

Abstract
It has been 10 years since the introduction of modern transposon-insertion sequencing (TIS) methods, which combine genome-wide transposon mutagenesis with high-throughput sequencing to estimate the fitness contribution or essentiality of each genetic component in a bacterial genome. Four TIS variations were published in 2009: transposon sequencing (Tn-Seq), transposon-directed insertion site sequencing (TraDIS), insertion sequencing (INSeq) and high-throughput insertion tracking by deep sequencing (HITS). TIS has since become an important tool for molecular microbiologists, being one of the few genome-wide techniques that directly links phenotype to genotype and ultimately can assign gene function. In this Review, we discuss the recent applications of TIS to answer overarching biological questions. We explore emerging and multidisciplinary methods that build on TIS, with an eye towards future applications.
Subject terms: Bacterial genetics, Bacterial genetics, Mutagenesis, High-throughput screening, Mobile elements
In this Review, several experts discuss progress in the decade since the development of transposon-based approaches for bacterial genetic screens. They describe how advances in both experimental technologies and analytical strategies are resulting in insights into diverse biological processes.
Introduction
Transposon-insertion sequencing (TIS) methods combine large-scale transposon mutagenesis with next-generation sequencing to estimate the essentiality and/or fitness contribution of each genetic feature in a bacterial genome simultaneously. A strength of TIS is that experiments are performed with pooled transposon libraries, which allows direct linkage of phenotype to genotype in a high-throughput manner. Ultimately TIS aims to elucidate the function of each genomic feature and is therefore a critical tool to help interpret the mounting levels of genome sequencing data being generated. TIS methods can be sensitive enough to detect even minor changes in mutant fitness but also, with sufficient density, precise enough to be able to assay not only genes but also intergenic regions, promoter regions and essential protein domains within coding regions. Four variations on the TIS method were published in 2009: transposon sequencing (Tn-Seq)1, transposon-directed insertion site sequencing (TraDIS)2, insertion sequencing (INSeq)3 and high-throughput insertion tracking by deep sequencing (HITS)4. Since then, TIS has become a valuable tool in our molecular biology toolkit, whose full utility is still being explored.
The basic TIS workflow is summarized in Fig. 1. Briefly, it begins with construction of a saturated mutant library (Fig. 1A) by introducing a randomly inserting transposon, commonly a Tn5 or mariner transposon, into a strain of interest often by transformation or conjugation. The goal is to create a population of bacteria where each cell carries a single transposon insertion in the genome, and when cells are pooled together, each genetic component is disrupted multiple times at different sites. By direct sequencing of the transposon-flanking regions of the initial library, potential essential features can be identified as those that do not tolerate insertions. Alternatively, the library can be subjected to a selective condition, for instance antibiotic stress (Fig. 1B), to query non-essential features involved in survival and growth within that environment. Such conditionally important components are defined by insertions whose frequency significantly changes in the population during the selection, determined by sequencing before and after selection. Genomic features that have disruptive transposon insertions with a decrease in frequency over experimental selection are assumed to be important for fitness in the test conditions; such features could include antibiotic resistance genes during antibiotic selection or virulence factors in an infection model. Features where insertions show an increase in frequency are assumed to have a disadvantageous effect in the test conditions, including negative regulators of fitness-enhancing features, or metabolically costly systems that are not necessary in those conditions.
Fig. 1. Basic TIS method overview.

A | Creation of the transposon-insertion sequencing (TIS) library has four steps. The first is to create random transposon (Tn) mutants (part Aa). The horizontal black lines and arrows represent the host’s genomic DNA (gDNA) and the coding regions of genes are marked ‘X’, ‘Y’ and ‘Z’. The horizontal blue line is the transposon containing an antibiotic resistance (AbR) selection marker and bounded by inverted repeats, shown in green. When the transposon inserts itself into the gDNA, the disruption in a gene (gene Y in this example) is shown by a red cross. The second step is to select and pool mutants (part Ab). The red cross represents a single mutation in each cell; these cells are selected, for instance on antibiotic-containing agar plates, and pooled and DNA is extracted. The third step is fragmentation, addition of adaptors and PCR amplification (part Ac). Fragmentation (vertical dashed lines) can be enzymatic or by shearing (depending on the version of TIS). Sequencing adaptors (yellow rectangles) are then added, and primers (purple arrows) P1 and P2 are used for PCR amplification. Step 4 is sequencing and mapping (part Ad). Sequences out from the transposon end (using primer P3) are mapped onto the reference genome and the transposon insertion point is determined (vertical red arrow) and mapped for each mutant. Genes that cannot tolerate insertions (gene Z in this example) will not have any TIS reads mapped. B | Challenging the TIS library — in this example with an antibiotic, colistin (bottom row), compared with an untreated control (top row) (data from ref.42). The vertical lines denote the density of insertions at each insertion site, and red and blue denote forward or reverse insertion direction, respectively. Below are the predicted genes, in light blue. The first gene (usq) has equivalent numbers of insertions in the treated and untreated samples and thus has no effect on fitness in colistin. The next gene (truA) has relatively more insertions in the treated sample compared with the control (its mutants have increased fitness in colistin) and thus is considered a sensitivity gene. The next gene (dedA) is an experimentally confirmed resistance gene42 and it has decreased insertions in the treated sample (mutants have decreased fitness in colistin). The last two genes (accD_1 and folC) have no insertions in either the treated sample or the untreated sample and are thus considered essential for growth.
There are four major TIS versions that differ in various steps of their sequencing procedures (see ref.5 for more detail on these variations). For example, the way DNA undergoes fragmentation for library preparation differs: Tn-Seq and INSeq use the type II restriction enzyme MmeI to yield uniform-length shorter reads, which can remove PCR amplification bias, whereas TraDIS and HITS use random-sized shearing via sonication, which can have the advantage of improved transposon mapping owing to longer reads. Similarly, Tn-Seq and INSeq exclusively use the mariner transposon, which inserts itself into thymine–adenine dinucleotide (TA) sites but otherwise does not have a sequence preference, and the others have the flexibility that they can use any transposon, but they commonly use Tn5 as it is commercially available and does not have a insertion site bias. After fragmentation, various adaptors are added, and transposon–genome junctions are amplified and sequenced with a sequencing primer facing out of either the transposon or the adaptor. Finally, mapping of the adjacent genomic DNA allows the exact position of each transposon in the bacterial genome to be determined with use of appropriate bioinformatic tools (see Developments in TIS data analysis).
Since the last comprehensive reviews on TIS5,6 in 2013, a range of exciting and multidisciplinary methods that build on TIS have emerged to answer increasingly complex biological questions. These advances include scaling TIS analysis to hundreds of different conditions using high-throughput phenotyping, the use of machine learning to predict bacterial survival outcomes and combining TIS with cutting-edge techniques from single-cell analysis (droplet Tn-Seq (dTn-Seq)) to fluorescence sorting (TraDISort). Analysis tools have also evolved to cope with this increase in complexity of TIS studies. Lastly, a broad range of in vitro and in vivo applications of TIS have been implemented in pathogenic, commensal and environmental bacteria in the past decade. In this Review, we discuss these exciting developments and applications of TIS and present our vision for TIS into the future. We refer readers to previous reviews5–7 for detailed information on the design of TIS experiments, including choice of transposon and statistical impacts of experimental parameters, comparisons of TIS method variations, limitations of standard TIS and details on applications before 2013.
Advances and extensions of TIS methods
Over the past decade, TIS methods have been developed to incorporate other technologies and techniques to answer complex biological questions in creative ways. These include physical separation and sorting of individual mutant cells, using inducible promoters to study essential genes and scaling of current techniques to simultaneously screen multiple environments and different species, facilitating pan-organism analysis (Fig. 2).
Fig. 2. Extensions to the TIS method.

a | Physical separation of mutant populations based on motility. This includes assaying genes for motility by inoculating transposon-insertion sequencing (TIS) mutants on an agar plate (yellow circle and beige spot) and separating the inner mutant pool (less motile) from the outer mutant pool (more motile). b | In density–TraDISort, mutant populations can be separated into top, middle and bottom fractions (shown by horizontal orange bands) on the basis of their increased or decreased cellular density using a Percoll gradient and centrifugation. c | Separation of single mutants using fluorescence (TraDISort). The mutant pool is treated with the fluorescent marker ethidium bromide (EthBr) and subjected to fluorescence-activated cell sorting, where each cell is sorted with use of a laser (horizontal red line) on the basis of its fluorescence (shown as green), reporting on efflux activity. d | Encapsulation, growth and sorting with microfluidics of single mutants in droplets for droplet transposon sequencing (dTn-Seq). Each single mutant with different growth rates (in the schematic on the left, low, medium and high levels of growth are represented as blue, orange and green background colours, respectively) will grow independently within its own droplet (grey circle), eliminating the effects of interactions between mutants. A final sorting step, based on cell fluorescence or microscopy, can also be added. Alternatively, cell-containing droplets (blue droplet on the right) can undergo multiple layers of re-encapsulation, so that an encapsulated mutant can be encapsulated within another droplet containing a different cell (shown in yellow; this can be another mutant, another bacterial cell or a host cell) and signals can freely diffuse between the layers (shown as red bolts) to allow cell interactions to be investigated by sorting those cell combinations that have altered fitness and grow at different rates, or those that can be separated by sorting based on markers, such as alterations of cell morphology observed with a microscope.
Beyond growth-based selection approaches
A major recent advance of TIS is based on the ability to separate mutants by their physical characteristics, rather than solely on the basis of growth. The simplest forms of this have adapted classical microbiological assays to the massive multiplexing made possible by TIS (Fig. 2a). For example, motility genes can be assayed by ‘racing’ mutant libraries across agar plates and comparing mutants in the inner population (less motile) with those in the outer population (more motile). This approach has been applied to Escherichia coli ST131 (ref.8) and Pseudomonas aeruginosa PA14 (ref.9), leading to the identification of known motility genes, such as those encoding common bacterial motors (flagella, fimbriae and pili), in addition to new candidates. Similarly, density–TraDISort10 combines TraDIS and density gradient centrifugation to separate mutants on the basis of their density (Fig. 2b) and identify genes involved in bacterial capsule production, which is a major virulence factor for many pathogens. In this study, 78 genes underlying capsule production were identified across two clinically relevant Klebsiella pneumoniae strains10.
The application of cell sorting to TIS has led to the development of techniques that progress from bulk separation to separation of single cells. One such application is TraDISort, which combines fluorescence-activated cell sorting (FACS) and TraDIS11 and sorts single cells on the basis of fluorescence. TraDISort has used the cytosolic concentration of ethidium bromide (EthBr), a fluorescent DNA intercalating agent, as a marker for altered efflux activity (Fig. 2c). For instance, mutants with insertions in efflux pump genes, such as amvA, had reduced ability to remove ethidium bromide from the cell, resulting in an overall higher level of fluorescence. By contrast, mutants such as the amvA repressor (amvR), had increased efflux and lower fluorescence. A similar approach used a fluorescent reporter to separate heterogeneous populations of Mycobacterium tuberculosis12, which uncovered lamA, a gene of previously unknown function that reduced overall heterogeneity in the population by decreasing asymmetric polar growth. Similarly, FAST-INSeq was developed to identify regulators of typhoid toxin production, with use of FACS of Salmonella enterica subsp. enterica serovar Typhi-infected macrophages with a fluorescent reporter for toxin expression13. Lastly, Tn-FACSeq was used to identify genes from Bdellovibrio bacteriovorus, a bacterial predator, that are important for attachment to Vibrio cholerae14. These types of fluorescence-based technique could be extended further, for instance to examine bacterial responses to other fluorescent (or fluorescently tagged) compounds, to other fluorescent reporter constructs or simply using FACS to differentiate mutants with altered cell size.
