

Abstract
Inflammasomes are intracellular signaling complexes that are assembled in response to a variety of pathogenic or physiologic stimuli to initiate inflammatory responses. Ubiquitously present lipopolysaccharides (LPS) in Gram-negative bacteria induce NLRP3 inflammasome activation that requires caspase-11. We have recently demonstrated that interferon regulatory factor 8 (IRF8) was dispensable for caspase-11–mediated NLRP3 inflammasome activation during LPS transfection; however, its role in Gram-negative bacteria–mediated NLRP3 inflammasome activation remains unknown. Herein, we found that IRF8 promotes NLRP3 inflammasome activation in murine bone marrow-derived macrophages (BMDMs) infected with Gram-negative bacteria such as C. rodentium, E. coli, or P. aeruginosa mutant strain ΔpopB. Moreover, BMDMs deficient in IRF8 showed substantially reduced caspase-11 activation and gasdermin-D cleavage, which are required for NLRP3 inflammasome activation. Mechanistically, IRF8-mediated phosphorylation of IRF3 was required for Ifnb transcription, which in turn triggered the caspase-11–dependent NLRP3 inflammasome activation in the infected BMDMs. Overall, our findings suggest that IRF8 promotes NLRP3 inflammasome activation during infection with Gram-negative bacteria.
Keywords: IRF8, Interferon consensus sequence-binding protein (ICSBP), interferon regulatory factor (IRF), NLR, caspase-1, caspase-11, gasdermin D, NLRP3, non-canonical inflammasome, cell death, Gram-negative bacteria, type I interferon, infection, immunity, infectious diseases, IRF3, ISG, GBPs
INTRODUCTION
Inflammasomes are molecular platforms that are assembled in response to a variety of pathogenic or physiologic stimuli to initiate activation of inflammatory caspases resulting in cytokine production and cell death. While some inflammasomes have defined ligands, the NLRP3 inflammasome serves as a global sensor of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) (1). The recognition of PAMPs and DAMPs by NLRP3 requires two steps: priming and activation. The priming step leads to the transcriptional upregulation of NLRP3 and pro-IL-1β; the activation step then leads to NLRP3 oligomerization and initiates the assembly of the NLRP3 inflammasome (2). However, non-canonical NLRP3 activation requires caspase-11 (in mice) or caspase 4/5 (in humans) and occurs strictly in response to the Gram-negative bacteria-derived PAMP lipopolysaccharide (LPS) (3, 4). Cytosolic LPS activates caspase-11, leading to the cleavage of gasdermin D (GSDMD), which is sufficient to induce pyroptosis (5, 6). Processing of the proinflammatory cytokines IL-1β and IL-18 is still dependent on subsequent NLRP3 inflammasome activation downstream of caspase-11 activation. Thus, successful cytoplasmic delivery of LPS, via intracellular infection with Gram-negative bacteria, delivery of bacterial outer membrane vesicles (OMV), or direct transfection, is a fundamental requirement for non-canonical NLRP3 inflammasome activation (7).
Interferon (IFN) signaling has been recognized as a central regulator of inflammasome activation during bacterial infections. In particular, type I IFN priming is required for non-canonical NLRP3 inflammasome activation during infection with Gram-negative bacteria (8). We and others have previously discovered that the TLR4-TRIF axis regulates caspase-11 expression and non-canonical NLRP3 inflammasome activation during infection with enteropathogens such as Escherichia coli and Citrobacter rodentium (9–11). However, bone marrow-derived macrophages (BMDMs) deficient in TRIF or the IFN-α/β receptor (IFNAR) undergo pyroptosis at a rate similar to that of wild-type (WT) BMDMs transfected with LPS, suggesting that TRIF/IFN signaling is dispensable for non-canonical NLRP3 inflammasome activation by LPS transfection (4). IFN-inducible guanylate-binding proteins (GBPs) and immunity-related GTPase family member b10 (IRGB10) also contribute to non-canonical NLRP3 inflammasome activation by liberating Gram-negative bacteria from pathogen-containing vacuoles and disrupting the structural integrity of the bacteria; this process ultimately releases LPS into the cytoplasm allowing detection by caspase-11 (12–14).
Type I IFN induction in dendritic cells is greatly enhanced by IFN regulatory factor 8 (IRF8) via the prolongation of recruitment of basal transcription machinery to the IFN promoters during viral infection (15). Similarly, the concerted activation of IRF8 and IRF3 in human monocytes regulates IFN-β production in response to LPS or viral infection (16), suggesting IRF8 is a critical contributor to the rapid and abundant type I IFN production in immune cells. Our recent study found that IRF8 is not required for non-canonical NLRP3 inflammasome activation during LPS transfection (17). However, given that IRF8 plays an important role in IFN-β production, we hypothesized that IRF8 would be required for Gram-negative bacteria–mediated NLRP3 inflammasome activation.
MATERIALS AND METHODS
Mice
Irf8–/– (17), Stat1–/– (18), Casp11–/– (14), Irgb10–/– (14), and Nlrp3–/– (14) mice have been described previously. Six to eight-week-old male and female mice were used in this study. Mice were bred at St. Jude Children’s Research Hospital. Animal studies were conducted according to the protocols approved by the St. Jude Institutional Animal Care and Use committee.
Bacterial culture
Citrobacter rodentium (51549, American Type Culture Collection), Escherichia coli (11775, American Type Culture Collection), and the isogenic mutant of Pseudomonas aeruginosa strain, ΔpopB (provided by Dr. Brent Berwin, Department of Microbiology and Immunology, Geisel School of Medicine at Dartmouth, Lebanon), were inoculated into Luria-Betrani (LB) media (3002–031, MP Biomedicals) and incubated overnight under aerobic conditions at 37°C. Bacteria were sub cultured (1:25) into fresh LB media for 3 h at 37°C to generate log-phase bacteria for infection.
Cytokine analysis
Cytokines were measured by performing multiplex ELISA (Millipore, MCYTOMAG-70K), ELISA for IL-18 (Invitrogen, BMS618–3) according to the manufacturer’s instructions (19), or ELISA for IFN-β (BioLegend, 439407) according to the manufacturer’s instructions.
Cell culture and stimulation
BMDMs were cultured for 6 d in in DMEM (Thermo Fisher Scientific, 11995–073) supplemented with 1% non-essential amino acids (Thermo Fisher Scientific, 11140–050), 10% FBS (Biowest, S1620), 30% medium conditioned by L929 mouse fibroblasts, and 1% penicillin and streptomycin (Thermo Fisher Scientific, 15070–063). BMDMs in antibiotic-free medium were seeded onto 12-well plates at a density of 1 × 106 cells per well, followed by overnight incubation. For bacterial infection, the following conditions were used: E. coli and C. rodentium at 1, 10, or 20 MOI for 16 h of incubation (for activation of caspases); ΔpopB at 10, 20, or 50 MOI for 16 h of incubation (for activation of caspases); and E. coli, C. rodentium, and ΔpopB at 10 MOI for indicated time of incubation (for signaling assessment). Cell culture supernatants were collected for enzyme-linked immunosorbent assays (ELISAs).
Bacterial phagocytic uptake
BMDMs were grown in 12-well plates and infected with 20 MOI of E. coli, C. rodentium, and ΔpopB at 37°C. Two h after infection, cells were washed three times with PBS. Cells were then lysed in 0.1% Triton X-100. The lysates were then serially diluted and plated on LB agar plates. The colonies were counted after 18 h of growth at 37°C.
