

Abstract
Purpose of the Review
Osteoarthritis (OA) is an aging-associated and injury-induced joint disease characterized by cartilage degradation, bone sclerosis, and persistent low-grade inflammation in the joint. Aging and injury are triggers of joint pathological changes mediated by pro-inflammatory factors, some of which are secreted by white adipose tissue. Adipokines including adiponectin, leptin, resistin, chemerin, IL-6, and TNF-α are major players not only during inflammation but also in metabolic regulation of joint cells including chondrocytes, osteoblasts, osteoclasts as well as mesenchymal stem cells. The purpose of this review is to summarize the signal transduction pathways of adipokines in the articular joint to provide new information on potential targets for intervention of OA.
Recent Findings
The risk of knee osteoarthritis is associated with adipokine gene polymorphism. While the infrapatellar fat pad is a major source of adipokines in knee synovial fluid, adipocytes also accumulate in the bone marrow during aging and obesity. Adipokines can act as SASPs (senescence associated secretory phenotype factors) that participate in cellular senescence of chondrocytes, but they also regulate energy metabolism impacting bone remodeling. Thus, adipokines are closely related to the metabolic syndrome and degenerative pathological changes in cartilage and bone during OA.
Summary
Modulating the effects of adipokines on different cell types in the intra-articular joint will be a promising new option for OA intervention.
Keywords: Adipokine, Osteoarthritis, SASPs, Chondrocytes, Bone cells
Introduction
Osteoarthritis (OA) is a chronic disease leading to cartilage degradation, synovial inflammation, subchondral bone remodeling, and the formation of osteophytes. Obesity is a common metabolic syndrome caused by excess fat accumulation in the body due to disorders of fat metabolism. Previous research has demonstrated a strong association between knee OA and obesity [1–5]. Obesity not only causes increased weight bearing by the joint but also enhances adipokine production by the adipose tissue, thereby contributing to inflammatory or autoimmune diseases [6, 7].
Adipocytes, the major cellular component of adipose tissue, have the ability to synthesize and release physiologically active molecules such as adiponectin, leptin, resistin, and chemerin, as well as inflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor (TNF-α) [8]. These secretory products of adipocytes are named adipokines. While the infrapatellar fat pad (IFP) is a major source of adipokines in knee synovial fluid, adipocytes also accumulate in other joint fat pads and the bone marrow during aging and obesity (Fig. 1). IFP is also in close contact with the synovium, into which blood vessels and immune cells can infiltrate (Fig. 1). Thus, IFP can be viewed not only as a structural cushion to absorb shock in the knee joint but also an endocrine or paracrine tissue to regulate inflammation in the joint. Although adipocytes are major sources of adipokines, some adipokines are also synthesized by other resident joint cells, including chondrocytes, synoviocytes, osteoblasts, stromal cells, macrophages, and immune cells (Table 1). Furthermore, adipokine receptors are present in many types of joint cells, thus forming a complex regulatory network of cellular “adipo-interaction” [8]. Thus, adipokines may contribute to synovial inflammation, matrix metalloproteinase (MMP) production, cartilage degeneration, and bone remodeling during OA pathogenesis (Fig. 2).
Fig. 1.
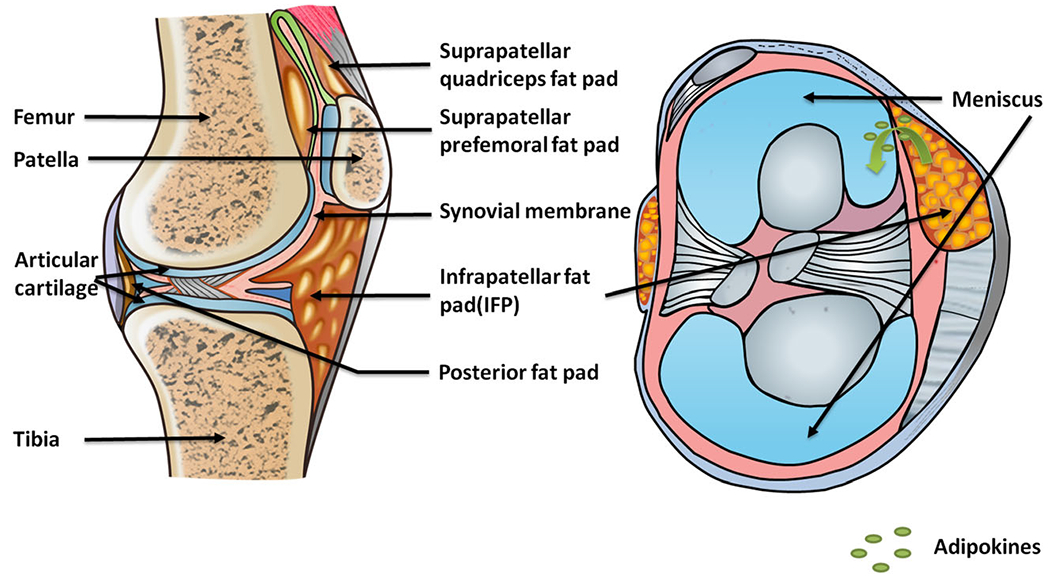
The relative position of fat pads in the knee joint. There are multiple fat pads in the knee joint, including infrapatellar, posterior, suprapatellar prefemoral, and suprapatellar quadriceps. All of them contain adipose tissues, which may serve both structural and regulatory roles. The regulatory roles are mainly achieved by signaling of adipokines that are secreted by adipocytes
Table 1.
