

Abstract
Understanding the different genetic landscape between lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) is important for understanding the underlying molecular mechanism, which may facilitate the development of effective and precise treatments. Although previous studies have identified a number of differentially expressed genes (DEGs) responsible for lung cancer, it is unknown which of these genes are causal. The present study integrated DNA methylation, RNA sequencing, clinical characteristics and survival outcomes of patients with LUAD and LUSC from The Cancer Genome Atlas. DEGs were first identified using edgeR by comparing tumor and normal tissue, and differentially methylated probes (DMPs) were assessed using ChAMP. Candidate genes for further time-to-event instrumental variable analysis were selected as the intersecting genes between DEGs and the genes including DMP CpG sites within the transcription start site (TSS1500), with DMPs in TSS1500 region being the instrumental variables. Extensive sensitivity analyses were conducted to assess the robustness of the results. The present study identified 906 DEGs for LUAD, among which 538 also had DMPs in the TSS1500 region. In addition, 1,543 DEGs were identified for LUSC, among which 1,053 also had DMPs in the TSS1500 region. Time-to-event instrumental variable analysis detected eight potential causal genes for LUAD survival, including aryl hydrocarbon receptor nuclear translocator like 2, semaphorin 3G, serum deprivation-response protein, chloride intracellular channel protein 5, LIM zinc finger domain containing 2, epithelial membrane protein 2, carbonic anhydrase 7 and LOC116437. The results also identified that phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor 2 may be a potential causal gene for LUSC. Therefore, the results of the present study suggested that there was molecular heterogeneity between these two lung cancer subtypes. Such analysis framework can be extended to other cancer genomics research.
Keywords: lung cancer survival, omics integration, causal gene, methylation, instrumental variable analysis
Introduction
Lung cancer (LC) remains the most commonly diagnosed cancer type worldwide, with 11.6% of total cancer cases, and is the leading cause of cancer mortality, accounting for 18.4% of the total cancer-associated mortalities (1). Non-small cell lung carcinoma (NSCLC) accounts for ~80% of all LC types, with adenocarcinoma (LUAD) and squamous cell carcinoma (LUSC) being the two major histological types (2). LUAD and LUSC have different cells of origin, location within the lung and growth patterns, and can develop and progress via different molecular mechanisms (3–6). Understanding the molecular mechanisms underlying the progression and survival of LUAD and LUSC is essential, and identifying the genetic difference between them may facilitate development of suitable and precise treatment strategies (6–8). Previous studies have demonstrated that differentially expressed genes (DEGs) serve an important role in the progression of both LUAD and LUSC (9–11). Gantenbein et al (12) have identified that upregulation of eukaryotic translation initiation factor 6 in NSCLC is associated with poor overall survival in LUAD, but not in LUSC. Qu et al (13) have demonstrated that interleukin-6 prevents the initiation, but enhances the progression of LC in a mouse model. Immunohistochemical analysis by Huang et al (14) has revealed that p16 protein expression is associated with poor prognosis in LUSC.
However, previous studies have mainly focused on the single level-omic analysis, such as differential gene expression analysis, and primarily examined association rather than the causal relationship between gene expression and LC survival (15). While the establishment of the potential causal relationship is key for precise treatment of LC, it is difficult to conduct causal inference in observational studies due to bias, which results from reverse causation and unobserved confounding factors (16). A powerful statistical tool to examine the causal relationship between the modifiable exposure, such as gene expression, and the outcome variable of interest (such as LC survival) is instrumental variable analysis (IVA) (17–20). IVA uses specific instrumental variables to estimate and test the causal effect of the exposure variable of interest on the outcome variable, under the assumptions that the instrumental variables are strongly associated with the exposure (21). Furthermore, the instrumental variable is independent of the confounders between the exposure and the outcome, and the instrumental variable influences the outcome only through the exposure (22). Therefore, determining suitable instrumental variables is highly important in IVA (23).
Generally, gene expression measured at the transcript level affects clinical outcome or disease progression more directly compared with gene methylation measured at the DNA/epigenetics level (24–27). Biologically, for one specific gene, methylation sites within the unique function of transcript start site [e.g., within 1,500 bps ahead of a transcription start site (TSS), but not including the 200 bps ahead of the TSS (TSS1500)] can downregulate its expression, and deregulated expression can further influence survival outcome (28,29). In addition, deregulated methylation and gene expression level and event are time sequential (29). Previous studies have illustrated a correlation between DNA methylation in the gene promoter region and gene expression (30,31). However, in instrumental variable analysis, more instruments can provide higher power than compared with fewer instruments; TSS1500 regions include more CpG sites than TSS200, and thus CpG sites in the TSS1500 region of one gene can be selected as instrumental variables to explore the potential causal relationship between gene expression and cancer survival outcome. DNA methylation is a key epigenetic factor that regulates gene expression, which has been described in several multi-omics integrative analyses in cancer research (32–34).