Immunoblot analysis
BMDM cell lysates and culture supernatants were combined in caspase lysis buffer (containing protease inhibitors, phosphatase inhibitors, 10% NP-40, and 25 mM DTT) and sample loading buffer (containing SDS and 2-mercaptoethanol) for immunoblot analysis of caspase-1 (20). For immunoblot analysis of signaling, supernatants were removed, and BMDMs were washed once with PBS, then lysed in RIPA buffer and sample loading buffer (containing SDS and 2-mercaptoethanol). Proteins were separated by electrophoresis through 8–12% polyacrylamide gels. Following electrophoretic transfer of proteins onto PVDF membranes (Millipore, IPVH00010), nonspecific binding was blocked by incubation with 5% skim milk, then membranes were incubated with primary antibodies: anti–caspase-1 (#AG-20B-0042, AdipoGen), anti–caspase-11 (#NB120–10454, Novus Biologicals), anti-GSDMD (#Ab209845, Abcam), anti-NLRP3 (#AG-20B-0014, AdipoGen), anti-apoptosis associated speck-like protein containing a caspase activation and recruitment domain (ASC) (#AG-25B-006-C100, AdipoGen), anti–pro-IL-1β (#12507, Cell Signaling Technology [CST]), anti-phospho (p)-TBK1 (#5483, CST), anti-total (t)-TBK1 (#3504, CST), anti–p-IRF3 (#37829S, CST), anti–t-IRF3 (#4302, CST), anti–p-IKKε (#8766, CST), anti–t-IKKε (#3416, CST), anti-GAPDH (#5174, CST), anti-IRF1 (#8478, CST), anti-GBP2 (#27299–1-AP, ProteinTech), anti-GBP5 (#13220–1-AP, ProteinTech), anti–p-STAT1 (#7649, CST), anti–t-STAT1 (#14994, CST), anti–caspase-3 (#9662, CST), anti-cleaved caspase-3 (#9661, CST), anti–caspase-7 (#9492, CST), anti-cleaved caspase-7 (#9491, CST), anti–caspase-8 (AG-20T-0138-C100, AdipoGen), anti-cleaved caspase-8 (#8592, CST), anti-IRGB10 rabbit serum raised against recombinant full-length IRGB10 (1:10,000 dilution) (21), and anti-IRF8 (#A5798, ABclonal). Membranes were then washed and incubated with the appropriate horseradish peroxidase (HRP)–conjugated secondary antibodies (1:5,000 dilution; Jackson Immuno Research Laboratories, anti-rabbit [111–035-047], anti-mouse [315–035-047], and anti-rat [112–035-003]) for 1 h. Proteins were visualized by using Luminata Forte Western HRP Substrate (Millipore, WBLUF0500).
Real-time cell death analysis
Real-time cell death assays were performed using a two-color IncuCyte zoom-in incubator imaging system (Essence Biosciences). BMDMs were seeded into 12-well plates and incubated at 37°C overnight. The next day, following bacterial infection, 20 nM of the cell-impermeable DNA-binding fluorescent dye Sytox Green (S7020; Life Technologies) was added, and the resulting images were analyzed using the software package supplied with the Incucyte imager, which allows precise analysis of the number of Sytox Green-positive cells present in each image. Experiments were conducted using a minimum of three separate wells for each experimental condition and a minimum of six image fields per well. Dead cell events were counted based on Sytox Green staining and plotted using GraphPad Prism v6.0 software.
Real-time (RT-PCR) analysis
For bacterial stimulation, the following conditions were used: E. coli, C. rodentium, and ΔpopB at 10 MOI for 2, 4, and 6 h. RNA was extracted using TRIzol (Thermo Fisher Scientific, 15596026) according to the manufacturer’s instructions. The isolated RNA was reverse-transcribed into cDNA by using a First-Strand cDNA Synthesis Kit (Applied Biosystems, 4368814). Real-time quantitative PCR was performed on an ABI 7500 RT-PCR instrument by using 2× SYBR Green (Applied Biosystems, 4368706) and the appropriate primers. RT-PCR primer sequences are detailed in Table 1.
Table 1.
Real-time qPCR primer sequences
| Gene | Primer Sequence |
|---|---|
| Ifnb | Forward: 5′-GCCTTTGCCATCCAAGAGATGC-3′ |
| Reverse: 5′-ACACTGTCTGCTGGTGGAGTTC-3′ | |
| Hprt | Forward: 5′-CTCATGGACTGATTATGGACAGGAC-3′ |
| Reverse: 5′-GCAGGTCAGCAAAGAACTTATAGCC-3′ |
Statistical analysis
GraphPad Prism v6.0 software was used for data analysis. Data are shown as mean ± SEM. Statistical significance was determined by t tests (two-tailed) for two groups and ANOVA (with Dunnett’s multiple comparisons test, Sidak’s multiple comparisons test, or Tukey’s multiple comparisons test) for three or more groups. P values less than 0.05 were considered to be statistically significant.
RESULTS
IRF8 promotes NLRP3 inflammasome activation by Gram-negative bacteria
Non-canonical NLRP3 inflammasome activation is observed in response to a number of Gram-negative bacteria due to the ubiquitous presence of LPS in their outer membrane (3). Both C. rodentium and E. coli are considered classical triggers of caspase-11–mediated NLRP3 inflammasome activation (9). Additionally, our recent findings show that the P. aeruginosa mutant strain ΔpopB can also engage the non-canonical NLRP3 inflammasome (22). To investigate the role of IRF8 in regulating Gram-negative bacteria-mediated non-canonical NLRP3 inflammasome activation, we first infected WT and Irf8–/– BMDMs with E. coli and analyzed caspase-1 activation. We observed reduced caspase-1 activation in Irf8–/– BMDMs compared with WT BMDMs (Figure 1A and S1A). The decreased production of IL-18 and IL-1β in E. coli–infected Irf8–/– BMDMs provided further evidence for reduced caspase-1 activation in Irf8–/– BMDMs (Figure 1B). In line with previous reports (9), caspase-1 activation and cytokine production in E. coli–infected WT BMDMs were dependent on both NLRP3 and caspase-11 (Figure 1B and S1A). Another feature of non-canonical NLRP3 inflammasome signaling is caspase-11 activation inducing pyroptosis without requiring downstream NLRP3 (23). Cell death analysis showed reduced cell death in BMDMs lacking IRF8 compared with WT BMDMs after infection with E. coli (Figure 1C and 1D). Casp11–/– BMDMs but not Nlrp3–/– BMDMs were protected from pyroptotic cell death (Figure 1C and 1D). Similar phenomena were observed in Irf8–/– BMDMs following infection with C. rodentium and ΔpopB (Figure 1E–H, S1B, S1C, and S2). However, release of inflammasome-independent cytokines KC, TNF, and IL-10 following bacterial infections was not impaired in the absence of IRF8 (Figure S3). Altogether, these results suggest that IRF8 contributes to caspase-11–mediated NLRP3 inflammasome activation and pyroptosis during infection with Gram-negative bacteria.
Figure 1. IRF8 promotes NLRP3 inflammasome activation by Gram-negative bacteria.