Population-independent mutant assays
In traditional TIS approaches, mutant fitness is measured within the context of the entire mutant population. However, the true fitness of a mutant can be obscured when it is grown in the presence of other mutants. For instance, TIS cannot report on the effect of secreted products or other ‘common goods’ that act beyond the cell containing the mutation, or similarly mutants that suffer from density dependence. Recently, on the basis of advances in single-cell analysis, dTn-Seq was developed to address these issues15. dTn-Seq sorts single mutants by combining microfluidics with TIS, encapsulating individual transposon mutants in growth-medium-in-oil droplets, facilitating isolated growth of mutants free from the influence of the population (Fig. 2d). dTn-Seq experiments showed that in Streptococcus pneumoniae 1–3% of mutants have altered fitness when grown in isolation; some mutants may grow faster or slower in isolation compared with their growth measured in a traditional TIS screen. To highlight its versatility, dTn-Seq has been applied to investigate hypercompetence, processing of host glycoproteins, defence against host immune factors and microcolony formation15. Moreover, dTn-Seq is compatible with microscopy and FACS-based screening, and by reloading droplets into a microfluidic device, multilayer encapsulations can be achieved. Such droplets consist of multiple layers containing different mutants, other bacterial species or even host cells, between which communication signals can freely diffuse, thereby facilitating investigations of interbacterial and bacteria–host cell interactions15 (Fig. 2d). Although these droplets cannot easily be applied to in vivo animal models, any interesting phenotypes that arise from dTn-Seq screens, including host–microorganism interaction mediators, can be directly confirmed in cell culture assays and/or in vivo in animal models using targeted mutants.
Assaying function of essential genes and gain-of-function screens
One limitation of standard TIS is that only non-essential genes can be assayed, as essential genes, by definition, do not tolerate insertions. A handful of studies have overcome this by using gain-of-function screens that use libraries of transposons with outward-facing promoters to facilitate gene overexpression and repression (Fig. 3A). Monitoring the change in frequency of transposons that induce the expression of downstream genes, including essential genes, during selection can identify phenotypes that may not be evident from gene disruption (Fig. 3B). This idea is not new; for example the TnAraOut method, developed in 2000 (ref.16), used transposons containing the arabinose inducible promoter PBAD to screen V. cholerae for essential antibiotic targets. Various approaches, where an outward facing promoter is engineered into a transposon system, have been developed to assay essential genes, for example in Caulobacter crescentus17 and Staphylococcus aureus18,19. Recently, this approach was combined with traditional TIS, resulting in the TraDIS-Xpress package20 (previously known as TraDIS+ (ref.21)). TraDIS-Xpress uses an inducible PBAD promoter facing out of a Tn5 in E. coli, in addition to detailed transposon-mediated inactivation data, to query all genes. It was successfully applied to identify both essential and non-essential genes affecting tolerance to various concentrations of the biocide triclosan, and differential responses to bactericidal and bacteriostatic concentrations were found. A high-throughput method for gain-of-function assays was recently developed, dual-barcoded shotgun expression library sequencing (Dub-Seq), where barcoded overexpression libraries of E. coli were mapped, barcoded and used to assign gene function in 52 experimental conditions on the basis of mutant fitness changes due to increased gene dosage22. To control expression in gain-of-function screens, some studies17,20 used inducible promoters on a single transposon, which can have the advantage (over using multiple transposons) of allowing high library density and reducing insertion bias. Other studies used constitutive promoters with different strengths on either barcoded or different types of transposons, which has the strategic advantage that different gene dosages can be assayed in the same culture18,19.
Fig. 3. TIS to assay the functions of essential genes.

A | An inducible promoter (right-angled red arrow), such as PBAD, is positioned facing out of each transposon (Tn) to overexpress all genes, including essential genes, so that their function can be assayed. Orange bars indicate transposon-induced transcription on top of wild-type expression (grey bars). B | In traditional transposon-insertion sequencing (TIS) approaches, essential genes can be identified as those that cannot tolerate insertions (part Ba). In gain-of-function screens, when transposons in the transposon pool are induced, for example with arabinose (part Bb), high expression of essential gene Z is achieved. When selection is applied, involvement of essential genes in the condition can now be assayed (part Bc) by monitoring relative differences in the number of transposons that influence expression levels. In this example, after exposure to the condition and sequencing out of the transposon, an increase in the number of transposon insertions that increase gene Z’s expression is observed. Therefore, mutants that overexpress gene Z have increased fitness within the overall population during selection, indicating that gene Z’s expression is beneficial in that condition. All other features are as in Fig. 1.
Scaling up TIS using high-throughput phenotyping
Although it is possible to apply the original TIS protocols at scale, the multistep library preparations involved can become increasingly costly when one is dealing with hundreds of samples. One solution to this problem, random barcode transposon-site sequencing (RB-Tn-Seq)23 introduces a random DNA barcode into each transposon. An initial conventional TIS approach is used to determine the insertion site associated with each barcode, and then a single-step PCR barcode amplicon can be directly sequenced in future experiments to track changes in mutant frequencies24, substantially speeding up screening. For instance, one recent upscaled study applied RB-Tn-Seq to 32 different bacterial strains across 129 conditions, and identified a large variety of leads for gene function25. A second problem in scaling TIS to large collections of bacteria is that optimized transposon delivery vectors often do not exist for non-model organisms. The ‘magic pool’ approach accelerates the optimization process using pools of transposon vectors, each of which has a different combination of upstream sequences (promoters and ribosome-binding sites) and antibiotic resistance markers as well as a random DNA barcode sequence, which allows quick measurement of vector efficiency during mutagenesis26.
Developments in TIS data analysis
A typical analysis protocol for TIS data can be summarized as follows: after the splitting of sequencing reads on the basis of their multiplexing barcode, any transposon or adaptor sequences are removed, reads are mapped to an annotated reference genome and the unique position of the transposon and relative insertion coverage (that is, number of reads) are recorded, processed and presented to calculate the effect of each transposon on fitness (for example, growth or survival). Over the past 10 years, several bioinformatics pipelines and protocols have been developed for this purpose (Table 1). These TIS tools include Web-based applications27,129, stand-alone graphical applications29 and command-line toolkits30–32. All of these tools implement variations on both gene essentiality and conditional fitness analyses, although they differ in the details of preprocessing and read alignment, normalization techniques, and statistical models or tests used. Often choices made in the experimental protocol can impact the appropriateness of a particular analysis procedure as much as any theoretical issue; for instance, many hidden Markov model and sliding window approaches to defining essential regions in the absence of annotation are applicable only to mariner transposon studies, as the assumption of a uniform insertion probability at TA sites simplifies the underlying statistical model. Many of these core issues were addressed in a 2016 review on design and analysis of TIS experiments7, but recent TIS developments have pushed analysis methods in new directions.
Table 1.
Transposon-insertion sequencing data analysis tools
| Name | Interface | Unique features | Transposon | URL | Ref. |
|---|---|---|---|---|---|
| INSeq pipeline | CL | Basic mapping pipeline | Any | https://www.nature.com/articles/nprot.2011.417 | 103 |
| ESSENTIALS | Web | Essentiality and conditional analysis; corrects for genomic location biases | Any | http://bamics2.cmbi.ru.nl/websoftware/essentials/essentials_start.php | 27 |
| ARTIST/conARTIST | CL | HMM-based mapping for fitness loci; sampling-based bottleneck correction | mariner | https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1004782#s4 | 30 |
| TRANSIT | GUI | Bayesian, HMM and resampling-based tests | mariner | https://orca1.tamu.edu/essentiality/transit/ | 29 |
| TraDIS Toolkit | CL | Empirical essentiality analysis; comparative analysis with RNA-seq tools | Any | https://github.com/sanger-pathogens/Bio-Tradis | 31 |
| MAGenTA | CL or Web | Fitness as growth rate calculations; bottleneck correction using neutral loci; sliding window analyses | Any | https://vanopijnenlab.github.io/MAGenTA/ | 28 |
| Aerobio | CL | Fitness as growth rate calculations; bottleneck correction using neutral loci; sliding window analyses; ability to switch to RNA-seq, Term-Seq and WGS analyses | Any | https://github.com/jsa-aerial/aerobio | 32 |
| AlbaTraDIS | CL | Multicondition and gene activation analysis based on TraDIS Toolkit | Any | https://github.com/quadram-institute-bioscience/albatradis | 38 |
CL, command-line (*NIX-based) utility; GUI, stand-alone graphical user interface; HMM, hidden Markov model; RNA-seq, RNA sequencing; Term-Seq, 3′-end RNA sequencing; Web, Web-based graphical user interface; WGS, whole-genome sequencing.
One major development of TIS analysis methods is in dealing with infection dynamics and particularly the effects of bottlenecks, which are transient reductions in population size during the course of the experiment (see later). While these have been dealt with in analyses using normalization based on changes in neutral loci33 or subsampling30, two interesting new approaches to this problem were proposed recently. In the first, principal component analysis (PCA) is performed on log fold changes in mutant abundance across replicate infection experiments34. Examination of the principal components recovered can then identify linear combinations of the changes across replicates that separate genes consistently across experiments, providing a score for association of any particular gene with survival in infection and eliminating the contribution of spurious stochastic changes. A second approach has adopted the zero-inflated negative binomial (ZINB) distribution to model transposon insertion counts35. The ZINB distribution is a mixture of a logit distribution, which captures the probability of detection of a data point, and the negative binomial, which captures the overdispersion generally observed in sequencing data. This distribution has attracted attention recently in the analysis of single-cell RNA sequencing (RNA-seq), as it provides a natural mechanism for capturing technical dropout of transcripts36. Similarly, by fitting the logit component of the ZINB genome-wide, this approach can correct for differences in library saturation between conditions arising either in library creation or due to bottlenecking.
A second development is the move from simple condition–control comparisons to the simultaneous investigation of large suites of conditions. For instance, the PCA and ZINB methods highlighted above demonstrated their effectiveness using combinations of existing datasets, identifying commonalities in genes required by different Vibrio strains in infection37 or response to a panel of antibiotics in M. tuberculosis35, respectively. A striking example of such a data analysis pipeline, named AlbaTraDIS38 was developed for the TraDIS-Xpress study examining triclosan tolerance (see ref.20 and the TraDIS-Xpress discussions earlier) and uses sliding window analyses integrating all available information to predict all genes and promoter regions involved during selection.
