

Abstract
Eukaryotic gene expression is regulated not only by genomic enhancers and promoters, but also by covalent modifications added to both chromatin and RNAs. Whereas cellular gene expression may be either enhanced or inhibited by specific epigenetic modifications deposited on histones (in particular, histone H3), these epigenetic modifications can also repress viral gene expression, potentially functioning as a potent antiviral innate immune response in DNA virus-infected cells. However, viruses have evolved countermeasures that prevent the epigenetic silencing of their genes during lytic replication, and they can also take advantage of epigenetic silencing to establish latent infections. By contrast, the various covalent modifications added to RNAs, termed epitranscriptomic modifications, can positively regulate mRNA translation and/or stability, and both DNA and RNA viruses have evolved to utilize epitranscriptomic modifications as a means to maximize viral gene expression. As a consequence, both chromatin and RNA modifications could serve as novel targets for the development of antivirals. In this Review, we discuss how host epigenetic and epitranscriptomic processes regulate viral gene expression at the levels of chromatin and RNA function, respectively, and explore how viruses modify, avoid or utilize these processes in order to regulate viral gene expression.
Subject terms: Viral infection, Virus-host interactions
In this Review, Tsai and Cullen discuss how host epigenetic and epitranscriptomic processes regulate viral gene expression at the levels of chromatin and RNA function, respectively, and explore how viruses modify, avoid or utilize these processes in order to promote their replication or persistence.
Introduction
Although all human cells in a given individual contain the same genome, they differ widely in the genes they express and, hence, in their biological properties. It is therefore well established that gene expression is not determined simply by the sequence information that is encoded into genomic DNA, but rather is subject to multiple levels of control. This Review discusses two complementary regulatory mechanisms that act, respectively, at the DNA and RNA levels. In the case of the host-cell genome, the level of expression of specific genes is regulated not only by flanking promoters and enhancers but also by how accessible the genes embedded within cellular chromatin are to transcription factors. This accessibility in turn is regulated by processes that include DNA methylation, histone remodelling, alternative histone variant usage and the deposition of modifications on histone tails, collectively referred to as epigenetic gene regulation1,2 (Box 1). Similarly, the function of the mRNAs transcribed from a given gene is regulated not only by their nucleotide sequence but also by a range of covalent modifications that are added to individual nucleotides, the most prevalent of which is methylation of the N6 position of adenosine (m6A) (refs3,4). These RNA modifications — which have been reported to regulate the stability, the translation and even the immunogenicity of RNA molecules — are referred to as epitranscriptomic modifications.
Epigenetic and epitranscriptomic gene regulation likely initially evolved as means of regulating cell growth and differentiation. However, they are also relevant to several important diseases, including cancer, in which both processes have been reported to be dysregulated5–7. Their importance for the regulation of viral gene expression has only recently begun to emerge. As obligate intracellular parasites, viruses misappropriate parts of the host-cell machinery in order to allow the expression of viral genes. This dependence on cellular gene regulation pathways can, however, also lead to viral gene expression being subject to host repression. Indeed, epigenetic repression has been proposed to function as a form of antiviral restriction used by host cells as an innate immune defence against DNA viruses, in a tug of war that is only revealed when viral countermeasures are experimentally removed8. Alternatively, viruses may repurpose epigenetic repression in order to suppress their own gene expression, as a way to establish latent infections and prevent the production of immunogenic viral proteins9. By contrast, epitranscriptomic modifications generally enhance the function of viral mRNAs, and both DNA and RNA viruses have therefore evolved to maximize the level of these modifications on their transcripts.
In this Review, we discuss epigenetic repression mechanisms, including histone tail modifications and alternative histone variants that are loaded onto viral DNA upon entry into host-cell nuclei and how viruses avoid or even utilize this repression to enhance viral replication or persistence. We then explore the epitranscriptomic RNA modifications, including methylation and acetylation, found on viral transcripts, and how they enhance viral gene expression or help evade innate immune detection. We highlight the importance of non-encoded regulatory information found on viral chromatin and RNA and discuss how the regulatory pathways that are influenced by this information could serve as novel targets for antiviral therapies.
Box 1 Relationship of chromatin architecture and histone modifications to gene expression.
