

Abstract
Intrinsically disordered proteins (IDPs) play important roles in biology, but little is known about the energetics of their inter-residue interactions. Methods that have been successfully applied to analyze the energetics of globular proteins are not applicable to the fluctuating partially ordered ensembles populated by IDPs. A combined computational experimental strategy is introduced for analyzing the energetic role of individual residues in the free state of IDPs. The approach combines experimental measurements of the binding of wild-type and mutant IDPs to their partners with alchemical free energy calculations of the structured complexes. This data allows quantitative information to be deduced about the free state via a thermodynamic cycle. The approach is validated by the analysis of the effects of mutations upon the binding free energy of the ovomucoid inhibitor third binding domain to its partners and is applied to the C-terminal domain of the measles virus nucleoprotein, a 125-residue IDP involved in the RNA transcription and replication of measles virus. The analysis reveals significant inter-residue interactions in the unbound IDP and suggests a biological role for them. The work demonstrates that advances in force fields and computational hardware have now led to the point where it is possible to develop methods which integrate experimental and computational techniques to reveal insights that cannot be studied using either technique alone.
Graphical Abstract
Introduction
Intrinsically disordered proteins (IDPs) lack stable secondary and tertiary structure due to their low content of bulky hydrophobic residues and their high content of polar and charged residues1–2. While they do not fold into well-defined globular structures in isolation3–4, IDPs populate ensembles ranging from expanded states with little residual structure to ensembles which are more compact and contain residual secondary and tertiary interactions and they often fold upon binding to their partners 5–10. IDPs play important roles in biology. Structural characterization of IDPs in their uncomplexed state and of the denatured state of globular proteins show that neither are true random-coils, instead, they often contain transient secondary structure and long-range interactions11–20. Inter-residue interactions in the unbound state of IDPs could modulate their binding affinity and other interactions, and thus modulate their biological activity. Moreover, long-range interactions in IDPs and in unfolded proteins may play an important role in controlling the propensity to aggregate and thus preventing protein misfolding diseases12–13, 21,22. In general, conservation of IDP sequences is believed to be lower than for the sequences of globular proteins, suggesting that there may be an overall lower free-energy cost to mutations in the free state of IDPs23. Progress has been made in characterizing the structural properties of IDPs and in defining relationships between sequence and conformational properties, but much less is known about the energetics of inter-residue interactions in IDPs2, 24–26, and quantitative analysis of the inter-residue energetics of IDPs is still absent.
The contribution of a residue to the energetics of the folded state of a protein is traditionally obtained by mutagenesis and unfolding free energy measurements, but such a strategy cannot be applied to IDPs. In principle, it is also possible to estimate the energetics of a residue in a globular protein via molecular modelling if the structure of the protein is resolved in atomic level27–28. Structural ensembles of IDPs can be obtained from ensemble fitting using experimental observables such as Rg and NMR as constraints and the ensembles used as input to potential energy functions to estimate energies. However, the ensembles derived are not fully deterministic as the experimental observables are not sufficient to uniquely define highly conformationally heterogeneous IDPs and the energetics obtained are not always reliable. Molecular dynamics (MD) simulations are becoming popular for studying the structural propensities of IDPs as newly developed force fields are being trained not only for globular proteins, but also for IDPs29–31. While many current force fields have shown good performance at folding proteins32–34, their accuracy for IDPs are still not as well established and many force fields have had difficulty reproducing global properties of protein unfolded states35. Simulations of IDPs also require considerably more sampling than simulations of folded proteins because of their flat free energy landscape, increasing computational cost. Larger IDPs are currently not suitable for MD simulations in explicit solvent so implicit solvent models must be used, which can be less accurate. This inability to accurately model the disordered state of IDPs has hampered the application of MD to study IDP energetics.
Here, we describe a strategy to quantitatively analyze the energetic effects of mutations upon the free state of IDPs and to define the energetic contribution made by individual residues in the free state. The approach combines experimental structural and energetic information on complexes of IDPs with their binding partners and alchemical free energy calculations of the bound state to deduce the properties of the unbound state via a thermodynamic cycle. The strategy is validated using mutants of the turkey ovomucoid inhibitor third binding domain (OMTKY3) and its binding partners and is then applied to the C-terminal domain of the measles virus nucleoprotein (NTAIL), a 125-residue IDP involved in the RNA transcription and replication of measles virus. NTAIL undergoes binding and folding upon encounter with the X domain (XD) of its phosphoprotein binding partner36. Residues 486 to 502 of NTAIL form a so-called α-helical Molecular Recognition Element (αMoRE) and fold into a stable helix upon binding to XD. SAXS studies have shown that NTAIL has a radius of gyration, Rg, of 27.5 ± 0.7 Å which is significantly less than the predicted Rg for a random coil with the same length as NTAIL, 35 – 38 Å37, suggesting inter-residue interactions lead to compaction in the free state. A recent study of the effect of the truncation of the disordered N-terminal region of NTAIL on the binding energetics support this hypothesis. A non-monotonic dependence of binding strength on truncation length was observed38, indicating that residues in the disordered N-terminal region of NTAIL contribute to the binding affinity of NTAIL and XD. This work also suggested that the folding of NTAIL upon binding is impeded by the disordered regions flanking the αMoRE. However, the molecular basis of these effects is unclear and the mechanisms behind these unexplained observations cannot be determined using conventional methods. We quantify the effect of three mutations in the unbound state of NTAIL and show that the residues participate in long-range interactions in the unbound state and that these interactions can modulate the binding of NTAIL to its partner. Interactions in the free state are predicted to reduce the affinity of NTAIL for XD, thereby providing a mechanism for tuning binding affinity and biological function. Our approach allows quantitative analysis of these interactions without the need to fully model the IDP ensemble.
This work illustrates that, as computational results become more and more reliable due to advances in force fields and computing hardware, it is possible to develop accurate and precise hybrid methods which rely on experiments and calculations simultaneously to reveal insights that cannot be studied by conventional methods.
Methods
Free energy calculations were performed using non-softcore thermodynamic integration (TI) implemented in Amber as described in detail in the supporting information (see SI). Minimization and equilibration under constant pressure were conducted to heat up and relax the X-ray structures. Production runs were conducted using the implementation of GPU-accelerated thermodynamic integration, pmemdGTI39–43, under constant volume (see SI). Details of the calculations, including the methods used to account for any changes in protonation state involving mutation of charged residues and details of the analysis of the data are provided in the supporting information.