As TIS experiments become more complex and the results are combined with other data types, tools for visualization and data delivery are becoming increasingly important. For example, a recent study integrating expression and fitness data from the HIV-associated Salmonella enterica subsp. enterica serovar Typhimurium strain D23580 (ref.39) included a Dalliance-based browser40, allowing readers to directly interrogate the data themselves and providing a valuable community resource. Platforms for easily providing this kind of interactive interface are beginning to emerge, such as ShinyOmics41, a Web-based application for rapid collaborative exploration of omics data, including TIS, RNA-seq and proteomics date, which allows comparisons between datasets, PCA and simple network analysis. As datasets accumulate and automation increases throughput, such integrative analysis approaches will become increasingly important.
Key biological applications of TIS
Since its development, TIS has been used in a range of in vitro studies as well as in vivo infection models. Here we summarize how the development of TIS has facilitated the investigation of key biological questions, with a focus on studies with implications for human health.
Identifying genes and networks involved in antibiotic resistance
The emergence of antibiotic resistance is a major global health problem, exacerbated by a lack of development of new antibiotics. TIS is well equipped to infer the relative impact that disrupting each genomic feature has on antibiotic sensitivity (Fig. 4) and can contribute to developing a better understanding of how resistance emerges, as well as guide the development of new strategies to target resistant bacteria. Traditional TIS experiments performed by culturing transposon libraries with inhibitory but sublethal concentrations of antibiotics for several generations have been used to define a comprehensive non-essential gene complement involved in intrinsic resistance for many clinically important pathogens, including the notorious ESKAPE species (Enterococcus faecium, S. aureus, K. pneumoniae, Acinetobacter baumannii, P. aeruginosa and Enterobacter species)18,42–44.
Fig. 4. Mapping complex genotype–phenotype relationships.

a | A gene–antibiotic network for three antibiotics (based on refs54,119). Each node (circle) depicts a gene, whereas each edge indicates a negative (solid grey line), neutral (dashed grey line) or positive (solid red line) effect on fitness between the genes in the presence of antibiotics as determined by transposon-insertion sequencing (TIS). All three antibiotics — penicillin G (PeniG), vancomycin (Vanco) and daptomycin (Dapto) — affect cell wall integrity but TIS uncovers a wide variety of genes involved, many of which are not direct targets of the antibiotic. In this case, the unknown gene Q is likely to be involved in peptidoglycan synthesis/membrane integrity on the basis of the function of other genes with similar fitness profiles. b | By mutation of gene Q and construction of a transposon library in this mutant background, genetic interactions can be identified (based on concepts from ref.33). In this case, the genes uncovered in the mutant background, as depicted in the gene interaction map (GIM), further support a role in peptidoglycan synthesis, that it may function in the cell membrane and that it may be controlled by a particular regulator. Additionally, in vivo TIS data with the mutant library performed in healthy mice and mice depleted of neutrophils (neut−) indicate that gene Q is needed to establish lung infection but is dispensable in the absence of neutrophils. Adapted from ref.54, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/).
These studies have shown that while antibiotics may have specific targets (for example, in cell wall synthesis, DNA replication or protein synthesis), the bacterial response to antibiotics is actually distributed across the genome. For example, fluoroquinolones target topoisomerase IV and DNA gyrase, which are essential enzymes involved in DNA replication. Although most TIS experiments cannot assay these targets directly owing to their inherent essentiality, TIS profiles generated under fluoroquinolone exposure implicate other genes involved in DNA replication and repair, such as recN and xseA33,45. While these genes are not direct fluoroquinolone targets, they contribute to intrinsic resistance as part of a secondary effect; for example, fluoroquinolones trigger DNA damage, which activates DNA repair. In general, antibiotic TIS profiles for each antibiotic tested, and each organism screened, show a role for genes beyond those related to the primary target, indicating the importance of genes with diverse functions, including amino acid and carbohydrate metabolism, energy generation, transport and regulation42,43,46–51. This finding underlines that although we have a limited view of how an antibiotic inhibits a bacterial cell, TIS can be used to uncover this complex, multifactorial process. As a result, TIS profiles have been demonstrated to be effective in determining the mechanism of action of novel antibiotics52,53. Creating profiles for multiple similar conditions can help to direct attention to genes with unknown function that are important under all of these conditions and thereby help to identify leads that may assist in uncovering their function (Fig. 4b and see Integrating TIS with other genomic approaches to predict complex traits). Moreover, TIS profiles can also uncover opportunities to sensitize a bacterium to a drug, facilitating the design of secondary or helper drugs42,54.
Investigating virulence genes, host adaptation and vaccine development
Interrogating the genomic requirements for pathogens to cause disease has been a major motivation in the development of large-scale reverse genetics approaches (Fig. 5). For instance, the development of early transposon screens in Salmonella Typhimurium55,56 provided key evidence for the discovery of major virulence factors. TIS has made it much easier and faster to screen bacterial pathogens virulence factors. These have included in vitro assays, such as capsule production10,57, growth in serum58 and colicin resistance and sensitivity in E. coli59, as well as sporulation in Clostridioides difficile60. An early in vivo screen used retrospective TIS on existing samples to examine Salmonella Typhimurium genes involved in infection of three farm animals (chickens, cows and pigs61,62) plus mice, and found multiple conserved virulence genes. Other examples include an analysis of the virulence genes of Legionella pneumophila using both cell culture and mouse models63, survival of Streptococcus pyogenes in human saliva64, survival of A. baumannii in a bloodstream infection mouse model65, survival of Streptococcus equi in horse blood or hydrogen peroxide66, the demonstration that oxidative stress resistance enhances V. cholerae host adaptation in a mouse model67, survival of Burkholderia cenocepacia in a Caenorhabditis elegans host68, Streptococcus mutans infection in an oral rodent model69 and the building of a targeted sublibrary of type IV secreted proteins in the intracellular pathogen Coxiella burnetii using INSeq, which was subsequently screened for vacuole formation in human HeLa cells70.
Fig. 5. Bottleneck and realism trade-off in TIS infection models.

Different animal models of infection (top) can induce strong bottleneck effects (bottom) in infecting bacterial populations, which can confound transposon-insertion sequencing (TIS) analysis. This bottleneck effect is particularly pronounced in models where bacteria must overcome barrier defences (for example oral infection models of Salmonella enterica subsp. enterica serovar Typhimurium). Organoid models can provide a complex environment while limiting bottlenecks, but technical limitations in culture may limit the total population size screened. Finally, cell culture screens can be scaled to arbitrary sizes, allowing screening of extremely large collections of mutants, but they often provide insight into only a particular aspect of disease.
Although TIS has streamlined the process of generating and monitoring mutant libraries, challenges remain in applying the technique to infection models. A primary concern is the effect of bottlenecks, which can be quantified experimentally by measuring the loss of neutral markers33, for instance using the wild-type isogenic tagged strains (WITS) method71. Bottleneck effects should be considered computationally (see earlier) and can, at least partially, be avoided by careful consideration of the infection model. The size and temporal structure of a bottleneck are often specific to the particular infection model, and can be influenced by a range of factors, including physical barriers, nutrient availability and competition with the native microbiota72. Whereas mild bottleneck effects can be partially compensated for during analysis, major bottlenecks can irreversibly bias an experiment that does not account for them. In these cases, the surviving mutants represent the subset of bacteria that happened to pass some barrier to infection, rather than being representative of all mutants that could, leading to skewed representation and a lack of reproducibility between experimental replicates. The wide variety of infection models developed to study Salmonella Typhimurium provide an example of how model choice can affect the design of a transposon screen (Fig. 5). For the most realistic infection models based on infection through the gut epithelium, bottlenecks can be severe73,74, limiting transposon analysis to small pools of tens to hundreds of mutants61,62. Intraperitoneal inoculation can bypass this major bottleneck in a mouse model, which allowed screening of ~10,000 mutants in a single animal75; however, the results are uninformative with regard to gastrointestinal disease. Finally, in the case of cell culture models, such as the macrophage model that captures a key challenge to the development of systemic salmonellosis55, the only constraint on library complexity is the number of cells available for infection, allowing efficient screening of very large mutant populations (~106)39. Similar trade-offs between realism and library complexity are likely to exist for many infection models, particularly those that involve bacterial penetration of barrier defences.
Experimental challenges notwithstanding, TIS applied to animal models has also proved useful in identifying and understanding vaccine targets. For example, screening of a S. pneumoniae TIS library in a ferret transmission model, describing the fitness landscape of genes during mammalian transmission, yielded valuable and translatable data. Targeted deletion confirmed that key TIS hits (putative C3-degrading protease CppA, iron transporter PiaA and competence regulatory histidine kinase ComD) significantly decreased transmissibility. Importantly, maternal vaccination with recombinant PiaA and CppA alone or in combination blocked transmission from mother to offspring and was more effective than capsule-based vaccines76. In a second example, a mouse sickle cell disease (SCD) model coupled with TIS identified a set of pneumococcal virulence genes specific to hosts with SCD. Not only did these factors point to aspects of SCD pathophysiology, but they also showed that the protective capacity of antigens can be different in the healthy versus the SCD population, highlighting the importance of understanding bacterial pathogenesis in the context of common comorbidities77.
Assaying functional components of mobile genetic elements
Mobile genetic elements, including plasmids, transposable elements and bacteriophages, are important players in interspecies and intraspecies gene transfer and are heavily implicated in the spread of antibiotic resistance and virulence determinants. Plasmids are notoriously difficult to study with screening approaches, owing to their independent replication systems and capacity to regulate copy number. TIS has been used to identify genes involved in maintenance of IncA/C plasmids in E. coli, and the results were then developed into an IncA/C plasmid typing scheme78. Additionally, TIS has also been used to demonstrate the involvement of a type IV secretion system in conjugation of an IncP plasmid in Edwardsiella piscicida79.
Bacteriophages (‘phages’) are important mobile genetic elements that have been used for bacterial typing for decades and can greatly influence bacterial pathogenesis through the transduction of pathogenicity islands. Additionally, phage therapy to treat resistant bacterial infections is experiencing a resurgence as an alternative to antibiotics. TIS is able to identify essential host factors that mediate or hinder bacteriophage infection. For example, challenge of an E. coli O157 TraDIS library with T4 and T7 bacteriophages identified new host genes involved in both bacteriophage resistance (for example, sspA, encoding stringent starvation protein A) and susceptibly (for example, the sap operon)80. Similarly, experiments with bacteriophages specific to particular capsules have allowed the identification of not only modifiers of bacteriophage resistance81, but also genes responsible for capsule expression57.
Uncovering essential genes and the influence of the pan-genome
One of the first uses of TIS was to define the essential genes for survival for a plethora of bacterial species, including human pathogens such as Porphyromonas gingivalis82, B. cenocepacia83 and Yersinia pseudotuberculosis84, animal pathogens such as S. equi85, plant pathogens such as Pseudomonas syringae86, the model organism E. coli K12 (ref.87) or commensal gut bacteria such as Bifidobacterium breve88. These valuable essential gene datasets gained from TIS studies have been shown to correlate well with existing phenotypic gene essentiality data, such as from the single-gene E. coli knockout library (Keio University) 89, and these can not only be interpreted to understand basic functioning of the cell but can also provide vital information for identifying potential novel drug targets.