The genomic DNA of cells is bound by basic proteins called histones, with every ~147 bp of DNA wrapped around a core octamer containing two molecules apiece of the histones H2A, H2B, H3 and H4, to form chromatin (see the figure). Although chromatin can protect genome integrity, histones can also hinder the accessibility of genomic DNA for transcription. This accessibility is a function of the degree of compaction of the histones bound to that genomic DNA, with open regions being referred to as euchromatin, whereas tightly compacted regions of DNA are referred to as heterochromatin. Chromatin compaction constitutes a form of gene regulation not directly encoded in the DNA nucleotide sequence itself; this is termed epigenetic gene regulation, as it is the molecular mechanism underlying epigenetic inheritance127. Epigenetic gene regulation can take the form of DNA modifications, histone tail modifications, chromatin remodelling and alternative histone subunits (see the figure). DNA modifications mainly involve CpG methylation, in which a methyl group is added to deoxycytidine residues located in CG-rich genomic regions. CpG methylation is primarily associated with repressive heterochromatin. Histone tail modifications — including methylations, acetylations, phosphorylations and ubiquitylations — are the best understood form of epigenetic regulation. Examples have been found on all four canonical histones (H2A, H2B, H3 and H4). H3 and H4 acetylations are associated with active euchromatin, whereas methylations are more diverse in function. For example, H3 lysine 9 and lysine 27 tri-methylation (H3K9me3 and H3K27me3, respectively) mark heterochromatin, whereas H3K4me3 marks are found on actively transcribed euchromatin128. Chromatin remodelling involves ATP-dependent histone chaperones that may slide, evict or load histones, modulating the local density of histones on the chromatin. Finally, chromatin accessibility can be modulated through the use of alternative histones, such as the H2A histone variant in place of H2, or the H3.3 histone variant as a replacement for H3.1 (ref.129). The canonical H3 histone variants H3.1 and H3.2 are deposited by the histone chaperone CAF-1 during DNA replication. However, histones can be displaced by transcription, and they need to be replaced by the histone chaperone complex HIRA, which deposits the replacement histone H3 variant H3.3. Thus, H3.3 previously was thought to mark transcriptionally active chromatin. However, H3.3 later was found also to be loaded by an alternative histone chaperone complex consisting of a heterodimer of Daxx and ATRX, which loads H3.3 onto repressed heterochromatic regions such as pericentromeric regions and telomeres, suggesting that different chaperones may load histone H3.3 with alternative histone tail marks onto either actively transcribed or repressed chromatin25,130,131.
Epigenetic repression and its avoidance
Incoming viral DNA molecules are sensed by target cells and are rapidly loaded with histones bearing heterochromatic marks, thus inhibiting viral gene expression. How cells distinguish viral DNA from their own remains undefined, though epigenetic repression of foreign DNA seems mainly to be orchestrated by proteins associated with the pro-myelocytic leukaemia nuclear bodies (PML-NBs) and by the innate immune DNA sensor interferon-inducible protein 16 (IFI16)8,10–12. Here we discuss how herpesviruses, hepatitis B virus (HBV) and retroviruses can be epigenetically repressed and how these viruses can disrupt or avoid repression in order to initiate productive infections. We also discuss how epigenetic repression can be misappropriated to establish viral latent infections.
PML-NB-mediated epigenetic repression
Epigenetic suppression of viral DNA is frequently associated with nuclear protein aggregates called PML-NBs or nuclear domain 10 (ND10) (Fig. 1a). PML-NBs consist of cage-like structures constructed of PML proteins, which contain other effector proteins that together appear as punctate foci when visualized by immunofluorescence microscopy13–15. Several DNA viruses, including Simian virus 40, adenovirus type 5, herpes simplex virus 1 (HSV-1) and human cytomegalovirus (HCMV), localize to and establish viral replication sites adjacent to PML-NBs, suggesting that this structure is at least spatially, if not functionally, related to viral replication16. Through the recruitment of cofactors, PML-NBs can be associated with many cellular functions, including antiviral defence and transcriptional repression. The expression of the PML-NB components Sp100 and PML increases in response to interferon signalling, and PML-NB components including PML, Sp100, Daxx and ATRX suppress viral replication13,17–20. Daxx and Sp100 also have transcriptional repressive functions, likely through recruiting co-repressors such as HP1 and HDAC II21–24. Moreover, Daxx and ATRX can load the histone variant H3.3 onto heterochromatic regions25,26. All of the functions above implicate PML-NBs as subnuclear hubs of the epigenetic repression of viral DNA.