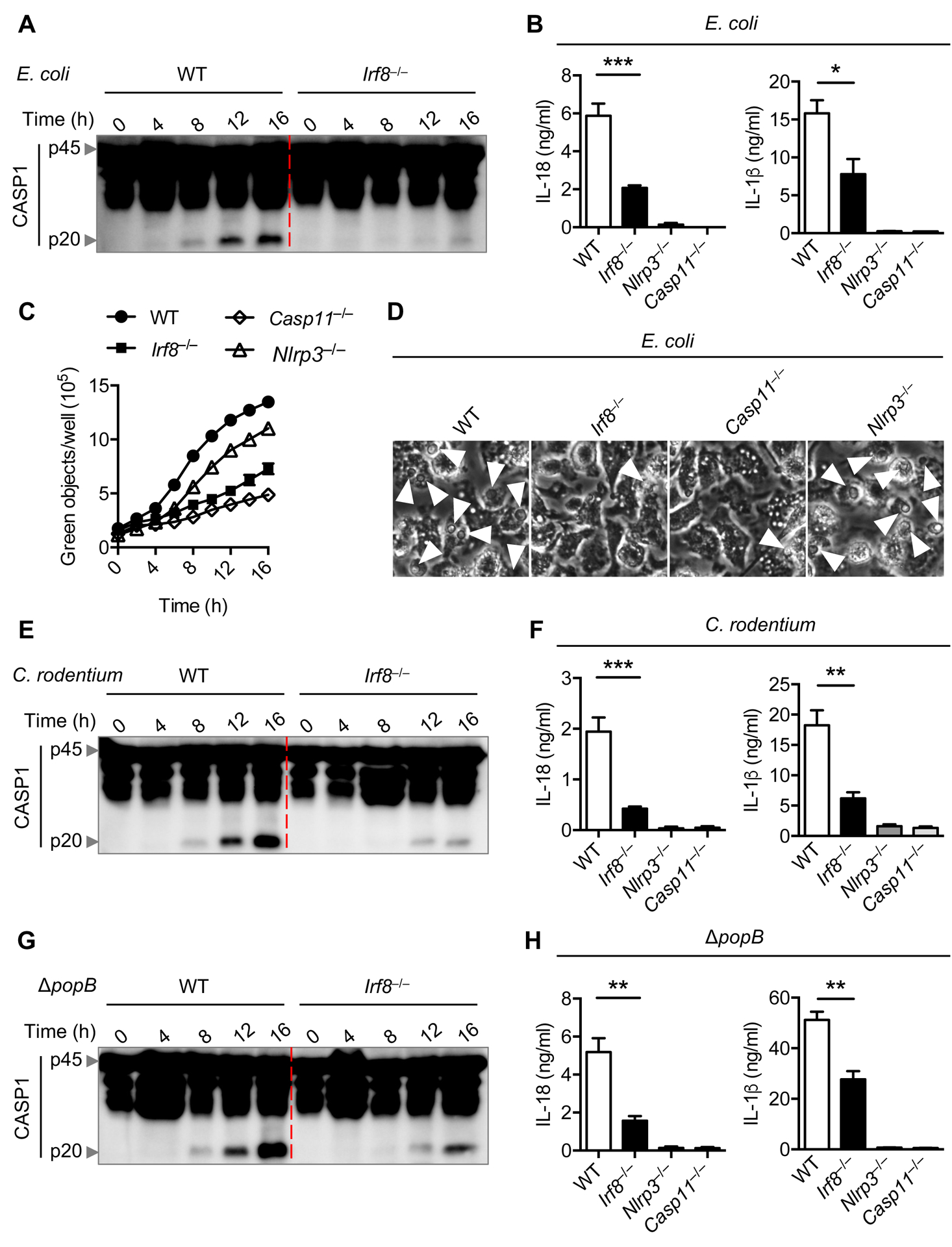
(A) Immunoblot analysis of pro-caspase-1 (CASP1) (p45) and cleaved CASP1 (p20) in wild-type (WT) or Irf8–/– bone marrow-derived macrophages (BMDMs) infected with E. coli (multiplicity of infection [MOI], 20) for the indicated time. (B) Assessment of IL-18 and IL-1β release by ELISA in BMDMs 16 h after E. coli infection. (C) Real time cell death analysis by Incucyte and SYTOX green staining in BMDMs infected with E. coli for the indicated time. (D) Representative images of BMDMs under light microscopy after E. coli infection for 16 h. The arrows indicate pyroptotic cells. (E) Immunoblot analysis of pro-CASP1 (p45) and cleaved CASP1 (p20) in WT or Irf8–/– BMDMs infected with C. rodentium (MOI, 20) for the indicated time. (F) Assessment of IL-18 and IL-1β release by ELISA in BMDMs 16 h after C. rodentium infection. (G) Immunoblot analysis of pro-CASP1 (p45) and cleaved CASP1 (p20) in WT or Irf8–/– BMDMs infected with P. aeruginosa ΔpopB (MOI, 50) for the indicated time. (H) Assessment of IL-18 and IL-1β release by ELISA in BMDMs 16 h after P. aeruginosa ΔpopB infection. *P < 0.05, **P < 0.01, and ***P < 0.001 (one-way ANOVA with Dunnett’s multiple comparisons test). Data are representative of three (A, D, E, and G) experiments or are from three (B, C, F, and H) independent experiments (mean ± SEM).
Changes in bacterial uptake can affect inflammasome activation (24). To understand whether the reduced inflammasome activation observed in Irf8–/– BMDMs during infection could be because of decreased bacterial uptake, cells were infected with E. coli, C. rodentium, or ΔpopB, and bacterial uptake was quantified at 2 h post infection. We found similar bacterial uptake in BMDMs from both genotypes, suggesting that IRF8 promotes inflammasome activation without affecting bacterial uptake (Figure S3). Overall, our results indicate that IRF8 is required for optimal non-canonical NLRP3 inflammasome activation in cells infected with Gram-negative bacteria.
IRF8 regulates GSDMD activation, but not other components of the NLRP3 inflammasome
Bacterial infection activates Toll-like receptor (TLR) signaling to upregulate the expression of NLRP3 and pro-IL-1β, which potentiates NLRP3 inflammasome activation (2). Failure to upregulate NLRP3 in the absence of IRF8 may explain the reduced NLRP3 inflammasome activation observed in Irf8–/– BMDMs during bacterial infection. To investigate whether there is a priming defect in Irf8–/– BMDMs, we analyzed the protein expression of inflammasome components during E. coli, C. rodentium, or ΔpopB infection. The protein expression of NLRP3, ASC, and pro-IL-1β was not impaired in Irf8–/– BMDMs, demonstrating that the priming signal required for NLRP3 inflammasome activation was not compromised in the absence of IRF8 (Figure 2A–C). The similar induction of NLRP3 was further supported by comparable activation of NF-κB between WT and Irf8–/– BMDMs during infection with E. coli (Figure 2D). Additionally, upregulation and activation of caspase-11 during infection are crucial for non-canonical NLRP3 inflammasome activation. We observed similar upregulation of pro-caspase-11 expression in WT and Irf8–/– BMDMs (Figure 2A–C). While the expression of GSDMD was similar between WT and Irf8–/– BMDMs, there was a reduction in the cleavage of GSDMD in Irf8–/– BMDMs during the bacterial infection (Figure 2A–C), suggesting that IRF8 acts upstream of GSDMD activation.
Figure 2. IRF8 regulates gasdermin D activation.
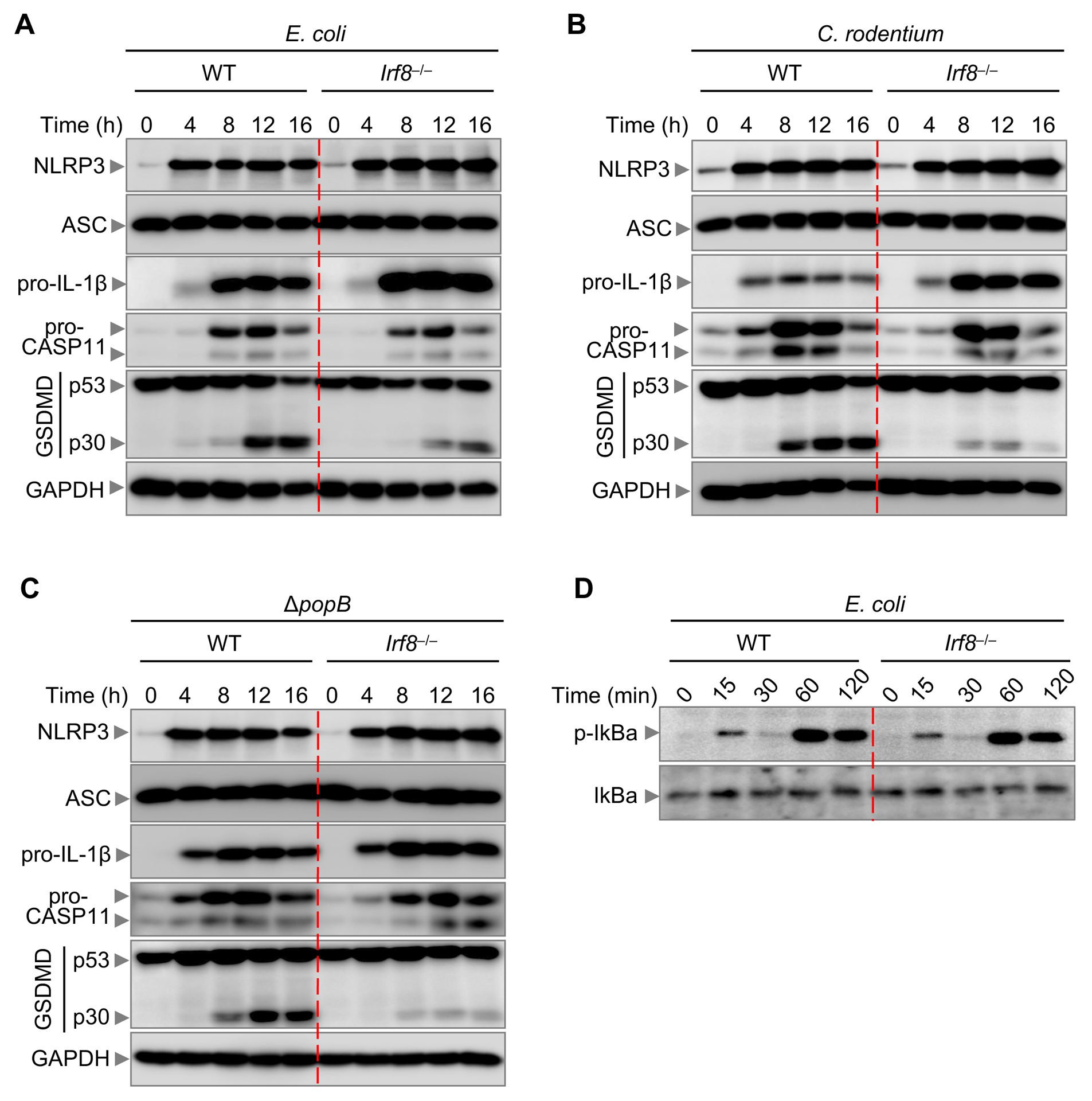
(A) Immunoblot analysis of NLRP3, apoptosis associated speck-like protein containing a caspase activation and recruitment domain (ASC), pro-IL-1β, caspase-11 (CASP11), and gasdermin D (GSDMD) in wild-type (WT) or Irf8–/– bone marrow-derived macrophages (BMDMs) infected with E. coli (multiplicity of infection [MOI], 10) for the indicated time. (B) Immunoblot analysis of NLRP3, ASC, pro-IL-1β, CASP11, and GSDMD in WT or Irf8–/– BMDMs infected with C. rodentium (MOI, 10) for the indicated time. (C) Immunoblot analysis of NLRP3, ASC, pro-IL-1β, CASP11, and GSDMD in WT or Irf8–/– BMDMs infected with P. aeruginosa ΔpopB (MOI, 10) for the indicated time. (D) Immunoblot analysis of phospho (p)-IκBα and IκBα in WT or Irf8–/– BMDMs infected with E. coli (MOI, 10) for the indicated time. GAPDH was used as an internal control. Data are representative of three independent experiments.