Once TIS has been implemented in a species it is often straightforward to create libraries in related strains. This flexibility facilitates functional exploration of a species’s pan-genome and how genetic background can affect phenotype. This kind of in-depth investigation can ultimately help to uncover how bacterial species, particularly diverse species that include both pathogens and non-pathogens, can become harmful or antibiotic resistant, for example via horizontal transfer of pathogen-associated genetic material. TIS in nine strains of P. aeruginosa determined the core essential genome in five media, and highlighted that essentiality of some genes depends on genomic context90. The influence of genetic background on phenotype is further illustrated by examples that highlight how genes involved in responding to antibiotic stress can be strain specific. For example, screening of two S. pneumoniae isolates for genes that are important for intrinsic resistance to antibiotics from three classes showed that on average only ~50% of the responsive genes are common between strains. Investigation of the underlying reasons for this variability showed that network architecture, including regulatory pathways that direct competence, are wired in a strain-specific manner, thereby making responses strain specific54. A recent study probed five diverse strains of S. aureus for daptomycin resistance mediators and identified several core pathways consistently involved across strains, including the lipoteichoic acid pathway, as well as factors that varied with strain diversity, such as the cell envelope18. Furthermore, in A. baumannii a single gyrA resistance allele results in preferential poisoning of topoisomerase IV by ciprofloxacin, leading to large alterations in the fitness landscape of insertion mutants compared with a wild-type gyrA background. This altered background triggers the activation of prophage and quickly leads to the emergence of ciprofloxacin-resistant clones91. In M. tuberculosis, loss-of-function mutations in katG can result in isoniazid resistance. However, TIS experiments have shown that several clinical strains have an increased requirement for katG compared with the reference strain H37Rv92. This variability underscores how genome variation can affect adaptive solutions and highlights the importance of extending TIS to clinical isolates that may have a very different genetic background to laboratory strains.
Understanding metabolism, the response to environmental factors and microorganism–microorganism interactions
Despite decades of accumulating genome sequences in public databanks, many protein-coding genes remain unannotated or carry inaccurate annotations, particularly in non-model and difficult-to-culture organisms. Often even the conditions under which a gene contributes to survival are unknown, leading to a serious roadblock in any attempt at molecular characterization. TIS can help us understand how bacterial cells experience the changing environments encountered in nature, by providing comprehensive profiling of mutant phenotypes93. Specifically, recent TIS studies have identified fitness determinants during energy-limited growth in P. aeruginosa94 and outlined how several bacterial species synthesize amino acids95. Others have identified genes in E. coli that promote survival during exposure to ionizing radiation96, genes in Salmonella Typhi that allow adaptation to survival in water97 and genes involved in desiccation stress in Salmonella Typhimurium98. TIS approaches have also uncovered how bacteria interact with plants or deal with soil environments99. For instance, TIS was used to identify genes required for growth of the soft-rot pathogenic bacterium Dickeya dadantii in chicory plants100 or genes of Pantoea stewartia that are essential for survival in planta to provide insights into how it causes wilt disease in corn101. As the breadth of examined stresses increases, shared adaptations to diverse stress conditions are emerging.
In a massively upscaled example of assaying genes for adapting to changing environments in parallel, a recent study demonstrated the utility of this approach by applying RB-Tn-seq to 32 diverse bacteria in ~200 conditions; this work assigned a phenotype to more than 11,000 uncharacterized proteins, with ~2,000 of these functional annotations demonstrated to be conserved across organisms25. A similar study investigated the major human gut commensal and obligate anaerobe Bacteroides thetaiotaomicron across 492 conditions and identified genes involved in metabolism and bile tolerance102. Together, these two studies demonstrate how TIS can be applied broadly across organisms and deeply within an organism to extract leads for future molecular characterization.
TIS approaches can also directly answer questions with implications for human health; for instance, how does the gut microbiota affect drug metabolism and toxicity? Using the robotics-driven TIS mutant array approach pioneered by the INSeq method3,103, where individual mutants are mapped through combinatorial pooling, a comprehensive library of ~1,300 B. thetaiotaomicron mutants were selected and incubated with the antiviral drug brivudine. Drug metabolites were then measured by mass spectrometry, and individual B. thetaiotaomicron genes required for the production of a hepatotoxic metabolite were identified. Follow-up studies in gnotobiotic mice confirmed the in vivo relevance of this toxin production pathway104, illustrating the power of this approach in understanding drug–microbiota interactions.
In the wild, microorganisms rarely live planktonically in isolation, but are constantly interacting with other microorganisms and forming communities, either by chance as in wound co-infections or as part of a stable ecosystem as in the mammalian gut. TIS provides an opportunity to understand these microorganism–microorganism interactions, as the genetic response of one bacterium to other bacteria can be recorded. Numerous studies have shown that the fitness effects of gene disruption can depend critically on the presence of other community members. For instance, co-infection has been shown to alter the bacterial fitness landscape in wound models, such as with the opportunistic pathogens Streptococcus gordonii and Aggregatibacter actinomycetemcomitans105. Furthermore, studies of co-infection with P. aeruginosa and S. aureus in mouse surgical wounds showed that ~25% of S. aureus genes that are essential during co-infection are no longer needed during single-species infection (Fig. 6a). Furthermore, single mutants, such as those of the community-dependent essential gene udk, encoding a uridine kinase, were confirmed to influence levels of co-infection but not monoinfection in vivo106. Interaction studies have even been extended to predatory relationships, such as in studies of B. bacteriovorus that identified genes required for predation of V. cholerae during planktonic and biofilm growth using Tn-Seq libraries of both the prey107 and the predator14. Similarly, bacterial genes that influence infection with viruses have been examined by TIS in numerous bacterial species57,80,81,108. The mechanisms of bacterial interactions within communities have also been investigated by screening for effectors of type VI protein secretion systems (T6SSs). T6SSs are conserved bacterial defence mechanisms that deliver toxins to neighbouring cells through a contact-dependent mechanism, killing those that lack immunity proteins (Fig. 6b). New toxin and immunity genes have been identified through TIS in V. cholerae109 and P. aeruginosa110. Together, these studies have provided insight into a wide range of relationships that shape the microbial environment.
Fig. 6. TIS to assay microorganism–microorganism interactions.

a | Transposon-insertion sequencing (TIS) of Staphylococcus aureus (SA) mutants alone (orange circles) compared with coculture of SA and Pseudomonas aeruginosa (PA) wild-type strain (green rods) can identify community-dependent essential genes that are needed only during coculture. Other features are as in Fig. 1. b | Using TIS to identify type VI secretion system (T6SS) toxin immunity pairs by growing cells with close cell-to-cell contact, and performing TIS so that genes involved in protection of T6SS-depending killing (depicted as a yellow bolt) can be detected. In this example, genes in PA encoding immunity proteins (Tsi; orange diamond) that protect from neighbour killing by toxins (Tse; red square) become essential only in a T6SS-active (retS; bottom panel) background but not in the inactive (H1_retS; top panel) cellular background.
Integrating TIS with other genomic approaches to predict complex traits
Whereas TIS has been most commonly used to make simple associations between environments and genetic components, it can also uncover more complex relationships. No genomic element, gene or pathway exists in isolation; rather they are connected through intricate networks, resulting in specific organismal properties and, ideally, an appropriate response when disturbed. One layer of these networks is gene regulation, including the non-coding genome. By combining saturated TIS libraries with expression profiling, one can identify functional non-coding RNAs (ncRNAs). For example, RNA-seq can be used to map out expression units across the entire genome and indicate whether non-coding/intergenic regions display significant levels of transcription. In turn, a parallel TIS experiment with insertions in these regions can then be used to associate a phenotype with the disrupted ncRNA. This approach was used in S. pneumoniae, and yielded 89 ncRNAs, more than half of which had not been identified previously, and several could be associated through TIS in vivo data as being critical for virulence111. A follow-up study used different RNA-seq techniques to map the full transcriptional landscape of the S. pneumoniae virulent type strain TIGR4 (ref.112). This resulted in identification of many non-coding regulatory regions, which could be associated with a phenotype through integration of TIS data from different environments. Another comprehensive TIS-based regulatory study in Neisseria meningitidis identified 288 genes and small ncRNAs needed for colonization of human epithelial and/or endothelial cells113.
Mapping of genetic interactions, which quantify fitness dependencies between genes, can be used to build genetic interaction networks and infer regulatory relationships, pathway structures or leads for gene function114. Genetic interactions can be identified by creating a TIS library in a query gene deletion background and screening for genes whose fitness deviates from the multiplicative fitness of the individual mutants (Fig. 7). The most obvious example of a genetic interaction is synthetic lethal interaction, where two individual mutants have no or little fitness effect but abolish growth when combined, and can occur if two gene products perform redundant essential functions. A variety of such interactions exist, which imply different types of relationships between components (reviewed in ref.114). Genetic interaction networks can be combined with in vivo studies to further inform gene function in the host. For example, if a gene involved in intrinsic resistance to cell wall-targeting antibiotics is important only in healthy mice but not when certain immune components are missing, this indicates that the gene product is not only a resistance factor but is also potentially visible to the immune system (Fig. 4). Several studies have successfully used a genetic interaction approach with TIS in S. pneumoniae, including to uncover regulatory dependencies for catabolite control protein A, how a potassium uptake system and a subpathway for nasopharyngeal colonization are regulated and how the protease ClpP is involved in competence1,33,54. Additionally, cell division components such as CozE in S. pneumoniae were identified by use of pbp1A as a query gene, revealing that CozE directs the activity of Pbp1A to the midcell plane, where it promotes zonal cell elongation115. Alternatively, a query gene/pathway can be inhibited by a drug or inhibitor, as has been done in the case of wall teichoic acid biosynthesis in S. aureus, and then screened with TIS for synthetic lethal interactions116. This study connected wall teichoic acids with other pathways, including cell-envelope D-alanylation, and peptidoglycan and lipoteichoic acid synthesis116. Other applications of note include investigating the role of the quorum-sensing and virulence regulator LasR in different P. aeruginosa infection models117, and assigning function to uncharacterized genes in M. tuberculosis118.
Fig. 7. Integrating TIS with RNA-seq data.