Fig. 1. Epigenetic repression of viral DNA.

Upon entry into the cell nucleus, the DNA of many viruses initiates the replication process adjacent to subnuclear structures called pro-myelocytic leukaemia nuclear bodies (PML-NBs). However, PML-NBs are an aggregation site for many heterochromatic repression proteins, which load repressive heterochromatin onto viral DNA that shuts down viral transcription. In the absence of viral de-repression factors, viral episomal DNA lacks active histone marks (shown as green histones with the active marks H3Ac, H4Ac and H3K4me3). a | Several PML-NB components — including PML itself, Sp100, Smc5, Smc6, Daxx and ATRX — are involved in the epigenetic repression of viruses, with the Daxx–ATRX complex having specifically been found to load the histone variant H3.3 bearing repressive marks onto viral DNA, leading to the accumulation of heterochromatic marks and blocked transcription (DNA is shown associated with red histones with the repressive marks H3K9me3 and H3K27me3). b | The innate immune DNA sensor IFI16 drives an alternative mechanism of antiviral epigenetic repression, promoting repressive methylations on histone tail H3K9. c | Specifically for the retrovirus murine leukaemia virus, NP220 and the human silencing hub (HUSH) complex have also been shown to deposit heterochromatic marks on unintegrated viral DNA.
IFI16-mediated epigenetic repression
Invading viral DNA can also be detected by the innate immune DNA sensor IFI16 (Fig. 1b). IFI16 contains an oligomerizing pyrin domain (PYD) and two double-stranded DNA (dsDNA)-binding HIN200 domains, and it oligomerizes on histone-free segments of dsDNA longer than 70 bp27. IFI16 was initially characterized as a DNA sensor that induces interferon-β through the signalling factors STING, TBK1 and IRF3 (ref.28), and it was shown to repress gene expression from transfected plasmids as well as from viruses including herpesviruses, papillomaviruses and HBV11,29–32. Interestingly, IFI16 suppression of HSV-1 and Kaposi sarcoma-associated herpesvirus (KSHV) transcription involves the deposition of suppressive H3K9me3 marks in an interferon-independent manner, with IFI16 directly recruiting H3K9 methyltransferases to KSHV DNA11,29,33. Although IFI16 has a key role in the epigenetic repression of viral DNA, it should be noted that IFI16 function is likely closely connected to the inhibition by PML-NBs, as IFI16 is known to interact with PML-NB components34.
Herpesviruses
Epigenetic repression of viruses is best understood for herpesviruses (Fig. 1). Herpesvirus DNA is packaged into virions ‘naked’ (that is, free of histones)35–37. Upon release into the nucleus, viral DNA is detected and then rapidly loaded with histones harbouring suppressive marks, as a form of innate immune response. Upon infection of fibroblasts by the alphaherpes virus HSV-1, within 2 h viral DNA is loaded with histones, mainly consisting of Daxx–ATRX and HIRA-loaded H3.3 with repressive H3K9me3 marks38–41. HSV-1 disarms this repression in two stages. First, the viral protein VP16, which is packaged into the tegument layer of incoming virions, recruits host proteins, including host-cell factor 1 (HCF-1) and octamer-binding factor (Oct-1), in order to form a complex that recruits the histone demethylases lysine-specific demethylase 1 (LSD1) and Jumonji domain 2 (JMJD2) family members as a means to remove repressive H3K9 marks from viral immediate early promoters42,43 (Fig. 2a). This VP16 complex also recruits the methyltransferases Set1 and MLL1, which deposit H3K4me3 activating marks on the histone H3 bound to viral DNA44. This allows expression of the immediate early viral protein ICP0, which orchestrates the second stage of de-repression. ICP0, an E3 ubiquitin ligase, induces the ubiquitylation and degradation of the PML-NB components PML and Sp100 (Fig. 2b). Once these structural components of PML-NBs are degraded, Daxx and ATRX are dispersed away from viral replication sites, de-repressing regions of the viral genome not already activated by VP16. Indeed, ICP0 promotes the gradual removal of repressive histone marks H3K9me3 and H3K27me3 from the entire viral DNA genome, ensuring a productive infection45. In parallel, ICP0 has also been implicated in the degradation of IFI16 (Fig. 2c). IFI16 suppresses gene expression from ICP0-deficient mutants of HSV-1, with IFI16 directly binding viral DNA, promoting the deposition of repressive H3K9me3 marks and the removal of active H3K4me3 marks11,29. However, the role of ICP0 in counteracting IFI16 is somewhat controversial, with one study reporting that inhibition of HSV-1 gene expression by IFI16 occurs only in the absence of ICP0, whereas another study reported that IFI16 is also active against wild-type HSV-1 (refs11,29). This discrepancy may be partially explained by the finding that ICP0-mediated degradation of IFI16 is efficient in primary cells yet less so in transformed cell lines such as HeLa46. Regardless, IFI16 clearly represents a key component of the host-cell viral-DNA recognition machinery47.
