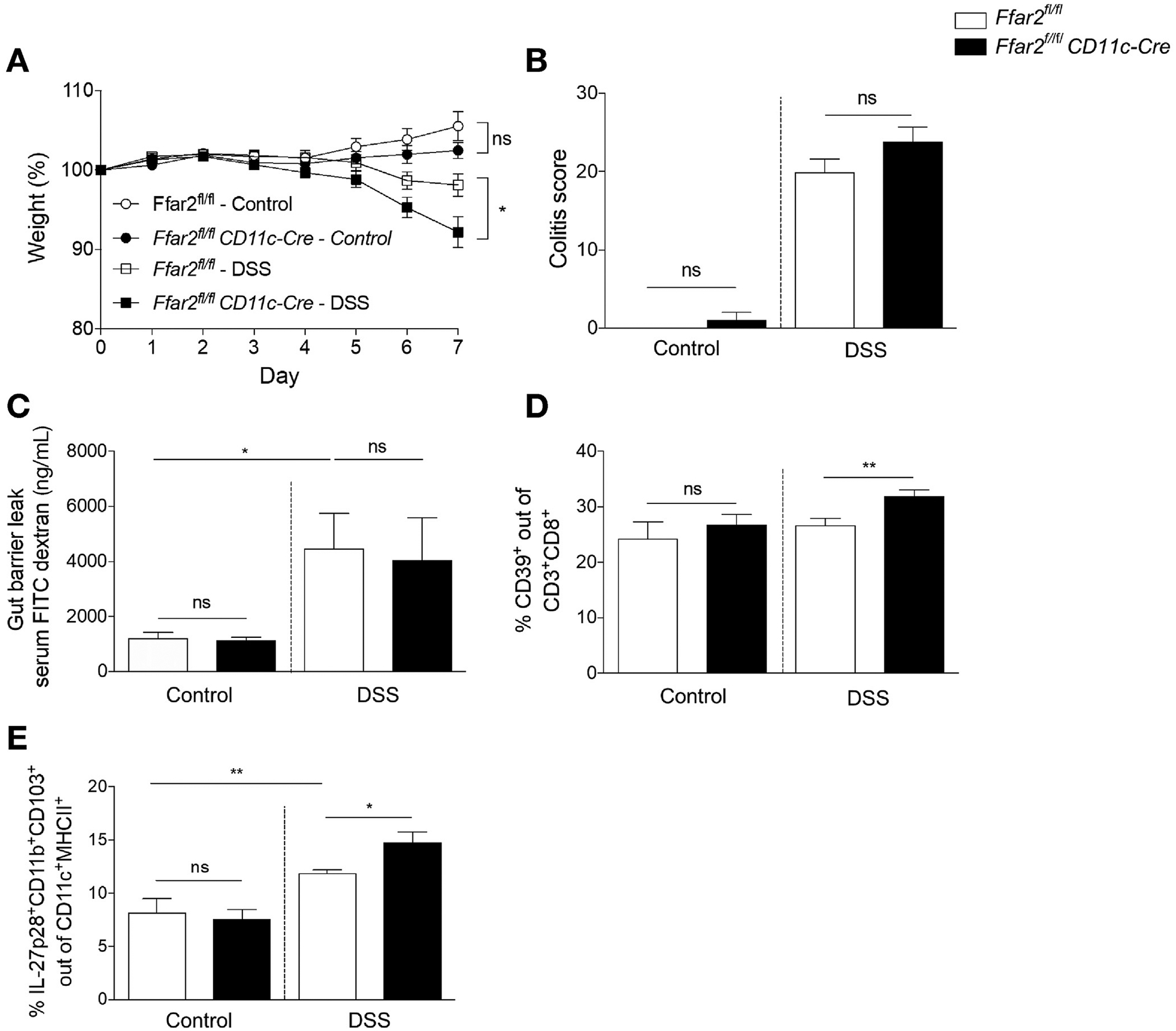
FFAR2 deficiency in DCs and gut barrier disruption increase CD39+CD8+ T cells and IL27+ DCs. (A) Percent weight loss of Ffar2fl/fl (N = 7) vs Ffar2fl/flCD11c-Cre (N = 6) and Ffar2fl/fl (N = 14) given DSS vs Ffar2fl/flCD11c-Cre (N = 14) mice, compared with day 0. (B) Histologic colitis scores for mice in (A). (C) FITC–dextran serum concentration (N = 9 per group). (D) Colon LP CD39+CD8+ cell frequency from control Ffar2fl/fl (N = 7) vs Ffar2fl/flCD11c-Cre (N = 7) and Ffar2fl/fl (N = 10) and Ffar2fl/flCD11c-Cre mice with DSS-induced colitis (N = 9). (E) Colon LP IL27+ DC frequency from Ffar2fl/fl (N = 7) vs Ffar2fl/fl CD11c-Cre (N = 7) and Ffar2fl/fl (N = 10) mice given DSSS and Ffar2fl/flCD11c-Cre mice (N = 9). Data from A–E are from 3 independent experiments and are plotted as mean ± SEM. *P < .05, **P < .01, Mann-Whitney U test. ns, not significant.