Summary of adipokine properties
| Cell source | Receptor | Form | Signaling pathway | Regulatory function | |
|---|---|---|---|---|---|
| Adiponectin | Adipocyte | AdipoR1 AdipoR2 T-cadherin |
Full-length adiponectin, globular adiponectin | PPARα, AMPK/m-TOR | Glucose and lipid metabolism |
| Leptin | Adipocyte, Chondrocyte | OB-R | Free form, Inactive form associated with receptor (OB-Re) | JAK/STAT ERK1/2, p38, JNK, PKC, SHP2/GRB2, PI3K/Akt | Food intake, energy consumption |
| Resistin | Adipocyte, osteoblast, osteoclast, chondrocyte, macrophage | TLR4 | Soc-3, STAT3, NF-KB, C/EBPβ | Insulin resistance, glucose homeostasis | |
| Chemerin | Adipocyte, immune cell | CMKLR1, ChemR23 | Precursor form, activation form | ERK1/2, Akt | Adipocyte differentiation, lipolysis, insulin resistance |
| IL-6 | Adipocyte, myocyte, osteoblast, macrophage | Membrane bound IL-6R, soluble IL-6R, gp130 | Trans-signaling, classic signaling | Liver regeneration, anti-inflammatory, pro-inflammatory | |
| TNF-alpha | Adipocyte, immune cell | TNF-R1, TNF-R2 | mTNF-α, sTNF-α | P38MAPK, NF-κB, NOTCH | Bone formation, induce inflammation process |
Fig. 2.
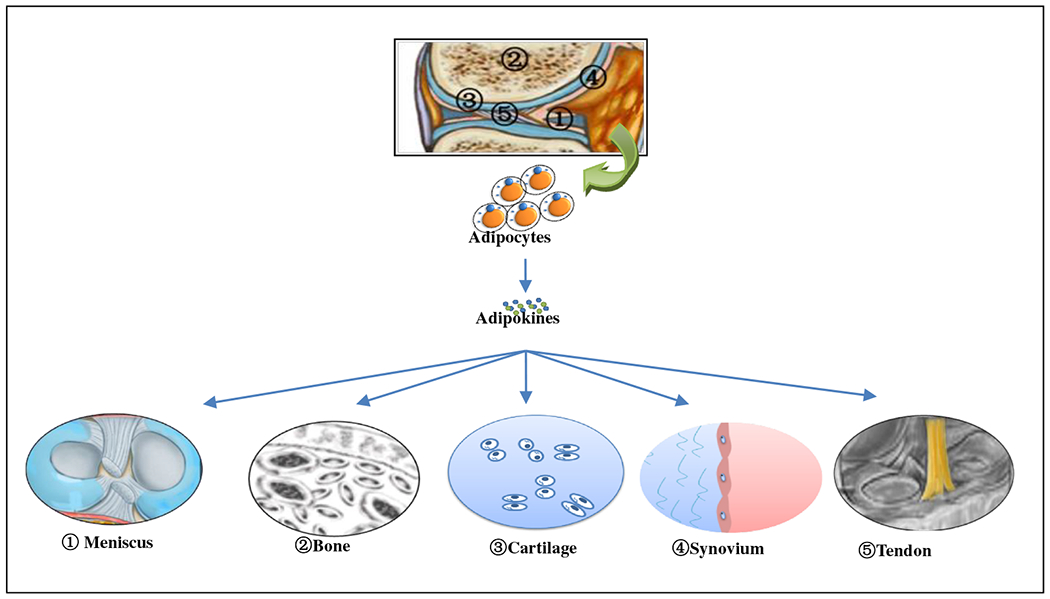
Adipokine signaling in the joint. Within the joint capsule, fad pads are in close proximity with other joint tissues, such as meniscus, bone, cartilage, synovium, and tendon. The infrapatellar fat pad secretes adipokines thereby affecting neighboring tissues biochemically. Infiltrating through synovial membrane, adipokines also become part of synovial fluid and affect cells at more distant sites in the joint. Since adipokine receptors are widely distributed in different types of cells, adipokine can regulate cellular functions of different joint tissues, including meniscus, bone, cartilage, synovium, and tendon
Osteoarthritis and Adipokines
The association between adipokines and OA is multi-factorial. Genetically, a SNP rs182052 in the ADIPOQ gene that encodes adiponectin may potentially modify individual susceptibility to knee OA in the Chinese population [9•]. Thus, a polymorphism in the adiponectin gene has been linked to OA. Biochemically, adiponectin is at higher levels in the circulation and in joint tissues of patients with rheumatoid arthritis (RA) and OA, and thus it is considered as a biomarker of arthritis [6, 7, 10]. Functionally, adipokines play a regulatory role affecting the metabolic balance in the joints through regulation of cytokine, chemokines, matrix-degrading enzymes, and cell growth and differentiation factors [11]. Chronologically, OA is associated with aging. The total amount of fat in the IFP is increased in aged people, compared with younger people, leading to the increase of adipokines during aging [12, 13]. In addition, joint cells undergo senescence induced by replicative exhaustion, telomere shortening, oxidative stress, and chromatin damage during OA [14]. One prominent feature of senescent cells is that they express senescence-associated secretory phenotype factors (SASPs), which include pro-inflammatory cytokines [15]. Adipokines, including some of the cytokines defined as true SASPs such as IL-6 and TNF-α, are up-regulated in OA synovial fluid [16], readily infiltrate into cartilage, and activate the degenerative cascade [12, 13]. Different types of adipokines and their roles in OA are described as follows, with emphasis on adiponectin as a prototypical adipokine.
Adipokine
Adiponectin
Structure
Adiponectin (APN), also called Acrp30, AdipoQ, ApM1, and GBP28, is a hormone secreted exclusively by adipocytes [17, 18]. Human adiponectin gene spanned 17 kb on chromosome 3q27, consisting of three exons and two introns. Adiponectin protein is a 28–30 kDa collagen-like protein containing 244 amino acids. The basic structure of adiponectin is that of a trimer [19]. The heterotrimers consist of a full-length adiponectin (fAd) or a smaller globular adiponectin (gAd) generated by proteolytic cleavage [20]. Adiponectin can form a wide range of multimers from low molecular weight (trimers), medium molecular weight (hexamers) to high molecular weight (HMW) multimers connected by disulfide bonds at the amino terminus [19].
