

Abstract
Ghrelin is a hormone that is secreted by the stomach during fasting periods and acts through its receptor, the growth hormone secretagogue 1a (GHSR1a), to promote food intake and prevent hypoglycemia. As such, GHSR1a is an important regulator of energy and glucose homeostasis and a target for the treatment of obesity. Here, we showed that the accessory protein MRAP2 altered GHSR1a signaling by inhibiting its constitutive activity, as well as by enhancing its G protein–dependent signaling and blocking the recruitment and signaling of β-arrestin in response to ghrelin. In addition, the effects of MRAP2 on the Gαq and β-arrestin pathways were independent and involved distinct regions of MRAP2. These findings may have implications for the regulation of ghrelin function in vivo and the role of MRAP2 in energy homeostasis. They also show that accessory proteins can bias signaling downstream of GPCRs in response to their endogenous agonist.
One-sentence summary:
An accessory protein suppresses β-arrestin–dependent signaling and constitutive activity of the ghrelin receptor.
Editor’s summary:
Skewing signaling with an accessory protein
Ghrelin is a peptide that is secreted by the stomach during fasting to promote food intake. The accessory protein MRAP2 interacts with the ghrelin receptor GHSR1a and is important for the orexigenic effects of ghrelin. Rouault et al. found that MRAP2 promoted biased signaling downstream of ghrelin-mediated activation of GHSR1a by inhibiting β-arrestin recruitment to the receptor and potentiating Gαq/11-dependent signaling. Furthermore, MRAP2 suppressed ligand-independent activity of GHSR1a, which is naturally high. These results show that accessory proteins can bias GPCR signaling and, for GHSR1a, limit its constitutive activity.
Introduction
Ghrelin is a key regulator of energy and glucose homeostasis. Ghrelin is secreted by the stomach during low-energy states (1) to enable survival by promoting the sensation of hunger and preventing hypoglycemia (2). Ghrelin acts through its receptor, the growth hormone secretagogue receptor 1a (GHSR1a) in hypothalamic neurons to promote food intake in humans (3) and rodents (4). Animals lacking active ghrelin (5) or GHSR1a (6) are resistant to diet-induced obesity and are unable to maintain a normal blood concentration of glucose when placed on a calorie-restricted diet (7,8). The latter is due to a loss of ghrelin-stimulated growth hormone release from the anterior pituitary which in turn stimulates liver glucose output. GHSR1a stimulation results in G-protein activation and β-arrestin recruitment; however, GHSR1a also displays high constitutive activity (9). A naturally occurring mutation in the gene encoding human GHSR1a that eliminates its constitutive activity was identified in probands with short stature (10), suggesting a possible role for the basal activity of GHSR1a in development and somatic growth. We previously showed that the actions of ghrelin in the hypothalamus, more specifically in orexigenic agouty-related peptide (AGRP) neurons, requires the melanocortin receptor accessory protein 2 (MRAP2) (11). MRAP2 is a single transmembrane protein displaying dual topology (12,13) that regulates several GPCRs involved in the control of energy homeostasis, including the melanocortin-4 receptor (14,15), the prokineticin receptor 1 (16,17), and the orexin receptor 1 (18). MRAP2 is expressed in the hypothalamus and its deletion causes obesity in rodents (15). We showed that MRAP2 interacts with GHSR1a and potentiates ghrelin-stimulated Gαq signaling both in cell lines and in AGRP neurons (11). Furthermore, ghrelin fails to induce food intake in Mrap2 knockout mice (11). Those results established MRAP2 as an essential partner of GHSR1a for optimal receptor signaling and for the orexigenic effect of ghrelin.
Whereas the role of MRAP2 in regulating ghrelin-stimulated Gαq –dependent signaling has been established, the effect of MRAP2 on other important aspects of GHSR1a signaling is unclear. In this study, we evaluated the role of MRAP2 in regulating ghrelin binding, Gαq/11 protein coupling, β-arrestin recruitment, and GHSR1a constitutive activity. We showed that whereas MRAP2 potentiated GHSR1a signaling through Gαq/11, it inhibited β-arrestin recruitment and β-arrestin–mediated signaling. We also showed that the effects of MRAP2 on Gαq/11 and β-arrestin signaling were independent and could be functionally separated with different MRAP2 mutants. We also showed that MRAP2 inhibited the constitutive activity of GHSR1a, suggesting that in cells in which MRAP2 is expressed, such as AGRP neurons, the agonist-independent activity of GHSR1a may be minimal. Together, these findings further demonstrate that MRAP2 is a critical endogenous regulator of GHSR1a function and identify previously uncharacterized roles of MRAP2 in the regulation of β-arrestin recruitment and GHSR1a constitutive activity.
Results
MRAP2 potentiates GHSR1a signaling and inhibits its constitutive activity.
To determine the concentration dependency of MRAP2 effect on GHSR1a signaling, Chinese Hamster Ovary (CHO) cells were transfected with a constant amount of receptor-coding plasmid and either empty vector or increasing concentration of plasmid coding for MRAP2. CHO cells were used because they do not express endogenous MRAP2 and thus represent a naïve system. Transfected cells were stimulated with different concentrations of ghrelin in the presence of lithium, to allow accumulation of inositol-1 phosphate (IP1), before measuring the concentration of accumulated IP1 as a reporter of inositol-3 phosphate (IP3) production (hereafter, IP is used to refer to either IP1 or IP3). Signal over baseline was calculated and whereas ghrelin caused only a slight increase in accumulated IP in cells expressing GHSR1a alone, MRAP2 enhanced the response to ghrelin in a dose-dependent manner (Fig. 1A). At a ratio of receptor to MRAP2 plasmid of 1/20, ghrelin caused a 6 fold increase in accumulated IP over baseline (Fig. 1A). High constitutive activity is one of the best-known features of GHSR1a expressed in heterologous systems. Mutation of Ala204 to a glutamate residue (A204E) in the receptor causes a loss of constitutive activity and results in short stature in humans (10). For those reasons we tested the effect of MRAP2 on the constitutive activity of GHSR1a. First, we compared the IP3 response to ghrelin in cells expressing GHSR1a alone or with MRAP2 as described earlier. However, rather than measuring the fold increase in accumulated IP compared to baseline, we expressed results as a percentage of the maximal response to ghrelin in cells lacking MRAP2. Whereas in the absence of MRAP2 the response of GHSR1a to ghrelin was small, basal IP concentration was high (67% of the maximal response) (Fig. 1B). In the presence of MRAP2, the basal IP concentration was greatly diminished (15% of maximal signal) but the ghrelin response was potentiated (Fig. 1B). This result suggests that MRAP2, in addition to increasing the response to ghrelin, inhibits the constitutive activity of GHSR1a. To further test this hypothesis, we transfected cells with increasing amounts of the GHSR1a coding plasmid in the presence or absence of a constant concentration of MRAP2 plasmid. Cells were then incubated with lithium without agonist for 1h to accumulate IP produced as a consequence of the constitutive activity of GHSR1a. In cells lacking MRAP2, we observed a linear correlation between the amount of receptor transfected and IP accumulation, thus confirming that GHSR1a has high constitutive activity. In the presence of MRAP2, IP accumulation was low and did not increase with GHSR1a expression, thus further demonstrating that MRAP2 inhibits the constitutive activity of GHSR1a (Fig. 1C).
Fig. 1. MRAP2 potentiates the ghrelin-stimulated and inhibits the constitutive activity of GHSR1a.
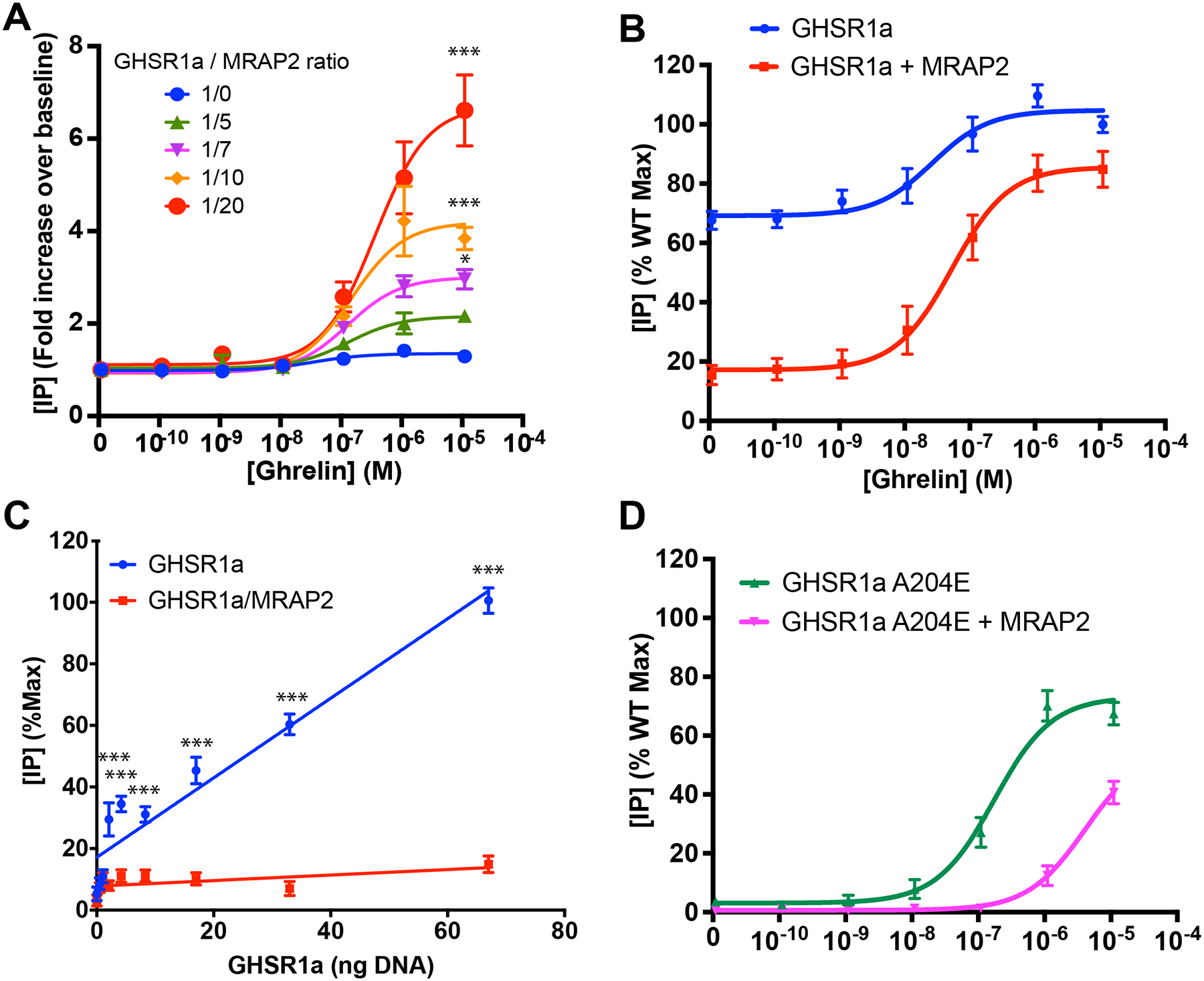
(A) Ghrelin-stimulated IP accumulation in CHO cells expressing GHSR1a with empty vector or increasing amount of MRAP2. Results are shown in fold increase over baseline and represent the mean + SEM of 3 independent experiments performed in triplicate. (B) Ghrelin-stimulated IP accumulation in CHO cells expressing GHSR1a with empty vector or MRAP2 at a 1/20 ratio. Results are shown as the percentage of the maximal response in cells expressing GHSR1a and empty vector and represent the mean + SEM of 3 independent experiments performed in triplicate. (C) Measurement of IP accumulation over 1 hour in the presence of lithium and absence of agonist in cells transfected with increasing amount of GHSR1a and either empty vector or MRAP2. Empty vector and MRAP2 concentration were kept constant. Data represent the mean + SEM of 3 independent experiments performed in triplicate. (D) Ghrelin-stimulated IP accumulation in CHO cells expressing GHSR1a(A204E) with empty vector or MRAP2. Measurement of IP3 production over 1 hour in the presence of lithium and absence of agonist in cells transfected with GHSR1a and empty vector, MRAP2 or the indicated MRAP2 mutant. Error are mean +/− SEM. Data represent the results of at least 3 independent experiments. Statistical analysis was done using multiple T-test (one unpaired T-test per condition). ***p<0.001, *p<0.05.