Results
A Thermodynamic Cycle for Analyzing the Energetics of the Free State of IDPs
We calculate the free energy changes caused by mutations in IDPs by taking a detour through a thermodynamic cycle (Fig. 1). The measurable binding free energy of the wild-type and mutant IDP are combined with high-level TI calculations on the folded complexes to define the energetics of the free state of the IDP. The approach relies on a thermodynamic cycle, in which the values of three branches of the cycle define the value of the fourth (Fig. 1). For example, the effect of changing an alanine to a glycine in the unbound state of an IDP, ΔGfree, can be obtained by ΔGfree = ΔGbind - ΔG’bind + ΔGcom. ΔGbind and ΔG’bind are the experimental binding free energies of the mutant and wildtype respectively. ΔGcom is the free energy change caused by mutation of Ala to Gly in the complex and is calculated using alchemical free energy calculations. The effect of the mutation in a capped tripeptide or a small fragment, in which the mutated residue is in the middle of the fragment and flanked by the same amino acids found next to the site in the full-length IDP, also needs to be calculated to provide a reference state and is denoted as ΔGfrag. The capped tripeptide is not meant to represent the unbound state of IDP as it ignores secondary structure and long-range interactions. Rather the capped tripeptide is just a necessarily bookkeeping device that accounts for purely local interactions and for the fact that the different sized side chains will make different interactions with water. It is important to reiterate that the capped tripeptide is not used as a model of the IDP, rather the differences in the values of ΔGfree and ΔGfrag denoted as ΔΔGinter, quantitatively defines the energetic effect of nonlocal interactions that are not present in the peptide model.
Figure 1.

Illustration of the approach used to deduce the energetics of the free IDP. The thermodynamic cycle describes the binding of a wild-type IDP and a mutant IDP to its partner. The capped tripeptide has the same residues adjacent to the mutation site as found in the full protein. ΔGbind and ΔG’bind are the binding free energies measured by experiment for wildtype and mutant respectively (red text). ΔGcom and ΔGfrag are calculated using alchemical free energy calculations (blue text). The value for ΔGfree is obtained from ΔGfree = ΔGbind - ΔG’bind + ΔGcom. The effect of secondary structure and long-range interactions on the mutations is obtained from ΔΔGinter = ΔGfree - ΔGfrg. ΔΔGinter and ΔGfree (green text) cannot be measured by either experiments or calculations alone but can be obtained by combining the experimental and computational measurements.
In principle, the effect of mutations on interactions in the unbound state of IDPs could be directly calculated using alchemical free energy calculations, but such calculations are practically impossible due to the dynamic nature of the free state which leads to insufficient sampling. The approach developed here circumvents this issue by bridging the complex state and the free state using the binding free energies measured by experiments such as isothermal titration calorimetry (ITC), differential scanning calorimetry (DSC), binding kinetic experiments and others. The experimental binding free energies (ΔGbind and ΔG’bind) provide relationships between the phase space of the bound and free state of the IDP. This allows the calculated free energy change in the complex state (ΔGcom) to be combined with experimental binding data to deduce the free energy changes in the free state (ΔGfree)
Another Interpretation of the Approach
From another perspective, this approach can be interpreted as a method which obtains the effects of mutations on the free state of IDPs by deconvoluting the experimentally measured binding free energy changes. The values of ΔGbind and ΔG’bind are the binding affinities of the wild-type and mutant measured experimentally using techniques such as ITC, DSC, binding kinetic experiments and others. The value of ΔGbind - ΔG’bind represents the effect of the mutation on the binding affinity of the IDP and its binding partner. However, ΔGbind - ΔG’bind is a convolution of the effect of the mutation on both the complex and free state of the IDP. This is analogous to protein folding where ΔGmutation contains contributions from the folded and unfolded states. If the effect of the mutation on the free state is to be quantified, ΔGbind - ΔG’bind must be deconvoluted. In our approach, ΔGbind - ΔG’bind is deconvoluted by calculating the effect of mutation on the complex state (ΔGcom) using free energy calculations. The deconvolution leads to the effect of the mutation on the free state (ΔGfree) of the IDP via a thermodynamic cycle. The resulting value of ΔGfree is another convolution of the purely local and long-range interactions disrupted by the mutation. The calculation using the capped tripeptide (ΔGfrag), which only describes the local interactions, allows further deconvolution of ΔGfree so that the long-range interactions disrupted by the mutation (ΔΔGinter) can be quantified. One practical consideration is that some IDPs retain disordered tails in the bound state. If necessary, the disordered regions in the bound state can be truncated during the calculation of ΔGcom to increase computational efficiency provided that the disordered regions do not alter the conformation or energetics of the structured regions under investigation in the complex. This is likely to be valid for residues which are buried in the binding interface and sequestered from solvent since they are protected from transient contacts with any disordered segments.
IDP Complexes and Mutation Sites that are Suitable for the Approach
Many IDPs retain a certain degree of so-called “fuzziness” upon binding to their partners and are relatively dynamic even in the bound state. These IDPs may remain partially or even fully flexible upon binding and they can adopt multiple binding conformations44–48. Similar to the free state of IDPs, calculations of ΔGcom for this type of IDPs are challenging due to the vast conformational ensembles that need to be sampled. However, some IDPs that form a “fuzzy” complex with one of their partners may form a rigid complex with a different partner48. Since the energetics of the free state of IDPs are independent of their binding partners and their structures in the complex state, our approach can be applied to any IDP as long as it forms a rigid complex with one of its natural binding partners or an engineered binding partner and the binding free energies can be measured experimentally.
For IDPs that form both structured and disordered regions upon binding, it is easiest to study the residues that belong to the structured regions since large segments of disordered regions are too computationally expensive to model directly. Thus, truncated IDP complexes, with only the structured region, may be used during the calculation of ΔGcom provided certain conditions are met. This approximation requires that the disordered regions do not form strong interactions with the structured regions and do not alter the conformations of the structured regions of the complexes. Moreover, analysis of surface mutations should be avoided if using truncated IDP complexes as they may form transient contacts with the disordered regions in experiments that will be missing during the calculations of ΔGcom using truncated models. There is a significantly smaller possibility of forming direct contacts between buried residues and residues in the disordered regions, so the effect of transient interactions with the disordered regions on these buried residues can be more safely ignored. For the example of the NTAIL/XD complex studied here, residues 486 to 502 of NTAIL form the αMoRE and fold into a stable helix upon binding to XD, but the regions preceding (401–485) and following (503–525) the αMoRE remain disordered upon binding, do not contact the structured region and make at most a small contribution to binding enerergetics5, 36, 49–54.