When caspase-11 recognizes bacterial LPS, it oligomerizes and becomes activated, which in turn cleaves GSDMD to generate the pyroptogenic 30 kD N-terminal fragment of GSDMD (5, 6, 25). Therefore, reduced caspase-11 activation can result in reduced GSDMD cleavage. We observed reduced caspase-11 activation in Irf8–/– BMDMs compared with WT BMDMs during E. coli infection (Figure 3A). Recently caspase-8 has also been shown to induce GSDMD cleavage during Yersinia infection (26). We observed reduced activation of caspase-8 and downstream executioner caspases, caspase-3 and −7, in Irf8–/– BMDMs compared with WT BMDMs after infection with E. coli. Similar findings were observed upon infection with C. rodentium and ΔpopB (Figure 3B and 3C). Altogether, these results suggest that IRF8 enhances caspase-11 and caspase-8 cleavage to induce GSDMD activation.
Figure 3. IRF8 regulates both CASP11 and CASP8 activation.
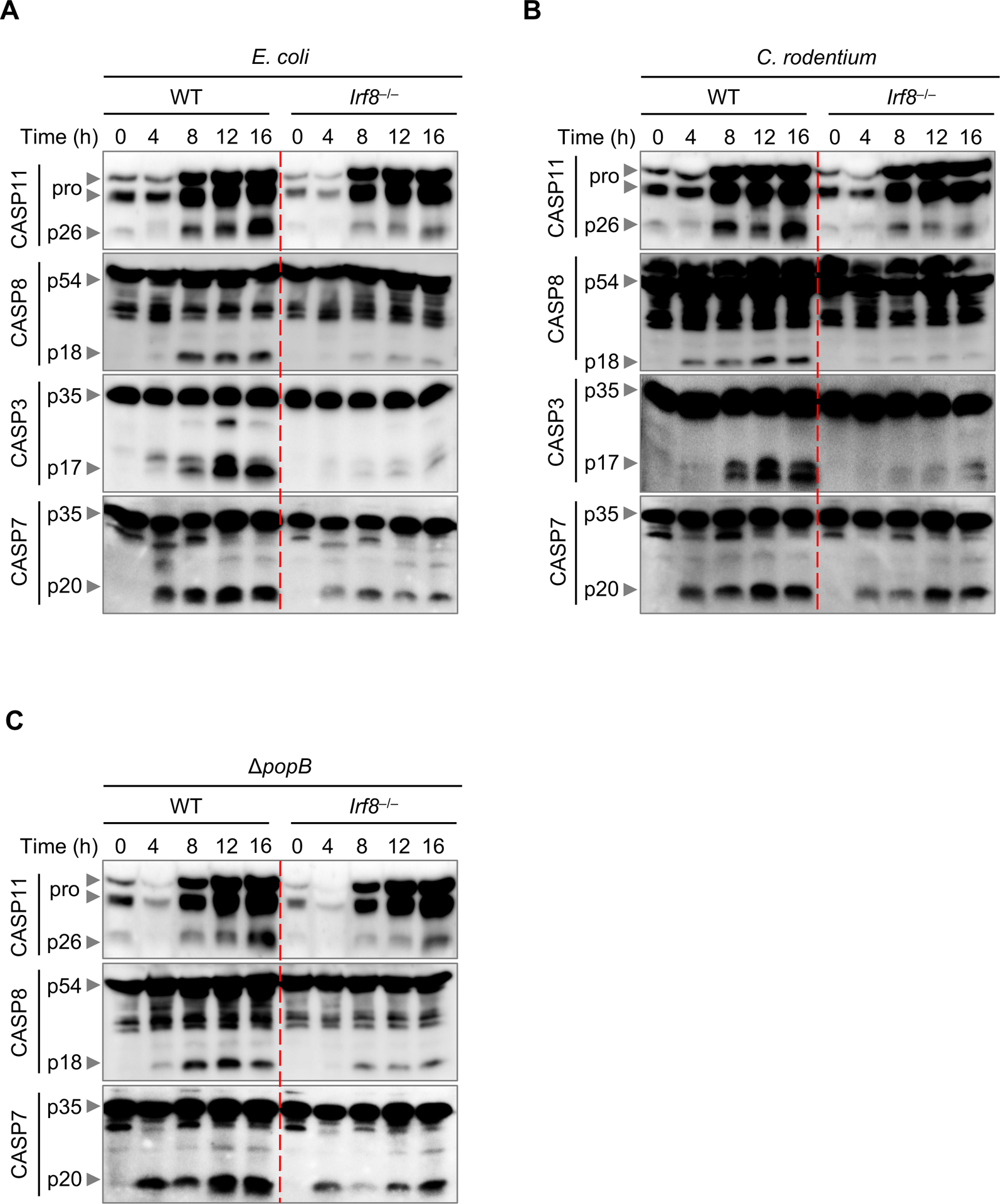
(A) Immunoblot analysis of pro- and cleaved caspase-11 (CASP11), caspase-8 (CASP8), caspase-3 (CASP3), and caspase-7 (CASP7) in WT or Irf8–/– bone marrow-derived macrophages (BMDMs) infected with E. coli (multiplicity of infection [MOI], 20) for the indicated time. (B) Immunoblot analysis of pro- and cleaved CASP11, CASP8, CASP3, and CASP7 in WT or Irf8–/– BMDMs infected with C. rodentium (MOI, 20) for the indicated time. (C) Immunoblot analysis of pro- and cleaved CASP11, CASP8, and CASP7 in WT or Irf8–/– BMDMs infected with P. aeruginosa ΔpopB (MOI, 50) for the indicated time. Data are representative of three independent experiments.