MRAP2 inhibits signaling downstream of GHSR1a(A204E)
To test whether MRAP2 potentiated ghrelin signaling downstream of the GHSR1a(A204E) mutant in a manner similar to its effects on WT GHSR1a, we measured ghrelin-stimulated IP3 production in cells expressing GHSR1a(A204E) with empty vector or MRAP2. Surprisingly, we found that, in contrast to the potentiating effects of MRAP2 on GHSR1a, MRAP2 decreased the potency of ghrelin 23 fold in cells expressing the mutant receptor (Fig. 1D). This inhibitory effect of MRAP2 on GHSR1a(A204E) could indicate that this mutation changes the conformation of the receptor in a way that alters its interaction with MRAP2.
MRAP2 inhibits ghrelin-stimulated β-arrestin recruitment to GHSR1a
MRAP2 potentiates ghrelin-stimulated IP3 production (11), thus showing that MRAP2 enhances GHSR1a Gαq–dependent signaling. Here, we tested whether this potentiating effect of MRAP2 also applied to ghrelin-stimulated β-arrestin recruitment. To this end, CHO cells were transfected with plasmids encoding GHSR1a and mCherry-β-arrestin2 in the presence or absence of plasmid encoding MRAP2. The cells were imaged after treatment with vehicle or 100 nM ghrelin for 10 min. Whereas ghrelin stimulated the translocation of mCherry-β-arrestin2 to the plasma membrane in cells lacking MRAP2, translocation of β-arrestin was not detected in cells expressing MRAP2 (Fig. 2A). This result suggests that MRAP2 inhibits ghrelin-stimulated β-arrestin recruitment to GHSR1a. To quantitatively measure the degree to which MRAP2 blocked β-arrestin recruitment, we used NanoBiT, a split luciferase recombination assay that monitors protein-protein interactions in live cells. We fused the two complementary fragments of the nanoluc luciferase, LgBiT and SmlBiT, to the C-terminus of 2HA-GHSR1a (GHSR1a N-terminally tagged with 2 hemagglutinin tags) and the N terminus of β-arrestin2, respectively (Fig. 2B). CHO cells were transfected with plasmids expressing GHSR1a-LgBiT and SmlBiT-β-arrestin2 in the presence or absence of plasmid expressing MRAP2. Consistent with the fluorescent microscopy experiments, the NanoBiT based assay demonstrated that ghrelin elicited the rapid recruitment of β-arrestin to GHSR1a in cells lacking MRAP2. In contrast, cells expressing MRAP2 showed a significant reduction in ghrelin-stimulated β-arrestin recruitment (about 35% of control without MRAP2) (Fig. 2C). To verify that the decrease in β-arrestin recruitment observed was not due to a decrease in surface density of the receptor, we measured surface 2HA-GHSR1a-LgBiT expression by cell-ELISA in parallel dishes transfected with the same plasmids. Detection of surface 2HA-GHSR1a-LgBiT with an HA antibody in fixed, non-permeabilized cells showed that MRAP2 did not significantly alter surface receptor density (Fig. 2D). Additionally, the recruitment of β-arrestin to GHSR1a depended on the ghrelin concentration and MRAP2 inhibited the recruitment of β-arrestin recruitment to GHSR1a at every concentration of ghrelin tested (Fig. 2E).
Fig. 2. MRAP2 inhibits β-arrestin recruitment to GHSR1a.
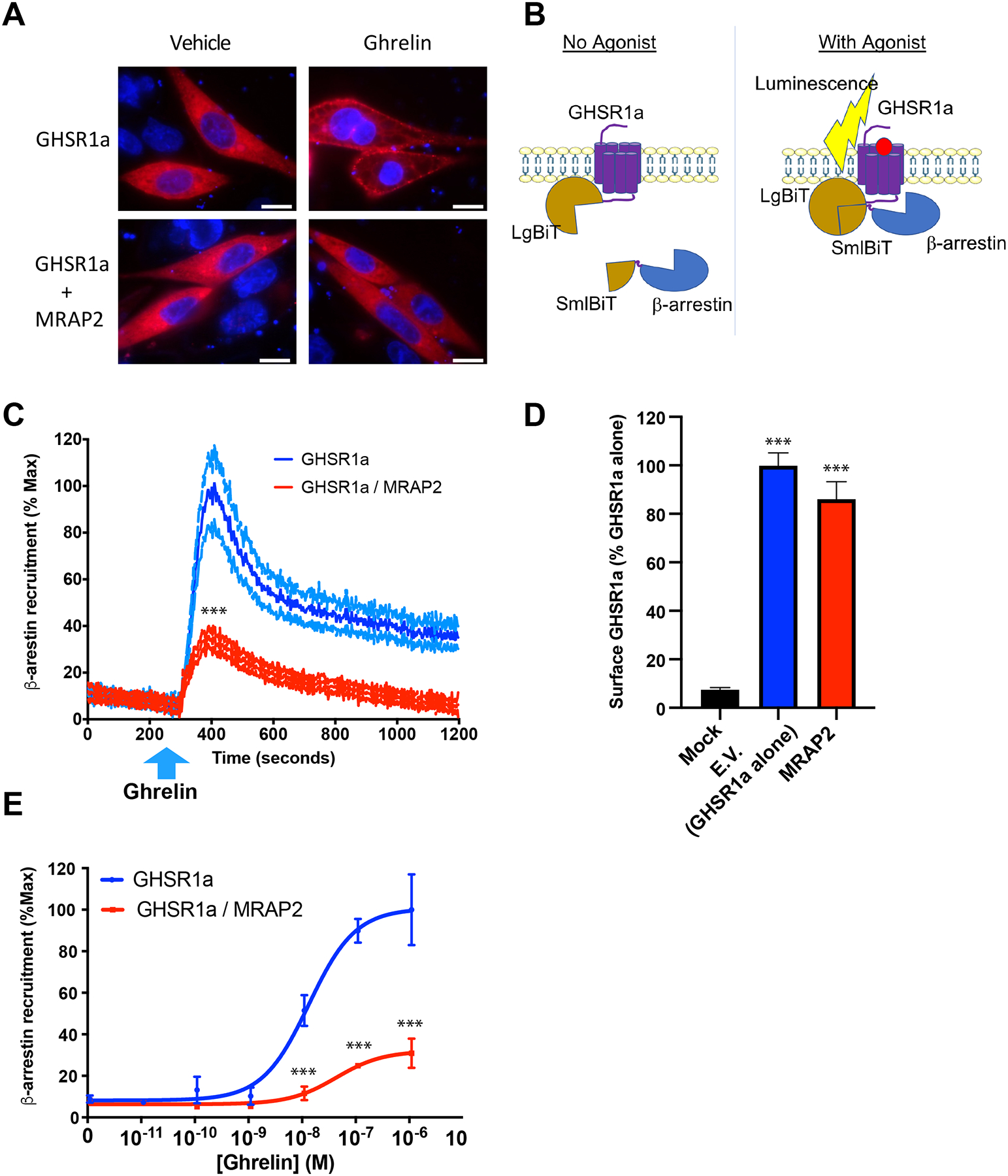
(A) CHO cells transfected with plasmids encoding 3HA-GHSR1a and mCherry-β-arrestin2 with or without plasmid encoding MRAP2 were analyzed by immunofluorescence microscopy. Micrographs of live cells were taken 10 min after stimulation with vehicle or 100 nM ghrelin. mCherry signal is shown in red and nuclei stained with Hoechst are shown in blue. Scale bars are 10 μm. Images are representative of 3 separate experiments. (B) Schematic of the NanoBiT based β-arrestin recruitment assay. Cells express GHSR1a-LgBiT and SmlBiT-β-arrestin. Agonist-stimulated recruitment of β-arrestin allows the recombination of a functional NanoLuc enzyme, resulting in measurable luminescent signal. (C) Kinetics of ghrelin-stimulated β-arrestin recruitment in cells expressing GHSR1a alone (blue) or with MRAP2 (red). The blue arrow indicates the time at which ghrelin was injected. Data are the means +/− SEM of 3 independent experiments performed in triplicate. T-test comparing the area under the curve was used for statistical analysis. (D) Surface density of GHSR1a measured by fixed-cells ELISA. Data are the means +/− SEM of 3 independent experiments performed in triplicate and significance was measured by one-way ANOVA. (E) Concentration-response curve of ghrelin-stimulated β-arrestin recruitment to GHSR1a in the presence or absence of MRAP2. Statistical analysis of the differences at each ghrelin concentrations were measured by T-test. Data are means +/− SEM of three independent experiments performed in triplicate. ***P < 0.001.