Ovomucoid Inhibitor Protease Interactions Provide an Excellent System to Validate the Approach
The accurate estimation of ΔGinter depends on precise and accurate free energy calculations since they are combined with experimental binding affinities to make predictions about the free state. Thus, it is necessary to evaluate critically the accuracy of alchemical free energy calculations and define their limitations. In a previous study, we successfully reproduced the experimentally measured effects of Gly-to-D-Ala substitutions on the unfolding free energy changes in eight proteins using TI calculations with a root-mean-square error of 0.23 kcal/mol28. In this study, using the same force field and solvent model, we further validated the accuracy of our free energy calculations on a more relevant system involving calculations of protein-protein binding energetics. We tested the ability of our protocol to reproduce the experimental binding free energy of OMTKY3 to its target proteases using TI calculations (Fig. 2). This is an excellent model system: high-resolution structures of the free and bound states are available and precise thermodynamic binding data has been reported for multiple mutations55–57. For the complexes between OMTKY3 and Streptomyces griseus proteinase B (SGPB) (Fig. 2), high-resolution crystal structures for all substitutions at position 18 of OMTKY3 have been reported except Met18 and Cys1856–57. The perturbations to the structures of the complexes caused by mutation at position 18 of OMTKY3 are minimal. For the 10 variants studied here (OMTKY3/Leu18, Ala18, Gly18, Asn18, Asp18, Val18, Thr18, Ser18, Phe18 and Tyr18), the root-mean-square deviations for the backbone coordinates are at most 0.158 Å (PDB codes listed in SI). The small differences in structures minimize the complexity of the free energy calculations on the bound state of the SGPB/OMTKY3 complex. Furthermore, the mutation site is only partially buried in the complex interface which allows the efficient exchange of water around the side chain during the free energy calculations of complexes. This avoids any complications that might arise from having different numbers of waters in the interface for different side chains. In the unbound state, OMTKY3 is highly stable and the mutation site is located in a short loop with the side chain fully exposed to solvent. These factors make SGPB/OMTKY3 an ideal system for testing the accuracy of free energy calculations. Calculations were also conducted on a Leu-to-Ala mutation in the complex of a different protease, subtilisin Carlsberg (CARL), with OMTKY3 to check if the calculation is sensitive enough to reproduce context-dependent ΔΔG values (reported experimental values of 2.95 kcal/mol for SGPB/OMTKY3 and 0.33 kcal/mol for CARL/OMTKY3) for Leu-to-Ala mutations58. This is important because it provides a significant test of context depend effects and because free energy changes for two Leu-to-Ala mutations in NTAIL are studied later. The difference between ΔΔGexp = (ΔGbind - ΔG’bind) and ΔΔGcalc = (ΔGfree - ΔGcom), see free energy cycle in Fig. 2, provides a rigorous test of the accuracy of the free energy calculations.
Figure 2.

Free energy cycle of L-to-A mutations in the binding of SGPB and OMTKY3. Ribbon structures represent SGPB (pink), OMTKY3 (blue) and the SGPB/OMTKY3 complex. Leu18 (top) and Ala18 (bottom) of OMTKY3 are shown in stick format. ΔG values in red are binding free energies measured by experiment55. ΔG values in blue are free energies calculated using TI.
The Amber force field ff14SB59 and the TIP3P60 water model were used for the TI calculations61–62, with the implementation of GPU-accelerated thermodynamic integration, using pmemdGTI39. In order to test the convergence of the TI calculations, two independent TI calculations were carried out for each X-to-Y substitution in the SGPB/OMTKY3 complex. One calculation started with the structure of SGPB/OMTKY3-X18, and the other calculation started with the structure of SGPB/OMTKY3-Y18. A total of 22 independent TI calculations were carried out.
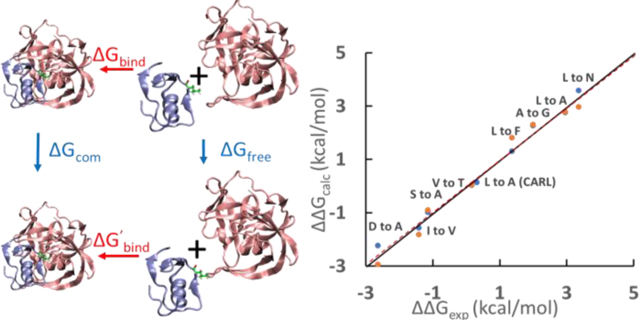
The calculations show excellent agreement with experiments. All numerical values are provided in Table S2. The mutation involving Asp18 require proper accounting of any changes in the pKa of Asp18 in the complex and free states. The free state pKa is available from NMR measurements43 and the bound state pKa has been estimated from experimental pKa dependent binding free energies63. Details are provided in the SI. The root-mean-square error between ΔΔGcalc and ΔΔGexp for all mutations was 0.86 kcal/mol (Fig. 3). V-to-A, Y-to-F and S-to-C have more significant errors, with deviations of 1.30 ~ 2.11 kcal/mol respectively from the experimental results, while all other mutations have errors below 0.45 kcal/mol. The probable causes of the larger errors in V-to-A, S-to-C and Y-to-F mutations are discussed in detail in the SI. Briefly, the issue with the S-to-C mutation is likely due to the Lennard-Jones parameters used for sulfur in Amber ff14SB force field. The issue with Y-to-F mutation may be due to a bound water. The V-to-A mutation is an example of a β-branched residue being changed to a non β-branched residue. We have observed problems with other substitutions of this type and believe it may reflect poor transferability between β-branched residue and non β-branched residue in the force field. Excluding these three apparent outliers reduces the root-mean-square error to only 0.27 kcal/mol and gives an even stronger correlation between ΔΔGcalc and ΔΔGexp with slope = 1.00 and R2 = 0.98, p < 10-13.
Figure 3.

Scatter plot of experimental (ΔΔGexp) and calculated (ΔΔGcalc) ΔΔG values for the binding of SGPB with different OMTKY3 variants. ΔΔGexp = ΔGbind -ΔG’bind. ΔΔGcalc = ΔGfree - ΔGcom. The point labeled CARL is for the binding of subtilisin Carlsberg with OMTKY3 variants. Blue and orange dots indicate calculated ΔΔG values calculated using different starting structures. For example, the value for the blue dot for A to G was calculated using the structure of SGPB/OMTKY3-Ala18 (PDB code 1SGP) and the value for the orange dot for A to G was calculated using the structure of SGPB/OMTKY3-Gly18 (PDB code 1SGQ). There is no structure for subtilisin Carlsberg/OMTKY3-Ala18 so only one ΔΔG values using the structure of subtilisin Carlsberg/OMTKY3-Leu18 (PDB code 1R0R) was calculated. Similarly, only one ΔΔG value for S-to-C mutation in SGPB/OMTKY3 was calculated. Three outliers, V-to-A, S-to-C and Y-to-F are labeled as crosses. The solid line represents ΔΔGexp = ΔΔGcalc. The dashed red line indicates the results of the linear regression of the data (y=0.86x+0.31, R2=0.82 p<10−9).