Fig. 2. Viral strategies to avoid epigenetic repression.

Viruses can counteract epigenetic repression in multiple ways in order to enrich for histones bearing active marks (shown as green histones with H3Ac, H4Ac and H3K4me3 marks) and prevent the loading of histones with repressive marks (red histone variant H3.3 bearing the repressive mark H3K9me3). a | The herpes simplex virus 1 (HSV-1) protein VP16 associates with the host factors HCF-1 and Oct-1, with HCF-1 recruiting the host H3K9 demethylases LSD1 and JMJD2 to remove repressive marks, along with the H3K4 methyltransferases Set1 and MLL1 to deposit active marks on viral DNA. b | Several viral proteins target pro-myelocytic leukaemia nuclear body (PML-NB) components in order to avoid epigenetic repression. HSV-1 ICP0 and human cytomegalovirus (HCMV) IE1 induce the degradation of PML and Sp100 (degraded proteins shown in light grey with dotted outlines). Epstein–Barr virus (EBV) BNRF1 disassembles the Daxx–ATRX histone chaperone complex, whereas HCMV pp71 degrades Daxx. Hepatitis B virus (HBV) protein HBx induces the degradation of Smc5 or Smc6. c | HSV-1 can also induce the degradation of IFI16, an alternative antiviral epigenetic repression factor, with ICP0 implicated as being involved in this degradation. d | Retroviruses avoid epigenetic repression by integrating their DNA into host euchromatin, where it can no longer be identified as foreign DNA. MLV, murine leukaemia virus.
The betaherpesvirus HCMV expresses a different tegument protein, pp71, that degrades Daxx and mislocalizes ATRX, ensuring the presence of open chromatin on viral DNA bearing H3Ac and minimal H3K9me3 marks48–50 (Fig. 2b). Similarly, the gammaherpesvirus Epstein–Barr virus (EBV) packages the tegument protein BNRF1, which localizes to PML-NBs and disassembles the histone chaperone complex Daxx–ATRX by binding Daxx and displacing ATRX51. This prevents the complex from depositing repressive forms of histone variant H3.3 on EBV DNA, ensuring the presence of active histone marks on the first-activated viral promoter (Wp)52,53. Thus, it is clear that herpesviruses have evolved in such a way to package factors into the tegument of viral particles that inhibit the epigenetic repression of viral DNA by components of host-cell PML-NBs (Fig. 1). Although it is less clear how IFI16 is inhibited by other herpesviruses, HCMV has been reported to encode a number of factors that block IFI16 activity and/or induce the cytoplasmic mislocalization of IFI16 (refs54–56).