MRAP2-mediated inhibition of β-arrestin recruitment is not required for the potentiation of G protein signaling downstream of GHSR1a
β-arrestin is involved in the desensitization and internalization of GPCRs. For this reason, we tested whether the inhibition of β-arrestin recruitment by MRAP2 was responsible for the potentiation of G protein signaling downstream of GHSR1a. To this end we generated a β-arrestin1/β-arrestin2 double knockout CHO cell line using CRISPR/Cas9 technology (Fig. 3A). Deletion of the targeted regions was verified by PCR-based amplification of the gene fragments containing the sequences to be excised. As expected, deletion of exon 5 of Arrb1 (which encodes β-arrestin1) and exons 4 to 6 of Arrb2 (which encodes β-arrestin2) resulted in lower amplicon sizes corresponding to the targeted excisions (Fig. 3B). These deletions were also verified by DNA sequencing. To test whether the potentiating effect of MRAP2 on ghrelin-stimulated IP production was due to a decrease in β-arrestin recruitment, both wild-type (WT) CHO cells and β-arrestin1/2 KO CHO cells were transfected with plasmid encoding GHSR1a with or without plasmid encoding MRAP2. The transfected cells were treated with increasing concentrations of ghrelin in the presence of lithium before the concentration of accumulated IP produced was measured. In WT CHO cells lacking MRAP2, ghrelin elicited a small response (about a 50% increase in IP abundance compared to baseline). In contrast, response in cells expressing MRAP2 was 6-fold over baseline (Fig. 3C), consistent with MRAP2 potentiating GHSR1a signaling. Surprisingly, we obtained a similar result in experiments with β-arrestin1/2 KO CHO cells (Fig. 3D), thus suggesting that inhibition of β-arrestin recruitment is not required for MRAP2 to potentiate Gαq–dependent signaling by GHSR1a.
Fig. 3. Regulation of G protein– and β-arrestin–dependent signaling pathways by MRAP2.
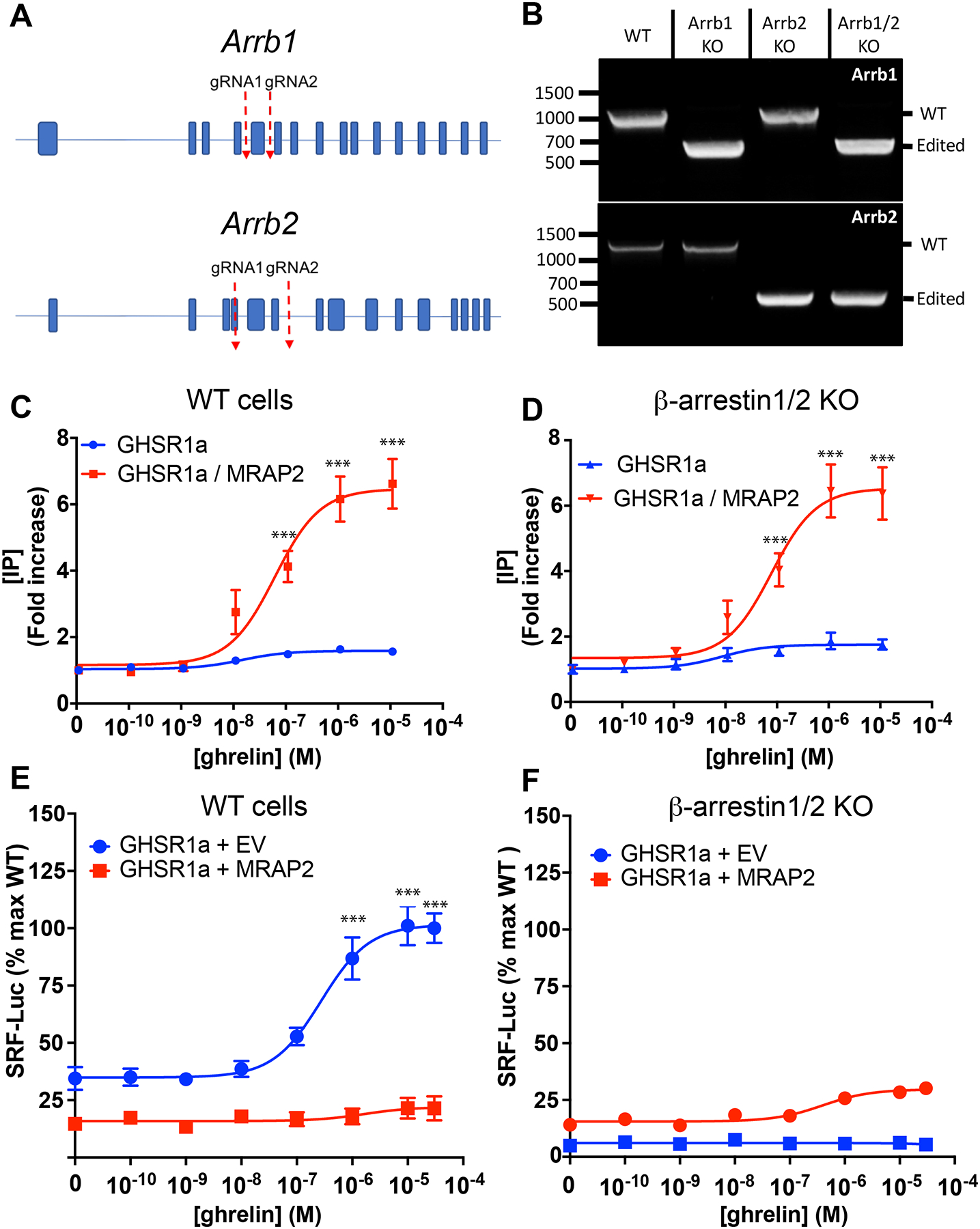
(A) Schematic of CRISPR/Cas9 targeted deletion for Arrb1 and Arrb2 genes. (B) PCR validation of β-arrestin1 KO, β-arrestin2 KO and β-arrestin 1/β-arrestin 2 double KO cell lines. (C and D) Concentration response curves of ghrelin-stimulated IP3 production in WT (C) and β-arrestin 1/2 KO (D) cells transfected with GHSR1a with or without MRAP2. Data represent the mean +/− SEM of 3 independent experiments performed in triplicate. (E and F) Concentration response curves of ghrelin-stimulated RhoA activation in WT (E) and β-arrestin1/2 KO (F) CHO cells transfected with SRF-RE-Luc reporter, GHSR1a and either empty vector or MRAP2. Statistical analysis of the differences at each ghrelin concentrations were measured by T-test. Data represent the mean +/− SEM of 3 independent experiments performed in triplicate, ***p<0.001.
MRAP2 inhibits β-arrestin–dependent GHSR1a signaling
Recruitment of β-arrestin to GHSR1a results in the activation of the small guanosine triphosphatase (GTPase) RhoA, and mutations in GHSR1a that impair its ability to recruit β-arrestin also decrease ghrelin-stimulated RhoA activation (19). Consequently, we reasoned that the inhibition of ghrelin-stimulated β-arrestin recruitment by MRAP2 should result in inhibition of RhoA activation. To test this notion, we used a luminescent reporter driven by SRF-RE, a modified serum responsive element activated by members of the RhoA GTPase family (20). CHO cells were transfected with plasmids encoding SRF-RE-Luc and GHSR1a and either empty vector or plasmid encoding MRAP2. Treatment with ghrelin caused a concentration-dependent increase in RhoA activation in cells expressing GHSR1a alone (Fig. 3E). In the presence of MRAP2, ghrelin-stimulated RhoA activation was blocked (Fig. 3E), thus suggesting that the inhibition of β-arrestin recruitment by MRAP2 also results in the inhibition of signaling downstream of β-arrestin. To verify that ghrelin-dependent stimulation of RhoA required β-arrestin, the same experiment was performed in β-arrestin1/2 KO cells, which showed that ghrelin largely failed to activate RhoA regardless of whether MRAP2 was present (Fig. 3F). These results confirm that RhoA is activated by ghrelin through a β-arrestin–dependent pathway and that MRAP2 prevents the recruitment and signaling of β-arrestin downstream of GHSR1a.
Potentiation of GHSR1a signaling requires the N-terminal domain containing residues 34–43, the transmembrane domain and C-tail of MRAP2.
To identify the region of MRAP2 that was important for the potentiation of GHSR1a signaling, we generated a series of MRAP2 deletion mutants and chimeras. These include four deletions in the N-terminal region (Δ2–9, Δ10–23, Δ24–33 and Δ34–43), a chimeric MRAP2 in which the transmembrane domain was replaced by the transmembrane domain of RAMP3 (MRAP2-RTM), and a chimeric MRAP2 in which the entire C-terminal domain was replaced by the C-terminal domain of MRAP1 (N2TM2C1), which does not share any sequence homology in this region (Fig. 4A). MRAP2 and mutants were all tagged with 3XFlag at the C-terminus and were expressed at similar amounts (Fig. 4B).
Fig. 4. Identification of the regions of MRAP2 required for the potentiation of G-protein–dependent GHSR1a signaling.