IRF8 promotes IFN-β production for NLRP3 inflammasome activation by Gram-negative bacteria
Type I IFN production and signaling through the IFN receptor are required for non-canonical NLRP3 inflammasome activation by Gram-negative bacteria (10). Given that IRF8 regulates rapid and abundant type I IFN production in immune cells (15, 16), we hypothesized that IRF8-mediated IFN-β production during infection with Gram-negative bacteria could regulate non-canonical inflammasome activation. To test this, we infected WT and Irf8–/– BMDMs with E. coli, C. rodentium, and ΔpopB and determined the gene expression and protein production levels of IFN-β. We observed reduced IFN-β transcription and protein production in Irf8–/– BMDMs compared with WT BMDMs, demonstrating that IRF8 regulates IFN-β production during infection with Gram-negative bacteria (Figure 4A–D). Likewise, we stimulated WT and Irf8–/– BMDMs with LPS or poly(I:C) and measured the amount of IFN-β released in cell supernatants. As expected, Irf8–/– BMDMs produced less IFN-β compared with WT BMDMs (Figure 4E). To confirm whether impaired non-canonical inflammasome activation in Irf8–/– BMDMs was a consequence of reduced IFN-β production, we supplemented WT and Irf8–/– BMDMs with recombinant IFN-β and infected them with E. coli, C. rodentium, or ΔpopB. We observed that IFN-β supplementation restored caspase-1 and GSDMD cleavage and IL-18 and IL-1β production in Irf8–/– BMDMs to levels similar to those in WT BMDMs (Figure 4F, 4G, and S4) suggesting that IRF8-regulated IFN-β production facilitates non-canonical inflammasome activation during Gram-negative bacterial infection.
Figure 4. IRF8 enhances IFN-β production and IFN-β supplementation rescues inflammasome activation in Irf8–/– BMDMs.
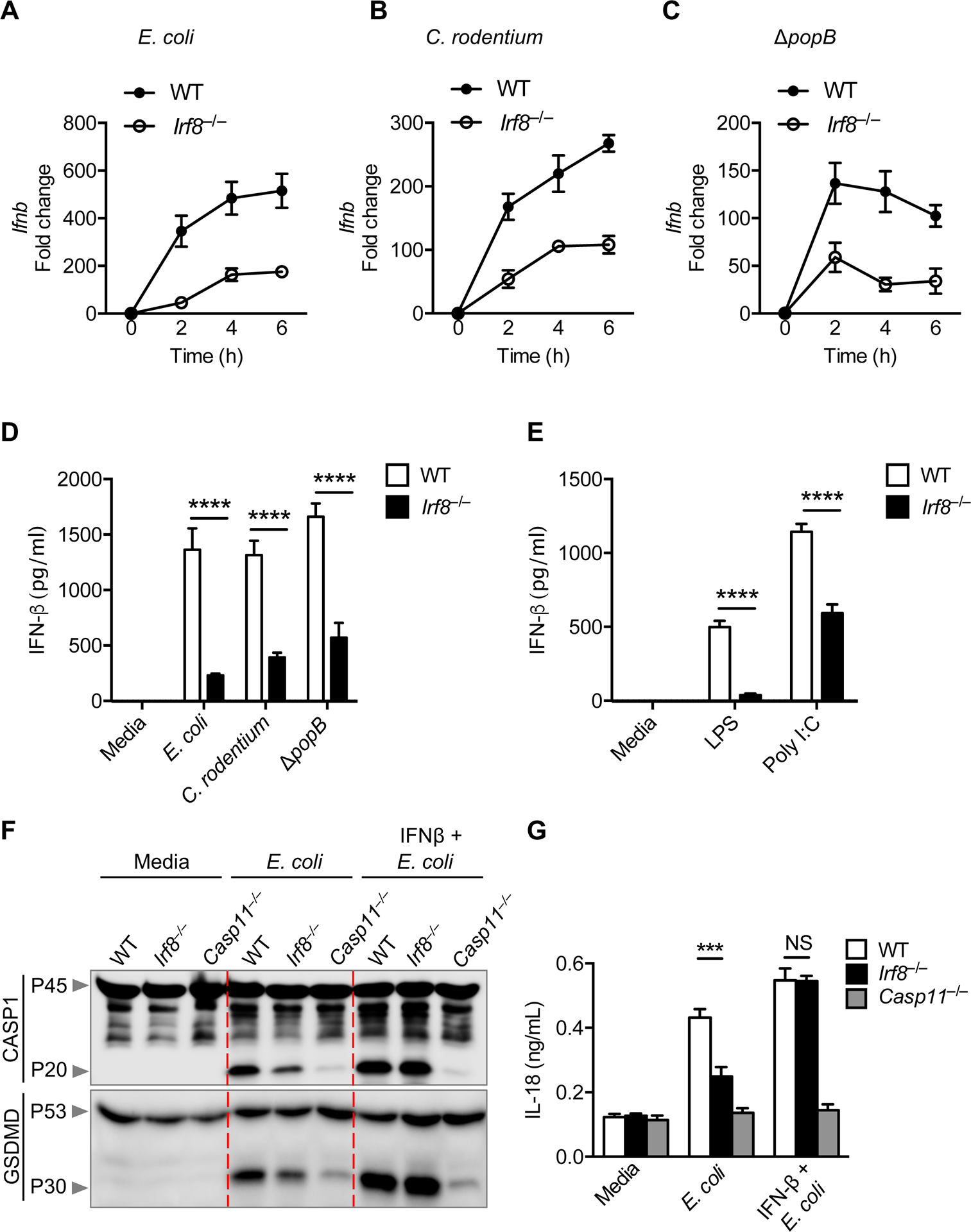
(A–C) The expression of Ifnb in wild-type (WT) or Irf8–/– bone marrow-derived macrophages (BMDMs) at 2, 4, and 6 h after infection with E. coli (multiplicity of infection [MOI], 10), C. rodentium (MOI, 10), or P. aeruginosa ΔpopB (MOI, 10). (D) Assessment of IFN-β release by ELISA in WT or Irf8–/– BMDMs 12 h after E. coli (MOI, 10), C. rodentium (MOI, 10), or P. aeruginosa ΔpopB (MOI, 10) infection. (E) Assessment of IFN-β release by ELISA in WT or Irf8–/– BMDMs 9 h after stimulation with LPS (100 ng/mL) or poly(I:C) (10 μg/mL) in the media. (F,G) Immunoblot analysis of pro- and cleaved caspase-1 (CASP1) and gasdermin D (GSDMD), and the assessment of IL-18 release in BMDMs pre-treated with IFN-β (400 U/mL) for 4 h followed by infection with E. coli (MOI, 20) for an additional 16 h. NS, not significant; ***P < 0.001 and ****P < 0.0001 (D and E, one-way ANOVA with Sidak’s multiple comparisons test and G, two-way ANOVA with Tukey’s multiple comparisons test). Data are representative of three (F) experiments or are from three (A–E and G) independent experiments (mean ± SEM).
The TLR4/TRIF signaling axis triggers upregulation of IFN-β via the IKKε and TBK1 signaling network that induces phosphorylation of IRF3 (27). This causes IRF3 dimerization and translocation to the nucleus, where it binds to the IFN-β promoter to stimulate IFN-β transcription (27). The reduced IFN-β transcription and protein production in Irf8–/– BMDMs could be the result of reduced activation of the molecules in the signaling cascade for IFN-β production. To test whether IRF8 regulates signaling steps upstream of IFN-β transcription during infection with Gram-negative bacteria, we infected WT and Irf8–/– BMDMs with E. coli or C. rodentium measured the kinetics of IKKε, TBK1, and IRF3 phosphorylation. We observed reduced phosphorylation of IRF3, but not IKKε and TBK1, in Irf8–/– BMDMs compared with WT BMDMs (Figure 5), demonstrating that IRF8 mediated the activation of IRF3 to trigger IFN-β gene upregulation during infection with Gram-negative bacteria. Overall, these data suggest that IRF8 controls IFN-β production during infection with Gram-negative bacteria to allow non-canonical inflammasome activation.
Figure 5. IRF8 activates IRF3 for IFN-β production.