In the present study, the aim to was to integrate DNA methylation (level 3), RNA sequencing (RNA-seq; level 3), clinical characteristics and survival outcome of patients with LUAD and LUSC from The Cancer Genome Atlas (TCGA). Differentially expressed genes (DEGs) and differentially expressed methylation positions (DMPs) were identified using tumor and normal tissue from patients with LUAD and LUAC. Furthermore, DMP CpG sites in the TSS1500 and DEG were paired by gene, and the regulatory association between them was assessed to identify candidate gene sets for subsequent time-to-event IVA, which was used to establish the potential causal effect of gene expression on LUAD and LUSC survival, and to investigate the different genetic difference between LUAD and LUSC. Various sensitivity analyses, including the weak instrumental association test, the heterogeneity among instrumental variables (IVs) and leave-one-out cross validation (LOOCV) analysis, were conducted to ensure the robustness for modeling misspecifications, and to improve the vailidity of the results.
Materials and methods
Software
R (version 3.6.1; http://www.R-project.org/) was used to conduct data processing and statistical analysis (35). edgeR (version 3.26.8) (36,37) and ChAMP (version 2.14.0) (38) were used with default settings for DEG and DMP analysis respectively. An R package TwoSLSanalysis, which is available on GitHub (https://github.com/LULIU1816/TwoSLSanalysis), was used to implement the time-to-event IVA.
Data collection and processing
Gene expression, RNA-Seq and the corresponding clinical data of patients with NSCLC were obtained from TCGA (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). Data were downloaded with published software TCGA-Assembler (version 2.0; http://www.compgenome.org/TCGA-Assembler) (39) and TCGAbiolinks (version 3.9; http://bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html) (40,41). DNA methylation was measured with Infinium HumanMethylation450 BeadChip (Illumina, Inc.) with 485,577 CpG sites, among which 84,242 methylation sites were located on the TSS1500. Gene expression was detected using the Illumina HiSeq2000 RNA Sequencing platform (Illumina, Inc.) with 20,502 transcripts.
To identify DEGs and DMPs, the methylation and gene expression data were used from paired tumor and normal tissue. In total, data from 50 pairs for LUSC and 57 pairs for LUAD were matched for DEG analysis, and data from 40 pairs for LUSC and 29 pairs for LUAD were obtained for DMP or different methylation region (DMR) analysis. For IVA, methylation, gene expression and clinical information (demographic characteristics, survival and treatment information) were downloaded from 504 patients with LUSC and 522 patients with LUAD. Information included age, sex and pack-years smoked (PYS) as covariates, as these have previously been reported to be associated with the survival of patients with LC (42,43). PYS was calculated by multiplying the average number of packs of cigarettes smoked per day by the number of years a person has smoked, which reflected smoking extent and history. Overall survival (OS) was regarded as the survival outcome and was defined as the time from diagnosis to death, and mortality was the censoring variable. Patients with missing PYS, survival time or methylation and gene expression information were excluded. In addition, 287 patients with LUSC and 280 patients with LUAD were included in the time-to-event IVA. The flow chart of all data processing and analysis is presented in Fig. 1.
Figure 1.
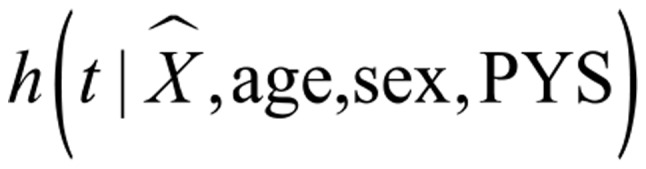
Flow chart of data processing and analysis. LUAD and LUSC followed the same process. First, the candidate gene sets were selected from overlapping DEGs and DMPs in TSS1500. Second, in stage I of IVA, the predicted expression value for each gene X was obtained by regressing the gene expression on the corresponding CpGs in TSS1500 with adjusted age, sex and PYS. In stage II of IVA, the potential causal effect was calculated by directly inputting the predicted gene expression value X into the hazard model with adjustments for age, sex and PYS. LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; DEGs, differentially expressed genes; DMPs, differentially methylated probes; IVA, instrumental variable analysis; PYS, pack-years smoked; TSS1500, 200-1,500 bp upstream of a transcription start site.
Identification of DEGs
The edgeR package was used to select the DEGs (36,37). Read count and reads per kilobase per million mapped reads matrix tables were extracted from classified TCGA RNA-Seq data to assess the DEGs. The trimmed mean of M-values method was used for normalization (44). In addition, the exact test, based on the quantile-adjusted conditional maximum likelihood methods (45), was used to define DEGs. Using previously described methods (46), the present study identified DEGs under the criteria that the absolute value of log2 fold-change (log-FC) of expression was >2 and the false discovery rate (FDR) was <0.05.