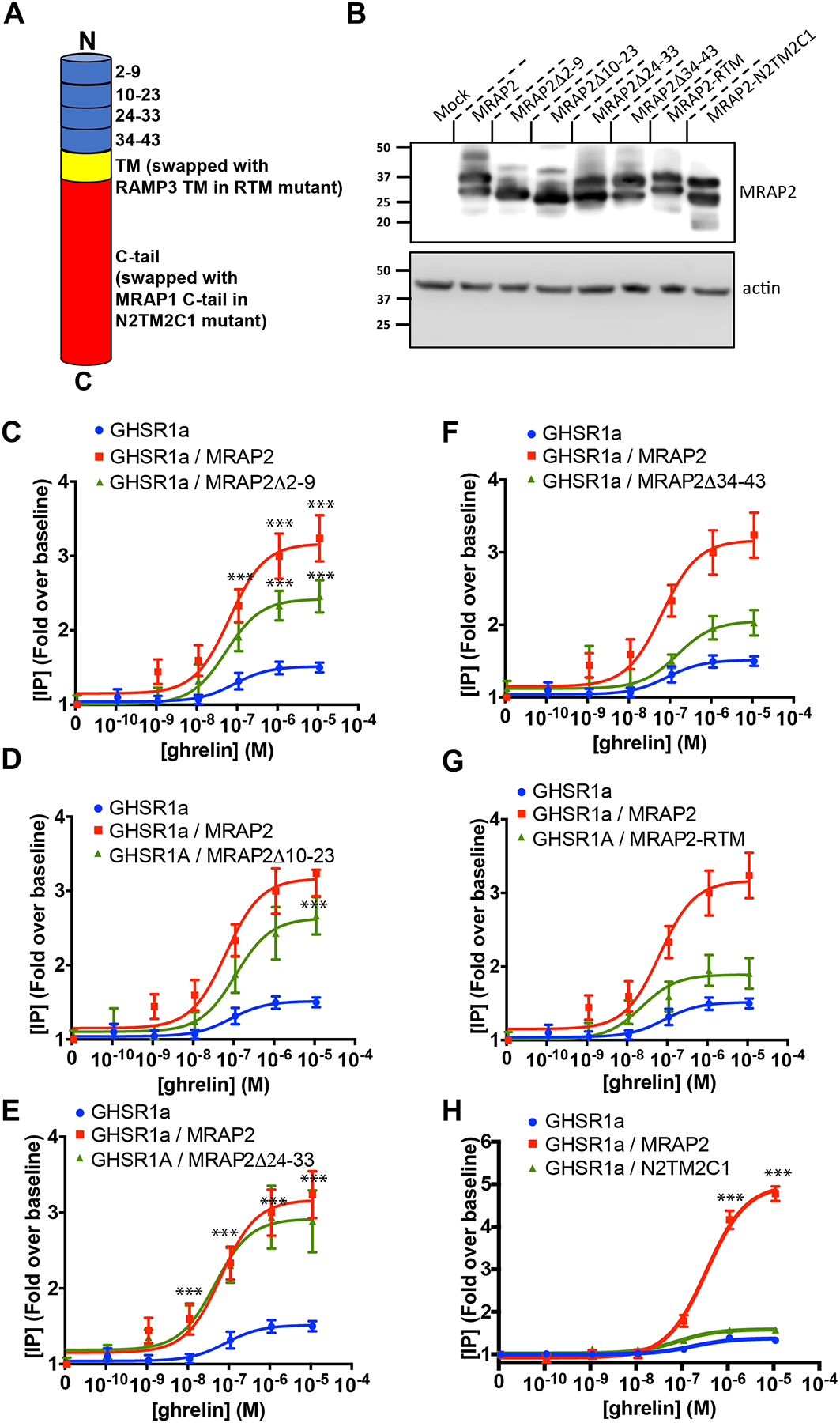
(A) Schematic representation of the different MRAP2 mutants generated (TM= transmembrane domain). (B) Western blot detection of MRAP2 mutants from lysates of transfected CHO cells (n=3 independent experiments). (C to H) Ghrelin-stimulated IP accumulation in CHO cells transfected with GHSR1a and empty vector, MRAP2 or the indicated MRAP2 mutant. Statistical analysis of the differences at each ghrelin concentrations were measured by T-test. Data represent the mean +/− SEM of 3 independent experiments performed in triplicate, ***p<0.001.
To test the ability of the various MRAP2 mutants to potentiate GHSR1a signaling, each MRAP2 mutant was transfected with GHSR1a before measuring ghrelin-stimulated IP3 production. We found that deletion of regions between amino acids 2 to 23 only caused a small reduction (25% for Δ2–9, 18% for Δ10–23) in the ability of MRAP2 to potentiate GHSR1a signaling (Fig. 4C and 4D) and that deletion of region 24–33 did not impair MRAP2 function (Fig. 4E). In contrast, loss of the N-terminal region 34–43 (Fig. 4F), the transmembrane domain (Fig. 4G) or the C-tail (Fig. 4H), almost completely abolished the potentiation of GHSR1a signaling by MRAP2, thus suggesting that those elements are essential for MRAP2 to enhance GHSR1a signaling.
Inhibition of β-arrestin recruitment to GHSR1a requires the C-tail of MRAP2 but not the residues 34–43 of the N-terminal region.
Using the same MRAP2 mutants, we tested the regions of MRAP2 that were required for the inhibition of β-arrestin recruitment to GHSR1a. To this end, cells were transfected with GHSR1a-LgBiT and SmlBiT- β-arrestin2 in the presence of each of the MRAP2 mutants. Cells were also transfected with empty vector or MRAP2 for comparison. Deletion of N-terminal residues 2–9 (Fig. 5A) and 10–23 (Fig. 5B) did not impair the ability of MRAP2 to inhibit β-arrestin recruitment. Deletion of the residues 24–33 (Fig. 5C) largely abolished the effect of MRAP2 on β-arrestin recruitment. MRAP2Δ34–43 inhibited β-arrestin recruitment (Fig. 5D) and replacing the transmembrane domain only caused a partial loss in MRAP2 activity (Fig. 5E). Finally, replacing the C-tail of MRAP2 by the C-tail of MRAP1 completely abolished the inhibition of b-arrestin recruitment by MRAP2 (Fig. 5F). To more accurately determine the effect of MRAP2 mutations on β-arrestin recruitment, we measured the surface density of HA-tagged GHSR1a and Flag-tagged MRAP2 with ELISAs. Surface expression of MRAP2 and mutants were different; however, the mutant depicting the lowest expression (MRAP2Δ10–23) (Fig. S1) retained full inhibitory activity on β-arrestin recruitment, suggesting that MRAPs were in excess. Several MRAP2 mutants, but not WT, significantly altered the surface expression of GHSR1a compared to control (Fig. 5G). Normalization of β-arrestin recruitment data according to the corresponding surface GHSR1a density revealed that the transmembrane domain and the C-terminal region of MRAP2 were required (Fig. 5H). In contrast to the IP results, MRAP2Δ34–43 displayed no defect in the inhibition of β-arrestin recruitment. Furthermore, while the mutant MRAP2Δ24–33 significantly decreased β-arrestin recruitment, its ability to do so was significantly impaired compared to WT MRAP2 (Fig. 5H), thus suggesting a role for residues 24–33 in MRAP2-mediated inhibition of β-arrestin recruitment. Based on these results, we conclude that the transmembrane and the C-terminal domains of MRAP2 are essential, and the N-terminal region 24–33 contribute for the inhibition β-arrestin recruitment to GHSR1a. Whereas the C-terminal tail of MRAP2 is important for both the potentiation of Gαq-dependent signaling and the inhibition of β-arrestin recruitment, the N-terminal regions involved in these two functions are distinct, thus further indicating that the regulation of Gαq signaling and β-arrestin recruitment by MRAP2 are independent functions.
Fig. 5. Identification of MRAP2 regions required for the inhibition of β-arrestin recruitment to GHSR1a.
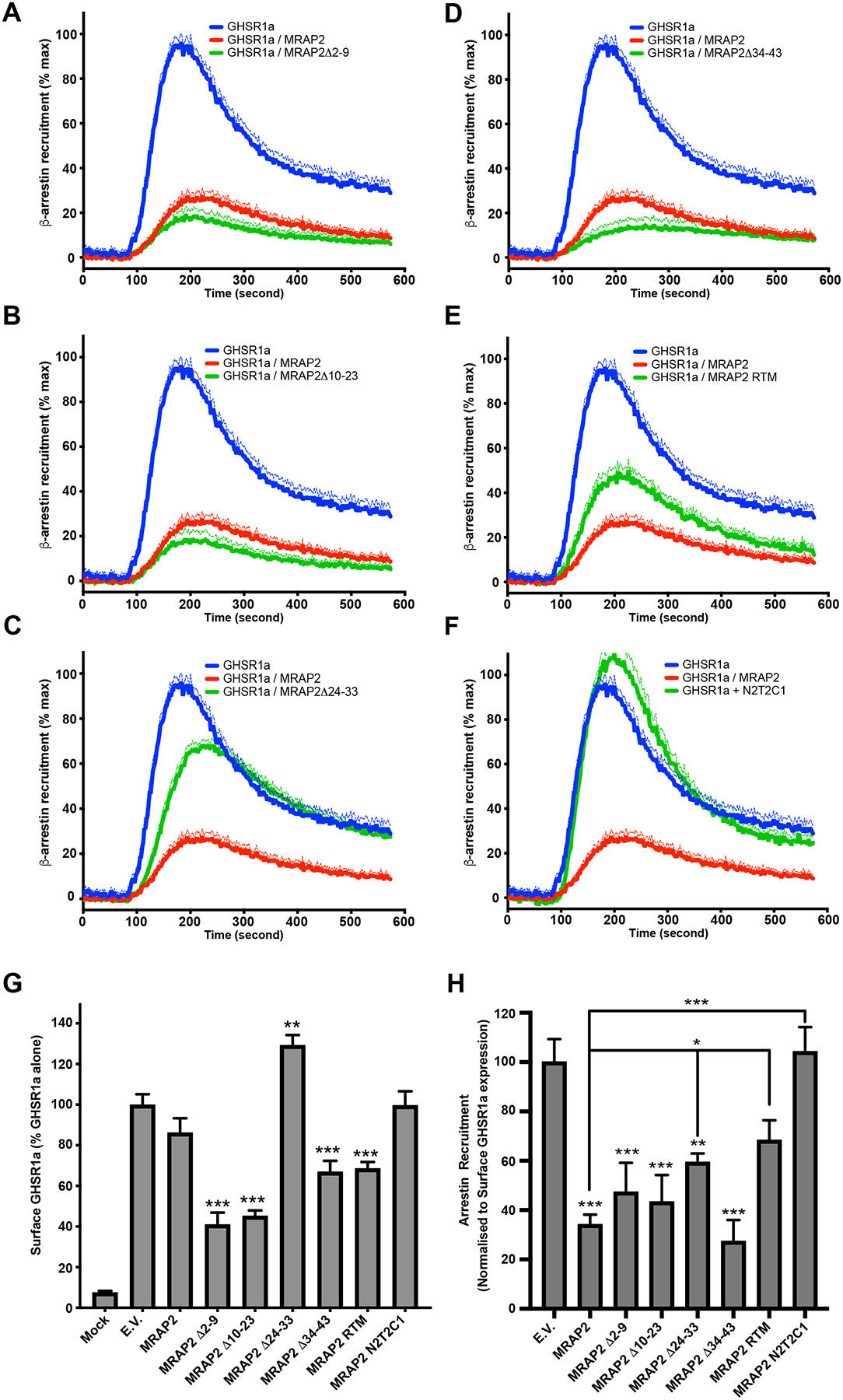
(A to F) Kinetics of ghrelin-stimulated β-arrestin recruitment to GHSR1a in CHO cells transfected with GHSR1a-LgBiT, SmlBiT-β-arrestin2 and empty vector, MRAP2 or the indicated MRAP2 mutant. Statistical analysis of the differences at each ghrelin concentrations were measured by T-test. Data represent the mean +/− SEM of 3 independent experiments performed in triplicate. (G) Surface density of GHSR1a measured by fixed-cell ELISA in each of the condition tested for β-arrestin recruitment. Statistical analysis was done using one-way ANOVA. Data represent the mean +/− SEM of 3 independent experiments performed in triplicate. (H) Area under the curve of β-arrestin recruitment normalized to the surface expression of GHSR1a for each condition tested. Statistical analysis was done using one-way ANOVA. Data are shown as mean +/− SEM of the percentage of normalized β-arrestin recruitment in the absence of MRAP2. *p<0.05, **p<0.01, ***p<0.001.