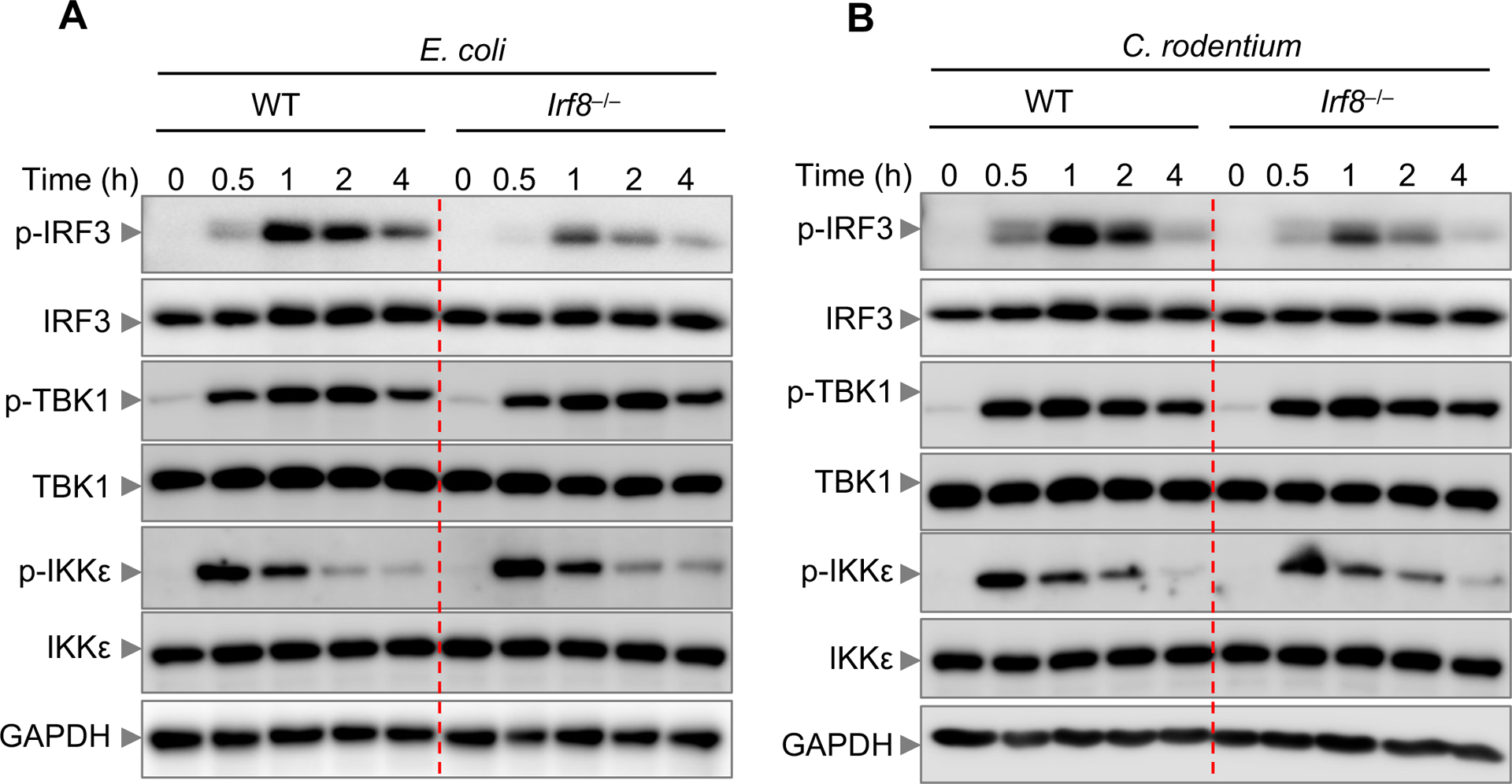
(A,B) Immunoblot analysis of phospho (p)-IRF3, IRF3, p-TBK1, TBK1, p-IKKε, and IKKε in wild-type (WT) or Irf8–/– bone marrow-derived macrophages (BMDMs) infected with E. coli (multiplicity of infection [MOI], 10) and C. rodentium (MOI, 10) for the indicated time. GAPDH was used as an internal control. Data are representative of three experiments.
IRF8 promotes NLRP3 inflammasome activation independently of GBPs and IRGB10
IFN-inducible proteins, especially GBPs and IRGB10, are known to activate caspase-11 by targeting and destroying pathogen-containing vacuoles to release bacterial LPS, thereby assembling the NLRP3 inflammasome (14). Given that IRF8 regulates IFN-β production (Figure 4), we hypothesized that IRF8-regulated GBPs and IRGB10 induction contribute to NLRP3 inflammasome activation. To test this, we determined the kinetics of IRF1, GBP2, GBP5, and IRGB10 induction in WT and Irf8–/– BMDMs during infection with E. coli and C. rodentium. We found that IRF1, GBP2, GBP5, and IRGB10 were similarly induced in both genotypes (Figure 6A and 6B), suggesting that the reduced inflammasome activation observed in Irf8–/– BMDMs was not associated with GBPs or IRGB10. Reduced IFN-β production in Irf8–/– BMDMs would affect activation of STAT1, which is a central hub for the IFN signaling pathway. As expected, we observed defective STAT1 activation in Irf8–/– BMDMs (Figure 6C). STAT1 has previously been shown to be required for GBP2 and GBP5 induction to activate the AIM2 inflammasome during Francisella infection (18). However, we observed similar kinetics of GBP2 and GBP5 induction in Stat1–/– BMDMs during Gram-negative bacterial infection (Figure 6D). Despite having similar GBP2 and GBP5 expression, Stat1–/– and Irf8–/– BMDMs showed reduced caspase-1 cleavage during C. rodentium infection (Figure 6E), suggesting that IRF8 and STAT1 regulate NLRP3 inflammasome activation by Gram-negative bacteria independently of GBPs and IRGB10.
Figure 6. IRF8 does not regulate GBPs and IRGB10 for NLRP3 inflammasome activation.