Identification of DMPs
ChAMP package (https://www.bioconductor.org/packages/release/bioc/vignettes/ChAMP/inst/doc/ChAMP.html) was used to identify the DMPs (41). ChAMP is an integrated analysis pipeline that includes functions for filtering low-quality probes based on detection P-values, chromosomal location, presence of single nucleotide polymorphisms in the probe sequence and cross-hybridization, adjustment for Infinium I and II probe design, batch effect correction used singular value decomposition, detecting DMPs, identifying DMRs and detection of copy number aberrations (41,47). LUAD- and LUSC-DMPs (P<0.05) were obtained from 485,577 CpGs after quality control and normalization.
Time-to-event IVA
Traditional two-stage regression was used to perform the time-to-event IVA. For one candidate gene, the instruments were the corresponding CpGs in the region of TSS1500 obtained by DMPs, and thus the number of IVs was gene-specific. Predicted gene expression value in the first stage was obtained by treating the differential methylation CpGs in the TSS1500 region as instrumental variables. In the second stage, the Cox regression model was run with the predicted gene expression used as the independent variable. The model used was as follows:
| (I) |
| (II) |
where Z is the methylation value of the TSS1500 region of a specific gene, and X is the predicted expression value of the specific gene. The present study defined the linear regression of gene expression on CpGs in the TSS1500 region, age, sex and PYS in model I. α1 is a px1 vector denoting the effect of CpGs on gene expression, p is the number of the instrumental variables of one specific gene. In model II, is a hazard function determined by the the predicted gene expression value X and covariates age, gender and PYS. h0(t) is the baseline hazard function. The prediction gene expression value X was directly plugged into the Cox model, and the parameter β1 represented the potential causal effect of gene expression on LC survival. A false discovery method was used to adjust multiple testing, and the threshold of FDR-q value was set to 0.15 (48). In addition, proportional hazards assumption was diagnosed by testing the correlation between the Schoenfeld residuals and survival time, with zero correlation indicating that the Cox model was valid (49).
Sensitivity analyses
Various sensitivity analyses were conducted to ensure the robustness for modeling misspecifications and to ensure the results were valid. Specifically, F statistic was used to test the weak instrumental bias. In addition, the I2-statisic was calculated to test the heterogeneity among instrumental variables, and leave-one-out cross validation (LOOCV) analysis was used to test whether one single instrumental variable may have a strong causal effect on gene expression. Weak association between instrumental variables and gene expression is observed if the F-statistics is <10, and heterogeneity among instrumental variables may exist when I2-statisic is >50% (50–52).
Results
Descriptive statistics
The demographic characteristics of the 567 patients with NSCLC are presented in Table I. For the 280 patients with LUAD, the median age was 67 years, and the proportion of female patients was 52.14%. The median PYS was 36.5 packs/year, and the median survival time was 216 months, with a 24.29% censoring rate. For the 287 patients with LUSC, the median age was 69 years, and the proportion of female patients was 26.13%. The median PYS was 50 packs/year, and the median survival time was 224 months, with a 29.90% censoring rate. No significant differences were observed in survival time (P=0.37), vital status (P=0.13) or history of other malignancy distributions (P=0.08) between LUAD and LUSC. However, age (P=0.007), sex (P=3.77×10−10), race (P=0.01), PYS (P=3.02×10−8), Kras gene analysis indicator (P=9.27×10−7) and epidermal growth factor receptor mutation status (P=1.38×10−7) were signifcantly different between LUAD and LUSC.
Table I.
Demographic and clinical characteristics for study populations.
| Variable | LUAD N=280 | LUSC N=287 | P-value |
|---|---|---|---|
| Age, median years (interquartile range) | 67.00 (13.25) | 69.00 (11.00) | 0.01a |
| Sex, n (%) | 3.77×10−10 | ||
| Female | 146 (52.14) | 75 (26.13) | |
| Male | 134 (47.86) | 212 (73.87) | |
| Ethnicity, n (%) | 0.01 | ||
| Asian | 2 (0.71) | 3 (1.05) | |
| Black or African American | 29 (10.36) | 19 (6.62) | |
| White | 226 (80.71) | 218 (75.96) | |
| Unknown | 23 (8.21) | 47 (16.38) | |
| Pack-years smoked, median (interquartile range) | 36.50 (30.00) | 50.00 (33.87) | 3.02×10−8a |
| Survival time, median months (interquartile range) | 216.00 (69.00) | 244.00 (975.00) | 0.37a |
| Dead, n (%) | 68 (24.29) | 87 (30.31) | 0.13 |
| History of other malignancy, n (%) | 0.08 | ||
| No | 227 (81.07) | 249 (86.76) | |
| Yes | 53 (18.93) | 38 (13.24) | |
| Kras gene analysis indicator, n (%) | 9.27×10−7 | ||
| No | 149 (53.21) | 205 (71.43) | |
| Yes | 38 (13.57) | 10 (3.48) | |
| Unknown | 93 (33.21) | 72 (25.09) | |
| EGFR mutation status, n (%) | 1.38×10−7 | ||
| No | 124 (44.29) | 186 (64.81) | |
| Yes | 49 (17.5) | 16 (5.57) | |
| Unknown | 107 (38.21) | 85 (29.62) |
P<0.05, Wilcoxon rank-sum test. LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; EGFR, epithelial growth factor receptor.