Regions of MRAP2 necessary for its inhibitory action on β-arrestin recruitment to GHSR1a are also important for ghrelin-mediated RhoA activation.
Because the activation of RhoA by ghrelin is mediated through β-arrestin, we hypothesized that mutations in MRAP2 that impair its ability to inhibit β-arrestin recruitment would also result impair its ability to block RhoA activation. We used the SRF-RE-Luciferase reporter to test this hypothesis. Expression of MRAP2Δ2–9 (Fig. 6A) and MRAP2Δ10–23 (Fig. 6B) inhibited ghrelin-stimulated RhoA activation to the same extent as WT MRAP2. Deletion of residues 24–33 reduced RhoA activation by 50% (Fig. 6C). MRAP2Δ34–43 abolished ghrelin-stimulated RhoA activation (Fig.6D). Replacement of the transmembrane domain resulted in a 66% decrease in activity (Fig. 6E). Finally, replacement of the C-tail of MRAP2 with the C-tail of MRAP1 abolished the ability of MRAP2 to inhibit ghrelin-stimulated RhoA activation (Fig. 6F). The similar effects of the mutants on the RhoA activity and β-arrestin recruitment provide further support for the notion that ghrelin-stimulated RhoA activation is mediated by β-arrestin.
Fig. 6. Identification of MRAP2 regions required for the inhibition of ghrelin-stimulated RhoA activation.
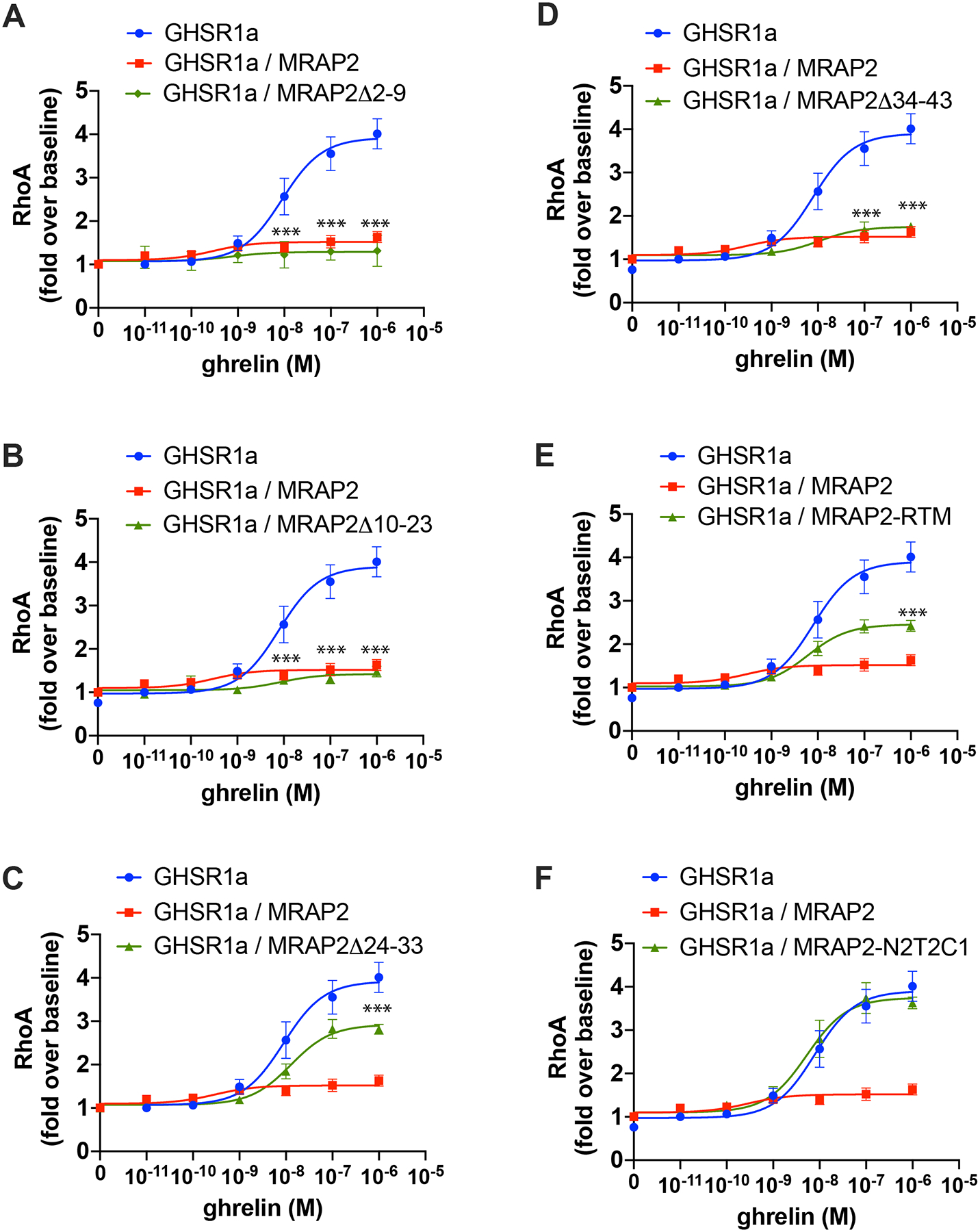
(A to F) Luminescence reporting ghrelin-stimulated RhoA activity in CHO cells expressing the SRF-RE reporter, GHSR1a and either empty vector, MRAP2 or the indicated MRAP2 mutant. Statistical analysis of the differences at each ghrelin concentrations were measured by T-test. Data are shown in fold increase over baseline and are mean +/− SEM of 3 experiments performed in triplicate. ***p<0.001.
MRAP2 does not increase the affinity of GHSR1a for ghrelin or potentiate the GEF activity of GHSR1a
To determine whether the increase in ghrelin efficacy observed in cells expressing MRAP2 was due to an increase in the affinity of the receptor for ghrelin, we performed a radioligand-receptor competition binding assay, which showed that expression of MRAP2 did not affect the affinity of GHSR1a for ghrelin (Fig. 7A). This result suggests that the potentiation of GHSR1a signaling by MRAP2 is not due to an increase in ghrelin binding affinity.
Fig. 7. MRAP2 does not increase ghrelin affinity or Gαq protein activation.