Adiponectin Receptors
Three adiponectin receptors have been identified: adiponectin receptors 1 and 2 (AdipoR1 and AdipoR2) and a small adiponectin receptor, T-cadherin [21, 22]. AdipoR1 is expressed abundantly in liver, skeletal muscle, macrophages, and hypothalamus, while AdipoR2 is expressed in liver, white adipose tissue (WAT), and vasculature. AdipoR1 is a high-affinity receptor for globular adiponectin and a low-affinity receptor for full-length adiponectin, whereas AdipoR2 is an intermediate-affinity receptor for both full-length and globular adiponectin [21, 23–26]. T-cadherin is a unique cell adhesion molecule anchored to the cell surface membrane through a glycosylinositol (GPI) moiety. The extracellular portion of T-cadherin contains five ectodomains, having the ability to bind the physiological high-molecular weight (HWM) APN isoforms in vitro.
Adiponectin Signaling Pathways
AdipoR1 and AdipoR2 are receptors for globular and full-length adiponectin that mediate antidiabetic effects. Adiponectin binding to AdipoR1 activates peroxisome proliferation-activated receptor (PPARα), AMPK and p38MAPK [27]. While PPARα activation is involved in adiponectin-stimulated fatty-acid oxidation, AMPK and p38MAPK may be involved in adiponectin-stimulated fatty-acid oxidation and glucose uptake [22]. The signaling pathway activated by adiponectin binding to T-cadherin is still not clear.
Adiponectin Function
As an anti-diabetic and anti-atherogenic adipokine, adiponectin plays important roles in the regulation of glucose and lipid metabolism [8, 21, 22]. APN concentrations in the circulation of lean healthy individuals range between 5 and 30 μg/ml and reduced in obese and insulin-resistant human subjects and in animal models of metabolic syndrome [21, 22]. The reduction of plasma adiponectin levels by genetic or nutritional factors is one of the important causes of type 2 diabetes. Adiponectin reduces glucose content in tissues and up-regulates insulin signaling. This insulin-sensitizing effect of adiponectin seems to be mediated by increased fatty-acid oxidation through activation of AMP kinase and PPARα [27].
Role of Adiponectin in OA Pathogenesis
In obese patients, OA is a comorbidity with diabetes, cardiovascular, and respiratory diseases [1]. Obesity may contribute to OA pathogenesis in two ways, by increasing joint load and mechanical wear of the joints, and by promoting systemic and local inflammation. Body composition studies demonstrated a direct association of obesity with the risk of knee OA, suggesting that weight loss strategies for knee OA should focus on reducing excess fat tissue [2, 3]. Partial or full IFP resection has been used to reduce knee pain resulting from IFP inflammation and impingement during total knee arthroplasty [28]. The clinical outcome of IFP resection in OA patients is controversial [28, 29]. For successful IFP resection, it is necessary to assess the pathology of the IFP before surgical intervention. Therefore, the development of pharmacological intervention to address IFP-derived inflammation and pain will require our understanding of the functions of adipokines such as adiponectin.
Adiponectin may have anti-inflammatory effects on chondrocytes, thereby protecting cartilage from degeneration. Adiponectin treatment of primary chondrocytes up-regulates tissue inhibitor of metalloproteinase-2 and down-regulates IL-1β-mediated MMP13 gene expression. Treatment of rat articular chondrocytes with globular adiponectin (gAd) induces autophagy by AMPK/m-TOR activation and attenuates H2O2-induced apoptosis [30•]. Decreased serum adiponectin is also associated with aseptic loosening 10 years after total hip replacement [31]. Thus, patients with high serum adiponectin have a longer durability of joint replacement than patients with low levels of adiponectin.
Adiponectin promotes osteogenic differentiation of human bone marrow stem cells via the Wnt/β-catenin pathway [32]. Adiponectin increases the expression of osteogenic markers, including osteocalcin, alkaline phosphatase, and Runx2 and decreases PTEN, which suppresses osteoblast activity and bone mineral density [33]. Furthermore, adiponectin regulates bone metabolism by inhibiting osteoclastic differentiation and promoting osteoblastic commitment through APPL1/phosphoinositide 3-kinase (PI3K)/Akt signaling pathway in vitro and in vivo [34, 35].
However, during RA, adiponectin aggravates bone erosion by promoting the production of osteopontin which recruits osteoclasts in synovial tissue [36•]. Adiponectin inhibits osterix and mineralization capacity in RA-derived-primary human osteoblasts and increases IL-8 secretion in osteoclasts and their bone resorptive activity [37]. Although the pro-inflammatory effect of adiponectin in RA seems contrary to its anti-inflammatory property in OA, this could be due to the differences in concentrations of this adipokine occurring under these disease conditions. Alternatively, the differential effects of adiponectin on cells could be due to the different inflammatory environment. The mechanisms underlying the dichotomous effect of adiponectin on OA and RA remain to be elucidated.
Leptin
Leptin is the most extensively studied adipokine so far. Leptin, a 16-kDa adipokine secreted by adipose tissue, is associated with the regulation of food intake, energy assumption, and immunity. Leptin deficiency causes obesity in mouse models. However, humans exhibit leptin resistance. Increasing leptin concentration in the human body fails to reduce food intake, energy consumption, or body weight.
Leptin and its receptor (OB-R) are also detected in human chondrocytes [38]. Leptin can promote proliferation and differentiation of growth plate chondrocytes [39]. Leptin can stimulate Col10a1in ATDC5 chondrocytes through the JAK/STAT and MAPK pathways [40]. Conversely, the growth plates of ob/ob mice show reduced type X collagen levels compared to the wild mice [41, 42]. Thus, leptin seems to be required for chondrocyte hypertrophy.
The concentrations of leptin are increased in cartilage and synovial fluid of OA patients [40]. Higher leptin mRNA levels were detected in end-stage knee OA cartilage compared to healthy joints [41], although there is no evidence at present of an association between serum leptin and cartilage damage or synovial inflammation. Chondrocytes from obese OA patients (BMI > 30 kg/m2) respond more intensely to leptin, possibly due to the elevated leptin level in the joint or the disruption of a leptin resistance mechanism in cartilage tissue [43]. Leptin can synergize with other pro-inflammatory and catabolic factors during OA. Leptin induces the production of IL-8, MMP-1, MMP-13, MMP-9, and MMP-3 in primary cultures of human chondrocytes and its concentration correlates with MMP-1 and MMP-3 in synovial fluid from OA patients [44, 45].