The precision of the method was tested by carrying out two independent calculations using different starting structures for each mutation (except L-to-A in CARL/OMTKY3 and C-to-S in SGPB/OMTKY3, see caption of Fig. 3). ΔΔGcalc values were essentially identical with an average absolute difference of 0.25 kcal/mol, indicating high precision and good convergence of the calculations. The ΔGfree values estimated by ΔGfree = ΔGbind - ΔG’bind + ΔGcom and the ΔGfree values directly calculated by TI calculations are compared in Table S3. Overall, the results demonstrate that the TI calculations are accurate and precise enough to be combined with experimental binding free energies as outlined in Fig. 1 with the exception of the mutations which lead to the outliers.
Application to the NTAIL Domain: Identification of Long-range Interactions
Having validated the approach, we next applied the strategy to the NTAIL domain. Residues 486 to 502 of NTAIL, form the so called α-helical Molecular Recognition Element (αMoRE), and fold into a stable helix upon binding to the XD, thereby forming an intermolecular four-helix bundle complex (Fig. 4)36, 49–50. The αMoRE of NTAIL has residual helicity in the unbound state, but the regions preceding (401–485) and following (503–525) αMoRE have much less residual structure5, 49, 51–54. The regions preceding (401–485) and following (503–525) the αMoRE remain disordered in the bound state and experimental data indicates that they do not form direct contacts with XD in the complex 5, 36, 49–54. In addition, ITC and surface plasmon resonance experiments indicate that the regions following the αMoRE of NTAIL make only minimal contributions to the binding between NTAIL and XD64–65. This data suggests that the disordered regions of NTAIL/XD complex do not alter the conformations of the structured region formed by the αMoRE and XD. Mutations of three residues which are buried in the complex, A494G, L495A and L498A, are examined in this study. A494, L495 and L498 are within the αMoRE region of NTAIL and located in the interface of NTAIL/XD complex (Fig. 4). Since A494, L495 and L498 are buried in the interface of NTAIL/XD complex, it is unlikely that the truncated disordered regions make contact with these residues in the complex.
Figure 4.

Ribbon representation of the NTAIL (486–504)/XD (458–506) complex from the X-ray structure (PDB code 1T6O). The X-ray structure includes the region shown in ribbons, and an artificial linker which was deleted in simulations and is not shown in this figure. Residue A494, L495 and L498 are shown in stick format. The disordered N and C-terminal regions of NTAIL were not included in the X-ray structure and MD simulations and are depicted schematically as thin lines.
The binding free energy changes (ΔGbind - ΔG’bind) caused by the A494G, L495A and L498A mutations have been reported66. The free energy changes caused by the mutations (ΔGcom) in the NTAIL/XD complex were calculated using the X-ray structure (PDB code 1T6O) and the protocol that we validated with the SGPB/OMTKY3 complexes. In order to check the convergence of the calculations of ΔGcom, two independent TI calculations for each mutation were conducted. One started with the X-ray structure which is the structure of the wild-type NTAIL/XD complex and proceeded toward the mutant. Only the structure of the wild-type NTAIL/XD complex is available, so the second starting structure was a model of the mutant complex. In silico mutations of A494G, L495A and L498A were applied to the X-ray structure of NTAIL/XD complex and a 100ns MD simulation for each mutant was conducted to relax any perturbation caused by the mutations. The TI calculations were performed starting with the structure from the last frame of the MD simulation of the mutant complex and proceeded in reverse to the wild-type complex. The forward and reverse calculations were in excellent agreement with differences ranging from only 0.06 to 0.36 kcal/mol (Table 1).
Table 1.
Free energy changes (kcal/mol) calculated for A494G, L495A and L498A mutations in the complex, free, capped tripeptide fragment and fully helical state of NTAIL. The calculated ΔG values are referenced to the values of their corresponding ΔGfrag. The original values are listed in the parenthesis. The original ΔG values have no physical meaning because they contain a force field dependent baseline. The referenced ΔG does have physical meanings as the force field dependent baseline effect is canceled.
| Forward ΔGcom | Backward ΔGcom | ΔGinter 1 | ΔGfrag 2 | ΔGhelix 2 | |
|---|---|---|---|---|---|
| A494G | 2.36 (−6.87) |
2.22 (−7.01) |
1.59±0.13 (−7.64±0.12) |
0 (−9.23±0.04) |
1.22±0.08 (−8.01±0.07) |
| L495A | 5.09 (23.75) |
4.73 (23.39) |
3.81±0.21 (22.47±0.21) |
0 (18.66±0.04) |
0.81±0.16 (19.47±0.15) |
| L498A | 2.26 (20.05) |
2.09 (19.87) |
0.77±0.16 (18.56±0.13) |
0 (17.79±0.09) |
0.42±0.10 (18.21±0.05) |
The uncertainties are calculated by combining the standard deviation of ΔGcom with the published standard deviation of (ΔGbind - ΔG’bind)66.
The uncertainties are the standard deviation of three independent runs of MD simulations with different starting velocities.
The free energy changes caused by the mutations in the free state of NTAIL (ΔGfree) are all significantly higher than the free energy changes in the capped tripeptide reference state (ΔGfrag) with differences ranging from 0.8 to 3.8 kcal/mol (Table 1). This indicates that the residues are involved in interactions in the free state which are not captured by the capped tripeptide reference state. In other words, these residues are involved in secondary structure or long-range interactions, or both in the free state.
Because the αMoREs region was experimentally found to have residual helicity in the free state of NTAIL5, 49, 51–54, we also examined whether the difference between ΔGfree and ΔGfrag could be explained by helicity rather than long-range interactions in the unbound (IDP) state. TI calculations were repeated using a fully helical conformation of unbound NTAIL (486–504) monomer (ΔGhelix). The structure was adopted from the PDB code 1T6O by deleting the XD domain and the artificial linker. If the contributions from the long-range interactions are negligible, then the free energy changes caused by the mutations in the free state of NTAIL (ΔGfree) should be between those of ΔGfrag and ΔGhelix depending on the fraction of helicity adopted in the free IDP. However, we found that ΔGfree values are still higher than the ΔGhelix values, especially for the L495A mutations. This argues that any propensity to adopt helical structure cannot explain the ΔGfree values, and that A494, L495 and L498 are all involved in favorable long-range interactions in the free state of NTAIL. This is especially notable at position 495, where the long-range interactions favor leucine over alanine.