Deletion of FFAR2 From Dendritic Cells Promotes Tumorigenesis After Colon Barrier Disruption
Our findings raised the question of whether there was a causal link between FFAR2 deficiency in DCs, increased CD8+ T-cell exhaustion in the colon, and tumorigenesis in conditional knockout (KO) mice. Thus, we bred Ffar2fl/flCD11c-Cre mice to ApcMin/+ mice and showed that DCs (both CD11c+CD11b+-CD103+ and -CD103–) from these conditional KO mice did not express FFAR2. Ffar2 mRNA was not detectable in tumor-associated macrophages and was at limit of detection in CD11b+Ly6C+ monocytes in ApcMin/+Ffar2fl/fl mice tumor-associated macrophages; therefore, monocytes were likely not responsible for any differences in CD8+ T cells observed in conditional KO mice (Supplementary Figure 6A).
We examined the CD8+ T-cell and DC phenotype in the colon LP from 3% DSS-treated ApcMin/+Ffar2fl/fl vs ApcMin/+ Ffar2fl/flCD11c-Cre mice 6 days after DSS to determine if changes in the gut occurred before macroscopic tumor development. We found an increase in the frequency of PD1+CD8+ T cells (Figure 5A) and IL27+ DCs (Figure 5B) in the colon LP of ApcMin/+Ffar2fl/flCD11c-Cre mice. ApcMin/+ Ffar2fl/flCD11c-Cre mice showed higher colon tumor number compared with ApcMin/+Ffar2fl/fl mice 17 days after DSS treatment (Figure 5C) but no difference in tumor burden without DSS treatment (Supplementary Figure 6B). Because colitis can contribute to tumorigenesis and loss of FFAR2 might increase the severity of colitis, it is possible that worse colitis could contribute