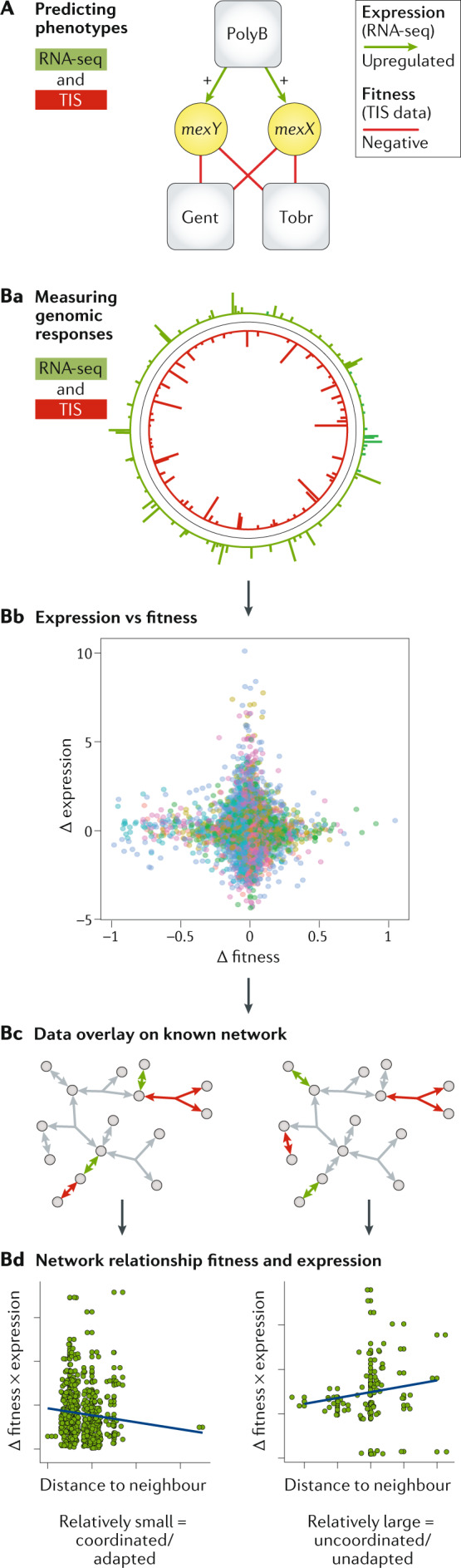
A | An example of combining RNA sequencing (RNA-seq; depicted in green throughout) and transposon-insertion sequencing (TIS; depicted in red throughout) to identify antagonistic interactions between the antibiotics polymyxin B (PolyB) and gentamicin (Gent) or tobramycin (Tobr) in Pseudomonas aeruginosa. TIS data indicate that genes mexY and mexX are involved in intrinsic resistance in P. aeruginosa to gentamicin and tobramycin, indicated by the red edges between the antibiotics and the genes. When these genes are disrupted by a transposon insertion, the bacterium becomes more sensitive to these antibiotics. Moreover, RNA-seq data reveal that polymyxin B induces expression of these genes, as indicated by the green arrows. This led to the hypothesis that polymyxin B, owing to its transcriptional activation of mexY and mexX, will make the bacterium less sensitive to either gentamycin or tobramycin. The study authors confirmed experimentally that these antibiotics work in an antagonistic manner48, which highlights the strength of probing response networks from different perspectives to extract biological meaning. B | Measurement of TIS and RNA-seq responses under the same conditions (part Ba) has shown that the transcriptional responses to a specific environment (Δ expression) are often not accurate predictors of gene deletion phenotypes, as expression and fitness do not correlate well (Δ fitness; part Bb)119. However, by overlaying these datasets over a known network (for example, a metabolic network; part Bc), network analyses can identify patterns between TIS and RNA-seq. In this example a small part of a metabolic network is depicted; grey circles are metabolites and arrows are genes encoding enzymes that mediate each reaction. Red arrows are phenotypically important genes in a specific environment identified by TIS, whereas green arrows are genes that change transcriptionally in the same environment, identified by RNA-seq. The distance between two genes in a metabolic network is the number of reactions between them, and can be calculated for all pairs across the network. In part Bd, the left network is an example where distances between pairs of fitness and expression changes are small, whereas the right network illustrates larger distances. An adapted response to an environment is characterized by fitness and expression changes that are relatively small (distance to neighbour) and correlated (Δ fitness × expression). Exposure to stress conditions to which a bacterium is not adapted leads to a loss in correlation between genes that change in transcription and those have a fitness effect (part Bd)119. Importantly, such associations can be used to make predictions on antibiotic susceptibility126. Part B is adapted with permission from ref.119, Elsevier.
Both TIS and RNA-seq data have shown that even relatively simple perturbations in bacteria (for example, changes in pH, or exposure to low-level antibiotics) trigger complex responses. There is value in combining these data sets; for example, combined TIS and RNA-seq antibiotic response data obtained from P. aeruginosa allowed predictions of antagonistic antibiotic combinations48 (Fig. 7A). Such observations suggest that TIS and RNA-seq may register distinct but complementary features of the underlying network architecture of the cell and illustrate how responses can be separated into at least two organizational levels (phenotypic and transcriptional). Remarkably, when expression and fitness are directly compared (Fig. 7Ba) there is often little correlation between them (Fig. 7Bb)119–121, with some notable exceptions, such as in some metabolic pathways120 and classical virulence factors39. This discordance has long been known in yeast122–124, and suggests that the majority of transcriptional regulation is not optimized by selection125.
Considering RNA-seq and TIS in the context of the underlying cellular network can clarify their relationship119 (Fig. 7Bc). When responding to an environmental stimulus to which a bacterium is adapted (for instance nutrient depletion), the majority of fitness and expression changes occur at short distances from each other within the metabolic network, with 80% of genes with fitness changes connected by two or fewer metabolic reactions to genes with expression changes, and 93% within a three-reaction radius119 (Fig. 7Bd). Furthermore, the correlation of fitness and expression changes decreases with distance in the network119. This indicates that fitness and expression changes are not only colocated within the network but are of a magnitude comparable to those of their neighbours. These relationships can disappear when a bacterium responds to an environment to which it is not adapted, for instance an antibiotic119 (Fig. 7Bb). This suggests that the apparently paradoxical lack of correlation between fitness and expression measurements can be in part understood through network models that incorporate regulatory and genetic relationships, which could aid drug target predictions and genetic network engineering. Quantifying the degree of disruption a stimulus creates in a bacterium’s transcriptional network has already resulted in accurate predictions of fitness, antibiotic sensitivity and drug mechanism of action126.
Conclusions and future perspectives
Considerable advances and extensions of TIS have been made since its introduction in 2009, many of which are highlighted in this Review. As a result of advances in cell sorting and microfluidics, TIS has been adapted to examine phenotypes on the level of single mutant cells11,15,127. TIS has also demonstrated its practical utility, for example, in the development of new vaccine candidates76,77 and new antibiotics or helper drug targets42. Lastly, genetic interaction approaches are beginning to build networks that map out the complex relationships between genetic components within the cell, and these could be further extended by combining two transposons into a single genome, by combining TIS and CRISPR-based transcriptional interference (CRISPRi) or by scaling approaches that simultaneously mutagenize interacting organisms128.
Furthermore, the vast majority of TIS studies to date have been performed in bacterial species, owing to this ease of genetic manipulation. For those species in which TIS works well, it is a powerful technique that can provide high volumes of valuable genotype–phenotype linkage data on a fine scale. Over the next decade, we hope to see an expansion in the types of organisms assayed by TIS methods. Encouragingly, several related TIS-like methods have been developed for use in mammalian, fungal, parasite and archaeal backgrounds. Profiles of TIS applied in the two model yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe identified essential genes, genes involved in rapamycin resistance and factors that contribute to the formation of heterochromatin129–131. Other examples include determining the essential genes of the archaeal species Sulfolobus islandicus132 and Methanococcus maripaludis133, phenotypic interrogation via tag sequencing (PhITSeq) in haploid human cells to assign gene function134,135, quantitative insertion site sequencing (QISeq) in mice using piggyBac and Sleeping Beauty transposons to screen them for cancer-related genes136,137, QI-Seq in Plasmodium falciparum, using the piggyBac transposon to determine essential genes138 and barcode analysis by sequencing (BarSeq) in yeast24,139. Moreover, we expect to see the biological questions answered with TIS to become increasingly complex. Such applications will only enhance the utility and breadth of TIS approaches in the future.
The analogous functional genomics method of pooled CRISPRi screening, which silences genes in a targeted fashion and uses single-guide RNAs and catalytically dead Cas proteins (first demonstrated with dCas9 (ref.140)), has been successfully applied to numerous bacterial species since the development of mobile CRISPRi systems141–145. CRISPRi has some advantages over TIS, primarily that silencing is directly targetable to regions of interest, which can reduce the complexity of the assay and thus the amount of sequencing reads required, and it can allow knockdown of any coding regions, for instance essential genes142, which traditional TIS cannot. However, CRISPRi requires design, synthesis and cloning libraries of sgRNAs, which can be technically challenging, and understanding the impact of off-target effects or differences in sgRNA efficiency can add complications during implementation and analysis. By contrast, the execution of TIS requires no prior specific knowledge of the genetic make-up of an organism, and owing to its more random nature, TIS can uncover unexpected or novel genes, can potentially assay transcriptionally inactive regions of the genome and can be precise enough to interrogate specific regions within the transcriptional units, such as essential protein domains38. Modifications of both technologies can assay the effects of gene overexpression and suppression through complementary approaches. Functional genomics screening techniques such as TIS and CRISPRi can suffer from similar shortfalls, namely that deciphering detailed mechanistic insight from the large datasets generated can be difficult to automate. For effective data analysis, research groups will have to pool data, resources and expertise to construct holistic workflows that can manage this complexity. To this end, we expect to see in an increase in data sharing platforms, such as the newly established TIS depository TraDIS-vault, the viewer available for the invasive Salmonella Typhimurium strain D23580 (ref.39) or interactive data visualization platforms, such as ShinyOmics41.
Looking forward, we predict that TIS methods will be applied to answer increasingly complex and diverse biological questions. For this expansion, we must move past the straightforward, homogeneously grown laboratory assays to more sophisticated ones that better mimic ecological states that occur in nature. One major limitation of TIS is that it is available only for use in easily culturable and genetically tractable species, which represent only the minority of total bacterial and archaeal species146. A key challenge will be to develop tools to allow the recalcitrant microbes of medical, industrial and environmental importance to be assayed. TIS-inspired methods, such as the ‘magic pool’ approach to optimizing transposon delivery in non-model organisms26, are already beginning to address this. A driving factor will be massive upscaling in the numbers of conditions and bacterial strains that can be simultaneously screened, building on RB-Tn-Seq25 and similar approaches. As these methods push beyond model strains, we will increasingly gain insight into how the genetic diversity within pan-genomes is shaped and maintained. This migration away from one-dimensional genotype versus phenotype experiments will require consideration of the larger genetic network and particularly interactions between genetic background and fitness (see Scaling up TIS using high-throughput phenotyping). This will involve the application of machine learning, modelling and network analyses to integrate and extract knowledge from accumulating TIS datasets. Eventually, and in combination with other postgenomic functional data, this approach will increasingly enable us to move from describing the genetic architecture of the cell to predicting future behaviours119,147. Collectively, these developments illustrate that the journey of TIS is far from over, with many exciting paths yet to be explored.
Acknowledgements
The authors acknowledge funding from various sources that supported the composition of this article: Australian Research Council Discovery Early Career Research Award fellowship DE180100929 and Australian National Health and Medical Research Council grant APP1159752 to A.K.C.; US National Institutes of Health grants U01AI124302, R01AI110724 and R21AI117247 to T.v.O.; a bayresq.net Bavarian research network grant to L.B.; UK Medical Research Council grant G1100100/1 to J.P.; Australian National Health and Medical Research Council grants GNT1060895 and GNT1120298 to I.T.P.; and US National Institutes of Health grant R35GM118159 to A.L.G.