Discussion
In this work, we have validated a hybrid strategy to measure quantitatively the free energy changes caused by mutations in the free state of IDPs, which cannot be measured directly using conventional experimental or computational techniques. A possible limitation of the strategy is that the IDP must form a structured complex with its receptor and many IDPs remain “fuzzy” upon binding44–48. However, since the energetics of the free state of an IDP are independent of its binding partner, this approach can still be applied to the IDP if it folds upon binding to one a different natural binding partner or an engineered molecule. Another potential limitation is that for IDPs that remain partly disordered upon binding, the truncation of the disordered regions, which is ideal for the calculations of ΔGcom, is only rigorous if the disordered regions have minimal interactions with the structured regions. Ideally, the residues of interest will be buried in the complex to avoid transient long-range interactions with disordered regions of the bound complex that complicate the modeling. Fortunately, residues in the binding interface are usually buried and these are often of major interest since residues in the interface normally contribute the most to the binding affinity of a complex. Studying their interactions in the unbound IDP can provide quantitative information about the properties at the free state and about the possible roles of any free state interactions in the regulation of biological activity.
Our free energy calculations on SGPB/OMTKY3 complexes show that the free energy changes caused by most mutations can be reproduced with high precession and within a small error. Although the outliers V-to-A, C-to-S and Y-to-F were identified in our calculations, the validation included L, I, F, and A, which are the four most commonly observed amino acids in the binding interface between IDPs and their binding partners67–69. On average, about 40% of the residues in the binding interface are one of these four residues69. This indicates that the approach should be broadly applicable to folding upon binding IDPs. Since the approach relies on accurate calculations of free energy, the sampling convergence of the free energy calculations must be considered carefully70. For example, buried mutations may cause displacement of interior water which usually has a slow relaxation beyond the timescale of MD simulations71. However, new algorithms have been developed to address the limitations of sampling in free energy calculations72–74. We believe that as the reliability of these calculations continues to improve, the impact of our approach will increase.
Our analysis shows that A494, L495 and L498, which form part of the binding interface of NTAIL and XD, participate in favorable long-range interactions in the free state of NTAIL that modulate the binding affinity of NTAIL to XD. The strength of the long-range interactions made by A494 and L498 in the free state of NTAIL range from 0.3 to 1.6 kcal/mol depending on the helicity of A494 and L498. If NTAIL were to exist as a true random coil in its free state, then L495 would be 4.9 kcal/mol more favorable than A495 in the complex of NTAIL/XD. Even if L495 is in a fully helical structure in the free state of NTAIL, L495 is predicted to be > 4.1 kcal/mol more favorable than A495 in the complex state. However, the experimentally measured binding free energy change indicates that L495 is only 1.1 kcal/mol more favored than A495 in the complex. The difference can be explained by favorable long-range interactions experienced by L495 in the free state of NTAIL, which compete with the binding interactions involving L495. Stated differently, the interactions in the free state modulate the binding energetics and are predicted to reduce affinity. This provides an important mechanism for tuning binding affinity. The insights revealed by our approach correlate well with prior observations on NTAIL and illustrate that long-range interactions between the flanking disordered regions and the αMoRE can modulate the overall dimensions of NTAIL and the NTAIL/XD binding free energy.
It may be surprising that the interactions in NTAIL appear to favor L495 more than A495 by as much as 3.0 ~ 3.8 kcal/mol. However, if such interactions are responsible for inhibiting the aggregation of proteins22, 75, then they should be reasonably strong. In addition, the full free energy contribution is very unlikely to arise from a pairwise interaction, instead it is highly likely that the residue in question makes interactions with more than one other residue in the free state, or that the mutation alters the unfolded state and thereby modulates multiple other interactions.
The identified long-range interactions identified in NTAIL may play a biological role. Dynamic binding and breaking of the nucleocapsid and polymerase are necessary to ensure the transcription and replication of the RNA encapsulated by nucleocapsids and the affinity needs to be tuned for optimal activity. Increasing and decreasing the binding affinity of NTAIL/XD both lead to a reduction in transcription activity and viral growth76–77, so a balanced interaction between NTAIL and XD is crucial. The long-range interactions involving A494, L495 and L498 in the free state attenuate the binding affinity to reach an optimal efficiency of transcription and replication. This provides a clear example of how interactions within the free state of an IDP can tune biological activities.
Conclusions
This work offers a general methodology for assessing the energetic contributions of individual residues in IDPs to the energetics of the free state of IDPs. The approach allows the identification of residues that participate in long-range interactions and thus may modulate binding affinity, but avoids the difficulties associated with MD simulations of free IDPs. The approach described here is complimentary to methods that generate ensemble level descriptions of residual structure and transient contacts in the free state of IDPs as it provides information about the energetics of specific residues in the free state. Combining energetic information with structural descriptions will provide more insight into the properties of IDPs.
Many IDPs become structured upon binding78–79, thus the strategy is expected to be broadly applicable and will become even easier to apply as computing power continues to increase. The work with the OMTKY3 and its binding partners also provides a rigorous evaluation of the accuracy and precision of TI calculations performed using the Amber ff14SB force field. The work also illustrates how IT calculations can be used to help validate force fields. This work demonstrates that advances in force fields and computing hardware have now led to the point where it is possible to develop novel methods which integrate experimental and computational techniques to reveal insights that cannot be studied by using either technique alone. The interactions and effects revealed by the analysis presented here could not be deduced from experiment and computation in isolation.
Supplementary Material
Acknowledgment
The authors gratefully acknowledge Koushik Kasavajhala, Chuan Tian and Kellon Belfon for their administration of computational resources.
Funding sources. This work was supported by NIH grants GM107104 to CS and GM GM078114 to DPR, an NSF grant MCB-1330259 to DPR and an NSF Petascale Computational Resource (PRAC) Award from the NSF (OCI-1036208) to CS. We gratefully acknowledge support from Henry and Marsha Laufer. J.Z. was supported in part by a fellowship from the Laufer Center.