Leptin regulates bone metabolism via both central and peripheral nerve pathways. In the central pathway, leptin binds to its hypothalamic receptors and signals to osteoblasts through β2 adrenergic receptors [46]. It activates two molecular cascades, c-myc, which inhibits osteoblast proliferation, and the PKA/ATF4/RANKL pathway, which enhances bone resorption of osteoclasts [47]. In the peripheral pathway, leptin binds to Ob-Rb on human mesenchymal stem cells (MSCs), enhancing proliferation and differentiation into osteoblasts [48]. Thus, leptin may cause chondrocyte hypertrophy and degeneration and excessive ossification during OA.
Resistin
The gene of resistin, encoding a 12.5-kDa adipokine protein, is located on chromosome 19p13.2. While the major source of resistin is white adipose tissue, resistin is also expressed by macrophages in humans. Resistin is related to metabolic disease and inflammation, since reduction of the levels of resistin leads to enhanced insulin sensitivity and glucose homeostasis. The local concentration of resistin is increased during OA, which is probably due to the resident macrophage cells in synovium [49].
Resistin is also expressed by osteoblasts, osteoclasts, and chondrocytes. There is a synergistic regulation between resistin and several cytokines, as well as chemokines. Resistin induces the expression of cytokines and chemokines in chondrocytes. Treatment of mouse cartilage with resistin leads to increased IL-6 and IL-8 level. Resistin may stimulate bone remodeling by promoting osteoblast proliferation and osteoclast differentiation. Resistin stimulates proliferation of pre-osteoblastic MC3T3-E1 cells [50]. Transfection of RAW264.7 cells with resistin cDNA stimulates osteoclastogenesis by enhancing NF-κB promoter activity [50]. Thus, resistin may promote OA pathogenesis by stimulating proinflammatory cytokines in cartilage and bone remodeling. However, the role of resistin in OA onset and progression in vivo remains to be determined.
Chemerin
Chemerin, a 14-kDa protein, is expressed mainly in white adipose tissue. The chemerin level in synovial fluid correlates with body mass index (BMI) and OA severity [51]. It is secreted as a 163 amino acid inactive form and then activated by cleavage in the C-terminus [52]. Chemerin is associated with energy metabolism that mediates glucose uptake, lipolysis, and adipocyte differentiation. Chemerin stimulates phosphorylation of ERK and MAPKs, signals that are involved in lipid synthesis in adipocytes, thereby promoting adipocyte maturation. In turn, adipocytes express higher levels of chemerin after differentiation.
Chemerin receptors contain CMKLR1 and ChemR23, which are expressed mainly in immune cells. Chemerin is a chemoattractant protein, which stimulates chemotaxis of dendritic cells and macrophages to the inflammatory site. For example, chemerin may recruit immune cells (including macrophages) in synovium as part of the inflammatory cascade in the pathogenesis of OA. Obesity patients have lager amount of chemerin in serum and the serum level of chemerin is correlated to OA severity [51].
Chemerin can be detected in cartilage and synovial fluid, while ChemR23 is also expressed in human chondrocytes [53]. In human chondrocytes, chermerin enhances the production of proinflammatory cytokines, as well as MMPs [53], although chemerin usually reduces the production of proinflammatory cytokines in other types of cells. Chemerin regulates bone metabolism through mediating testosterone production and the balance between osteoblasts and osteoclasts differentiation [54]. Although inhibiting CMKLR1 stimulated expression of osteoblast markers in primary MSCs in vitro [55], its deficiency in vivo resulted in lower bone mass in male mice [54]. Thus, chemerin may stimulate inflammation, cartilage degeneration, and ossification. However, its role in OA pathogenesis remains to be determined in vivo.
IL-6
IL-6, a glycosylated protein of 21–28 kDa, has four-α-helix bundle structure in an up-up-down-down topology. The IL-6 family contains other members including IL-11, leukemia inhibitory factor (LIF), and oncostatin M (OSM). The IL-6 receptors include soluble IL-6 receptor (sIL-6R), membrane bounding receptor (mbIL-6R), and IL-6 family common receptor gp130, which exists as inactive dimers at the cell membrane [56, 57]. Two IL-6 signaling pathways exist. The classical signaling pathway involves IL-6 binding to mbIL-6R, which only a few cell types express, and activation of JAK/STAT, ERK, and PI3K in cells. The trans-signaling pathway involves binding of the IL-6/sIL-6R complexes to constitutively expressed receptor gp130, which then transduces IL-6 signals.
As one of the most prominent SASPs, IL-6 is expressed in adipose and muscle tissues especially during aging. It is involved in metabolic diseases such as obesity and other inflammatory diseases. IL-6−/− mice develop late onset obesity [58]. Specific blockade of IL-6 trans-signals by spg130Fc affects insulin sensitivity and glucose tolerance [59]. High levels of IL-6 are associated with the increased risk of knee OA [60]. The patellar fat pad secretes adipokines, including IL-6, which infiltrates into cartilage via synovial fluid [12, 13]. IL-6 mediates suppression of aggrecan (Acan) and induction of Mmp13 gene expression by Notch in chondrocytes [61]. IL-6/sIL-6R trigger osteoclast formation and bone resorption by inducing RANKL expression via JAK/STAT signaling [62, 63]. Taken together, there is solid evidence that IL-6 regulates metabolism and OA progression. Thus, IL-6 or IL-6 receptor should be considered as a therapeutic target for obesity-related OA.