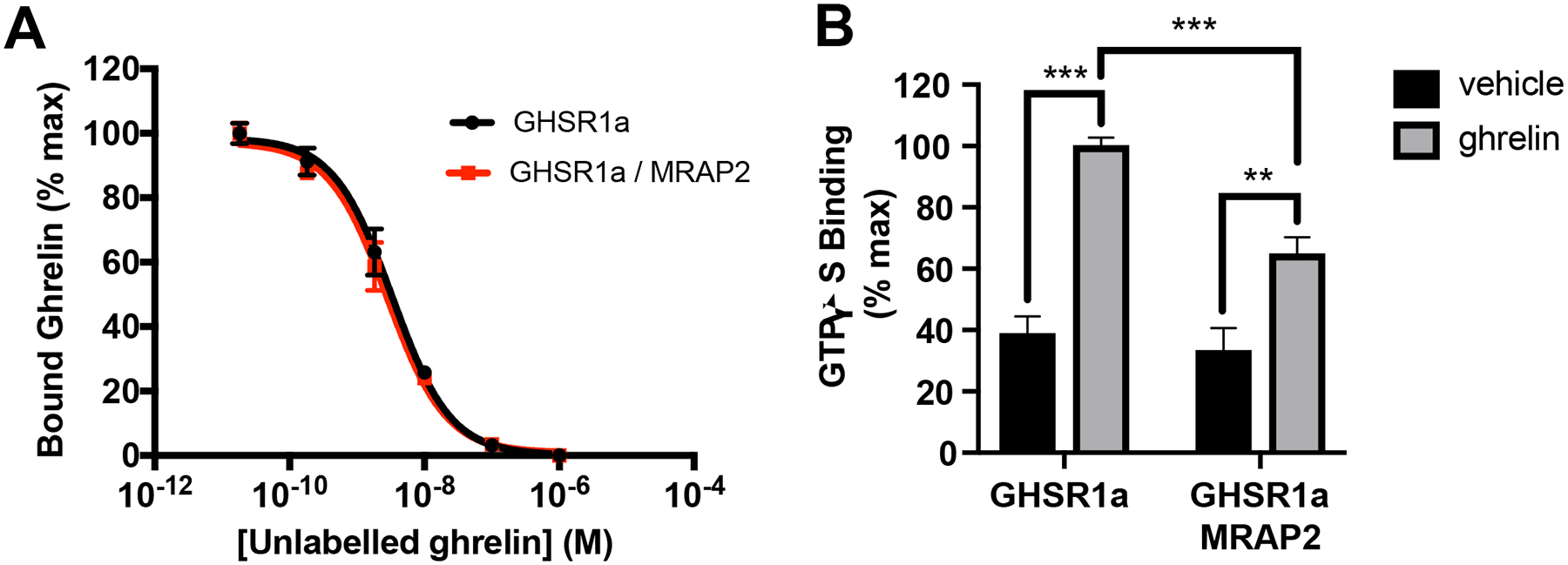
(A) 125I-Ghrelin competition binding assay in CHO cells transfected with GHSR1a and empty vector or MRAP2. Non-specific binding from mock transfected cells was subtracted and results were normalized to the maximal binding for each condition. Data shown are the mean +/− SEM of 3 independent experiments performed in triplicate. (B) GTPγS binding assay in membranes prepared from CHO cells transfected with GHSR1a and empty vector or MRAP2. Binding values obtained with membranes from mock transfected cells was subtracted and results were normalized to GHSR1a expression measured by ELISA. Statistical analysis was done using one-way ANOVA. Results shown represent the data attained from 3 independent experiments. Error bar are SEM. ***p<0.001, **p<0.01
Agonist-activated GPCRs facilitate the exchange of GDP for GTP in the Gα subunit of G proteins. Because MRAP2 potentiates the G protein signaling pathway downstream of GHSR1a we tested whether MRAP2 promotes the GHSR1a-mediated guanine nucleotide exchange of Gαq. [35S]GTPγS binding assays were performed with membranes from CHO cells expressing GHSR1a in the presence or absence of MRAP2. Ghrelin promoted [35S]GTPγS binding to GHSR1a, as expected, and MRAP2 did not potentiate this process. In fact, a small inhibition of [35S]-GTPγS binding was measured in membranes from cells expressing MRAP2, suggesting that the potentiation of GHSR1a signaling by MRAP2 was not caused by an increase in the GEF activity of GHSR1a (Fig. 7B). The decrease in GEF activity in the presence of MRAP2 may suggest that other intracellular factors, which are lost during the membrane preparation, are important for the normal activity of the GHSR1a / MRAP2 complex.
Discussion
GPCRs are responsible for many physiological functions and consequently are the largest family of proteins targeted by pharmacological drugs. However, GPCR agonists tend to have side effects due to the multiple signaling pathways activated downstream of the targeted receptor. In some cases, ligands that selectively activate one or a subset of signaling pathways downstream of their receptor target, which are known as biased agonists, sometimes display improved therapeutic profiles compared to classical orthosteric agonists (21,22). For example, biased agonists of the μ-opioid receptor can decrease nociception through activation of the G protein-dependent pathway while limiting unwanted, β-arrestin–mediated side effects, such as constipation and respiratory distress (23,24). Whereas many biased ligands have been developed for several receptors, there are few identified examples of endogenous proteins that promote GPCR signaling bias. One example is the receptor activity modifying protein (RAMP) family which modifies the selectivity of some GPCRs for Gα subunits (25,26). Here, we showed that MRAP2 biased GHSR1a signaling toward Gαq-dependent pathways rather than β-arrestin–dependent signaling. MRAP2 expression induced an increase in ghrelin-stimulated IP3 production, a decrease in ghrelin-stimulated β-arrestin recruitment to the receptor, and an almost complete loss of ghrelin-stimulated RhoA activation, a small GTPase that is largely activated through β-arrestin downstream of GHSR1a (19). The requirement of β-arrestin for ghrelin to activate RhoA was further demonstrated here by the inability of ghrelin to stimulate RhoA in β-arrestin1/2 KO cells. However, the inhibition of β-arrestin recruitment by MRAP2 was not required for MRAP2 to potentiate GHSR1a signaling, suggesting that the potentiation of GHSR1a by MRAP2 is not simply due to a decrease in β-arrestin-mediated desensitization of the receptor. Indeed, MRAP2 enhanced GHSR1a signaling to the same extent in cells expressing or lacking β-arrestins. Further establishing the independence of MRAP2-mediated regulation of Gαq/11 and β-arrestin, we identified mutations in MRAP2 that potentiated ghrelin-stimulated IP3 production with only a limited effect on β-arrestin recruitment and that inhibited β-arrestin recruitment without substantially enhancing the Gαq/11-dependent pathway. We found that deletion of β-arrestins did not increase GHSR1a signaling through Gαq/11, which suggests that β-arrestin interacts with GHSR1a preferentially in the tail rather than core conformation, thus not restricting G-protein access to the receptor (27).
The signaling bias downstream of GHSR1a caused by MRAP2 implies that, depending on the cellular distribution and abundance of MRAP2, ghrelin could elicit different cellular signaling outcomes and physiological consequences. Whereas the orexigenic function of ghrelin is mediated through the activation of Gαq/11 in AGRP neurons (28) and requires MRAP2 (11), the physiological role of β-arrestin signaling downstream of GHSR1a is not yet understood. Based on our results, we expect that transgenic animal models expressing strategically mutated MRAP2 would be valuable in understanding the physiological role of β-arrestin signaling downstream of GHSR1a in vivo.
We have previously shown that MRAP2 regulates several GPCRs including the melanocortin-4 receptor (14), prokineticin receptors 1 and 2 (16) and the orexin receptor 1 (18). However, the N-terminal region of MRAP2 involved in potentiating GHSR1a signaling is distinct from the regions of MRAP2 required for the regulation of the prokineticin receptor 1 and orexin receptor 1 (18), thus suggesting that different mechanisms and specific interactions may exist between MRAP2 and its GPCR targets.
Ghrelin is important in regulating energy and glucose homeostasis, especially during energy deficit. During starvation, ghrelin action on the hypothalamus promotes food intake and its action on the pituitary and pancreas prevents hypoglycemia (2). Ghrelin also promotes gastric emptying (29). Consequently, the ghrelin receptor is a promising target for the treatment of obesity (30) and diabetic gastroparesis (31). Small molecule antagonists of the ghrelin receptor developed for the treatment of obesity cause an unexpected increase in food intake (32,33) which may be due to intrinsic partial agonism (28). In contrast, generating compounds that target the interface of MRAP2 and GHSR1a may produce ghrelin inhibitors that do not bind the orthosteric site of GHSR1a and lack partial agonism activity, thus resulting in decreased food intake and weight loss. Additionally, understanding the regulation of GHSR1a signaling by MRAP2 in different tissues and the specific physiological actions of each downstream pathway is likely to improve future drug discovery efforts that target GHSR1a.
The physiological role for the constitutive activity of GHSR1a is not well understood, although the identification of a mutation (A204E) in GHSR1a that causes a loss of agonist-independent signaling in individuals with familial short stature has implicated the constitutive activity of GHSR1an in somatic growth. In this study, we confirmed that this mutation substantially decreased the constitutive activity of GHSR1a. In contrast to the WT receptor, GHSR1a(A204E) signaling was inhibited by MRAP2. This finding implies that in individuals with the GHSR1a(A204E) mutation, in which the abundance of MRAP2 is likely unaltered, both the constitutive activity of the receptor and its response to ghrelin are impaired, raising the question of which alteration causes the short stature. Additionally, because MRAP2 substantially decreases the constitutive activity of GHSR1a, agonist-independent signaling by the receptor, at least in cells that express MRAP2, may not play a role in vivo.
In conclusion, this study identifies MRAP2 as a modulator of GHSR1a signaling both by biasing ghrelin signaling and blocking the constitutive activity of the receptor. Determining how MRAP2 abundance is regulated in GHSR1a-expressing cells in vivo is thus important to accurately predict the cellular effect of ghrelin and may improve the design of therapies targeting GHSR1a.
Materials and Methods
Cell culture and transfection
CHO-K1 cells were cultured in DMEM/F-12 (ThermoFisher supplemented with 5% v/v of a 1/1 mix of calf bovine serum and fetal bovine serum and 1% penicillin-streptomycin. Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2. Cells were transfected with LipoD293 DNA Transfection Reagent from SignaGen laboratory (Cat. #: SL100668). The quantity of DNA for each plasmid varied based on the assay; however, the ratio receptor / accessory protein was constant and set at 1/20.
Plasmids
The 3HA-GHSR1a plasmid was obtained from the Missouri S&T cDNA Resource Center. The SRF-RE-Luciferase plasmid was purchased from Promega (plasmid #E1350). Plasmids coding for mutants of MRAP2 were previously described (18). Empty vector refers to an empty pcDNA3.1 plasmid (Life Technologies). The N2TM2C1 plasmid, a chimera comprising residues 1 through 63 from human MRAP2 (putative N-terminus and transmembrane domains) and residues 57 through 172 from human MRAPα, was generated using the NEBuilder HiFi DNA assembly kit (New England Biolabs, Ipswich MA). Oligonucleotides 5’tatagggagacccaagctggaattcATGTCCGCCCAGAGGTTAATTTC’3 and 5’tgtagagcaaCACAAAAAACATAAAAATCACGAAGACTG’3 (bases in upper case represent the sequence matching either MRAP2 or MRAPα) were used to generate the N2T2 portion, while oligonucleotides 5’gttttttgtgTTGCTCTACATGTCCTGG’3 and 5’gcttttaattaatctagaggcgcgccGCTCTGCAATTGAGAGGTC’3 were used to generate the C1 construct fragments respectively. Assembly was performed following the manufacturer’s recommendations, using pcDNA3.1(+) as vector backbone after digest with EcoRI and AscI. The resulting clone was confirmed by Sanger sequencing. A triple C-terminus Flag tag was later added by subcloning the N2T2C1 sequence into a previously constructed pcDNA3–3xFlag plasmid following standard cloning procedures.