Time-to-event IVA for LUSC and LUAD
The present study identified 1,543 DEGs in LUSC and 906 DEGs in LUAD (Tables SI and SII). A total of 9,799 differentially methylated genes were located in genes in the TSS1500 regions for LUAD (Table SIII), among which 538 also differed in gene expression. In addition, 12,283 differentially methylated CpGs were located in genes in the TSS1500 regions for LUSC (Table SIV), among which 1,053 also differed in gene expression. In total, 538 genes in LUAD and 1,053 genes for LUSC were regarded as candidate genes after overlapping the DGEs and DMPs in the TSS1500 region (Table SV) for the downstream time-to-event IVA in order to identify potential causal genes related to the survival of patients with LC (Tables SVI and SVII). The present study only included 476 genes for LUAD and 922 genes for LUSC after removing genes with missing methylation data. In addition, the proportional hazards assumption in the second stage was confirmed to be valid for the correlation between the Schoenfeld residuals and survival time.
The results of the present study identified eight significant potential causal genes for LUAD survival and one significant causal gene in LUSC using FDR-q<0.15 (Table II). The causal genes for LUAD were aryl hydrocarbon receptor nuclear translocator like 2 (ARNTL2, HR=1.037; 95% CI: 1.017–1.056; P=1.81×10−4; FDR-q=0.029), semaphorin 3G (SEMA3G, HR=0.632; 95% CI: 0.504–0.792; P=6.79×10−5; FDR-q=0.029), serum deprivation-response protein (SDPR, HR=0.980; 95% CI: 0.969–0.990; P=1.38×10−4; FDR-q=0.029), chloride intracellular channel protein 5 (CLIC5, HR=0.987; 95% CI: 0.980–0.995; P=7.39×10−4; FDR-q=0.070), LIM zinc finger domain containing 2 (LIMS2, HR=0.924; 95% CI: 0.884–0.967; P=6.22×10−4; FDR-q=0.070), epithelial membrane protein 2 (EMP2, HR=0.997; 95% CI: 0.995–0.999; P=2.52×10−3; FDR-q=0.150), carbonic anhydrase 7 (CA7, HR=1.34×10−9; 95% CI: 2.3×10−15−7.0×10−4; P=2.53×10−3; FDR-q=0.150) and LOC116437 (HR=0.141; 95% CI: 0.040–0.496; P=2.25×10−3; FDR-q=0.150). The causal gene for LUSC was phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor 2 (PREX2, HR=1.958; 95% CI: 1.450–2.644; P=1.16×10−5; FDR-q=0.011). All HR values were calculated with 10-unit increment of gene expression.
Table II.
Result of time-to-event instrument variable analysis for causal genes.
| A, LUAD | ||||||
|---|---|---|---|---|---|---|
| Gene | Chr | Position | IVs | HR (95% CI) | P-value | FDR |
| ARNTL2 | 12 | 27,485,787-27,578,746 | cg26165146 | 1.037 (1.017–1.056) | 1.81×10−4 | 0.029 |
| cg17367616 | ||||||
| cg01986577 | ||||||
| SEMA3G | 3 | 52,467,268-52,479,112 | cg25134747 | 0.632 (0.504–0.792) | 6.79×10−5 | 0.029 |
| SDPR | 2 | 191,834,310- 191,847,088 | cg10082589 | 0.980 (0.969–0.990) | 1.38×10−4 | 0.029 |
| cg18843739 | ||||||
| CLIC5 | 6 | 45,866,188- 46,048,085 | cg23716866 | 0.987 (0.980–0.995) | 7.39×10−4 | 0.070 |
| cg14339765 | ||||||
| cg09347495 | ||||||
| LIMS2 | 2 | 128,395,996-128,439,360 | cg07262244 | 0.924 (0.884–0.967) | 6.22×10−4 | 0.070 |
| cg14282137 | ||||||
| cg08385249 | ||||||
| cg23966569 | ||||||
| cg22542731 | ||||||
| EMP2 | 16 | 10,622,279- 10,674,539 | cg04339790 | 0.997 (0.995–0.999) | 2.52×10−3 | 0.150 |
| CA7 | 16 | 66,878,282-66,888,052 | cg10352418 | 1.34×10−9 (2.33×10−15−7.0×10−4) | 2.53×10−3 | 0.150 |
| cg06438797 | ||||||
| cg11258532 | ||||||
| cg00182273 | ||||||
| LOC116437 | 12 | 131,649,556-131,697,476 | cg20183756 | 0.141 (0.040–0.496) | 2.25×10−3 | 0.150 |
| cg03859668 | ||||||
| B, LUSC | ||||||
| Gene | Chr | Position | IVs | HR (95% CI) | P-value | FDR |
| PREX2 | 8 | 68,864,244-69,143,897 | cg13652336 | 1.958 (1.450–2.644) | 1.16×10−5 | 0.011 |
| cg16009633 | ||||||
| cg11549615 | ||||||
| cg05293738 | ||||||
| cg17747005 | ||||||
IVs, instrumental variables; Chr, chromosome; HR, hazard ratio; 95% CI, 95% confidence interval; FDR, false discovery rate; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma.