Glossary
- Transposon
A mobile genetic element that inserts itself into a genome and disrupts genes or genetic features at that site.
- Next-generation sequencing
DNA sequencing using a massively parallel platform that separates DNA templates on a flow cell and clonally amplifies clusters for sequencing.
- Genomic features
Every component of the genome that can be annotated as being a feature, whether it be a gene, coding RNA, non-coding RNA or promoter region.
- Fragmentation
Breaking up DNA into smaller pieces in order to be sequenced. This can be done by physical shearing methods, such as sonication, or enzymatic digestion.
- Fluorescence-activated cell sorting
(FACS). A specialized type of flow cytometry that separates cells, one cell at time, by their fluorescent characteristics on the basis of light scattering.
- Sliding window
A window of arbitrary length is set and events within that window are assayed. This window is then moved around the genome. This approach provides a more objective method of assessing the genome that is independent of annotation.
- Bottlenecks
When a population size is drastically reduced through stochastic processes and the surviving cells will make up the new population but will have reduced genetic diversity.
- Overdispersion
When data exhibit greater variability than would be expected under a statistical model. In the context of sequencing data, overdispersion is often used in reference to the negative binomial distribution, which can be understood as a generalization of the Poisson distribution that allows a larger variance relative to the mean.
- Reverse genetics
Determining the phenotypic effects of a genetic feature by altering the genetic feature and observing changes in the organism compared with a wild type. ‘Reverse’ refers to the genotype-to-phenotype mode of investigation, being opposite the classical phenotype-to-genotype genetic investigations (‘forward genetics’).
- Organoid
A three-dimensional, simplified replica of an organ derived from stem cells to realistically model the organ in vitro.
- Pan-genome
The complete set of genes in all strains within a species, in contrast to the core genome, which is the set of genes shared by all strains within a species.
- Essential genome
The complete set of genes and genetic features in a genome that are essential for a cell to survive and grow, the examplar of which are ‘housekeeping’ genes for core processes such as replication and division.
- Gnotobiotic
An environment for culturing microorganisms, such as an animal model, where all microorganisms are either defined or removed.
- Synthetic lethal
Where individual mutants have no or little fitness effect, but when two or more of these mutations are combined, this leads to arrest in cell growth or to cell death.
- Antagonistic antibiotic combinations
When the activity of an antibiotic combination is lower than would be predicted from the effects of the individual antibiotics.
Author contributions
A.K.C., L.B. and T.v.O. substantially contributed to the discussion of the content. All authors researched content for the article, contributed to writing and reviewed/edited the manuscript before submission.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information
Nature Reviews Genetics thanks B. Akerley, A. Deutschbauer and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
SalmonellaTyphimurium strain D23580: https://hactar.shef.ac.uk/D23580_2/
TraDIS-vault: https://tradis-vault.qfab.org/apollo/jbrowse/
Contributor Information
Amy K. Cain, Email: amy.cain@mq.edu.au
Tim van Opijnen, Email: vanopijn@bc.edu.
References
- 1.van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods. 2009;6:767–772. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langridge GC, et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 2009;19:2308–2316. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodman AL, et al. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe. 2009;6:279–289. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gawronski JD, et al. Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc. Natl Acad. Sci. USA. 2009;106:16422–16427. doi: 10.1073/pnas.0906627106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Opijnen T, Camilli A. Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nat. Rev. Microbiol. 2013;11:435–442. doi: 10.1038/nrmicro3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barquist L, Boinett CJ, Cain AK. Approaches to querying bacterial genomes with transposon-insertion sequencing. RNA Biol. 2013;10:1161–1169. doi: 10.4161/rna.24765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chao MC, Abel S, Davis BM, Waldor MK. The design and analysis of transposon insertion sequencing experiments. Nat. Rev. Microbiol. 2016;14:119–128. doi: 10.1038/nrmicro.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kakkanat A, Phan M-D, Lo AW, Beatson SA, Schembri MA. Novel genes associated with enhanced motility of Escherichia coli ST131. PLoS One. 2017;12:e0176290. doi: 10.1371/journal.pone.0176290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nolan LM, et al. A global genomic approach uncovers novel components for twitching motility-mediated biofilm expansion in Pseudomonas aeruginosa. Microb. Genomics. 2018 doi: 10.1099/mgen.0.000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorman MJ, Feltwell T, Goulding DA, Parkhill J, Short FL. The capsule regulatory network of Klebsiella pneumoniae defined by density-TraDISort. MBio. 2018;9:e01863-18. doi: 10.1128/mBio.01863-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hassan KA, et al. Fluorescence-based flow sorting in parallel with transposon insertion site sequencing identifies multidrug efflux systems in Acinetobacter baumannii. MBio. 2016;7:e01200-16. doi: 10.1128/mBio.01200-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rego EH, Audette RE, Rubin EJ. Deletion of a mycobacterial divisome factor collapses single-cell phenotypic heterogeneity. Nature. 2017;546:153–157. doi: 10.1038/nature22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fowler CC, Galán JE. Decoding a Salmonella typhi regulatory network that controls typhoid toxin expression within human cells. Cell Host Microbe. 2018;23:65–76. doi: 10.1016/j.chom.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duncan MC, et al. High-throughput analysis of gene function in the bacterial predator Bdellovibrio bacteriovorus. MBio. 2019;10:1–12. doi: 10.1128/mBio.01040-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thibault D, et al. Droplet Tn-Seq combines microfluidics with Tn-Seq for identifying complex single-cell phenotypes. Nat. Commun. 2019;10:5729. doi: 10.1038/s41467-019-13719-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Judson N, Mekalanos JJ. TnAraOut, a transposon-based approach to identify and characterize essential bacterial genes. Nat. Biotechnol. 2000;18:740–745. doi: 10.1038/77305. [DOI] [PubMed] [Google Scholar]
- 17.Christen B, et al. The essential genome of a bacterium. Mol. Syst. Biol. 2011;7:1–7. doi: 10.1038/msb.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coe KA, et al. Multi-strain Tn-Seq reveals common daptomycin resistance determinants in Staphylococcus aureus. PLoS Pathog. 2019;15:e1007862. doi: 10.1371/journal.ppat.1007862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santiago M, et al. A new platform for ultra-high density Staphylococcus aureus transposon libraries. BMC Genomics. 2015;16:1–18. doi: 10.1186/s12864-015-1361-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yasir M, et al. TraDIS-Xpress: a high-resolution whole-genome assay identifies novel mechanisms of triclosan action and resistance. Genome Res. 2020;30:1–11. doi: 10.1101/gr.254391.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yasir M, et al. A new massively-parallel transposon mutagenesis approach comparing multiple datasets identifies novel mechanisms of action and resistance to triclosan. bioRxiv. 2019 doi: 10.1101/596833. [DOI] [Google Scholar]
- 22.Mutalik VK, et al. Dual-barcoded shotgun expression library sequencing for high-throughput characterization of functional traits in bacteria. Nat. Commun. 2019;10:308. doi: 10.1038/s41467-018-08177-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wetmore KM, et al. Sequencing randomly bar-coded transposons. MBio. 2015;6:1–15. doi: 10.1128/mBio.00306-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson DG, Chen W, Storey JD, Gresham D. Design and analysis of Bar-seq experiments. G3. 2014;4:11–18. doi: 10.1534/g3.113.008565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price MN, et al. Mutant phenotypes for thousands of bacterial genes of unknown function. Nature. 2018;557:503–509. doi: 10.1038/s41586-018-0124-0. [DOI] [PubMed] [Google Scholar]
- 26.Liu H, et al. Magic pools: parallel assessment of transposon delivery vectors in bacteria. mSystems. 2018;3:1–17. doi: 10.1128/mSystems.00143-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zomer A, Burghout P, Bootsma HJ, Hermans PWM, van Hijum SAFT. Essentials: software for rapid analysis of high throughput transposon insertion sequencing data. PLoS One. 2012;7:1–9. doi: 10.1371/journal.pone.0043012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCoy KM, Antonio ML, van Opijnen T. MAGenTA: a galaxy implemented tool for complete Tn-Seq analysis and data visualization. Bioinformatics. 2017;33:2781–2783. doi: 10.1093/bioinformatics/btx320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeJesus MA, Ambadipudi C, Baker R, Sassetti C, Ioerger TR. TRANSIT - a software tool for Himar1 TnSeq analysis. PLoS Comput. Biol. 2015;11:1–17. doi: 10.1371/journal.pcbi.1004401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pritchard JR, et al. ARTIST: high-resolution genome-wide assessment of fitness using transposon-insertion sequencing. PLoS Genet. 2014;10:e1004782. doi: 10.1371/journal.pgen.1004782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barquist L, et al. The TraDIS toolkit: sequencing and analysis for dense transposon mutant libraries. Bioinformatics. 2016;32:1109–1111. doi: 10.1093/bioinformatics/btw022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anthony, J. S. & T. van O. Aerobio: an extensible full DAG streaming computation server with services and jobs for RNA-Seq, Tn-Seq, WG-Seq and Term-Seq. GitHubhttps://github.com/jsa-aerial/aerobio (2019).