Hepatitis B virus
Epigenetic repression by host factors also affects other nuclear DNA viruses. HBV, a hepadnavirus, causes chronic liver infections that can result in cirrhosis or hepatocellular carcinoma. The transcriptionally active form of viral DNA is covalently closed circular DNA (cccDNA). Epigenetic repression of cccDNA becomes apparent when the HBV X protein (HBx) is mutationally inactivated57. In this context, the structural maintenance of chromosome 5 and 6 (Smc5/6) proteins, which colocalize with PML-NBs, act as restriction factors for HBV transcription. In a striking parallel to herpesviruses, the HBx protein recruits the cellular E3 ubiquitin ligase DDB1 in order to induce the ubiquitylation and degradation of Smc5/6 (refs58–60) (Fig. 2b). Importantly, artificial depletion of the PML-NB components PML and Sp100 using RNA interference not only results in the nuclear redistribution of Smc6 but also allows the transcription of HBV cccDNA in the absence of HBx. Thus, HBV, like herpesviruses, disrupts PML-NBs as a means to circumvent the epigenetic silencing of viral DNA.
Retroviruses
As we argued above, host cells can recognize incoming viral DNA as targets for epigenetic silencing. Whereas herpesviruses and HBV use viral proteins to disperse or degrade PML-NB components and so prevent silencing, retroviruses appear to avoid proviral DNA silencing through the alternative strategy of chromosomal integration (Fig. 2d). In the absence of integrase function, the potency of epigenetic repression becomes apparent, as unintegrated retroviral DNA is poorly transcribed61,62. Retroviral particles contain RNA genomes, which are reverse transcribed in the cytoplasm. Like herpesviral DNA, retroviral DNA enters the nucleus ‘naked’ and is rapidly loaded with histones63,64. Specifically, unintegrated HIV-1 DNA is loaded with the histone variant H3.3 bearing the repressive H3K9me3 mark and only low levels of the active mark H3Ac. In the case of murine leukaemia virus (MLV), epigenetic repression requires the DNA-binding protein NP220, which recruits epigenetic repressors including the H3K9me3 methyltransferase SETDB1, along with the human silencing hub (HUSH) complex65, which has previously been identified as a repressor of endogenous retrotransposons and has been proposed to have a role in the epigenetic silencing of latent HIV-1 proviruses66 (Fig. 1c). However, the previous study65 also demonstrated that the HUSH complex has no role in silencing unintegrated HIV-1 proviruses; how these are targeted for epigenetic repression remains to be determined. It is also unclear how the repressive marks deposited on unintegrated proviral DNA are removed after integration; however, HIV-1 proviruses are known to preferentially integrate into actively transcribed chromatin, which may allow active chromatin marks of the surrounding region to spread to the proviral DNA67,68. Regardless, the ability of cells to effectively silence unintegrated retroviral DNA means that integrase inhibitors are currently among the most clinically effective antiretroviral drugs.
If unintegrated retroviral DNA is indeed epigenetically silenced, this implies that a factor that induces the transcription of unintegrated retroviral DNA should be able to rescue the replication of integrase-deficient (ΔIN) retroviruses. Indeed, this has recently been demonstrated. Specifically, the Tax protein encoded by human T cell leukaemia virus 1, which is a potent activator of the cellular NF-κB transcription factors RelA and RelB, was shown to rescue the robust replication of ΔIN HIV-1 in T cells69. This rescue correlated with the effective recruitment of RelA and RelB to the two NF-κB binding sites located in the HIV-1 enhancer element as well as with the acquisition of active, and the loss of repressive, epigenetic marks on unintegrated HIV-1 DNA. Thus, in the presence of Tax, ΔIN HIV-1 replicates as transcriptionally active DNA circles that are analogous to HBV cccDNAs.
Latent viral infections
Upon infection of certain cell types, viruses may be epigenetically repressed, resulting in a latent infection. The successful repression of viral DNA, while it prevents the killing of individual cells by lytic viral replication, does not necessarily reflect a clear victory for the host, as the establishment of latency can allow viruses to maintain long-term infections that avoid the host adaptive immune response yet revert to a lytic replication cycle at a later time. This is the case with latent infection of neurons by HSV-1, of B lymphocytes by EBV and even of resting T cells by HIV-1. Each of these examples of latent infection is associated with the addition of repressive epigenetic marks to viral DNA, including H3K9me3 and H3K27me3, and with low levels of active markers70. Interestingly, latent infection by HSV-1 in neuronal cells may be partly explained by VP16 and its host cofactor HCF-1 being sequestered in the cytoplasm and thus unable to de-repress nuclear viral DNA8,71. The fact that HCF-1 is uniquely localized to the cytosol in neurons but not in other cell types suggests that HSV-1 may have evolved to utilize the cytosolic localization of HCF-1 to identify cell types in which latency is the preferred strategy.