to tumorigenesis in these mice. However, we observed no difference in histology-based colitis scores from ApcMin/+Ffar2fl/flCD11c-Cre mice given DSS (Supplementary Figure 6C). These results suggest that loss of FFAR2 selectively in DCs affects T-cell exhaustion markers, DC phenotype, and tumor burden only when there is a breach in the gut barrier.
Figure 5.
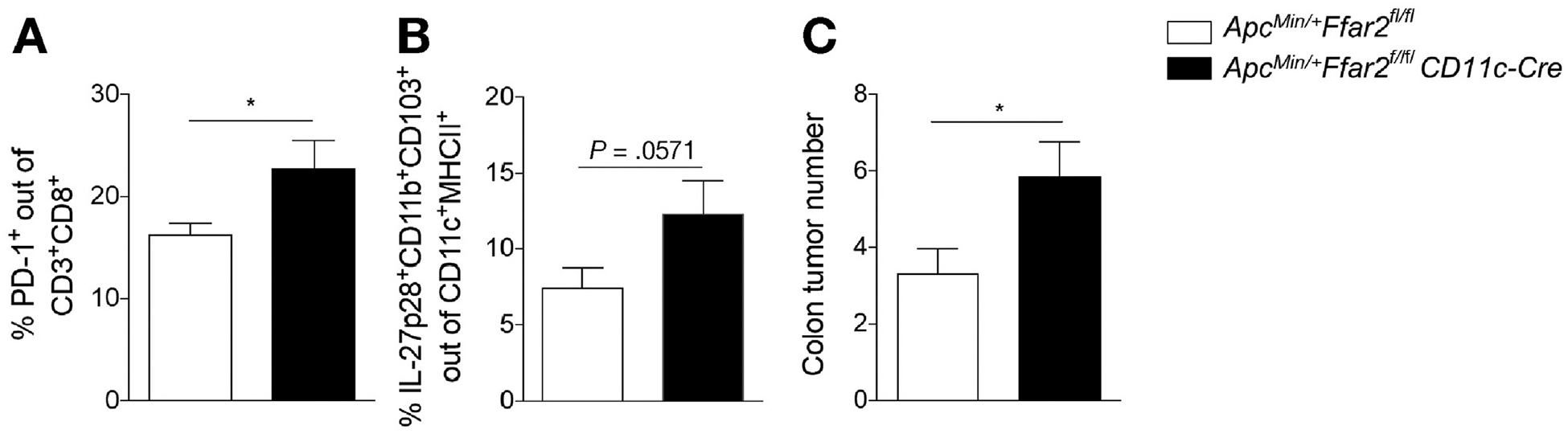
Conditional deletion of FFAR2 from DCs contributes to tumorigenesis after colon barrier disruption. (A) Colon LP PD1+CD8+ T-cell frequency from ApcMin/+Ffar2fl/fl (N = 4) mice given DSS vs ApcMin/+Ffar2fl/flCD11c-Cre (N = 4) mice given DSS and killed on day 10. (B) Colon LP IL27+ DC frequency from ApcMin/+Ffar2fl/fl (N = 4) vs ApcMin/+Ffar2fl/flCD11c-Cre (N = 4) mice given DSS and killed on day 10. (C) Tumor number in ApcMin/+Ffar2fl/fl (N = 9) vs ApcMin/+Ffar2fl/flCD11c-Cre (N = 7) mice given DSS and killed on day 21. Data from A and B are from 2 independent experiments. Data from C are from 3 independent experiments. Data from A–C are plotted as mean ± SEM. *P ≤ .05, Mann–Whitney U test.
FFAR2 Agonist Reduces Colon Tumor Burden and Alters Gene Expression Profile of Dendritic Cells
We verified that FFAR2 is expressed in human colon and rectal adenocarcinoma tissue at a similar level to normal tissue using the TCGA COAD and READ data sets (Figure 6A). Given that SCFAs can act via an FFAR2-independent (HDAC inhibition) mechanism, we studied the effects of an agonist against FFAR2.27,28 Using DSS-ApcMin/+ mice, we tested whether the