Tumor Necrosis Factor-Alpha (TNF-α)
TNF-α is a pro-inflammatory cytokine produced by macrophages, and also by adipocytes and vascular endothelial cells. TNF-α exists as two forms, membrane-bound (mTNF-α) and free soluble (sTNF-α) [64–66]. There are two TNF-α receptors, TNF-R1 (CD120a) and TNF-R2 (CD120b). While constitutively expressed TNF-R1 binds both sTNF-α and mTNF-α, TNF-R2 is inducible and binds mTNF-α only. TNF-R1 contains a death domain, which is similar to the death region of Fas protein. This intracellular region is essential for TNF-α-initiated apoptosis by providing the binding sites for accessory signaling proteins, including Fas-associated death segment binding protein (FADD), TNFR-1 associated death segment binding protein (TRADD), and TNFR-related factor 2 (TRAF-2). TNF-R1 signaling initiates the inflammatory response pathway by recruiting two different signaling complexes that activate NF-κB, ERK, and p38MAPK signaling pathways [67] and the FADD/pro-caspase-8 cell death pathway [68].
The critical role of TNF-α in OA pathogenesis is well established. TNF-α can be detected in synovial fluids of OA patients and the TNF-α level increases as the OA score increases [69, 70]. TNF-α induces the production of iNOS, COX-2, IL-6, and PGE2 [71–73], and inhibits the synthesis of type II collagen, proteoglycans, and proteoglycan-binding proteins [73, 74]. TNF-α regulates the process of bone remodeling involving both osteoclasts and osteoblasts. While TNF-α induces osteoclastogenesis via RANKL, it suppresses bone formation by inhibiting MSC differentiation into osteoblasts via Notch activation [75].
Conclusion and Future Direction
Adipokines are a newly emerging class of signaling molecules, produced by adipocytes and involved in regulating inflammation, bone and cartilage metabolism, and homeostasis. In the past, attentions were focused on the cytokines and chemokines produced by synovium and immune cells in the joint. However, in recent years, research has shown that OA, which was considered as a local disease, can be related to systemic metabolism disease such as obesity [4, 5]. The close association between OA and obesity strongly suggests that OA can be a systemic disease involving multiple tissues and triggered by inflammation and metabolic imbalance. OA is also associated with aging and injury, both of which induce inflammation, cell senescence, and SASPs that hamper tissue repair.
In the joint, the roles of cartilage, bone, ligament, muscle, and synovium in OA pathogenesis have been well studied. In contrast, less is known about the roles of adipose tissues in the joint. Understanding the properties of adipose tissues and derived cells becomes even more critical when adipose-derived stem cells are touted as a good source of joint tissue repair. The sources of adipose tissues come from two sites in the joint, fat pads such as the IFP and bone marrow. During aging and obesity, the fat content in adipose tissues increases, which triggers increased secretion of adipokines in the joint. Furthermore, the yellow bone marrow is developed, which enriches adipocytes by activating adipocyte lineage expansion [76•]. This is done at the expense of impairing osteogenic lineage in the long bone, resulting in bone dysfunction in the aged. An attractive hypothesis is that the metabolic syndrome in elderly people leads to increased body fat, inflammaging, and senescence of cells in articular tissues [77].
There are hundreds of adipokines and this review only cover several whose roles in bone and joint have been studied. These adipokines can be divided into two classes. The first class contains IL-6 and TNF-α, which are well-studied due to their synthesis by a variety of immune cells and are among the most important SASPs. Thus, the antagonists that neutralize their actions are some of the most successful drugs to treat arthritis. The second class contains adiponectin, leptin, resistin, and chemerin. In comparison to the first class of adipokines, these are more specifically synthesized by adipocytes although less well characterized for their roles in regulating joint health. For example, it is not known whether they can be classified as SASPs. However, their increased expression during aging and their synergistic effects in stimulating other SASPs such as IL-1, IL-6, and TNF-α make them excellent candidate SASPs.
These factors accumulate during aging and contribute to local inflammatory reactions in the joints. They can also cause changes the surrounding environment of the joint cells, thereby changing in the fate of the cells—inducing cell senescence. Therefore, attention to changes in the joint environment is important for the progress of OA research. It is important to note that chondrocytes are not the only cell type when studying the fate of cartilage-associated cells. There are also mesenchymal stem/progenitor cells in articular cartilage that undergo cell senescence [78•, 79••]. Studying the response of these cells to these new adipokines in the joint environment will be a new direction for the treatment of OA.
Injury-related OA induces the expansion of chondrocytes to repair cartilage lesion, which results in loss of quiescence of chondrocytes and increases the accumulation of cell cycle inhibitors (e.g., P16INK4A), as well as reactive oxygen species (ROS), leading eventually to senescence. Local clearance of senescent cells attenuates the development of the injury-induced OA and creates a pro-regenerative environment [80••]. However, the source of senescent cells accumulating in joint tissues after ACLT (anterior cruciate ligament transection) has not been defined [80••]. Cells other than synoviocytes and chondrocytes may contribute to inflammation, SASP, and joint degeneration. We suggest that joint adipose tissues may contribute to synovial inflammation, cartilage degeneration, bone remodeling, and OA pathogenesis by producing two classes of adipokines, including SASPs.
If the first class of adipokines can be used as a good example, anti-arthritis pharmaceuticals may be developed using the second class of adipokines as targets. It will be necessary, therefore, to characterize the properties of these adipokines in more detail, especially in OA animal models in vivo. Furthermore, while leptin, resistin, and chemerin seem to promote joint inflammation, adiponectin has dichotomous effects depending on its molecular forms and concentrations in the joint. Since adiponectin presents intriguing anti-inflammatory and pro-regenerative properties that are unique among adipokines, it warrants further investigation.
Acknowledgments
Funding Information Grant sponsor: NIGMS/NIH; Grant number: P30GM122732
Footnotes
Conflicts of Interest The authors declare that they have no conflict of interest.