Sensitivity analyses
F-statistics of all causal genes used to detect the weak instrumental bias were <10, which indicated a weak association between instruments and gene expression due to the small number of instrumental DMP sites within each gene (Table SVIII). Despite this, all the significant genes were still to be identified powerfully.
The present study performed a heterogeneity test to identify any instrumental outliers that may affect the results. The results demonstrated that the I2-statistics were 75.9, 78.2 and 63.1% for ARNTL2, CLIC5 and PREX2, respectively (Table SVIII). To address the heterogeneity among the IVs, instrumental outliers were removed; similar results were obtained when removing the instrumental outliers. In addition, LOOCV analysis identified no causal genes with outlier single instrument for both LUAD and LUSC (Table SVIII).
Discussion
The present study integrated DNA methylation, RNA-seq, clinical charancteristics and survival outcomes from TCGA to investigate the potentical causal relationship between gene expression and LUAD and LUSC survival, respectively.
The identified causal relationship between gene expression and survival of disease was robust with respect to the choice of statistical methods, and was assessed with various sensitivity analyses. Non-overlapping causal genes between LUAD and LUSC further highlighted the heterogeneity between these two subtypes of LC. From the two-stage time-to-event IVA, the present results indicated the potential causal role of ARNTL2, SEMA3G, SDPR, CLIC5, LIMS2, EMP2, CA7 and LOC116437 in LUAD survival, and PREX2 in LUSC survival. In addition, the present study identified pivotal regulatory genes, the expression levels of which were upregulated with poor survival, including PREX2 in LUSC and ARNTL2 in LUAD. Furthermore, several genes with downregulated expression levels associated with poor survival were identified, including SEMA3G, SDPR, CLIC5, LIMS2, EMP2, CA7 and LOC116437 in LUAD. The causal effect of gene expression and NSCLC suggested that these genes may be potential epigenetic therapeutic targets.
The majority of the potential causal genes identified in the present study have also been detected by previous studies, which have demonstrated a possible association with the prognosis in NSCLC. ARNTL2 drives metastatic self-sufficiency by orchestrating the expression of a complex pro-metastatic secretome, and high ARNTL2 expression predicts poor survival among patients with LUAD (53). In addition, SEMA3G is a potential transcription gene associated with cancer susceptibility candidate 9, and is significantly associated with the malignant progression of LUSC (54). A previous study using Oncomine and TCGA databases has demonstrated that low expression of CLIC5 is associated with poor overall survival after adjusting for age, sex and PYS (55). In addition, EPAS1, a transcription factor that serves a vital role in tumor progression, has been reported to directly regulate the LUAD-associated genes EMP2 and LIMS2 (56). It has been identified that upregulation of CA7 in tissues from resectable NSCLC is a biomarker of good prognosis (57). As LUAD is a major subtype of NSCLC, CA7 may have the same effect on LUAD. A xenograft study demonstrated that SDPR may elicit a metastasis suppressor function by directly interacting with ERK and have a limited pro-survival role (58). A previous study has reported that somatic alterations in PREX2 modulate the activity of immunomodulators, according to a significant overlap between the Master Regulator- and SYGNAL-PanImmune, which is associated with survival across all cancer types (59). Thus, upregulated PREX2 may lead to a short survival time, but this has not been identified in previous studies. In addition, there is no previous evidence that LOC116437 is the potential causal gene in NSCLC.
The analysis pipeline used in the present study can be considered as a gene-centered data integration method by combining multi-omics data with clinical information. One single level of genomic measurements can be insufficient to fully exploit the knowledge underlying the etiology of cancer prognosis. Based on the follow-up data from TCGA, gene expression was used as the exposure variable, and survival time was the censored outcome variable to avoid the reverse causation. For any one specific gene, DMP sites within the promoter region TSS1500 were used as instrumental variables, due to the biologically plausible assumption that CpG sites in TSS1500 must first regulate gene expression before affecting the survival. However, it may be necessary to include additional instrumental variables to increase the power of IVA. The present study only used DMPs within the functional region of TSS1500, rather than including DMPs within the gene body. Since DNA methylation in the gene body can be associated with survival outcome through changes in gene expression and some alternative mechanisms, these may possess the possibility of violating the instrumental variable assumptions (60). The present study performed extensive sensitivity analyses to ensure the robustness of the results and to prevent any possible model assumption violation in the IVA.