Footnotes
Supporting information
Detailed methods for the free energy calculations and MD simulations of OMTKY3 and the NTAIL protein in their complexed state and monomer states. Analysis of the origin of outliers in the TI calculations. PDB codes for the monomer and complexes of OMTKY3. Experimental and calculated ΔG values for the mutations of OMTKY3 in complex and monomer states. A diagram for the thermodynamic cycle of OMTKY3-ASP18 forming a complex with SGPB with the additional process of protonation included. A figure of the X-ray structures of SGPB/OMYKY3-Phe18 and SGPB/OMTKY3-Tyr18 showing an expanded view of the region near the residues
References
- 1.Romero P; Obradovic Z; Li X; Garner EC; Brown CJ; Dunker AK, Sequence complexity of disordered protein. Proteins 2001, 42 (1), 38–48. [DOI] [PubMed] [Google Scholar]
- 2.Das RK; Ruff KM; Pappu RV, Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr Opin Struct Biol 2015, 32, 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oldfield CJ; Dunker AK, Intrinsically disordered proteins and intrinsically disordered protein regions. Annu Rev Biochem 2014, 83, 553–584. [DOI] [PubMed] [Google Scholar]
- 4.Dyson HJ; Wright PE, Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 2005, 6 (3), 197–208. [DOI] [PubMed] [Google Scholar]
- 5.Jensen MR; Communie G; Ribeiro EA Jr.; Martinez N; Desfosses A; Salmon L; Mollica L; Gabel F; Jamin M; Longhi S, et al. , Intrinsic disorder in measles virus nucleocapsids. Proc Natl Acad Sci U S A 2011, 108 (24), 9839–9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dima RI; Thirumalai D, Asymmetry in the shapes of folded and denatured states of proteins. J Phys Chem B 2004, 108 (21), 6564–6570. [Google Scholar]
- 7.Muller-Spath S; Soranno A; Hirschfeld V; Hofmann H; Ruegger S; Reymond L; Nettels D; Schuler B, Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc Natl Acad Sci U S A 2010, 107 (33), 14609–14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tran HT; Mao A; Pappu RV, Role of backbone-solvent interactions in determining conformational equilibria of intrinsically disordered proteins. J Am Chem Soc 2008, 130 (23), 7380–7392. [DOI] [PubMed] [Google Scholar]
- 9.Brucale M; Schuler B; Samori B, Single-molecule studies of intrinsically disordered proteins. Chem Rev 2014, 114 (6), 3281–3317. [DOI] [PubMed] [Google Scholar]
- 10.Warner J. B. t.; Ruff KM; Tan PS; Lemke EA; Pappu RV; Lashuel HA, Monomeric huntingtin exon 1 has similar overall structural features for wild-type and pathological polyglutamine lengths. J Am Chem Soc 2017, 139 (41), 14456–14469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iesmantavicius V; Jensen MR; Ozenne V; Blackledge M; Poulsen FM; Kjaergaard M, Modulation of the intrinsic helix propensity of an intrinsically disordered protein reveals long-range helix-helix interactions. J Am Chem Soc 2013, 135 (27), 10155–10163. [DOI] [PubMed] [Google Scholar]
- 12.Meng W; Lyle N; Luan B; Raleigh DP; Pappu RV, Experiments and simulations show how long-range contacts can form in expanded unfolded proteins with negligible secondary structure. Proc Natl Acad Sci U S A 2013, 110 (6), 2123–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holehouse AS; Pappu RV, Collapse transitions of proteins and the interplay among backbone, sidechain, and solvent interactions. Annu Rev Biophys 2018, 47, 19–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohn JE; Millett IS; Jacob J; Zagrovic B; Dillon TM; Cingel N; Dothager RS; Seifert S; Thiyagarajan P; Sosnick TR, et al. , Random-coil behavior and the dimensions of chemically unfolded proteins. Proc Natl Acad Sci U S A 2004, 101 (34), 12491–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan HS; Dill KA, Polymer principles in protein structure and stability. Annu Rev Biophys Biophys Chem 1991, 20, 447–490. [DOI] [PubMed] [Google Scholar]
- 16.Roder H; Maki K; Cheng H, Early events in protein folding explored by rapid mixing methods. Chem Rev 2006, 106 (5), 1836–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borgia A; Zheng W; Buholzer K; Borgia MB; Schuler A; Hofmann H; Soranno A; Nettels D; Gast K; Grishaev A, et al. , Consistent view of polypeptide chain expansion in chemical denaturants from multiple experimental methods. J Am Chem Soc 2016, 138 (36), 11714–11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sherman E; Haran G, Coil-globule transition in the denatured state of a small protein. Proc Natl Acad Sci U S A 2006, 103 (31), 11539–11543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho JH; Raleigh DP, Mutational analysis demonstrates that specific electrostatic interactions can play a key role in the denatured state ensemble of proteins. J Mol Biol 2005, 353 (1), 174–185. [DOI] [PubMed] [Google Scholar]
- 20.Cho JH; Meng W; Sato S; Kim EY; Schindelin H; Raleigh DP, Energetically significant networks of coupled interactions within an unfolded protein. Proc Natl Acad Sci U S A 2014, 111 (33), 12079–12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han H; Weinreb PH; Lansbury PT Jr., The core Alzheimer’s peptide NAC forms amyloid fibrils which seed and are seeded by beta-amyloid: is NAC a common trigger or target in neurodegenerative disease? Chem Biol 1995, 2 (3), 163–169. [DOI] [PubMed] [Google Scholar]
- 22.Bertoncini CW; Jung YS; Fernandez CO; Hoyer W; Griesinger C; Jovin TM; Zweckstetter M, Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc Natl Acad Sci U S A 2005, 102 (5), 1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pritisanac I; Vernon RM; Moses AM; Kay JDF, Entropy and Information within Intrinsically Disordered Protein Regions. Entropy 2019, 21 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jensen MR; Zweckstetter M; Huang JR; Blackledge M, Exploring free-energy landscapes of intrinsically disordered proteins at atomic resolution using NMR spectroscopy. Chem Rev 2014, 114 (13), 6632–6660. [DOI] [PubMed] [Google Scholar]
- 25.Peran I; Holehouse AS; Carrico IS; Pappu RV; Bilsel O; Raleigh DP, Unfolded states under folding conditions accommodate sequence-specific conformational preferences with random coil-like dimensions. Proc Natl Acad Sci U S A 2019, 116 (25), 12301–12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruff KM; Pappu RV; Holehouse AS, Conformational preferences and phase behavior of intrinsically disordered low complexity sequences: insights from multiscale simulations. Curr Opin Struct Biol 2019, 56, 1–10. [DOI] [PubMed] [Google Scholar]