Compliance with Ethical Standards
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Spector TD. The fat on the joint: osteoarthritis and obesity. J Rheumatol. 1990;17(3):283–4. [PubMed] [Google Scholar]
- 2.Jin WS, Choi EJ, Lee SY, Bae EJ, Lee TH, Park J. Relationships among obesity, sarcopenia, and osteoarthritis in the elderly. J Obes Metab Syndr. 2017;26(1):36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Misra D, et al. Risk of knee osteoarthritis with obesity, sarcopenic obesity, and sarcopenia. Arthritis Rheum. 2019;71(2):232–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hochberg MC, Lethbridge-Cejku M, Scott WW Jr, Reichle R, Plato CC, Tobin JD. The association of body weight, body fatness and body fat distribution with osteoarthritis of the knee: data from the Baltimore Longitudinal Study of Aging. J Rheumatol. 1995;22(3): 488–93. [PubMed] [Google Scholar]
- 5.Felson DT. Relation of obesity and of vocational and avocational risk factors to osteoarthritis. J Rheumatol. 2005;32(6):1133–5. [PubMed] [Google Scholar]
- 6.Giles JT, van der Heijde DM, Bathon JM. Association of circulating adiponectin levels with progression of radiographic joint destruction in rheumatoid arthritis. Ann Rheum Dis. 2011. ;70(9):1562–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frommer KW, Zimmermann B, Meier FM, Schroder D, Heil M, Schaffler A, et al. Adiponectin-mediated changes in effector cells involved in the pathophysiology of rheumatoid arthritis. Arthritis Rheum. 2010;62(10):2886–99. [DOI] [PubMed] [Google Scholar]
- 8.Yamauchi T, Iwabu M, Okada-Iwabu M, Kadowaki T. Adiponectin receptors: a review of their structure, function and how they work. Best Pract Res Clin Endocrinol Metab. 2014;28(1):15–23. [DOI] [PubMed] [Google Scholar]
- 9.•.Jiang L, et al. Obesity, osteoarthritis and genetic risk: the rs182052 polymorphism in the ADIPOQ gene is potentially associated with risk of knee osteoarthritis. Bone Joint Res. 2018;7(7):494–500 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates genetic association of the ADIPOQ gene with knee OA.
- 10.Koskinen A, Juslin S, Nieminen R, Moilanen T, Vuolteenaho K, Moilanen E. Adiponectin associates with markers of cartilage degradation in osteoarthritis and induces production of proinflammatory and catabolic factors through mitogen-activated protein kinase pathways. Arthritis Res Ther. 2011;13(6):R184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halade GV, el Jamali A, Williams PJ, Fajardo RJ, Fernandes G. Obesity-mediated inflammatory microenvironment stimulates osteoclastogenesis and bone loss in mice. Exp Gerontol. 2011. ;46(1): 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ushiyama T, Chano T, Inoue K, Matsusue Y. Cytokine production in the infrapatellar fat pad: another source of cytokines in knee synovial fluids. Ann Rheum Dis. 2003;62(2):108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Presle N, Pottie P, Dumond H, Guillaume C, Lapicque F, Pallu S, et al. Differential distribution of adipokines between serum and synovial fluid in patients with osteoarthritis. Contribution of joint tissues to their articular production. Osteoarthr Cartil. 2006;14(7): 690–5. [DOI] [PubMed] [Google Scholar]
- 14.Mobasheri A, Matta C, Zákány R, Musumeci G. Chondrosenescence: definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas. 2015;80(3):237–44. [DOI] [PubMed] [Google Scholar]
- 15.Coppe JP, et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuchida AI, Beekhuizen M, ‘t Hart MC, Radstake TR, Dhert WJ, Saris DB, et al. Cytokine profiles in the joint depend on pathology, but are different between synovial fluid, cartilage tissue and cultured chondrocytes. Arthritis Res Ther. 2014;16(5):441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270(45):26746–9. [DOI] [PubMed] [Google Scholar]
- 18.Nakano Y, Tobe T, Choi-Miura NH, Mazda T, Tomita M. Isolation and characterization of GBP28, a novel gelatin-binding protein purified from human plasma. J Biochem. 1996;120(4):803–12. [DOI] [PubMed] [Google Scholar]
- 19.Waki H, Yamauchi T, Kamon J, Ito Y, Uchida S, Kita S, et al. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J Biol Chem. 2003;278(41):40352–63. [DOI] [PubMed] [Google Scholar]
- 20.Waki H, Yamauchi T, Kamon J, Kita S, Ito Y, Hada Y, et al. Generation of globular fragment of adiponectin by leukocyte elastase secreted by monocytic cell line THP-1. Endocrinology. 2005;146(2):790–6. [DOI] [PubMed] [Google Scholar]
- 21.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423(6941):762–9. [DOI] [PubMed] [Google Scholar]
- 22.Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13(3):332–9. [DOI] [PubMed] [Google Scholar]
- 23.Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature. 2010;464(7293):1313–9. [DOI] [PubMed] [Google Scholar]
- 24.Kadowaki T, et al. Adiponectin and adiponectin receptors in obesity-linked insulin resistance. Novartis Found Symp. 2007;286:164–76 discussion 176-82, 200-3. [DOI] [PubMed] [Google Scholar]
- 25.Yamauchi T, Kadowaki T. Adiponectin receptor as a key player in healthy longevity and obesity-related diseases. Cell Metab. 2013;17(2):185–96. [DOI] [PubMed] [Google Scholar]
- 26.Zhu W, et al. Vascular effects of adiponectin: molecular mechanisms and potential therapeutic intervention. Clin Sci (Lond). 2008;114(5):361–74. [DOI] [PubMed] [Google Scholar]
- 27.Kanazawa I, et al. Adiponectin and AMP kinase activator stimulate proliferation, differentiation, and mineralization of osteoblastic MC3T3-E1 cells. BMC Cell Biol. 2007;8:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Beeck A, et al. Does infrapatellar fat pad resection in total knee arthroplasty impair clinical outcome? A systematic review. Knee. 2013;20(4):226–31. [DOI] [PubMed] [Google Scholar]
- 29.Pan F, et al. A longitudinal study of the association between infrapatellar fat pad maximal area and changes in knee symptoms and structure in older adults. Ann Rheum Dis. 2015;74(10):1818–24. [DOI] [PubMed] [Google Scholar]
- 30.•.Hu J, et al. Globular adiponectin attenuated H2O2-induced apoptosis in rat chondrocytes by inducing autophagy through the AMPK/mTOR pathway. Cell Physiol Biochem. 2017;43(1):367–82 [DOI] [PubMed] [Google Scholar]; This study demonstrates the effect of globular adiponection on chondrocytes.