However, the present study has certain limitations. First, similar to other IVA studies, the present study assumed a linear relationship between DMPs in the promoter region and the corresponding gene expression. While a linear relationship can be considered a first-order approximation to any non-linear relationship, modeling a linear relationship can be suboptimal in terms of power if the true relationship is non-linear. Second, the censored rate of TCGA cohort was relatively high. Considering the heterogeneity and various manifestations of NSCLC, the present results should be verified in larger samples to evaluate the findings among specific subgroups. Furthermore, the present results should be interpreted with caution among other populations. The analysis framework could be extended to other ethnicities to detect the possible differences. In addition, several studies have demonstrated that when the same dataset is used for the selection of IVs and the estimation of instrument-exposure effect, substantial selection bias occurs even if the selection threshold is very stringent (61,62). Therefore, further studies are required to investigate other independent samples to select IVs.
Supplementary Material
Acknowledgements
Not applicable.
Funding
This work was supported by The National Natural Science Foundation of China (grant nos. 81673272, 81703321 and 81872712), the Natural Science Foundation of Shandong Province (grant no. ZR2019ZD02) and the Young Scholars Program of Shandong University (grant no. 2016WLJH23).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
ZY and SY conceived the study. LL contributed to data analysis, with assistance from SY and ZY. SY and PZ contributed to the data interpretation. LL, SY and ZY wrote the manuscript with participation from all other authors. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC. IARC Press Oxford University Press (distributor); Lyon Oxford: 2004. Pathology and genetics of tumours of the lung, pleura, thymus and heart (WHO classification of tumours) [Google Scholar]
- 3.McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, Caporaso NE, Johansson M, Xiao X, Li Y, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet. 2017;49:1126–1132. doi: 10.1038/ng.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen M, Liu X, Du J, Wang XJ, Xia L. Differentiated regulation of immune-response related genes between LUAD and LUSC subtypes of lung cancers. Oncotarget. 2017;8:133–144. doi: 10.18632/oncotarget.13346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu B, Chen Y, Yang J. LncRNAs are altered in lung squamous cell carcinoma and lung adenocarcinoma. Oncotarget. 2017;8:24275–24291. doi: 10.18632/oncotarget.13651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Relli V, Trerotola M, Guerra E, Alberti S. Distinct lung cancer subtypes associate to distinct drivers of tumor progression. Oncotarget. 2018;9:35528–35540. doi: 10.18632/oncotarget.26217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 8.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 9.Jiang L, Zhu W, Streicher K, Morehouse C, Brohawn P, Ge X, Dong Z, Yin X, Zhu G, Gu Y, et al. Increased IR-A/IR-B ratio in non-small cell lung cancers associates with lower epithelial-mesenchymal transition signature and longer survival in squamous cell lung carcinoma. BMC Cancer. 2014;14:131. doi: 10.1186/1471-2407-14-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bosse Y, Amos CI. A decade of GWAS results in lung cancer. Cancer Epidemiol Biomarkers Prev. 2018;27:363–379. doi: 10.1158/1055-9965.EPI-16-0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Välk K, Vooder T, Kolde R, Reintam MA, Petzold C, Vilo J, Metspalu A. Gene expression profiles of non-small cell lung cancer: Survival prediction and new biomarkers. Oncology. 2010;79:283–292. doi: 10.1159/000322116. [DOI] [PubMed] [Google Scholar]
- 12.Gantenbein N, Bernhart E, Anders I, Golob-Schwarzl N, Krassnig S, Wodlej C, Brcic L, Lindenmann J, Fink-Neuboeck N, Gollowitsch F, et al. Influence of eukaryotic translation initiation factor 6 on non-small cell lung cancer development and progression. Eur J Cancer. 2018;101:165–180. doi: 10.1016/j.ejca.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Qu Z, Sun F, Zhou J, Li L, Shapiro SD, Xiao G. Interleukin-6 prevents the initiation but enhances the progression of lung cancer. Cancer Res. 2015;75:3209–3215. doi: 10.1158/0008-5472.CAN-14-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang CI, Taki T, Higashiyama M, Kohno N, Miyake M. p16 protein expression is associated with a poor prognosis in squamous cell carcinoma of the lung. Br J Cancer. 2000;82:374–380. doi: 10.1054/bjoc.1999.0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas ML, Marcato P. Epigenetic modifications as biomarkers of tumor development, therapy response, and recurrence across the cancer care continuum. Cancers (Basel) 2018;10(pii):E101. doi: 10.3390/cancers10040101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burgess S, Thompson DJ, Rees JMB, Day FR, Perry JR, Ong KK. Dissecting causal pathways using mendelian randomization with summarized genetic data: Application to age at menarche and risk of breast cancer. Genetics. 2017;207:481–487. doi: 10.1534/genetics.117.300191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright PG. The Tariff on Animal and Vegetable Oils. In: Moulton HG, editor. The Macmillan Company; New York, NY: 1928. [Google Scholar]