- 27.Dang LX; Merz KM; Kollman PA, Free-energy calculations on protein stability - Thr-157-] Val-157 mutation of T4 Lysozyme. J Am Chem Soc 1989, 111 (22), 8505–8508. [Google Scholar]
- 28.Zou J; Song B; Simmerling C; Raleigh D, Experimental and computational analysis of protein stabilization by Gly-to-d-Ala substitution: A convolution of native state and unfolded state effects. J Am Chem Soc 2016, 138 (48), 15682–15689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang LP; McKiernan KA; Gomes J; Beauchamp KA; Head-Gordon T; Rice JE; Swope WC; Martinez TJ; Pande VS, Building a more predictive protein force field: A systematic and reproducible route to AMBER-FB15. J Phys Chem B 2017, 121 (16), 4023–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmuller H; MacKerell AD Jr., CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 2017, 14 (1), 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robustelli P; Piana S; Shaw DE, Developing a molecular dynamics force field for both folded and disordered protein states. Proc Natl Acad Sci U S A 2018, 115 (21), E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindorff-Larsen K; Piana S; Dror RO; Shaw DE, How fast-folding proteins fold. Science 2011, 334 (6055), 517–520. [DOI] [PubMed] [Google Scholar]
- 33.Nguyen H; Maier J; Huang H; Perrone V; Simmerling C, Folding simulations for proteins with diverse topologies are accessible in days with a physics-based force field and implicit solvent. J Am Chem Soc 2014, 136 (40), 13959–13962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voelz VA; Bowman GR; Beauchamp K; Pande VS, Molecular simulation of ab initio protein folding for a millisecond folder NTL9(1–39). J Am Chem Soc 2010, 132 (5), 1526–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rauscher S; Gapsys V; Gajda MJ; Zweckstetter M; de Groot BL; Grubmuller H, Structural ensembles of intrinsically disordered proteins depend strongly on force field: A comparison to experiment. J Chem Theory Comput 2015, 11 (11), 5513–5524. [DOI] [PubMed] [Google Scholar]
- 36.Bourhis JM; Johansson K; Receveur-Brechot V; Oldfield CJ; Dunker KA; Canard B; Longhi S, The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res 2004, 99 (2), 157–167. [DOI] [PubMed] [Google Scholar]
- 37.Longhi S; Receveur-Brechot V; Karlin D; Johansson K; Darbon H; Bhella D; Yeo R; Finet S; Canard B, The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J Biol Chem 2003, 278 (20), 1863818648. [DOI] [PubMed] [Google Scholar]
- 38.Gruet A; Dosnon M; Blocquel D; Brunel J; Gerlier D; Das RK; Bonetti D; Gianni S; Fuxreiter M; Longhi S, et al. , Fuzzy regions in an intrinsically disordered protein impair protein-protein interactions. FEBS J 2016, 283 (4), 576–594. [DOI] [PubMed] [Google Scholar]
- 39.Lee TS; Hu Y; Sherborne B; Guo Z; York DM, Toward fast and accurate binding affinity prediction with pmemdGTI: An efficient implementation of GPU-accelerated thermodynamic integration. J Chem Theory Comput 2017, 13 (7), 3077–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee TS; Cerutti DS; Mermelstein D; Lin C; LeGrand S; Giese TJ; Roitberg A; Case DA; Walker RC; York DM, GPU-accelerated molecular dynamics and free energy methods in Amber18: Performance enhancements and new features. J Chem Inf Model 2018, 58 (10), 2043–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salomon-Ferrer R; Gotz AW; Poole D; Le Grand S; Walker RC, Routine microsecond molecular dynamics simulations with Amber on GPUs. 2. Explicit solvent particle mesh ewald. J Chem Theory Comput 2013, 9 (9), 3878–3888. [DOI] [PubMed] [Google Scholar]
- 42.Gotz AW; Williamson MJ; Xu D; Poole D; Le Grand S; Walker RC, Routine microsecond molecular dynamics simulations with Amber on GPUs. 1. Generalized born. J Chem Theory Comput 2012, 8 (5), 1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le Grand S; Gotz AW; Walker RC, SPFP: Speed without compromise-A mixed precision model for GPU accelerated molecular dynamics simulations. Comput Phys Commun 2013, 184 (2), 374380. [Google Scholar]
- 44.Kurzbach D; Schwarz TC; Platzer G; Hofler S; Hinderberger D; Konrat R, Compensatory adaptations of structural dynamics in an intrinsically disordered protein complex. Angew Chem Int Ed Engl 2014, 53 (15), 3840–3843. [DOI] [PubMed] [Google Scholar]
- 45.Nagulapalli M; Parigi G; Yuan J; Gsponer J; Deraos G; Bamm VV; Harauz G; Matsoukas J; de Planque MR; Gerothanassis IP, et al. , Recognition pliability is coupled to structural heterogeneity: a calmodulin intrinsically disordered binding region complex. Structure 2012, 20 (3), 522533. [DOI] [PubMed] [Google Scholar]
- 46.Beier A; Schwarz TC; Kurzbach D; Platzer G; Tribuzio F; Konrat R, Modulation of correlated segment fluctuations in IDPs upon complex formation as an allosteric regulatory mechanism. J Mol Biol 2018, 430 (16), 2439–2452. [DOI] [PubMed] [Google Scholar]
- 47.Tuttle LM; Pacheco D; Warfield L; Luo J; Ranish J; Hahn S; Klevit RE, Gcn4-mediator specificity is mediated by a large and dynamic fuzzy protein-protein complex. Cell Rep 2018, 22 (12), 3251–3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bignon C; Troilo F; Gianni S; Longhi S, Modulation of Measles virus NTAIL interactions through fuzziness and sequence features of disordered binding sites. Biomolecules 2018, 9 (1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gely S; Lowry DF; Bernard C; Jensen MR; Blackledge M; Costanzo S; Bourhis JM; Darbon H; Daughdrill G; Longhi S, Solution structure of the C-terminal X domain of the measles virus phosphoprotein and interaction with the intrinsically disordered C-terminal domain of the nucleoprotein. J Mol Recognit 2010, 23 (5), 435–447. [DOI] [PubMed] [Google Scholar]
- 50.Kingston RL; Hamel DJ; Gay LS; Dahlquist FW; Matthews BW, Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc Natl Acad Sci U S A 2004, 101 (22), 8301–8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belle V; Rouger S; Costanzo S; Liquiere E; Strancar J; Guigliarelli B; Fournel A; Longhi S, Mapping alpha-helical induced folding within the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by site-directed spin-labeling EPR spectroscopy. Proteins 2008, 73 (4), 973988. [DOI] [PubMed] [Google Scholar]