Latency establishment is an intricately regulated process that allows viruses to temporarily adopt an alternative viral state. Taking EBV as an example, upon infection of B lymphocytes, the virus first activates all eight latency genes to aid in the activation of naive B cells (type III latency), and then certain genes are switched off (type I/II latency), to minimize the number of viral proteins that are detectable by the immune system, with different latency types utilizing distinct viral promoters that are associated with active chromatin marks only when needed72–74. Meanwhile, lytic viral genes remain transcriptionally silent and are associated with heterochromatic markers during latency70,75. Thus, herpesviruses appear to have subverted what may have originally evolved as an innate antiviral defence mechanism (the epigenetic silencing of viral DNA) as a means to maintain viral latency by selectively silencing viral genes. In this way, herpesviruses can establish a long-lived viral reservoir that avoids detection by the host adaptive immune system.
In conclusion, viral episomal DNA can be epigenetically silenced by the host using mechanisms analogous to host heterochromatic gene repression, while DNA viruses including herpesviruses and HBV package and/or encode viral proteins that can overcome this suppression. Interestingly, the identified targets of viral de-repressor proteins have highlighted PML-NBs as a key structure in the epigenetic suppression of viral DNA. Although it remains enigmatic how PML-NBs differentiate between host and episomal viral DNA, one hypothesis suggests that IFI16 may act as the DNA sensor that recruits PML-NB components to viral DNA, because IFI16 associates with PML-NB components and because both IFI16 and PML-NBs are recruited to incoming HSV-1 DNA. By contrast, some reports suggest that IFI16 and PML-NBs represent distinct mechanisms34,76–78. Retroviruses appear to avoid epigenetic repression through integration of their DNA into euchromatic regions of the host-cell genome, as the epigenetic repression of unintegrated retroviral DNA closely mirrors that seen with mutant DNA viruses that are unable to mount the necessary countermeasures. Alternatively, when viruses invade host cells that are at least transiently non-permissive, viruses may utilize epigenetic suppression to enter a controlled, dormant state, thereby forming viral reservoirs that have so far proven impossible to eradicate.
Epitranscriptomic regulation
RNA transcripts are subject to a range of different covalent modifications at the single-nucleotide level, and over 100 such modifications have been identified, particularly on tRNAs and other non-coding RNAs (ncRNAs); several of these modifications have been shown to regulate ncRNA function3. The repertoire of epitranscriptomic modifications (primarily but not exclusively methylations) found on eukaryotic mRNAs is more limited than that found on ncRNAs, and functional data exist for only a fraction of these mRNA modifications4 (Table 1). For the purposes of this Review, we only discuss the four epitranscriptomic modifications that so far have been reported to affect viral gene expression — N6-methyladenosine (m6A), 5-methylcytidine (m5C), N4-acetylcytidine (ac4C) and 2′O-methylation of the ribose moiety of all four ribonucleosides (collectively Nm) (Fig. 3). Quantification of the levels of several different epitranscriptomic modifications on highly purified samples of the genomic RNA of both HIV-1 and the model animal retrovirus MLV has revealed that these viral RNAs contain far higher levels of m6A, m5C and Nm residues than do cellular mRNAs expressed in infected cells79,80. Specifically, m5C was detected at a level 14–30 times higher on these viral RNAs than on cellular poly(A)+ RNA; Nm levels were also 10–20 times higher, and m6A modifications were 2–10 times more prevalent. The level of ac4C was not examined in these two studies, but it was reported to be at a level comparable to that of m6A on HIV-1 transcripts in a third study that, despite having used virion RNA samples of unknown purity, obtained m6A and m5C levels similar to those in the previous reports81. Regardless, the very high prevalence of m6A, m5C, Nm and likely ac4C modifications on these viral RNAs indicates that viruses have evolved to maximize their addition to viral transcripts, which strongly suggests that these epitranscriptomic modifications are facilitating one or more steps in the viral replication cycle (Fig. 3).