- 18.Davies NM, Smith GD, Windmeijer F, Martin RM. Issues in the reporting and conduct of instrumental variable studies: A systematic review. Epidemiology. 2013;24:363–369. doi: 10.1097/EDE.0b013e31828abafb. [DOI] [PubMed] [Google Scholar]
- 19.Palmer TM, Sterne JA, Harbord RM, Lawlor DA, Sheehan NA, Meng S, Granell R, Smith GD, Didelez V. Instrumental variable estimation of causal risk ratios and causal odds ratios in Mendelian randomization analyses. Am J Epidemiol. 2011;173:1392–1403. doi: 10.1093/aje/kwr026. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Briesacher BA. Use of instrumental variable in prescription drug research with observational data: A systematic review. J Clin Epidemiol. 2011;64:687–700. doi: 10.1016/j.jclinepi.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Angrist JD, Imbens GW, Rubin DB. Identification of causal effects using instrumental variables. J Am Stat Assoc. 1996;91:444–455. doi: 10.1080/01621459.1996.10476902. [DOI] [Google Scholar]
- 22.Pearl J. Causality: Models, Reasoning, and Inference. In: Harvey A, editor. Cambridge University Press; Cambridge, New York: 2000. [Google Scholar]
- 23.Baser O. Too much ado about instrumental variable approach: Is the cure worse than the disease? Value Health. 2009;12:1201–1209. doi: 10.1111/j.1524-4733.2009.00567.x. [DOI] [PubMed] [Google Scholar]
- 24.Glinsky GV. Integration of HapMap-based SNP pattern analysis and gene expression profiling reveals common SNP profiles for cancer therapy outcome predictor genes. Cell Cycle. 2006;5:2613–2625. doi: 10.4161/cc.5.22.3498. [DOI] [PubMed] [Google Scholar]
- 25.Fabiani E, Leone G, Giachelia M, D'alo' F, Greco M, Criscuolo M, Guidi F, Rutella S, Hohaus S, Voso MT. Analysis of genome-wide methylation and gene expression induced by 5-aza-2′-deoxycytidine identifies BCL2L10 as a frequent methylation target in acute myeloid leukemia. Leuk Lymphoma. 2010;51:2275–2284. doi: 10.3109/10428194.2010.528093. [DOI] [PubMed] [Google Scholar]
- 26.Wang W, Baladandayuthapani V, Morris JS, Broom BM, Manyam G, Do KA. iBAG: Integrative bayesian analysis of high-dimensional multiplatform genomics data. Bioinformatics. 2013;29:149–159. doi: 10.1093/bioinformatics/bts655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Tayrac M, Le S, Aubry M, Mosser J, Husson F. Simultaneous analysis of distinct omics data sets with integration of biological knowledge: Multiple Factor Analysis approach. BMC Genomics. 2009;10:32. doi: 10.1186/1471-2164-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, Wu X, Wen L, Tang F, Huang Y, Peng J. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016;26:304–319. doi: 10.1038/cr.2016.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith AA, Huang YT, Eliot M, Houseman EA, Marsit CJ, Wiencke JK, Kelsey KT. A novel approach to the discovery of survival biomarkers in glioblastoma using a joint analysis of DNA methylation and gene expression. Epigenetics. 2014;9:873–883. doi: 10.4161/epi.28571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lokk K, Modhukur V, Rajashekar B, Märtens K, Mägi R, Kolde R, Koltšina M, Nilsson TK, Vilo J, Salumets A, Tõnisson N. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014;15:r54. doi: 10.1186/gb-2014-15-4-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saif I, Kasmi Y, Allali K, Ennaji MM. Prediction of DNA methylation in the promoter of gene suppressor tumor. Gene. 2018;651:166–173. doi: 10.1016/j.gene.2018.01.082. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Baggerly KA, Orouji E, Manyam G, Chen H, Lam M, Davis JS, Lee MS, Broom BM, Menter DG, et al. Gene-specific methylation profiles for integrative methylation-expression analysis in cancer research. https://doi org/10.1101/618033. [Apr 24;2019 ];bioRxiv. 2019 [Google Scholar]
- 33.Denis M, Tadesse MG. Evaluation of hierarchical models for integrative genomic analyses. Bioinformatics. 2016;32:738–746. doi: 10.1093/bioinformatics/btv653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Q, Shi X, Xie Y, Huang J, Shia B, Ma S. Combining multidimensional genomic measurements for predicting cancer prognosis: Observations from TCGA. Brief Bioinform. 2015;16:291–303. doi: 10.1093/bib/bbu003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Team RC. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 2019. https://www.R-project.org/ [Google Scholar]