- 52.Bischak CG; Longhi S; Snead DM; Costanzo S; Terrer E; Londergan CH, Probing structural transitions in the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by vibrational spectroscopy of cyanylated cysteines. Biophys J 2010, 99 (5), 1676–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morin B; Bourhis JM; Belle V; Woudstra M; Carriere F; Guigliarelli B; Fournel A; Longhi S, Assessing induced folding of an intrinsically disordered protein by site-directed spin-labeling electron paramagnetic resonance spectroscopy. J Phys Chem B 2006, 110 (41), 20596–20608. [DOI] [PubMed] [Google Scholar]
- 54.Wang Y; Chu X; Longhi S; Roche P; Han W; Wang E; Wang J, Multiscaled exploration of coupled folding and binding of an intrinsically disordered molecular recognition element in measles virus nucleoprotein. Proc Natl Acad Sci U S A 2013, 110 (40), E3743–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu W; Apostol I; Qasim MA; Warne N; Wynn R; Zhang WL; Anderson S; Chiang YW; Ogin E; Rothberg I, et al. , Binding of amino acid side-chains to S1 cavities of serine proteinases. J Mol Biol 1997, 266 (2), 441–461. [DOI] [PubMed] [Google Scholar]
- 56.Huang K; Lu W; Anderson S; Laskowski M Jr.; James MN, Water molecules participate in proteinase-inhibitor interactions: crystal structures of Leu18, Ala18, and Gly18 variants of turkey ovomucoid inhibitor third domain complexed with Streptomyces griseus proteinase B. Protein Sci 1995, 4 (10), 1985–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bateman KS; Anderson S; Lu W; Qasim MA; Laskowski M Jr.; James MN, Deleterious effects of beta-branched residues in the S1 specificity pocket of Streptomyces griseus proteinase B (SGPB): crystal structures of the turkey ovomucoid third domain variants Ile18I, Val18I, Thr18I, and Ser18I in complex with SGPB. Protein Sci 2000, 9 (1), 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horn JR; Ramaswamy S; Murphy KP, Structure and energetics of protein-protein interactions: the role of conformational heterogeneity in OMTKY3 binding to serine proteases. J Mol Biol 2003, 331 (2), 497–508. [DOI] [PubMed] [Google Scholar]
- 59.Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C, ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015, 11 (8), 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983, 79 (2), 926–935. [Google Scholar]
- 61.Case DA; Ben-Shalom IY; Brozell SR; Cerutti DS; Cheatham TE; Cruzeiro I,VWD; Darden TA; Duke RE; Ghoreishi D; Gilson MK, et al. , AMBER 2018. University of California, San Francisco, 2018. [Google Scholar]
- 62.Kaus JW; Pierce LT; Walker RC; McCammont JA, Improving the efficiency of free energy calculations in the Amber molecular dynamics package. J Chem Theory Comput 2013, 9 (9), 4131–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abul Qasim M; Ranjbar MR; Wynn R; Anderson S; Laskowski M Jr., Ionizable P1 residues in serine proteinase inhibitors undergo large pK shifts on complex formation. J Biol Chem 1995, 270 (46), 27419–27422. [DOI] [PubMed] [Google Scholar]
- 64.Blocquel D; Habchi J; Costanzo S; Doizy A; Oglesbee M; Longhi S, Interaction between the C-terminal domains of measles virus nucleoprotein and phosphoprotein: a tight complex implying one binding site. Protein Sci 2012, 21 (10), 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yegambaram K; Kingston RL, The feet of the measles virus polymerase bind the viral nucleocapsid protein at a single site. Protein Sci 2010, 19 (4), 893–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bonetti D; Troilo F; Toto A; Brunori M; Longhi S; Gianni S, Analyzing the folding and binding steps of an intrinsically disordered protein by protein engineering. Biochemistry 2017, 56 (29), 3780–3786. [DOI] [PubMed] [Google Scholar]
- 67.Teilum K; Olsen JG; Kragelund BB, Globular and disordered-the non-identical twins in protein-protein interactions. Front Mol Biosci 2015, 2, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meszaros B; Tompa P; Simon I; Dosztanyi Z, Molecular principles of the interactions of disordered proteins. J Mol Biol 2007, 372 (2), 549–561. [DOI] [PubMed] [Google Scholar]
- 69.Wong ET; Na D; Gsponer J, On the importance of polar interactions for complexes containing intrinsically disordered proteins. PLoS Comput Biol 2013, 9 (8), e1003192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chodera JD; Mobley DL; Shirts MR; Dixon RW; Branson K; Pande VS, Alchemical free energy methods for drug discovery: progress and challenges. Curr Opin Struct Biol 2011, 21 (2), 150160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maurer M; de Beer SB; Oostenbrink C, Calculation of relative binding free energy in the water-filled active site of oligopeptide-binding protein A. Molecules 2016, 21 (4), 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Deng Y; Roux B, Computation of binding free energy with molecular dynamics and grand canonical Monte Carlo simulations. J Chem Phys 2008, 128 (11), 115103. [DOI] [PubMed] [Google Scholar]
- 73.Luccarelli J; Michel J; Tirado-Rives J; Jorgensen WL, Effects of water placement on predictions of binding affinities for p38alpha MAP kinase Inhibitors. J Chem Theory Comput 2010, 6 (12), 3850–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ben-Shalom IY; Lin C; Kurtzman T; Walker RC; Gilson MK, Simulating water exchange to buried binding sites. J Chem Theory Comput 2019, 15 (4), 2684–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klein-Seetharaman J; Oikawa M; Grimshaw SB; Wirmer J; Duchardt E; Ueda T; Imoto T; Smith LJ; Dobson CM; Schwalbe H, Long-range interactions within a nonnative protein. Science 2002, 295 (5560), 1719–1722. [DOI] [PubMed] [Google Scholar]
- 76.Brunel J; Chopy D; Dosnon M; Bloyet LM; Devaux P; Urzua E; Cattaneo R; Longhi S; Gerlier D, Sequence of events in measles virus replication: role of phosphoprotein-nucleocapsid interactions. J Virol 2014, 88 (18), 10851–10863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bloyet LM; Brunel J; Dosnon M; Hamon V; Erales J; Gruet A; Lazert C; Bignon C; Roche P; Longhi S, et al. , Modulation of re-initiation of Measles virus transcription at intergenic regions by PXD to NTAIL binding strength. PLoS Pathog 2016, 12 (12), e1006058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wright PE; Dyson HJ, Linking folding and binding. Curr Opin Struct Biol 2009, 19 (1), 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dyson HJ; Wright PE, Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol 2002, 12 (1), 54–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.