- 31.Landgraeber S, Putz S, Schlattjan M, Bechmann LP, Totsch M, Grabellus F, et al. Adiponectin attenuates osteolysis in aseptic loosening of total hip replacements. Acta Biomater. 2014;10(1):384–93. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, et al. Adiponectin regulates BMSC osteogenic differentiation and osteogenesis through the Wnt/beta-catenin pathway. Sci Rep. 2017;7(1):3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu X, Chen T, Wu Y, Tang Z. Role and mechanism of PTEN in adiponectin-induced osteogenesis in human bone marrow mesenchymal stem cells. Biochem Biophys Res Commun. 2017;483(1): 712–7. [DOI] [PubMed] [Google Scholar]
- 34.Chen T, Wu YW, Lu H, Guo Y, Tang ZH. Adiponectin enhances osteogenic differentiation in human adipose-derived stem cells by activating the APPL1-AMPK signaling pathway. Biochem Biophys Res Commun. 2015;461(2):237–42. [DOI] [PubMed] [Google Scholar]
- 35.Wu Y, Tu Q, Valverde P, Zhang J, Murray D, Dong LQ, et al. Central adiponectin administration reveals new regulatory mechanisms of bone metabolism in mice. Am J Physiol Endocrinol Metab. 2014;306(12):E1418–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.•.Qian J, et al. Adiponectin aggravates bone erosion by promoting osteopontin production in synovial tissue of rheumatoid arthritis. Arthritis Res Ther. 2018;20, 26(1) [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates the role of adiponectin in bone erosion during rheumatoid arthritis.
- 37.Krumbholz G, Junker S, Meier FMP, Rickert M, Steinmeyer J, Rehart S, et al. Response of human rheumatoid arthritis osteoblasts and osteoclasts to adiponectin. Clin Exp Rheumatol. 2017;35(3): 406–14. [PubMed] [Google Scholar]
- 38.Otero M, Gomez RJ, Gualillo O. Synergistic induction of nitric oxide synthase type II: in vitro effect of leptin and interferon-gamma in human chondrocytes and ATDC5 chondrogenic cells. Arthritis Rheum. 2003;48(2):404–9. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Shao YY, Ballock RT. Leptin antagonizes peroxisome proliferator-activated receptor-gamma signaling in growth plate chondrocytes. PPAR Res. 2012;2012:756198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ben-Eliezer M, Phillip M, Gat-Yablonski G. Leptin regulates chondrogenic differentiation in ATDC5 cell-line through JAK/STAT and MAPK pathways. Endocrine. 2007;32(2):235–44. [DOI] [PubMed] [Google Scholar]
- 41.Simopoulou T, Malizos KN, Iliopoulos D, Stefanou N, Papatheodorou L, Ioannou M, et al. Differential expression of leptin and leptin’s receptor isoform (Ob-Rb) mRNA between advanced and minimally affected osteoarthritic cartilage; effect on cartilage metabolism. Osteoarthr Cartil. 2007;15(8):872–83. [DOI] [PubMed] [Google Scholar]
- 42.Kishida Y, Hirao M, Tamai N, Nampei A, Fujimoto T, Nakase T, et al. Leptin regulates chondrocyte differentiation and matrix maturation during endochondral ossification. Bone. 2005;37(5):607–21. [DOI] [PubMed] [Google Scholar]
- 43.Pallu S, Francin PJ, Guillaume C, Gegout-Pottie P, Netter P, Mainard D, et al. Obesity affects the chondrocyte responsiveness to leptin in patients with osteoarthritis. Arthritis Res Ther. 2010;12(3):R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vuolteenaho K, et al. Leptin enhances synthesis of proinflammatory mediators in human osteoarthritic cartilage-mediator role of NO in leptin-induced PGE2, IL-6, and IL-8 production. Mediat Inflamm. 2009;2009:345838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koskinen A, et al. Leptin enhances MMP-1, MMP-3 and MMP-13 production in human osteoarthritic cartilage and correlates with MMP-1 and MMP-3 in synovial fluid from OA patients. Clin Exp Rheumatol. 2011;29(1):57–64. [PubMed] [Google Scholar]
- 46.Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker KL, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111(3):305–17. [DOI] [PubMed] [Google Scholar]
- 47.Fu L, Patel MS, Bradley A, Wagner EF, Karsenty G. The molecular clock mediates leptin-regulated bone formation. Cell. 2005;122(5): 803–15. [DOI] [PubMed] [Google Scholar]
- 48.Astudillo P, et al. Increased adipogenesis of osteoporotic human-mesenchymal stem cells (MSCs) characterizes by impaired leptin action. J Cell Biochem. 2008;103(4):1054–65. [DOI] [PubMed] [Google Scholar]
- 49.Lee JH, Ort T, Ma K, Picha K, Carton J, Marsters PA, et al. Resistin is elevated following traumatic joint injury and causes matrix degradation and release of inflammatory cytokines from articular cartilage in vitro. Osteoarthr Cartil. 2009;17(5):613–20. [DOI] [PubMed] [Google Scholar]
- 50.Thommesen L, et al. Expression and regulation of resistin in osteoblasts and osteoclasts indicate a role in bone metabolism. J Cell Biochem. 2006;99(3):824–34. [DOI] [PubMed] [Google Scholar]
- 51.Huang K, du G, Li L, Liang H, Zhang B. Association of chemerin levels in synovial fluid with the severity of knee osteoarthritis. Biomarkers. 2012;17(1): 16–20. [DOI] [PubMed] [Google Scholar]
- 52.Zabel BA, Allen SJ, Kulig P, Allen JA, Cichy J, Handel TM, et al. Chemerin activation by serine proteases of the coagulation, fibrinolytic, and inflammatory cascades. J Biol Chem. 2005;280(41): 34661–6. [DOI] [PubMed] [Google Scholar]