- 36.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris TJ, Butcher LM, Feber A, Teschendorff AE, Chakravarthy AR, Wojdacz TK, Beck S. ChAMP: 450k chip analysis methylation pipeline. Bioinformatics. 2014;30:428–430. doi: 10.1093/bioinformatics/btt684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu Y, Qiu P, Ji Y. TCGA-assembler: Open-source software for retrieving and processing TCGA data. Nat Methods. 2014;11:599–600. doi: 10.1038/nmeth.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. TCGAbiolinks: An R/bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44:e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mounir M, Lucchetta M, Silva TC, Olsen C, Bontempi G, Chen X, Noushmehr H, Colaprico A, Papaleo E. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput Biol. 2019;15:e1006701. doi: 10.1371/journal.pcbi.1006701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janjigian YY, McDonnell K, Kris MG, Shen R, Sima CS, Bach PB, Rizvi NA, Riely GJ. Pack-years of cigarette smoking as a prognostic factor in patients with stage IIIB/IV nonsmall cell lung cancer. Cancer. 2010;116:670–675. doi: 10.1002/cncr.24813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peto J. That the effects of smoking should be measured in pack-years: Misconceptions 4. Brit J Cancer. 2012;107:406–407. doi: 10.1038/bjc.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson MD, Smyth GK. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics. 2008;9:321–332. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- 46.Crow M, Lim N, Ballouz S, Pavlidis P, Gillis J. Predictability of human differential gene expression. Proc Natl Acad Sci USA. 2019;116:6491–6500. doi: 10.1073/pnas.1802973116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian Y, Morris TJ, Webster AP, Yang Z, Beck S, Feber A, Teschendorff AE. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 2017;33:3982–3984. doi: 10.1093/bioinformatics/btx513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, Jiang P, Shen H, Aster JC, Rodig S, et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016;17:174. doi: 10.1186/s13059-016-1028-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grambsch PM, Therneau TM. Proportional Hazards Tests and Diagnostics Based on Weighted Residuals. Biometrika. 1994;81:515–526. doi: 10.1093/biomet/81.3.515. [DOI] [Google Scholar]
- 50.Staiger D, Stock JH. Instrumental variables regression with weak instruments. Econometrica. 1997;65:557–586. doi: 10.2307/2171753. [DOI] [Google Scholar]
- 51.Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–1163. doi: 10.1002/sim.3235. [DOI] [PubMed] [Google Scholar]
- 52.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brady JJ, Chuang CH, Greenside PG, Rogers ZN, Murray CW, Caswell DR, Hartmann U, Connolly AJ, Sweet-Cordero EA, Kundaje A, Winslow MM. An Arntl2-driven secretome enables lung adenocarcinoma metastatic self-sufficiency. Cancer Cell. 2016;29:697–710. doi: 10.1016/j.ccell.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao L, Guo YN, Zeng JH, Ma FC, Luo J, Zhu HW, Xia S, Wei KL, Chen G. The expression, significance and function of cancer susceptibility candidate 9 in lung squamous cell carcinoma: A bioinformatics and in vitro investigation. Int J Oncol. 2019;54:1651–1664. doi: 10.3892/ijo.2019.4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu W, Ouyang S, Zhou Z, Wang M, Wang T, Qi Y, Zhao C, Chen K, Dai L. Identification of genes associated with cancer progression and prognosis in lung adenocarcinoma: Analyses based on microarray from oncomine and the cancer genome Atlas databases. Mol Genet Genomic Med. 2019;7:e00528. doi: 10.1002/mgg3.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, Xie D, He Z, Zheng L. Integrated analysis reveals five potential ceRNA biomarkers in human lung adenocarcinoma. PeerJ. 2019;7:e6694. doi: 10.7717/peerj.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ilie MI, Hofman V, Ortholan C, Ammadi RE, Bonnetaud C, Havet K, Venissac N, Mouroux J, Mazure NM, Pouysségur J, Hofman P. Overexpression of carbonic anhydrase XII in tissues from resectable non-small cell lung cancers is a biomarker of good prognosis. Int J Cancer. 2011;128:1614–1623. doi: 10.1002/ijc.25491. [DOI] [PubMed] [Google Scholar]
- 58.Ozturk S, Papageorgis P, Wong CK, Lambert AW, Abdolmaleky HM, Thiagalingam A, Cohen HT, Thiagalingam S. SDPR functions as a metastasis suppressor in breast cancer by promoting apoptosis. Proc Natl Acad Sci USA. 2016;113:638–643. doi: 10.1073/pnas.1514663113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. The immune landscape of cancer. Immunity. 2018;48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koellinger PD, de Vlaming R. Mendelian randomization: The challenge of unobserved environmental confounds. Int J Epidemiol. 2019;48:665–671. doi: 10.1093/ije/dyz138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7(pii):e34408. doi: 10.7554/eLife.34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haycock PC, Burgess S, Wade KH, Bowden J, Relton C, Davey Smith G. Best (but oft-forgotten) practices: The design, analysis, and interpretation of Mendelian randomization studies. Am J Clin Nutr. 2016;103:965–978. doi: 10.3945/ajcn.115.118216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.