Generation of β-arrestin1 and β-arrestin2 knockout cells
To delete β-arrestin 1 and β-arrestin 2 in CHO cells, we designed guide RNAs targeting regions that would result in the excision of exon 5 from the Arrb1 gene (which encodes β-arrestin1) and exons 4 to 6 from the Arrb2 gene (which encodes β-arrestin2). gRNAs: Arrb1, TTGATACAACTCTTAGGAAG and CATTCTGGAAGCCTCACCTC, and Arrb2, AAGGTACTAGACCCAAGTCC and AAGACAAGGAGGTGTCCCCA. gRNA and trRNA (IDT, Coralville, IA) were annealed to form RNP and transfected with Cas9 protein using Lipofectamine CRISPRMAX (Thermo Fisher Scientific) following the manufacturer’s instructions. 48 hours after transfection, cells were diluted and plated in 96 well plates at a density of 1 cell per 3 wells. Single clones were allowed to grow to confluency. DNA from each clone was extracted using QuickExtract DNA Extraction Solution (Lucigen) and deletion of the targeted regions was assessed by PCR using the following primers: Arrb1, 5’-CCA TCA ATA AGG CTT GGC ACA GG-3’ and 5’-CAA CCC AGA ATC ATG GAG TCT TG-3’, and Arrb2, 5’-CTT GAC ATC CTC AGC TCA CCG TGT-3’ and 5’-TCT CAA AGT CGA CTC CAC AGG C-3’. Selected KO clones were then verified by sequencing and cultured for experiments.
SRF-RE luciferase assay
WT and β-arrestin1/2 KO CHO cells were plated in white opaque 96 well plates and transfected with SRF-RE, GHSR1a and either empty vector or MRAP2. The day after transfection, 60 μl of serum free media containing the indicated concentration of ghrelin was added. Cells were incubated at 37C for 4 hours. After incubation, 60 μl of substrate solution containing 200 mM Tris-HCl, 10 mM MgCl2, 300 μM ATP, 1% Igepal, 12.2 mM Coenzyme A; 30 μg/ml D-luciferin was added to each well and incubated 5 minutes at room temperature. Luminescence was measured on a Spectramax i3 (Molecular Devices).
125I-Ghrelin competition binding assay
[125I]-Ghrelin was purchased from Perkin Elmer. CHO cells seeded in six-well plates were transfected with 3HA-GHSR1a and either empty vector or MRAP2. The next day cells were rinsed once with PBS and incubated with [125I]Ghrelin (250,000 cpm/ml) in binding buffer (Dulbecco’s PBS with 20 mM HEPES, 0.1% BSA; pH 7.5) for 1 h at 37 °C. Nonspecific binding was determined in wells incubated with radioligand and 1 μM unlabeled Ghrelin and was subtracted. Dishes were placed on ice and washed three times with ice-cold saline to remove unbound labeled ligand. Cells were lysed with 0.1% SDS in PBS. Lysates were transferred to scintillation vials and 5 ml of scintillation fluid was added to each vial. Radioactivity was counted using a Perkin Elmer Tri-Carb 2800TR scintillation counter.
Inositol phosphate quantification
CHO cells were plated in a 6 well plate and transfected with the indicated plasmids. Cells were then lifted using TrypLE Express (Thermo Fisher Scientific) and resuspended in 500 μl of DMEM/F12. 7 μl of cell suspension was seeded per well in a white opaque 384-well plate (14,000 cells/well). 7 μl of agonist solution diluted in IP-ONE kit stimulation buffer containing lithium was then added to each well and incubated for 1 hour at 37 °C. Cells were then lysed and accumulated IP1 was measured by TR-FRET using the IP-ONE kit (Cisbio) following the manufacturer’s instruction. Readings were performed on a Spectramax® i3 (Molecular Devices). Each condition was run in triplicate and experiments were repeated independently at least 3 times.
Measurement of GHSR1a constitutive activity
To quantify the effect of MRAP2 on the constitutive activity of GHSR1a, CHO-K1 cells were transfected with 67, 33, 17, 8.3, 4.2, 2.1, 1, 0.5, and 0 ng of GHSR1a per well with 0.9 μg of empty vector or MRAP2. Cells were lifted with TrypLE, resuspended in DMEM/F12 and plated at a density of 14,000 cells/well in a 384-well plate. The IP-ONE assay was performed as described earlier but omitting the agonist. Each condition was run in triplicate and experiments were repeated independently at least 3 times.
β-arrestin recruitment
To measure β-arrestin2 recruitment to GHSR1a we used the NanoBiT protein-protein interaction assay (Promega). 3HA-GHSR1a was amplified by PCR and inserted between NheI and EcoRI sites in the vector containing the coding sequence for the LgBiT nanoluc fragment (pBiT1.2-C) to produce 3HA-GHSR1a-LgBiT. Similarly, the sequence for β-arrestin2 was inserted 3’ of the coding sequence for the SmlBiT nanoluc fragment in the pBiT2.2-N vector (Promega). Plasmids were validated by sequencing. CHO-K1 cells seeded in 6 well plates were transfected with HA-GHSR1a-LgBiT and SmBiT-β-arrestin2 with either empty vector or MRAP2. Cells were plated in a black 384-well plate (clear bottom), Nanobit substrate was added, and the plate was read with a FLIPR Tetra automated kinetic plate reader (Molecular Devices). A separate plate with agonist was prepared and placed in the FLIPR Tetra. Luminescence was measured in every well at a sampling rate of 4 to 5 seconds. After 5 minutes of recording without agonist to allow measurement of the basal luminescence signal, agonist was injected and luminescence was measured for an additional 15 minutes. Baseline signal was subtracted for each well. Each condition was run in triplicate and experiments were repeated independently at least 3 times.
Fixed cell enzyme-linked immunosorbent assay (ELISA)
CHO cells were plated in 12 well plates and triplicate wells were transfected with empty vector (mock), 3HA-GHSR1a-LgBiT, SmlBiT-β−arrestin2 and either empty vector or indicated MRAP2 construct (WT and mutant MRAP2 are C-terminally tagged with 3XFlag) using the same amount of DNA as for the β-arrestin recruitment assay. 24 hours after transfection, cells were rinsed with PBS and fixed for 10 min in 4% PFA. PFA was washed 3 times with PBS and cells were blocked with 5% non-fat dried milk in PBS for 1 hour at room temperature on a shaker. Cells were incubated with primary antibody (anti-HA 1/5000, anti-Flag 1/5000) in blocking buffer for 2 hours at room temperature on a shaker. Cells were washed 3 time for 5 min with PBS at room temperature on a shaker and incubated with secondary antibody (anti-mouse-HRP 1/5000) in blocking buffer for 1 hour at room temperature on a shaker. Cells were washed 3 time for 5 min with PBS at room temperature on a shaker. 200 μl of ELISA substrate (3,3′,5,5′-Tetramethylbenzidine, Sigma-Aldrich) was then added until blue color was visible, and the reaction was stopped with 200 μl 10% sulfuric acid. 300 ml of each sample was transferred to a 96 well plate and absorbance was measured at 450 nm using a Spectramax I3 plate reader.
Microscopy
CHO cells were plated in 6-well plates on glass coverslips and transfected with 3HA-GHSR1a, TdTomato-βarrestin2 and either empty vector or MRAP2. The next day, coverslips were transferred to an imaging chamber and placed on an inverted epifluorescence microscope (Olympus ix83). Images were acquired before and 10 minutes after addition of 100 nM Ghrelin. Experiments were repeated 3 times independently.
Western blotting
CHO cells transfected with the indicated plasmid were lysed in RIPA buffer containing protease inhibitors (Sigma-Aldrich P8340) for 30 minutes at 4⁰C. Lysates were centrifuged at 10,000 rpm for 10 minutes at 4⁰C and supernatants were transferred to new tubes and supplemented with LDS loading buffer and 5% v/v β-mercaptoethanol. Samples were boiled for 5 minutes, separated by SDS-PAGE and transferred to PVDF membranes. Membranes were blocked for 1 hour in 5% milk in PBST and incubated with mouse anti-FLAG M2 antibody (Sigma-Aldrich) at 1/5000 dilution overnight at 4⁰C on a shaker. Membranes were washed 3 times with PBST for 5 minutes and incubated with goat anti-Mouse IgG (H + L)-HRP Conjugate (Biorad #: 1706516) diluted 1/5000 in 5% milk PBST for 1 hour. Membranes were washed 3 times in PBST for 5 minutes, incubated with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific), and scanned using a C-DIGIT Blot Scanner (LI-COR).
GTPγS binding assay
CHO cells transiently transfected with GHSR1a alone and co-transfected with GHSR1a and MRAP2, were harvested, lysed in HE buffer [10 mM Hepes (pH 7.4), 1 mM EGTA, and protease inhibitor mixture (23 μg/mL phenylmethylsulfonyl fluoride, 21 μg/mL Nα-p-tosyl-L-lysine-chloromethyl ketone, 21 μg/mL L-1-p-tosylamino-2-phenylethyl-chloro ketone, 3.3 μg/mL leupeptin, and 3.3 μg/mL lima bean trypsin inhibitor)] using a Parr disruption vessel (Parr Instrument Co.). Cell debris were cleared by centrifugation at 1,500 × g. Membranes were recovered by centrifugation at 100,000 × g and washed by Dounce homogenization in HE buffer. Membranes were recentrifuged and Dounce-homogenized into membrane storage buffer [HE buffer with 12% (v/v) sucrose] and stored at −80 °C. Prepared GHSR1/MRAP2 membranes (10 μg/per assay point) were incubated for 5 min with 200 nM Gαq and 500 nM Gβ1γ2 in preincubation buffer (50 mM Hepes, pH 7.4, 1 mM DTT, 1 mM EDTA, 3 μg/mL BSA). For agonist assays, membranes were incubated with or without 10 μM Gherlin for 5 min then with G protein. Endpoint assays were initiated by addition of an equal volume of [35S]-GTPγS binding buffer [50 mM Hepes (pH 7.4), 1 mM DTT, 1 mM EDTA, 20 μM GDP, 3 μg/mL BSA, 10 mM MgCl2, 50 mM NaCl, 2 μM [35S]-GTPγS (20,000–50,000 cpm/pmol)]. Reactions were performed in triplicate, quenched after 30 minutes with 20 mM Tris, pH 7.7, 100 mM NaCl, 10 mM MgCl2, 1 mM GTP, and 0.08 (m/v) deionized polyoxyethylene 10 lauryl ether C12E10, then filtered onto Protran BA85 nitrocellulose filters (GE Healthcare). Filters were washed with 20 mM Tris, pH 7.7, 100 mM NaCl, and 2 mM MgCl2, dried, and subjected to liquid scintillation counting.