FFAR2 agonist could reduce colon tumor burden. Administration of the FFAR2 agonist reduced colon tumor number (Figure 6B) and decreased frequency of IL27+ DCs in tumors of ApcMin/+ treated with DSS (Figure 6C). These data show that activation of FFAR2 can independently regulate a population of tumorigenic IL27+ DCs and decrease colonic tumor burden.
Figure 6.
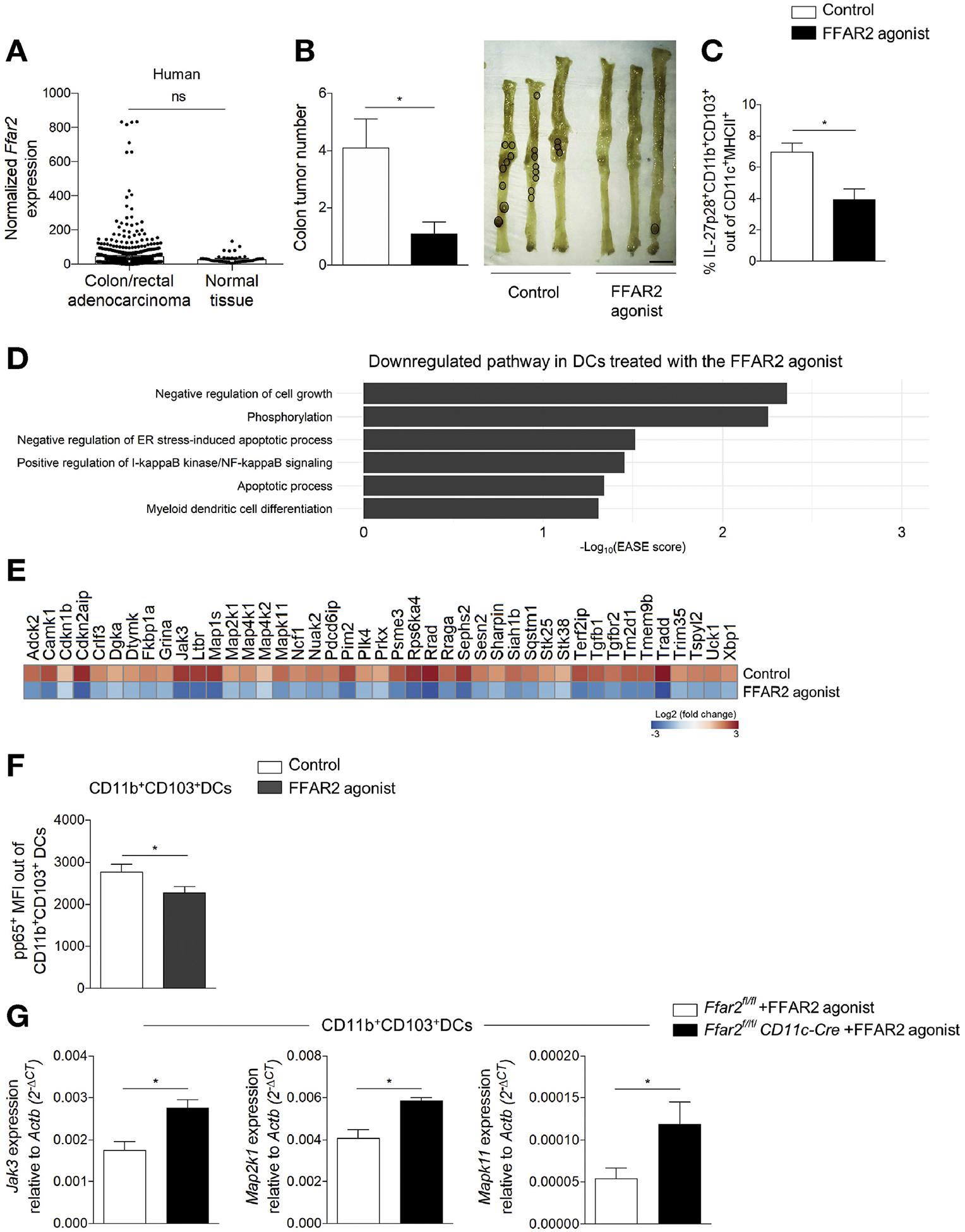
Agonist of FFAR2 reduces colon tumor burden and affects DCs. (A) Normalized FFAR2 expression in human colon and rectal adenocarcinoma tissue (N = 622) vs normal tissue (N = 51). (B) Tumor number (left) from ApcMin/+ mice with DSSinduced colitis fed a control (N = 9) or the FFAR2 agonist (N = 10). Representative tumor images (tumors circled, right). Scale bar, 1 cm. (C) Tumor frequency of IL27+ DCs from ApcMin/+ with DSS-induced colitis given the control (N = 4) or FFAR2 agonist (N = 5). (D) Differential expression analysis showing down-regulated pathways in DCs incubated with the FFAR2 agonist. (E) Heatmap of down-regulated genes from (D). (F) Activation of NF-κB (p65 subunit) in sorted MLN CD11b+CD103+ DCs (WT mice, N = 16) cultured with the FFAR2 agonist (10 μmol/L) for 30 minutes. (G) JAk3, Map2k1, and Mapk11 expression in sorted MLN CD11b+CD103+ DCs from Ffar2fl/fl (N = 12) vs Ffar2fl/flCD11c-Cre (N = 9) mice fed the FFAR2 agonist. Data in A are from the TCGA COAD and READ data set. Data from B–G are from ≥3 independent experiments and are plotted as mean ± SEM. *P < .05, Mann–Whitney U test (A–C), unpaired t test (F and G).