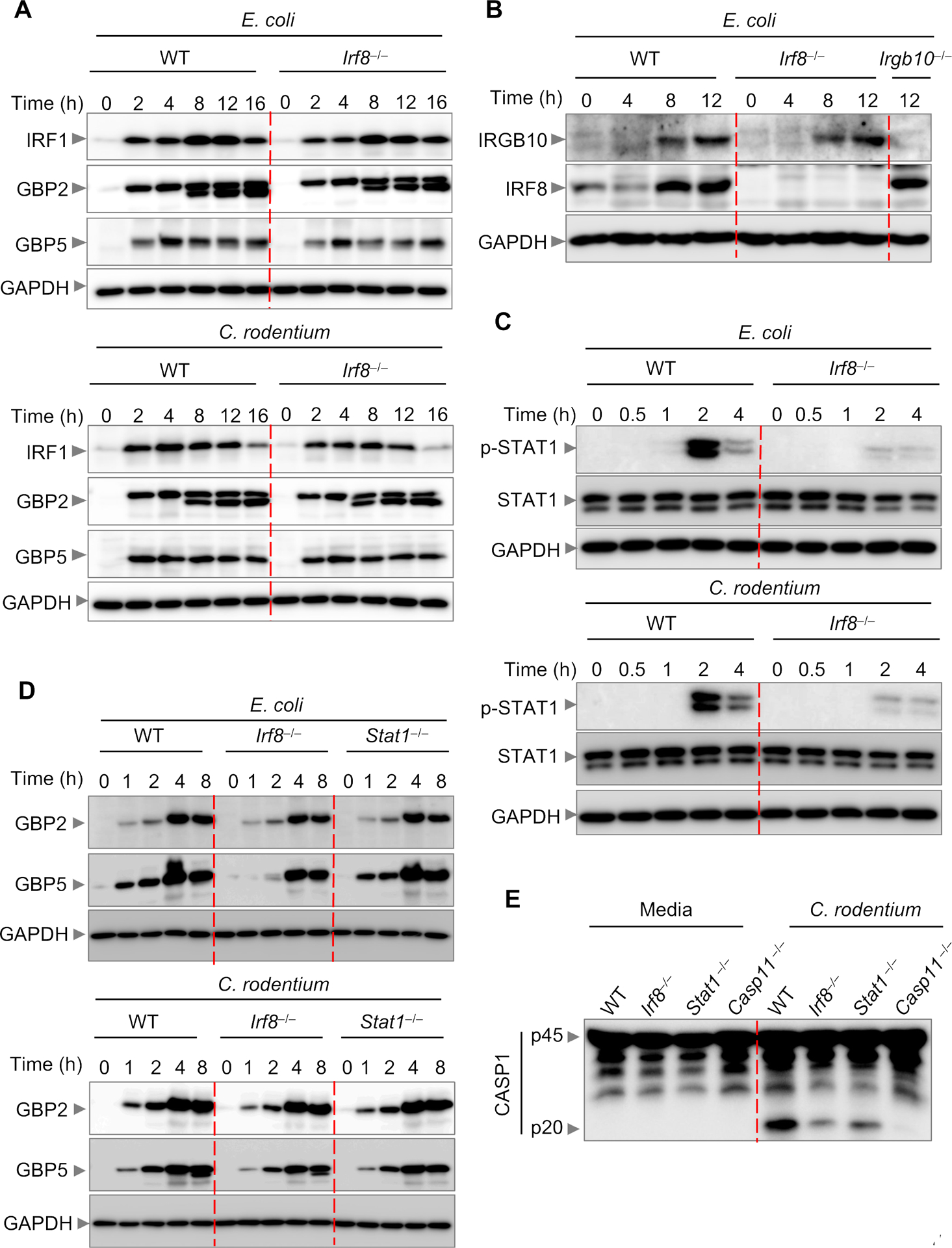
(A) Immunoblot analysis of interferon regulatory factor 1 (IRF1), guanylate binding protein 2 (GBP2), and GBP5 in wild-type (WT) or Irf8–/– bone marrow-derived macrophages (BMDMs) after E. coli (multiplicity of infection [MOI], 10) or C. rodentium (MOI, 10) infection. (B) Immunoblot analysis of IRGB10 and IRF8 in WT or Irf8–/– BMDMs after E. coli (MOI, 10) infection. (C) Immunoblot analysis of phospho (p)-STAT1 and STAT1 in WT or Irf8–/– BMDMs after E. coli (MOI, 10) or C. rodentium (MOI, 10) infection. (D) Immunoblot analysis of GBP2 and GBP5 WT, Irf8–/–, or Stat1–/– BMDMs after E. coli (MOI, 10) or C. rodentium (MOI, 10) infection. (E) Immunoblot analysis of pro-caspase-1 (CASP1) (p45) and cleaved CASP1 (p20) in WT, Irf8–/–, Stat1–/–, or Casp11–/– BMDMs infected with C. rodentium (MOI, 20) for 16 h. Data are representative of three independent experiments.
DISCUSSION
IRF8 plays a key role in NLRC4 inflammasome activation by regulating the transcription of genes encoding NAIPs and NLRC4 (17). However, IRF8 is dispensable for AIM2, pyrin, and NLRP3 inflammasome activation in response to classical triggers and LPS transfection. Here, we identified an additional role for IRF8 in promoting Gram-negative bacteria-mediated NLRP3 inflammasome activation. Deficiency of IRF8 in BMDMs resulted in reduced NLRP3 inflammasome activation, caspase-11 and GSDMD cleavage, and cell death. However, the expression of major components of the non-canonical NLRP3 inflammasome, including NLRP3, ASC, CASP11, and GSDMD, was not impaired in IRF8-deficient BMDMs. The supplementation of IFN-β in Irf8–/– BMDMs rescued the inflammasome activation, suggesting that reduced production of IFN-β in Irf8–/– BMDMs was the driving force for the attenuated inflammasome activation. We also found that IRF8 regulated IRF3 phosphorylation to control the production of IFN-β.
The inflammatory caspases caspase-1 and −11 and the apoptotic caspase caspase-8 are activated during microbial infections and are known to cleave GSDMD, a key executioner of pyroptosis (6, 26). The precise molecular mechanism for the activation of the NLRP3 inflammasome by caspase-11 is still unknown. Furthermore, caspase-8 also acts as an effector and regulator of NLRP3 inflammasome signaling (28). The decreased GSDMD cleavage and NLRP3 inflammasome activation in Irf8–/– BMDMs could be explained by the reduced activation of both caspase-11 and caspase-8. However, the crosstalk between these caspases in the regulation of the NLRP3 inflammasome during bacterial infections needs further investigation. Caspase-3 activation during bacterial infection is one of the consequences of caspase-8 activation. Caspase-3 can cleave GSDME, another member of the gasdermin family which has been shown to induce pyroptosis in certain cancer cells (29). Therefore, the reduced caspase-3 activation observed in Irf8–/– BMDMs could also be contributing to decreased pyroptotic cell death executed by GSDME.
The molecules involved in type I IFN production and IFN signaling promote NLRP3 inflammasome activation during Gram-negative bacterial infection, but not in response to LPS transfection (4). The supplementation of IFN-β in Trif–/– BMDMs rescues the reduced inflammasome activation in those cells. However, the detailed molecular mechanisms behind IFN-β enhancing NLRP3 inflammasome activation during bacterial infection is obscure. Several studies have suggested that members of the GBP and IRG families, which are induced during bacterial infection in an IFN-β–dependent manner, target and destroy pathogen-containing vacuoles to release bacterial LPS, resulting in caspase-11 activation and assembly of the NLRP3 inflammasome (14, 18). Despite reduced IFN-β production in Irf8–/– BMDMs, we did not observe differential induction of caspase-11, GBP2, GBP5, or IRGB10 expression between WT and Irf8–/– BMDMs. The pro-caspase-11 and GBP5 promoters contain NF-κB and STAT binding sites, and their expression is induced by NF-κB–activating ligands such as LPS and TNF (30–32). Similarly, both NF-κB and TRIF signaling contribute to GBP2 and IRGB10 induction (33). The Gram-negative bacteria not only induce type I IFN, but also robustly activate NF-κB signaling (14, 34). The similar IκBα phosphorylation, along with the similar expression of NLRP3, observed in WT and Irf8–/– BMDMs suggest that IRF8 does not regulate NF-κB signaling during the bacterial infection. The similar expression of caspase-11, GBP2, GBP5, and IRGB10 in the WT and Irf8–/– BMDMs infected with Gram-negative bacteria is therefore possibly a result of the similar activation of NF-κB signaling. Alternatively, the low level of IFN-β in Irf8–/– BMDMs may be sufficient to induce GBPs to the extent observed in WT BMDMs. Indeed, constitutive IFN has been shown to maintain the level of GBP expression required for the release of bacterial ligands upstream of pyroptosis (35). Therefore, it is likely that other inducible genes that are unique to the IFN signaling pathway promote NLRP3 inflammasome activation, which is further supported by the attenuated, but not abolished, caspase-11 and caspase-1 cleavage observed in Gbpchr3 cells (12, 14) during bacterial infection.
Previous studies have shown that IRF8 contributes to IFN-β production in murine dendritic cells and human monocytes (16). The scaffold complex consisting of IRF8 and PU.1 on the IFN-β promoter recruits IRF3 for the rapid induction of IFN-β transcription in human monocytes (16). Upon stimulation or infection, cytosolic IRF3 undergoes phosphorylation and dimerization, and then translocate into the nucleus to carry out its transcriptional activity. Here we found reduced phosphorylation of IRF3 in Irf8–/– BMDMs, which accounted for the decreased IFN-β production during bacterial infection. IRF3 phosphorylation relies on TBK1 activation. However, TBK1 was similarly phosphorylated in WT and Irf8–/– BMDMs, suggesting that IRF8 activates IRF3 without impairing the activation of TBK1. It is possible that IRF8 interacts with both TBK1 and IRF3, promoting the phosphorylation of IRF3 by TBK1. Indeed, IRF8 has been shown to be an interacting partner of IRF3 in human monocytes (16). Furthermore, a similar bridging concept has been demonstrated with STING and RIOK3 in the activation of the type I IFN pathway (36). The increased production of KC and expression of pro-IL-1β in Irf8–/– BMDMs could be a consequence of decreased production of type I IFN in those cells. Type I IFNs produced during Streptococcus pyogenes infection inhibit the production of IL-1β to prevent lethal systemic hyper-inflammation of soft tissue (37). Similarly, KC is upregulated in peritoneal macrophages from IFN−β null mice upon bacterial infection (38). Altogether, our findings provide insights into the role of IRF8 in mediating non-canonical inflammasome activation.