- 53.Berg V, Sveinbjornsson B, Bendiksen S, Brox J, Meknas K, Figenschau Y. Human articular chondrocytes express ChemR23 and chemerin; ChemR23 promotes inflammatory signalling upon binding the ligand chemerin(21-157). Arthritis Res Ther. 2010;12(6):R228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao H, et al. Chemokine-like receptor 1 deficiency leads to lower bone mass in male mice. Cell Mol Life Sci. 2019;76(2):355–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muruganandan S, Roman AA, Sinal CJ. Role of chemerin/CMKLR1 signaling in adipogenesis and osteoblastogenesis of bone marrow stem cells. J Bone Miner Res. 2010;25(2):222–34. [DOI] [PubMed] [Google Scholar]
- 56.Giese B, Roderburg C, Sommerauer M, Wortmann SB, Metz S, Heinrich PC, et al. Dimerization of the cytokine receptors gp130 and LIFR analysed in single cells. J Cell Sci. 2005;118(Pt 21): 5129–40. [DOI] [PubMed] [Google Scholar]
- 57.Tenhumberg S, et al. gp130 dimerization in the absence of ligand: preformed cytokine receptor complexes. Biochem Biophys Res Commun. 2006;346(3):649–57. [DOI] [PubMed] [Google Scholar]
- 58.Wallenius V, Wallenius K, Ahren B, Rudling M, Carlsten H, Dickson SL, et al. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med. 2002;8(1):75–9. [DOI] [PubMed] [Google Scholar]
- 59.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813(5):878–88. [DOI] [PubMed] [Google Scholar]
- 60.Greene MA, Loeser RF. Aging-related inflammation in osteoarthritis. Osteoarthr Cartil. 2015;23(11):1966–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zanotti S, Canalis E. Interleukin 6 mediates selected effects of Notch in chondrocytes. Osteoarthr Cartil. 2013;21(11):1766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tamura T, Udagawa N, Takahashi N, Miyaura C, Tanaka S, Yamada Y, et al. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc Natl Acad Sci U S A. 1993;90(24): 11924–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hashizume M, Hayakawa N, Mihara M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatology (Oxford). 2008;47(11):1635–40. [DOI] [PubMed] [Google Scholar]
- 64.Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell. 1988;53(1): 45–53. [DOI] [PubMed] [Google Scholar]
- 65.Tang P, Hung M, Klostergaard J. Human pro-tumor necrosis factor is ahomotrimer. Biochemistry. 1996;35(25):8216–25. [DOI] [PubMed] [Google Scholar]
- 66.Idriss HT, Naismith JH. TNF alpha and the TNF receptor super-family: structure-function relationship(s). Microsc Res Tech. 2000;50(3):184–95. [DOI] [PubMed] [Google Scholar]
- 67.Campbell J, et al. A novel mechanism for TNF-alpha regulation by p38 MAPK: involvement of NF-kappa B with implications for therapy in rheumatoid arthritis. JImmunol. 2004;173(11):6928–37. [DOI] [PubMed] [Google Scholar]
- 68.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2): 181–90. [DOI] [PubMed] [Google Scholar]
- 69.Farahat MN, Yanni G, Poston R, Panayi GS. Cytokine expression in synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Ann Rheum Dis. 1993;52(12):870–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ozler K, et al. Serum and knee synovial fluid matrixmetalloproteinase-13 and tumor necrosis factor-alpha levels in patients with late stage osteoarthritis. Acta Orthop Traumatol Turc. 2016;50(6):670–3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 71.El MF, et al. Contribution of H3K4 methylation by SET-1A to interleukin-1-induced cyclooxygenase 2 and inducible nitric oxide synthase expression in human osteoarthritis chondrocytes. Arthritis Rheum. 2011;63(1):168–79. [DOI] [PubMed] [Google Scholar]
- 72.Guerne PA, Carson DA, Lotz M. IL-6 production by human articular chondrocytes. Modulation of its synthesis by cytokines, growth factors, and hormones in vitro. J Immunol. 1990;144(2):499–505. [PubMed] [Google Scholar]
- 73.Seguin CA, Bernier SM. TNFalpha suppresses link protein and type II collagen expression in chondrocytes: role of MEK1/2 and NF-kappaB signaling pathways. J Cell Physiol. 2003;197(3):356–69. [DOI] [PubMed] [Google Scholar]
- 74.Xue J, Wang J, Liu Q, Luo A. Tumor necrosis factor-alpha induces ADAMTS-4 expression in human osteoarthritis chondrocytes. Mol Med Rep. 2013;8(6):1755–60. [DOI] [PubMed] [Google Scholar]
- 75.Zhao B TNF and bone remodeling. Curr Osteoporos Rep. 2017;15(3):126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.•.Ambrosi TH, et al. Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell. 2017;20(6):771–784.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates a role of bone marrow adipocytes in bone regeneration.
- 77.Lotz M, Loeser RF. Effects of aging on articular cartilage homeostasis. Bone. 2012;51(2):241–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.•.Fellows CR, et al. Characterisation of divergent progenitor cell sub-populations in human osteoarthritic cartilage: the role of telomere erosion and replicative senescence. Sci Rep. 2017;7:41421. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates part of the OA cartilage progenitor cells is senescent.
- 79.••.Jayasuriya, et al. Molecular characterization of mesenchymal stem cells in human osteoarthritis cartilage reveals contribution to the OA phenotype. Sci Rep. 2018;8(1):7044. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates a direct association of mesenchymal stem cells with OA disease phenotypes in human OA cartilage.
- 80.••.Jeon OH, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23(6):775–81 [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first study to show that clearance of senescent cells reduces PTOA.