We conducted unbiased transcriptional analyses of DCs exposed to the FFAR2 agonist using RNA sequencing. DCs incubated with the FFAR2 agonist reduced the expression of genes in pathways that control cell proliferation, phosphorylation, activation of nuclear factor κB (NF-κB), apoptosis, and differentiation compared with DCs not exposed to the agonist (Figure 6D). We identified specific down-regulated genes in DCs incubated with the FFAR2 agonist that are relevant to the pathway analysis (Figure 6E). Toll-like receptor 4 and/or IFNG signaling induce expression of IL27 in myeloid cells.45,46 Therefore, down-regulation of proteins involved in toll-like receptor 4 and IFNG signaling in cells exposed to the FFAR2 agonist, such as mitogen-activated protein kinase (MAPK11) and mitogen-activated protein kinase kinases (MAP2K1) (Figure 6E), is consistent with our observation that an agonist of FFAR2 reduces expression of IL27 in DCs. The observed increased in DC death in ApcMin/+Ffar2−/− tumors (Figure 3C) is consistent with the observed down-regulation of apoptotic pathways in cells incubated with the FFAR2 agonist (Figure 6D). Given that NF-κB signaling can up-regulate IL2747 and that the FFAR2 agonist reduces the gene expression associated with NF-κB activation in DCs (Figure 6D), we tested if the FFAR2 agonist regulates NF-κB phosphorylation in CD11b+CD103+ DCs, which are the major DC population producing IL27. Using a phospho–NF-κB p65 antibody, we found that the FFAR2 agonist decreased phosphorylated p65 mean fluorescence intensity level in sorted MLN CD11b+CD103+ DCs (Figure 6F). Additionally, for 3 genes that encode kinases implicated in IL27 regulation and that were down-regulated with the FFAR2 agonist in the RNA-sequencing studies, we further validated the effects of the FFAR2 agonist and confirmed their dependence on FFAR2 expression in CD11b+CD103+ DCs in vivo. Using Ffar2fl/flCD11c-Cre vs Ffar2fl/fl mice, we found that administration of the FFAR2 agonist increased Janus kinase 3 (JAK3), MAPK11, and MAP2K1 expression in sorted MLN CD11b+CD103+ DCs from Ffar2fl/flCD11c-Cre mice (Figure 6G). These data support the idea that compromised FFAR2 function in DCs alters signaling and inflammatory pathways and prevents apoptosis, resulting in a protumorigenic tissue microenvironment.
Discussion
The microbial metabolite-sensing receptor FFAR2 is critical for the regulation of immune cell function and the maintenance of gut homeostasis, and the loss of FFAR2 increases tumor burden in multiple models of tumorigenesis.12,13,15 Smoldering inflammation can potentiate tumorigenesis in the gut, and FFAR2 regulates intestinal inflammation. For example, FFAR2 deficiency increases mononuclear cell inflammatory cytokine levels and exacerbates colitis.48 Additionally, FFAR2 regulates antimicrobial peptide production from gut epithelial cells,21 enforcing gut barrier integrity. Activation of FFAR2, therefore, affects many epithelial and immune cells and prevents inflammation. FFAR2 agonists might therefore be developed for treatment of CRC.
We have identified a mechanism by which FFAR2 deficiency promotes tumorigenesis, by reducing gut barrier integrity, which increases tumor bacterial load, and altering DC expression of IL27 and phenotypes and functions of CD8+ T cells in tumors. Tumors then contain dying and overactivated DCs and exhausted CD8+ T cells, allowing the tumor to grow and spread. Although studies have shown an epithelial-dependent effect of FFAR2 signaling on tolerogenic functions of DCs,20 little is known about the effects of FFAR2 in colon or tumor DCs.
Phenotypes of DCs in tumors have been difficult to study.40 Although DCs are important in acquiring tumor antigens and inducing an antitumor response of T cells, our findings indicate that bacteria can activate DC subsets in the tumor microenvironment. We provide evidence that DCs that express FFAR2 prevent colon tumorigenesis, although other cells that express CD11c+ and FFAR2 might also regulate tumorigenesis in the colon. Studies are needed to determine whether FFAR2-deficient CD11b+CD103+ DCs acquire bacterial antigens and whether FFAR2 affects their acquisition, processing, and presentation of tumor antigen to elicit a productive antitumor adaptive immune response.
IL27 has changing and contradictory effects on tumorigenesis. IL27 can increase the CD8+ T-cell–mediated antitumor response and act directly on tumor cells to inhibit their proliferation.49 However, IL27 also regulates CD39 on DCs50 and T-regulatory cells,51 and induces IL10-producing type 1 regulatory T cells,52 to promote tumorigenesis. We show that production of IL27 by CD11b+CD103+ DCs prevents the antitumor response in the colon and correlates with markers of CD8+ T-cell exhaustion, including CD39.34,42 In ApcMin/+Ffar2−/− tumors, we observed not only increased CD39 but also other markers of exhausted CD8+ T cells, specifically, increased PD1 and LAG-3 and reduced levels of IFNG. We show that colon DCs, but not colon CD8+ T cells, express FFAR2; this information might be used to reduce T-cell exhaustion in colorectal tumors.