ELISA-based analysis of GHSR1a abundance in membranes
50 μl of the prepared membrane homogenates were plated in triplicate in a poly-D-lysine coated plates and spun at 2500 × g for 10 min at 4 °C. 100 ul of a solution of 2% w/v Glutaraldehyde in PBS was added and incubated 15 min at room temperature. Supernatant was removed and 100 ul of 0.1M glycine, 0.1% BSA in PBS was added to each well for 30 min at room temperature. Liquid was aspirated and wells were blocked in 5% w/v milk in PBS for 30 min at room temperature. Anti-HA antibody (1/2000) in 5% w/v milk was added to wells and incubated for 1h 30 min at room temperature. Wells were washed 3 times in PBS before adding secondary antibody (anti-mouse HRP, 1/5000) in 5% w/v milk for 45 min at room temperature. Wells were washed 3 times in PBS and 50 μl substrate (3,3’,5,5’-tetramethyl-benzidine, sigma #T0440) was added for 5 min at room temperature. Reactions were terminated by adding 50 μl of 10% v/v sulfuric acid and absorbance at 450 nm was measured on a spectramax i3 (molecular devices). Results from GTPγS binding assays were normalized to GHSR1a levels for each sample.
Supplementary Material
Acknowledgments:
We thank Roger D. Cone, Ph.D. and Jeffrey M. Zigman for insightful comments regarding this project.
Funding: This project was funded by NIH grant RO1DK115567-01 to X.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper.
References and Notes
- 1.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, and Kangawa K (1999) Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402, 656–660 [DOI] [PubMed] [Google Scholar]
- 2.Mani BK, and Zigman JM (2017) Ghrelin as a Survival Hormone. Trends Endocrinol Metab 28, 843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, Dhillo WS, Ghatei MA, and Bloom SR (2001) Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab 86, 5992. [DOI] [PubMed] [Google Scholar]
- 4.Wren AM, Small CJ, Ward HL, Murphy KG, Dakin CL, Taheri S, Kennedy AR, Roberts GH, Morgan DG, Ghatei MA, and Bloom SR (2000) The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology 141, 4325–4328 [DOI] [PubMed] [Google Scholar]
- 5.Kouno T, Akiyama N, Ito T, Okuda T, Nanchi I, Notoya M, Oka S, and Yukioka H (2016) Ghrelin O-acyltransferase knockout mice show resistance to obesity when fed high-sucrose diet. J Endocrinol 228, 115–125 [DOI] [PubMed] [Google Scholar]
- 6.Lee JH, Lin L, Xu P, Saito K, Wei Q, Meadows AG, Bongmba OY, Pradhan G, Zheng H, Xu Y, and Sun Y (2016) Neuronal Deletion of Ghrelin Receptor Almost Completely Prevents Diet-Induced Obesity. Diabetes 65, 2169–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao TJ, Liang G, Li RL, Xie X, Sleeman MW, Murphy AJ, Valenzuela DM, Yancopoulos GD, Goldstein JL, and Brown MS (2010) Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc Natl Acad Sci U S A 107, 7467–7472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li RL, Sherbet DP, Elsbernd BL, Goldstein JL, Brown MS, and Zhao TJ (2012) Profound hypoglycemia in starved, ghrelin-deficient mice is caused by decreased gluconeogenesis and reversed by lactate or fatty acids. J Biol Chem 287, 17942–17950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holst B, Cygankiewicz A, Jensen TH, Ankersen M, and Schwartz TW (2003) High constitutive signaling of the ghrelin receptor--identification of a potent inverse agonist. Mol Endocrinol 17, 2201–2210 [DOI] [PubMed] [Google Scholar]
- 10.Pantel J, Legendre M, Cabrol S, Hilal L, Hajaji Y, Morisset S, Nivot S, Vie-Luton MP, Grouselle D, de Kerdanet M, Kadiri A, Epelbaum J, Le Bouc Y, and Amselem S (2006) Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J Clin Invest 116, 760–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srisai D, Yin TC, Lee AA, Rouault AAJ, Pearson NA, Grobe JL, and Sebag JA (2017) MRAP2 regulates ghrelin receptor signaling and hunger sensing. Nat Commun 8, 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sebag JA, and Hinkle PM (2007) Melanocortin-2 receptor accessory protein MRAP forms antiparallel homodimers. Proc Natl Acad Sci U S A 104, 20244–20249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sebag JA, and Hinkle PM (2010) Regulation of G protein-coupled receptor signaling: specific dominant-negative effects of melanocortin 2 receptor accessory protein 2. Sci Signal 3, ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sebag JA, Zhang C, Hinkle PM, Bradshaw AM, and Cone RD (2013) Developmental control of the melanocortin-4 receptor by MRAP2 proteins in zebrafish. Science 341, 278–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asai M, Ramachandrappa S, Joachim M, Shen Y, Zhang R, Nuthalapati N, Ramanathan V, Strochlic DE, Ferket P, Linhart K, Ho C, Novoselova TV, Garg S, Ridderstrale M, Marcus C, Hirschhorn JN, Keogh JM, O’Rahilly S, Chan LF, Clark AJ, Farooqi IS, and Majzoub JA (2013) Loss of function of the melanocortin 2 receptor accessory protein 2 is associated with mammalian obesity. Science 341, 275–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaly AL, Srisai D, Gardner EE, and Sebag JA (2016) The Melanocortin Receptor Accessory Protein 2 promotes food intake through inhibition of the Prokineticin Receptor-1. Elife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rouault AAJ, Srinivasan DK, Yin TC, Lee AA, and Sebag JA (2017) Melanocortin Receptor Accessory Proteins (MRAPs): Functions in the melanocortin system and beyond. Biochim Biophys Acta Mol Basis Dis 1863, 2462–2467 [DOI] [PubMed] [Google Scholar]
- 18.Rouault AAJ, Lee AA, and Sebag JA (2017) Regions of MRAP2 required for the inhibition of orexin and prokineticin receptor signaling. Biochim Biophys Acta Mol Cell Res 1864, 2322–2329 [DOI] [PubMed] [Google Scholar]
- 19.Evron T, Peterson SM, Urs NM, Bai Y, Rochelle LK, Caron MG, and Barak LS (2014) G Protein and beta-arrestin signaling bias at the ghrelin receptor. J Biol Chem 289, 33442–33455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill CS, Wynne J, and Treisman R (1995) The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81, 1159–1170 [DOI] [PubMed] [Google Scholar]
- 21.Rankovic Z, Brust TF, and Bohn LM (2016) Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg Med Chem Lett 26, 241–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luttrell LM (2014) Minireview: More than just a hammer: ligand “bias” and pharmaceutical discovery. Mol Endocrinol 28, 281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, and Violin JD (2013) A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther 344, 708–717 [DOI] [PubMed] [Google Scholar]
- 24.Austin Zamarripa C, Edwards SR, Qureshi HN, Yi JN, Blough BE, and Freeman KB (2018) The G-protein biased mu-opioid agonist, TRV130, produces reinforcing and antinociceptive effects that are comparable to oxycodone in rats. Drug Alcohol Depend 192, 158–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weston C, Lu J, Li N, Barkan K, Richards GO, Roberts DJ, Skerry TM, Poyner D, Pardamwar M, Reynolds CA, Dowell SJ, Willars GB, and Ladds G (2015) Modulation of Glucagon Receptor Pharmacology by Receptor Activity-modifying Protein-2 (RAMP2). J Biol Chem 290, 23009–23022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weston C, Winfield I, Harris M, Hodgson R, Shah A, Dowell SJ, Mobarec JC, Woodlock DA, Reynolds CA, Poyner DR, Watkins HA, and Ladds G (2016) Receptor Activity-modifying Protein-directed G Protein Signaling Specificity for the Calcitonin Gene-related Peptide Family of Receptors. J Biol Chem 291, 21925–21944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cahill TJ 3rd, Thomsen AR, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, Huang LY, Kahsai AW, Bassoni DL, Gavino BJ, Lamerdin JE, Triest S, Shukla AK, Berger B, Little J. t., Antar A, Blanc A, Qu CX, Chen X, Kawakami K, Inoue A, Aoki J, Steyaert J, Sun JP, Bouvier M, Skiniotis G, and Lefkowitz RJ (2017) Distinct conformations of GPCR-beta-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci U S A 114, 2562–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mende F, Hundahl C, Plouffe B, Skov LJ, Sivertsen B, Madsen AN, Luckmann M, Diep TA, Offermanns S, Frimurer TM, Bouvier M, and Holst B (2018) Translating biased signaling in the ghrelin receptor system into differential in vivo functions. Proc Natl Acad Sci U S A 115, E10255–E10264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trudel L, Tomasetto C, Rio MC, Bouin M, Plourde V, Eberling P, and Poitras P (2002) Ghrelin/motilin-related peptide is a potent prokinetic to reverse gastric postoperative ileus in rat. Am J Physiol Gastrointest Liver Physiol 282, G948–952 [DOI] [PubMed] [Google Scholar]
- 30.Horvath TL, Castaneda T, Tang-Christensen M, Pagotto U, and Tschop MH (2003) Ghrelin as a potential anti-obesity target. Curr Pharm Des 9, 1383–1395 [DOI] [PubMed] [Google Scholar]
- 31.Alam U, Asghar O, and Malik RA (2010) Diabetic gastroparesis: Therapeutic options. Diabetes Ther 1, 32–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Costantini VJ, Vicentini E, Sabbatini FM, Valerio E, Lepore S, Tessari M, Sartori M, Michielin F, Melotto S, Bifone A, Pich EM, and Corsi M (2011) GSK1614343, a novel ghrelin receptor antagonist, produces an unexpected increase of food intake and body weight in rodents and dogs. Neuroendocrinology 94, 158–168 [DOI] [PubMed] [Google Scholar]
- 33.Halem HA, Taylor JE, Dong JZ, Shen Y, Datta R, Abizaid A, Diano S, Horvath TL, and Culler MD (2005) A novel growth hormone secretagogue-1a receptor antagonist that blocks ghrelin-induced growth hormone secretion but induces increased body weight gain. Neuroendocrinology 81, 339–349 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.