Supplementary Material
Key points.
IRF8 promotes NLRP3 inflammasome activation in response to Gram-negative bacteria.
IRF8 contributes to caspase-11 and caspase-8 cleavage to enhance cell death.
Phosphorylation of IRF3 mediated by IRF8 is required for the type I IFN response.
ACKNOWLEDGEMENTS
We thank A. Burton from St. Jude Children’s Research Hospital for technical support and R. Tweedell, PhD, for scientific editing.
This work was supported by funding from the National Institutes of Health grants CA163507, AR056296, AI124346, and AI101935 and by ALSAC to T.‐D.K.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Kanneganti TD 2018. The inflammasome starts rolling. Nat Rev Immunol 18: 483. [DOI] [PubMed] [Google Scholar]
- 2.Karki R, and Kanneganti TD. 2019. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat Rev Cancer 19: 197–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, and Dixit VM. 2011. Non-canonical inflammasome activation targets caspase-11. Nature 479: 117–121. [DOI] [PubMed] [Google Scholar]
- 4.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, and Dixit VM. 2013. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341: 1246–1249. [DOI] [PubMed] [Google Scholar]
- 5.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, and Dixit VM. 2015. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526: 666–671. [DOI] [PubMed] [Google Scholar]
- 6.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F. 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665. [DOI] [PubMed] [Google Scholar]
- 7.Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, and Rathinam VAK. 2016. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 165: 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Latz E, Xiao TS, and Stutz A. 2013. Activation and regulation of the inflammasomes. Nat Rev Immunol 13: 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurung P, Malireddi RK, Anand PK, Demon D, Vande Walle L, Liu Z, Vogel P, Lamkanfi M, and Kanneganti TD. 2012. Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J Biol Chem 287: 34474–34483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, and Fitzgerald KA. 2012. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150: 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, and Monack DM. 2012. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490: 288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, Roose-Girma M, Bumann D, Kayagaki N, Takeda K, Yamamoto M, and Broz P. 2014. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 509: 366–370. [DOI] [PubMed] [Google Scholar]
- 13.Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, Ernst RK, Yamamoto M, Miao EA, and Coers J. 2014. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci U S A 111: 6046–6051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, Frase S, Zhu Q, Malireddi RKS, Kuriakose T, Peters JL, Neale G, Brown SA, Yamamoto M, and Kanneganti TD. 2016. IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 167: 382–396 e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tailor P, Tamura T, Kong HJ, Kubota T, Kubota M, Borghi P, Gabriele L, and Ozato K. 2007. The feedback phase of type I interferon induction in dendritic cells requires interferon regulatory factor 8. Immunity 27: 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li P, Wong JJ, Sum C, Sin WX, Ng KQ, Koh MB, and Chin KC. 2011. IRF8 and IRF3 cooperatively regulate rapid interferon-beta induction in human blood monocytes. Blood 117: 2847–2854. [DOI] [PubMed] [Google Scholar]
- 17.Karki R, Lee E, Place D, Samir P, Mavuluri J, Sharma BR, Balakrishnan A, Malireddi RKS, Geiger R, Zhu Q, Neale G, and Kanneganti TD. 2018. IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 173: 920–933 e913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Man SM, Karki R, Malireddi RK, Neale G, Vogel P, Yamamoto M, Lamkanfi M, and Kanneganti TD. 2015. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat Immunol 16: 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma BR, Karki R, Lee E, Zhu Q, Gurung P, and Kanneganti TD. 2019. Innate immune adaptor MyD88 deficiency prevents skin inflammation in SHARPIN-deficient mice. Cell Death Differ 26: 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karki R, Man SM, Malireddi RKS, Gurung P, Vogel P, Lamkanfi M, and Kanneganti TD. 2015. Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe 17: 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steinfeldt T, Konen-Waisman S, Tong L, Pawlowski N, Lamkemeyer T, Sibley LD, Hunn JP, and Howard JC. 2015. Correction: Phosphorylation of Mouse Immunity-Related GTPase (IRG) Resistance Proteins Is an Evasion Strategy for Virulent Toxoplasma gondii. PLoS Biol 13: e1002199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balakrishnan A, Karki R, Berwin B, Yamamoto M, and Kanneganti TD. 2018. Guanylate binding proteins facilitate caspase-11-dependent pyroptosis in response to type 3 secretion system-negative Pseudomonas aeruginosa. Cell Death Discov 4: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He Y, Hara H, and Nunez G. 2016. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci 41: 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patankar YR, Lovewell RR, Poynter ME, Jyot J, Kazmierczak BI, and Berwin B. 2013. Flagellar motility is a key determinant of the magnitude of the inflammasome response to Pseudomonas aeruginosa. Infect Immun 81: 2043–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J. 2016. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, Berger SB, Gough PJ, Bertin J, Proulx MM, Goguen JD, Kayagaki N, Fitzgerald KA, and Lien E. 2018. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362: 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, and Maniatis T. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4: 491–496. [DOI] [PubMed] [Google Scholar]
- 28.Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, and Kanneganti TD. 2014. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 192: 1835–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, and Shao F. 2017. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547: 99–103. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen TT, Hu Y, Widney DP, Mar RA, and Smith JB. 2002. Murine GBP-5, a new member of the murine guanylate-binding protein family, is coordinately regulated with other GBPs in vivo and in vitro. J Interferon Cytokine Res 22: 899–909. [DOI] [PubMed] [Google Scholar]
- 31.Ramsauer K, Farlik M, Zupkovitz G, Seiser C, Kroger A, Hauser H, and Decker T. 2007. Distinct modes of action applied by transcription factors STAT1 and IRF1 to initiate transcription of the IFN-gamma-inducible gbp2 gene. Proc Natl Acad Sci U S A 104: 2849–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schauvliege R, Vanrobaeys J, Schotte P, and Beyaert R. 2002. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J Biol Chem 277: 41624–41630. [DOI] [PubMed] [Google Scholar]
- 33.Briard B, Karki R, Malireddi RKS, Bhattacharya A, Place DE, Mavuluri J, Peters JL, Vogel P, Yamamoto M, and Kanneganti TD. 2019. Fungal ligands released by innate immune effectors promote inflammasome activation during Aspergillus fumigatus infection. Nat Microbiol 4: 316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elewaut D, DiDonato JA, Kim JM, Truong F, Eckmann L, and Kagnoff MF. 1999. NF-kappa B is a central regulator of the intestinal epithelial cell innate immune response induced by infection with enteroinvasive bacteria. J Immunol 163: 1457–1466. [PubMed] [Google Scholar]
- 35.Liu BC, Sarhan J, Panda A, Muendlein HI, Ilyukha V, Coers J, Yamamoto M, Isberg RR, and Poltorak A. 2018. Constitutive Interferon Maintains GBP Expression Required for Release of Bacterial Components Upstream of Pyroptosis and Anti-DNA Responses. Cell Rep 24: 155–168 e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng J, De Jesus PD, Su V, Han S, Gong D, Wu NC, Tian Y, Li X, Wu TT, Chanda SK, and Sun R. 2014. RIOK3 is an adaptor protein required for IRF3-mediated antiviral type I interferon production. J Virol 88: 7987–7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castiglia V, Piersigilli A, Ebner F, Janos M, Goldmann O, Dambock U, Kroger A, Weiss S, Knapp S, Jamieson AM, Kirschning C, Kalinke U, Strobl B, Muller M, Stoiber D, Lienenklaus S, and Kovarik P. 2016. Type I Interferon Signaling Prevents IL-1beta-Driven Lethal Systemic Hyperinflammation during Invasive Bacterial Infection of Soft Tissue. Cell Host Microbe 19: 375–387. [DOI] [PubMed] [Google Scholar]
- 38.Perkins DJ, Rajaiah R, Tennant SM, Ramachandran G, Higginson EE, Dyson TN, and Vogel SN. 2015. Salmonella Typhimurium Co-Opts the Host Type I IFN System To Restrict Macrophage Innate Immune Transcriptional Responses Selectively. J Immunol 195: 2461–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.