Expression of Il27 mRNA was increased in human colon and rectal adenocarcinomas compared with nontumor tissues, and neutralization of IL27 reduced colon tumor burden in mice, so IL27 appears to support colorectal tumorigenesis. Further studies are needed to determine the most effective manner in which to target IL27 for the treatment or prevention of CRC. The efficacy of immunotherapy in patients with CRC has been limited compared to that with other tumors.53
An agonist of FFAR2 reduced the frequency of IL27+ DCs in colon tumors and the tumor burden in mice. Using RNA-sequencing analysis, we found that activation of FFAR2 down-regulates pathways that induce expression of IL27 in DCs, so FFAR2 might regulate expression of cytokines that control T-cell exhaustion. The potential beneficial effects of SCFA in preventing CRC, in the absence of mismatch repair defects, are far reaching and have been described previously.7–9 The mechanisms described range from FFAR2-dependent apoptosis of tumor cells54 to gut barrier integrity reinforcement.15 However, we provide evidence that FFAR2 signaling regulates DC control cytotoxic T-cell exhaustion. We found that FFAR2 affects mucosal barrier integrity, DCs, and CD8+ T cells to prevent colorectal carcinogenesis. FFAR2 agonists might be used, in tandem with nontraditional DC-targeted immune checkpoint inhibitors or neutralization of IL27, to treat or prevent CRC.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
Colorectal cancer (CRC) has been associated with alterations in intestinal microbes and their metabolites. Short-chain fatty acids are metabolites generated by intestinal microbes from dietary fiber. The free fatty acid receptor 2 (FFAR2), a receptor for short-chain fatty acids, affects the composition of the intestinal microbiome and might control development of CRC.
NEW FINDINGS
Loss of FFAR2 promoted colon tumorigenesis in susceptible mice by reducing gut barrier integrity, increasing bacteria in tumors, promoting exhaustion of CD8+ T cells, and over-activating dendritic cells, leading to their death. Antibodies against IL27 and agonists of FFAR2 reduced tumorigenesis in these mice.
LIMITATIONS
These studies were performed in mice.
IMPACT
Agonists of FFAR2 or inhibitors of IL27 might be developed for treatment of CRC.
Acknowledgments
The authors thank the members of the Garrett lab for their thoughtful discussion. We would also like to thank the Harvard T H Chan Gnotobiotic Center for Mechanistic Microbiome Studies. These studies were supported by RO1 CA154426 and Cancer Research UK’s Grand Challenge Initiative, C10674/A27140 (WSG), DK105653 (SL), National Institutes of Health under award number, R01DK104927-01A1 (BTL), The University of Chicago DR&TC P30DK020595 (BTL) and Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, VA merit 5I01BX003382-02 (BTL). We thank the BPF Next-Gen Sequencing Core Facility at Harvard Medical School for their instrumentation and expertise and L. Ricci for preparing the graphical abstract.
Abbreviations used in this paper:
- COAD
human colon adenocarcinoma data set
- CRC
colorectal cancer
- DC
dendritic cell
- DSS
dextran sulfate sodium
- FACS
fluorescence-activated cell sorter
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- FFAR
free fatty acid receptor
- IFNG
interferon gamma
- IL
interleukin
- KO
knockout
- LP
lamina propria
- MACS
magnetically activated cell sorter
- MLN
mesenteric lymph node
- NF-κB
nuclear factor κB
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- READ
human rectal adenocarcinoma data set
- rRNA
ribosomal RNA
- SCFA
short-chain fatty acid
- SEM
standard error of the mean
- TCGA
The Cancer Genome Atlas
- WT
wild type
Footnotes
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2019.12.027.