- 33.van Opijnen T, Camilli A. A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res. 2012;22:2541–2551. doi: 10.1101/gr.137430.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warr AR, et al. Transposon-insertion sequencing screens unveil requirements for EHEC growth and intestinal colonization. PLoS Pathogens. 2019;15:e1007652. doi: 10.1371/journal.ppat.1007652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramaniyam S, et al. Statistical analysis of variability in TnSeq data across conditions using zero-inflated negative binomial regression. BMC Bioinformatics. 2019;20:603. doi: 10.1186/s12859-019-3156-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van den Berge K, et al. Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol. 2018;19:24. doi: 10.1186/s13059-018-1406-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hubbard TP, D’Gama JD, Billings G, Davis BM, Waldor MK. Unsupervised learning approach for comparing multiple transposon insertion sequencing studies. mSphere. 2019;4:e00031-19. doi: 10.1128/mSphere.00031-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Page AJ, et al. AlbaTraDIS: comparative analysis of large datasets from parallel transposon mutagenesis experiments. bioRxiv. 2019 doi: 10.1101/593624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Canals R, et al. The fitness landscape of the African Salmonella Typhimurium ST313 strain D23580 reveals unique properties of the pBT1 plasmid. PLoS Pathog. 2019;15:e1007948. doi: 10.1371/journal.ppat.1007948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Down TA, Piipari M, Hubbard TJP. Dalliance: interactive genome viewing on the web. Bioinformatics. 2011;27:889–890. doi: 10.1093/bioinformatics/btr020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Surujon D, Van Opijnen T. ShinyOmics: vollaborative exploration of omics-data. BMC Bioinformatics. 2020;21:1–8. doi: 10.1186/s12859-020-3360-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jana B, et al. The secondary resistome of multidrug-resistant Klebsiella pneumoniae. Sci. Rep. 2017;7:42483. doi: 10.1038/srep42483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boinett CJ, et al. Clinical and laboratory-induced colistin-resistance mechanisms in Acinetobacter baumannii. Microb. Genom. 2019;5:e000246. doi: 10.1099/mgen.0.000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coe KA, et al. Comparative Tn-Seq reveals common daptomycin resistance determinants in Staphylococcus aureus despite strain-dependent differences in essentiality of shared cell envelope genes. bioRxiv. 2019 doi: 10.1101/648246. [DOI] [Google Scholar]
- 45.Geisinger E, Mortman NJ, Vargas-Cuebas G, Tai AK, Isberg RR. A global regulatory system links virulence and antibiotic resistance to envelope homeostasis in Acinetobacter baumannii. PLOS Pathog. 2018;14:e1007030. doi: 10.1371/journal.ppat.1007030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu W, et al. Chemical genetic interaction profiling reveals determinants of intrinsic antibiotic resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2017;61:e01334-17. doi: 10.1128/AAC.01334-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willcocks S, et al. Genome-wide assessment of antimicrobial tolerance in Yersinia pseudotuberculosis under ciprofloxacin stress. Microb. Genomics. 2019 doi: 10.1099/mgen.0.000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murray JL, Kwon T, Marcotte EM, Whiteley M. Intrinsic antimicrobial resistance determinants in the superbug Pseudomonas aeruginosa. MBio. 2015;6:1–10. doi: 10.1128/mBio.01603-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajagopal M, et al. Multidrug intrinsic resistance factors in Staphylococcus aureus identified by profiling fitness within high-diversity transposon libraries. MBio. 2016;7:e00950-16. doi: 10.1128/mBio.00950-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blake KL, O’Neill AJ. Transposon library screening for identification of genetic loci participating in intrinsic susceptibility and acquired resistance to antistaphylococcal agents. J. Antimicrob. Chemother. 2012;68:12–16. doi: 10.1093/jac/dks373. [DOI] [PubMed] [Google Scholar]
- 51.Gallagher, L. A., Shendure, J. & Manoil, C. Genome-scale identification of resistance functions in Pseudomonas aeruginosa using Tn-seq. MBio2, (2011). [DOI] [PMC free article] [PubMed]
- 52.Cain, A. K. et al. Transposon directed insertion-site sequencing (TraDIS) to elucidate the mode of action of the antimicrobial arenicin-3 (Arn-3). in 54th International Conference of Antimicrobial Agents and Chemotherapy (ICAAC) and Infectious Diseases Society of America (IDSA) (ASM, 2014).
- 53.Santiago M, et al. Genome-wide mutant profiling predicts the mechanism of a lipid II binding antibiotic. Nat. Chem. Biol. 2018;14:601–608. doi: 10.1038/s41589-018-0041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Opijnen T, Dedrick S, Bento J. Strain dependent genetic networks for antibiotic-sensitivity in a bacterial pathogen with a large pan-genome. PLoS Pathog. 2016;12:e1005869. doi: 10.1371/journal.ppat.1005869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fields PI, Swanson RV, Haidaris CG, Heffron F. Mutants of Salmonella Typhimurium that cannot survive within the macrophage are avirulent. Proc. Natl Acad. Sci. USA. 1986;83:5189–5193. doi: 10.1073/pnas.83.14.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hensel M, et al. Simultaneous identification of bacterial virulence genes by negative selection. Science. 1995;269:400–403. doi: 10.1126/science.7618105. [DOI] [PubMed] [Google Scholar]
- 57.Goh KGK, et al. Genome-wide discovery of genes required for capsule production by uropathogenic Escherichia coli. MBio. 2017;8:e01558-17. doi: 10.1128/mBio.01558-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCarthy AJ, Stabler RA, Taylor PW. Genome-wide identification by transposon insertion sequencing of Escherichia coli K1 genes essential for in vitro growth, gastrointestinal colonizing capacity, and survival in serum. J. Bacteriol. 2018;200:e00698-17. doi: 10.1128/JB.00698-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharp C, et al. O-antigen-dependent colicin insensitivity of uropathogenic Escherichia coli. J. Bacteriol. 2019;201:e00545-18. doi: 10.1128/JB.00545-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dembek M, et al. High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. MBio. 2015;6:1–13. doi: 10.1128/mBio.02383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaudhuri RR, et al. Comprehensive assignment of roles for Salmonella Typhimurium genes in intestinal colonization of food-producing animals. PLoS Genet. 2013;9:e1003456. doi: 10.1371/journal.pgen.1003456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vohra P, et al. Retrospective application of transposon-directed insertion-site sequencing to investigate niche-specific virulence of Salmonella Typhimurium in cattle. BMC Genomics. 2019;20:20. doi: 10.1186/s12864-018-5319-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shames SR, et al. Multiple Legionella pneumophila effector virulence phenotypes revealed through high-throughput analysis of targeted mutant libraries. Proc. Natl Acad. Sci. USA. 2017;114:E10446–E10454. doi: 10.1073/pnas.1708553114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu L, et al. Novel genes required for the fitness of streptococcus pyogenes in human saliva. mSphere. 2017;2:e00460-17. doi: 10.1128/mSphereDirect.00460-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Subashchandrabose S, et al. Acinetobacter baumannii genes required for bacterial survival during bloodstream infection. mSphere. 2016;1:e00013-15. doi: 10.1128/mSphere.00013-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Charbonneau ARL, et al. Identification of genes required for the fitness of Streptococcus equi subsp. equi in whole equine blood and hydrogen peroxide. Microb. Genomics. 2020 doi: 10.1099/mgen.0.000362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang H, et al. Hypermutation-induced in vivo oxidative stress resistance enhances Vibrio cholerae host adaptation. PLoS Pathog. 2018;14:1–22. doi: 10.1371/journal.ppat.1007413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wong Y-C, et al. Genetic determinants associated with in vivo survival of Burkholderia cenocepacia in the Caenorhabditis elegans model. Front. Microbiol. 2018;9:1118. doi: 10.3389/fmicb.2018.01118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shields RC, Zeng L, Culp DJ, Burne RA. Genomewide identification of essential genes and fitness determinants of streptococcus mutans UA159. mSphere. 2018;3:e00031-18. doi: 10.1128/mSphere.00031-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Crabill E, Schofield WB, Newton HJ, Goodman AL, Roy CR. Dot/Icm-translocated proteins important for biogenesis of the Coxiella burnetii-containing vacuole identified by screening of an effector mutant sublibrary. Infect. Immun. 2018;86:e00758-17. doi: 10.1128/IAI.00758-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grant AJ, et al. Modelling within-host spatiotemporal dynamics of invasive bacterial disease. PLoS Biol. 2008;6:e74. doi: 10.1371/journal.pbio.0060074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Abel S, Abel zur Wiesch P, Davis BM, Waldor MK. Analysis of bottlenecks in experimental models of infection. PLoS Pathog. 2015;11:e1004823. doi: 10.1371/journal.ppat.1004823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carter PB, Collins FM. The route of enteric infection in normal mice. J. Exp. Med. 1974;139:1189–1203. doi: 10.1084/jem.139.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maier L, et al. Granulocytes impose a tight bottleneck upon the gut luminal pathogen population during salmonella typhimurium colitis. PLoS Pathog. 2014;10:e1004557. doi: 10.1371/journal.ppat.1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lawley TD, et al. Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLoS Pathog. 2006;2:e11. doi: 10.1371/journal.ppat.0020011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rowe HM, et al. Bacterial factors required for transmission of streptococcus pneumoniae in mammalian hosts. Cell Host Microbe. 2019;25:884–891. doi: 10.1016/j.chom.2019.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carter R, et al. Genomic analyses of pneumococci from children with sickle cell disease expose host-specific bacterial adaptations and deficits in current interventions. Cell Host Microbe. 2014;15:587–599. doi: 10.1016/j.chom.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hancock SJ, et al. Identification of IncA/c plasmid replication and maintenance genes and development of a plasmid multilocus sequence typing scheme. Antimicrob. Agents Chemother. 2017;61:e01740-16. doi: 10.1128/AAC.01740-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Y, et al. Transposon insertion sequencing reveals T4SS as the major genetic trait for conjugation transfer of multi-drug resistance pEIB202 from Edwardsiella. BMC Microbiol. 2017;17:1–15. doi: 10.1186/s12866-017-1013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cowley LA, et al. Transposon insertion sequencing elucidates novel gene involvement in susceptibility and resistance to phages T4 and T7 in Escherichia coli O157. MBio. 2018;9:e00705-18. doi: 10.1128/mBio.00705-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pickard D, et al. A genomewide mutagenesis screen identifies multiple genes contributing to Vi capsular expression in Salmonella enterica serovar typhi. J. Bacteriol. 2013;195:1320–1326. doi: 10.1128/JB.01632-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klein BA, Duncan MJ, Hu LT. Defining essential genes and identifying virulence factors of Porphyromonas gingivalis by massively parallel sequencing of transposon libraries (Tn-seq) Methods Mol. Biol. 2015;1279:25–43. doi: 10.1007/978-1-4939-2398-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wong Y-C, et al. Candidate essential genes in Burkholderia cenocepacia J2315 identified by genome-wide TraDIS. Front. Microbiol. 2016;7:1288. doi: 10.3389/fmicb.2016.01288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Willcocks SJ, Stabler RA, Atkins HS, Oyston PF, Wren BW. High-throughput analysis of Yersinia pseudotuberculosis gene essentiality in optimised in vitro conditions, and implications for the speciation of Yersinia pestis. BMC Microbiol. 2018;18:46. doi: 10.1186/s12866-018-1189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Charbonneau ARLL, et al. Defining the ABC of gene essentiality in streptococci. BMC Genomics. 