References
- 1.Zhang K, Hornef MW, Dupont A. The intestinal epithelium as guardian of gut barrier integrity. Cell Microbiol 2015; 17:1561–1569. [DOI] [PubMed] [Google Scholar]
- 2.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140:883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Martel C, Franceschi S. Infections and cancer: established associations and new hypotheses. Crit Rev Oncol Hematol 2009;70:183–194. [DOI] [PubMed] [Google Scholar]
- 4.Kang M, Martin A. Microbiome and colorectal cancer: unraveling host-microbiota interactions in colitis-associated colorectal cancer development. Semin Immunol 2017;32:3–13. [DOI] [PubMed] [Google Scholar]
- 5.Kostic AD, Chun E, Meyerson M, et al. Microbes and inflammation in colorectal cancer. Cancer Immunol Res 2013;1:150–157. [DOI] [PubMed] [Google Scholar]
- 6.Tan J, McKenzie C, Potamitis M, et al. The role of short-chain fatty acids in health and disease. Adv Immunol 2014;121:91–119. [DOI] [PubMed] [Google Scholar]
- 7.Hansen L, Skeie G, Landberg R, et al. Intake of dietary fiber, especially from cereal foods, is associated with lower incidence of colon cancer in the HELGA cohort. Int J Cancer 2012;131:469–478. [DOI] [PubMed] [Google Scholar]
- 8.Schatzkin A, Mouw T, Park Y, et al. Dietary fiber and whole-grain consumption in relation to colorectal cancer in the NIH–AARP Diet and Health Study. Am J Clin Nutr 2007;85:1353–1360. [DOI] [PubMed] [Google Scholar]
- 9.Wang T, Cai G, Qiu Y, et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J 2012;6:320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roediger WE. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 1980;21:793– 798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corrêa-Oliveira R, Fachi JL, Vieira A, et al. Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunol 2016;5(4):e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan P, Skaer CW, Wang H-T, et al. Loss of free fatty acid receptor 2 enhances colonic adenoma development and reduces the chemopreventive effects of black raspberries in ApcMin/+ mice. Carcinogenesis 2017;38:86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sivaprakasam S, Gurav A, Paschall AV, et al. An essential role of Ffar2 (Gpr43) in dietary fibre-mediated promotion of healthy composition of gut microbiota and suppression of intestinal carcinogenesis. Oncogenesis 2016;5(6):e238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DonohoeDR HolleyD, Collins LB, et al. Agnotobioticmouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov 2014;4:1387–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim M, Friesen L, Park J, et al. Microbial metabolites, short-chain fatty acids, restrain tissue bacterial load, chronic inflammation, and associated cancer in the colon of mice. Eur J Immunol 2018;48:1235–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tazoe H, Otomo Y, Kaji I, et al. Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J Physiol Pharmacol 2008;59(Suppl 2):251–262. [PubMed] [Google Scholar]
- 17.Maslowski KM, Vieira AT, Ng A, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009; 461(7268):1282–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sina C, Gavrilova O, Förster M, et al. G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J Immunol 2009; 183:7514–7522. [DOI] [PubMed] [Google Scholar]
- 19.Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013;341(6145):569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan J, McKenzie C, Vuillermin PJ, et al. Dietary fiber and bacterial SCFA enhance oral tolerance and protect against food allergy through diverse cellular pathways. Cell Rep 2016;15:2809–2824. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y, Chen F, Wu W, et al. GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal Immunol 2018;11:752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vinolo MAR, Hatanaka E, Lambertucci RH, et al. Effects of short chain fatty acids on effector mechanisms of neutrophils. Cell Biochem Funct 2009;27:48–55. [DOI] [PubMed] [Google Scholar]
- 23.Kamp ME, Shim R, Nicholls AJ, et al. G protein-coupled receptor 43 modulates neutrophil recruitment during acute inflammation. PLoS One 2016;11(9):e0163750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakajima A, Nakatani A, Hasegawa S, et al. The short chain fatty acid receptor GPR43 regulates inflammatory signals in adipose tissue M2-type macrophages. PLoS One 2017;12(7):e0179696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu W, Sun M, Chen F, et al. Microbiota metabolite short-chain fatty acid acetate promotes intestinal IgA response to microbiota which is mediated by GPR43. Mucosal Immunol 2017;10:946–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson LR, Sanders JG, McDonald D, et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017;551(7681):457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puppa MJ, White JP, Sato S, et al. Gut barrier dysfunction in the ApcMin/+ mouse model of colon cancer cachexia. Biochim Biophys Acta 2011;1812:1601–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grivennikov SI, Wang K, Mucida D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/ IL-17-mediated tumour growth. Nature 2012; 491(7423):254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider MR, Dahlhoff M, Horst D, et al. A key role for E-cadherin in intestinal homeostasis and Paneth cell maturation. PLoS One 2010;5(12):e14325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thirunavukkarasan M, Wang C, Rao A, et al. Short-chain fatty acid receptors inhibit invasive phenotypes in breast cancer cells. PLoS One 2017;12(10):e0186334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014;15:317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brennan CA, Garrett WS. Gut microbiota, inflammation, and colorectal cancer. Annu Rev Microbiol 2016;70: 395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol 2019;234:8509–8521. [DOI] [PubMed] [Google Scholar]
- 34.Gupta PK, Godec J, Wolski D, et al. CD39 expression identifies terminally exhausted CD8+ T cells. PLoS Pathog 2015;11(10):e1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis 2015;6:e1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol 2019;37:457–495. [DOI] [PubMed] [Google Scholar]
- 37.Gulubova MV, Ananiev JR, Vlaykova TI, et al. Role of dendritic cells in progression and clinical outcome of colon cancer. Int J Colorectal Dis 2012;27:159–169. [DOI] [PubMed] [Google Scholar]
- 38.Legitimo A, Consolini R, Failli A, et al. Dendritic cell defects in the colorectal cancer. Hum Vaccin Immunother 2014;10:3224–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwiebs A, Friesen O, Katzy E, et al. Activation-induced cell death of dendritic cells is dependent on sphingosine kinase 1. Front Pharmacol 2016;7:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tran Janco JM, Lamichhane P, Karyampudi L, et al. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol 2015;194:2985–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu M, Mizoguchi I, Morishima N, et al. Regulation of antitumor immune responses by the IL-12 family cytokines, IL-12, IL-23, and IL-27. Clin Dev Immunol 2010; 2010:832454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Canale FP, Ramello MC, Núñez N, et al. CD39 expression defines cell exhaustion in tumor-infiltrating CD8+ T cells. Cancer Res 2017;78:115–128. [DOI] [PubMed] [Google Scholar]
- 43.Chihara N, Madi A, Kondo T, et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 2018;558(7710):454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fu C, Jiang A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Front Immunol 2018;9:3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu J, Guan X, Ma X. Regulation of IL-27 p28 gene expression in macrophages through MyD88- and interferon-γ-mediated pathways. J Exp Med 2007;204:141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdalla AE, Li Q, Xie L, et al. Biology of IL-27 and its role in the host immunity against Mycobacterium tuberculosis. Int J Biol Sci 2015;11:168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parada Venegas D, De la Fuente MK, Landskron G, et al. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel disease. Front Immunol 2019;10:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masui R, Sasaki M, Funaki Y, et al. G protein-coupled receptor 43 moderates gut inflammation through cytokine regulation from mononuclear cells. Inflamm Bowel Dis 2013;19:2848–2856. [DOI] [PubMed] [Google Scholar]
- 49.Li M-S, Liu Z, Liu J-Q, et al. The yin and yang aspects of IL-27 in induction of cancer-specific T-cell responses and immunotherapy. Immunotherapy 2015; 7:191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mascanfroni ID, Yeste A, Vieira SM, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol 2013;14:1054–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sekar D, Hahn C, Brüne B, et al. Apoptotic tumor cells induce IL-27 release from human DCs to activate Treg cells that express CD69 and attenuate cytotoxicity. Eur J Immunol 2012;42:1585–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Apetoh L, Quintana FJ, Pot C, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol 2010;11:854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dawood S The evolving role of immune oncology in colorectal cancer. Chin Clin Oncol 2018;7(2):17. [DOI] [PubMed] [Google Scholar]
- 54.Tang Y, Chen Y, Jiang H, et al. G-protein-coupled receptor for short-chain fatty acids suppresses colon cancer. Int J Cancer 2011;128:847–856. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All flow cytometry analysis was conducted using FlowJo 10.4.2 software (FlowJo, Ashland, OR).
All graphs and statistical analysis were generated using Prism 7 software (GraphPad Software, San Diego, CA).
The 16S rRNA gene amplicon survey data and RNA-sequencing data were submitted to the BioProject database, BioProject identification PRJNA550456. Data are available at http://www.ncbi.nlm.nih.gov/bioproject/550456.