2017;18:426. doi: 10.1186/s12864-017-3794-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mesarich CH, et al. Transposon insertion libraries for the characterization of mutants from the kiwifruit pathogen Pseudomonas syringae pv. actinidiae. PLoS One. 2017;12:e0172790. doi: 10.1371/journal.pone.0172790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goodall ECA, et al. The essential genome of Escherichia coli K-12. MBio. 2018;9:e02096-17. doi: 10.1128/mBio.02096-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ruiz L, et al. The essential genomic landscape of the commensal Bifidobacterium breve UCC2003. Sci. Rep. 2017;7:5648. doi: 10.1038/s41598-017-05795-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barquist L, et al. A comparison of dense transposon insertion libraries in the Salmonella serovars Typhi and Typhimurium. Nucleic Acids Res. 2013;41:4549–4564. doi: 10.1093/nar/gkt148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Poulsen BE, et al. Defining the core essential genome of Pseudomonas aeruginosa. Proc. Natl Acad. Sci. USA. 2019;116:10072–10080. doi: 10.1073/pnas.1900570116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Geisinger E, et al. The landscape of phenotypic and transcriptional responses to ciprofloxacin in Acinetobacter baumannii: acquired resistance alleles modulate drug-induced SOS response and prophage replication. MBio. 2019;10:1–19. doi: 10.1128/mBio.01127-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Carey AF, et al. TnSeq of mycobacterium tuberculosis clinical isolates reveals strain-specific antibiotic liabilities. PLoS Pathog. 2018;14:e1006939–e1006939. doi: 10.1371/journal.ppat.1006939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Deutschbauer A, et al. Towards an informative mutant phenotype for every bacterial gene. J. Bacteriol. 2014;196:3643–3655. doi: 10.1128/JB.01836-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Basta DW, Bergkessel M, Newman DK. Identification of fitness determinants during energy-limited growth arrest in Pseudomonas aeruginosa. MBio. 2017;8:e01170-17. doi: 10.1128/mBio.01170-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Price MN, et al. Filling gaps in bacterial amino acid biosynthesis pathways with high-throughput genetics. PLoS Genet. 2018;14:1–23. doi: 10.1371/journal.pgen.1007147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Byrne RT, Chen SH, Wood EA, Cabot EL, Cox MM. Escherichia coli genes and pathways involved in surviving extreme exposure to ionizing radiation. J. Bacteriol. 2014;196:3534–3545. doi: 10.1128/JB.01589-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kingsley RA, et al. Functional analysis of Salmonella Typhi adaptation to survival in water. Environ. Microbiol. 2018;20:4079–4090. doi: 10.1111/1462-2920.14458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mandal RK, Kwon YM. Global screening of Salmonella enterica serovar Typhimurium genes for desiccation survival. Front. Microbiol. 2017;8:1723. doi: 10.3389/fmicb.2017.01723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fabian BK, Tetu SG, Paulsen IT. Application of transposon insertion sequencing to agricultural science. Front. Plant Sci. 2020;11:291. doi: 10.3389/fpls.2020.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Royet K, Parisot N, Rodrigue A, Gueguen E, Condemine G. Identification by Tn-seq of Dickeya dadantii genes required for survival in chicory plants. Mol. Plant Pathol. 2019;20:287–306. doi: 10.1111/mpp.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Duong DA, Jensen RV, Stevens AM. Discovery of Pantoea stewartii ssp. stewartii genes important for survival in corn xylem through a Tn-Seq analysis. Mol. Plant Pathol. 2018 doi: 10.1111/mpp.12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu H, et al. Large-scale chemical-genetics of the human gut bacterium Bacteroides thetaiotaomicron. bioRxiv. 2019 doi: 10.1101/573055. [DOI] [Google Scholar]
- 103.Goodman AL, Wu M, Gordon JI. Identifying microbial fitness determinants by insertion sequencing using genome-wide transposon mutant libraries. Nat. Protoc. 2011;6:1969–1980. doi: 10.1038/nprot.2011.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, Goodman AL. Separating host and microbiome contributions to drug pharmacokinetics and toxicity. Science. 2019;363:eaat9931. doi: 10.1126/science.aat9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stacy A, Fleming D, Lamont RJ, Rumbaugh KP, Whiteley M. A commensal bacterium promotes virulence of an opportunistic pathogen via cross-respiration. MBio. 2016;7:e00782-16. doi: 10.1128/mBio.00782-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ibbersona CB, et al. Co-infecting microbes dramatically alter pathogen gene essentiality during polymicrobial infection. Nat. Microbiol. 2017;2:17079. doi: 10.1038/nmicrobiol.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Duncan MC, et al. Vibrio cholerae motility exerts drag force to impede attack by the bacterial predator Bdellovibrio bacteriovorus. Nat. Commun. 2018;9:4757. doi: 10.1038/s41467-018-07245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chatterjee A, et al. Parallel genomics uncover novel enterococcal-bacteriophage interactions. MBio. 2020;11:e03120-19. doi: 10.1128/mBio.03120-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fu Y, Waldor MK, Mekalanos JJ. Tn-seq analysis of vibrio cholerae intestinal colonization reveals a role for T6SS-mediated antibacterial activity in the host. Cell Host Microbe. 2013;14:652–663. doi: 10.1016/j.chom.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nolan LM, et al. Discovery of a pseudomonas aeruginosa type VI secretion system toxin targeting bacterial protein synthesis using a global genomics approach. bioRxiv. 2019 doi: 10.1101/733030. [DOI] [Google Scholar]
- 111.Mann B, et al. Control of virulence by small RNAs in Streptococcus pneumoniae. PLoS Pathog. 2012;8:34. doi: 10.1371/journal.ppat.1002788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Warrier I, et al. The Transcriptional landscape of Streptococcus pneumoniae TIGR4 reveals a complex operon architecture and abundant riboregulation critical for growth and virulence. PLoS Pathog. 2018;14:1–25. doi: 10.1371/journal.ppat.1007461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Capel E, et al. Comprehensive identification of meningococcal genes and small noncoding RNAs required for host cell colonization. MBio. 2016;7:e01173-16. doi: 10.1128/mBio.01173-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Costanzo M, et al. Global genetic networks and the genotype-to-phenotype relationship. Cell. 2019;177:85–100. doi: 10.1016/j.cell.2019.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fenton AK, El Mortaji L, Lau DTC, Rudner DZ, Bernhardt TG. CozE is a member of the MreCD complex that directs cell elongation in Streptococcus pneumoniae. Nat. Microbiol. 2016;2:16237. doi: 10.1038/nmicrobiol.2016.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Santa Maria JPJ, et al. Compound-gene interaction mapping reveals distinct roles for Staphylococcus aureus teichoic acids. Proc. Natl Acad. Sci. USA. 2014;111:12510–12515. doi: 10.1073/pnas.1404099111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lorenz A, et al. Importance of flagella in acute and chronic Pseudomonas aeruginosa infections. Environ. Microbiol. 2019;21:883–897. doi: 10.1111/1462-2920.14468. [DOI] [PubMed] [Google Scholar]
- 118.DeJesus MA, et al. Statistical analysis of genetic interactions in Tn-Seq data. Nucleic Acids Res. 2017;45:e93. doi: 10.1093/nar/gkx128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jensen PA, Zhu Z, van Opijnen T. Antibiotics disrupt coordination between transcriptional and phenotypic stress responses in pathogenic bacteria. Cell Rep. 2017;20:1705–1716. doi: 10.1016/j.celrep.2017.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Turner KH, Everett J, Trivedi U, Rumbaugh KP, Whiteley M. Requirements for Pseudomonas aeruginosa acute burn and chronic surgical wound infection. PLoS Genet. 2014;10:e1004518. doi: 10.1371/journal.pgen.1004518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Deutschbauer A, et al. Evidence-based annotation of gene function in Shewanella oneidensis MR-1 using genome-wide fitness profiling across 121 conditions. PLoS Genet. 2011;7:e1002385. doi: 10.1371/journal.pgen.1002385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Smith JJ, et al. Expression and functional profiling reveal distinct gene classes involved in fatty acid metabolism. Mol. Syst. Biol. 2006;2:2006.0009. doi: 10.1038/msb4100051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 124.Keren L, et al. Massively parallel interrogation of the effects of gene expression levels on fitness. Cell. 2016;166:1282–1294.e18. doi: 10.1016/j.cell.2016.07.024. [DOI] [PubMed] [Google Scholar]
- 125.Price MN, et al. Indirect and suboptimal control of gene expression is widespread in bacteria. Mol. Syst. Biol. 2013;9:660. doi: 10.1038/msb.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhu Z, et al. Entropy of a bacterial stress response is a generalizable predictor for fitness and antibiotic sensitivity. bioRxiv. 2019 doi: 10.1101/813709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Paulsen IT, Cain AK, Hassan KA. Physical enrichment of transposon mutants from saturation mutant libraries using the TraDISort approach. Mob. Genet. Elements. 2017;7:1–7. doi: 10.1080/2159256X.2017.1313805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.O’Connor TJ, Boyd D, Dorer MS, Isberg RR. Aggravating genetic interactions allow a solution to redundancy in a bacterial pathogen. Science. 2012;338:1440–1444. doi: 10.1126/science.1229556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lee SY, et al. Dense transposon integration reveals essential cleavage and polyadenylation factors promote heterochromatin formation. Cell Rep. 2020;30:2686–2698. doi: 10.1016/j.celrep.2020.01.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Michel AH, et al. Functional mapping of yeast genomes by saturated transposition. Elife. 2017;6:e23570. doi: 10.7554/eLife.23570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Guo Y, et al. Integration profiling of gene function with dense maps of transposon integration. Genetics. 2013;195:599–609. doi: 10.1534/genetics.113.152744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang C, Phillips APR, Wipfler RL, Olsen GJ, Whitaker RJ. The essential genome of the crenarchaeal model Sulfolobus islandicus. Nat. Commun. 2018;9:1–11. doi: 10.1038/s41467-018-07379-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sarmiento F, Mrázek J, Whitman WB. Genome-scale analysis of gene function in the hydrogenotrophic methanogenic archaeon Methanococcus maripaludis. Proc. Natl Acad. Sci. USA. 2013;110:4726–4731. doi: 10.1073/pnas.1220225110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Carette JE, et al. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat. Biotechnol. 2011;29:542–546. doi: 10.1038/nbt.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Carette JE, et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- 136.Friedrich MJ, et al. Genome-wide transposon screening and quantitative insertion site sequencing for cancer gene discovery in mice. Nat. Protoc. 2017;12:289–309. doi: 10.1038/nprot.2016.164. [DOI] [PubMed] [Google Scholar]
- 137.Rad R, et al. A conditional piggyBac transposition system for genetic screening in mice identifies oncogenic networks in pancreatic cancer. Nat. Genet. 2015;47:47–56. doi: 10.1038/ng.3164. [DOI] [PubMed] [Google Scholar]
- 138.Zhang M, et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science. 2018;360:eaap7847. doi: 10.1126/science.aap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Smith AM, et al. Quantitative phenotyping via deep barcode sequencing. Genome Res. 2009;19:1836–1842. doi: 10.1101/gr.093955.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu X, et al. High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol. Syst. Biol. 2017;13:931. doi: 10.15252/msb.20167449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Peters JM, et al. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell. 2016;165:1493–1506. doi: 10.1016/j.cell.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lee HH, et al. Functional genomics of the rapidly replicating bacterium Vibrio natriegens by CRISPRi. Nat. Microbiol. 2019;4:1105–1113. doi: 10.1038/s41564-019-0423-8. [DOI] [PubMed] [Google Scholar]
- 144.Rousset F, et al. Genome-wide CRISPR-dCas9 screens in E. coli identify essential genes and phage host factors. PLoS Genet. 2018;14:e1007749. doi: 10.1371/journal.pgen.1007749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang T, et al. Pooled CRISPR interference screening enables genome-scale functional genomics study in bacteria with superior performance. Nat. Commun. 2018;9:2475. doi: 10.1038/s41467-018-04899-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Steen AD, et al. High proportions of bacteria and archaea across most biomes remain uncultured. ISME J. 2019;13:3126–3130. doi: 10.1038/s41396-019-0484-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhu, Z., Surujon, D., Pavao, A., Bento, J. & van Opijnen, T. Forecasting bacterial survival-success and adaptive evolution through multi-omics stress-response mapping, network analyses and machine learning. bioRxiv10.1101/387910 (2018).