Table 1.
Epitranscriptomic RNA modifications that regulate virus replication
| RNA modification | Writer | Readers | Reported phenotypic effect |
|---|---|---|---|
| N6-methyladenosine (m6A) | METTL3 (N) |
YTHDC1 (N) YTHDC2 (C) YTHDF1 (C) YTHDF2 (C) YTHDF3 (C) |
Increased viral RNA and protein expression; avoidance of RIG-I activation |
| 5-methylcytidine (m5C) | NSUN2 (N) | Unknown | Increased viral mRNA translation |
| N4-acetylcytidine (ac4C) | NAT10 (N) | Unknown | Increased viral mRNA stability |
| 2ʹO-methylated nucleosides (Nm) | FTSJ3 (No) | Unknown | Avoidance of MDA5 activation |
C, cytoplasmic localization; N, nuclear localization; No, nucleolar localization.
Fig. 3. Epitranscriptomic modifications of viral RNA.

The indicated epitranscriptomic marks are N6-methyladenosine (m6A), 5-methylcytidine (m5C), N4-acetylcytidine (ac4C) and 2ʹO-methylated nucleosides (Nm), deposited respectively by a complex that includes the proteins METTL3, METTL14 and WTAP (among other cofactors), by NSUN2, by nuclear N-acetyltransferase 10 (NAT10) and by FTSJ3. All four of these epitranscriptomic marks have been reported to promote viral replication by affecting different steps in the viral replication cycle, either by upregulating viral mRNA stability or translation, or by preventing the detection of viral RNAs by host RNA-specific innate immunity factors, including RIG-I and MDA5. Of note, it is currently unknown whether RNA modifications have distinct functions when deposited on viral mRNA as opposed to viral RNA genomes, but this remains a possibility.
N6-methyladenosine
The most common epitranscriptomic modification found on eukaryotic cellular mRNAs is m6A, representing 0.2–0.4% of all adenosine residues4,82,83. As a result, m6A has been a major focus of epitranscriptomic research, resulting in the identification of methyltransferase-like 3 (METTL3) as the enzyme that adds m6A to mRNAs (Table 1). METTL3, which is referred to as the m6A ‘writer’, functions as part of a complex with several cofactors, including METTL14 and WTAP, and uses S-adenosylmethionine (SAM) as the methyl donor4,82 (Fig. 3). This complex is predominantly nuclear, and the addition of m6A to mRNAs has been reported to occur co-transcriptionally84. Once deposited, m6A residues can be detected by five m6A ‘readers’, including the predominantly nuclear protein YTHDC1 and the cytoplasmic factors YTHDC2, YTHDF1, YTHDF2 and YTHDF3. All five of these proteins contain a YTH domain, which directly binds m6A residues, and all five m6A readers appear to bind to all m6A sites equivalently. Two m6A demethylases (also known as ‘erasers’), called ALKBH5 and FTO, have also been reported, and it has been proposed that mRNA modification by m6A may be dynamic and reversible, though this claim remains controversial85,86.
Although the presence of m6A on viral transcripts was reported as long ago as the 1970s87,88, analysis of the effect of m6A residues on viral gene expression and replication only recently became technically feasible, with the development of techniques that allow epitranscriptomic modifications, including m6A, to be mapped to specific sites on RNAs83 (Box 2). Initially, we and others reported that m6A promotes HIV-1 gene expression and replication; knockdown of either METTL3 or the most highly expressed m6A reader, YTHDF2, inhibited HIV-1 gene expression, whereas knockdown of the ALKBH5 eraser or YTHDF2 overexpression promoted HIV-1 gene expression89,90. Subsequent studies from our laboratory and others — using a combination of gene knockdown, knockout and overexpression strategies — demonstrated that m6A residues also promote the replication of a range of other viruses, including influenza A virus (IAV), the picornavirus enterovirus 71, respiratory syncytial virus (RSV), human metapneumovirus (HMPV) and the polyomavirus SV40 (refs91–95). In the case of IAV, HMPV and RSV, viral variants carrying silent mutations that eliminated a subset of the mapped m6A sites but that left the underlying open reading frames unaffected were found to be not only attenuated in culture but also substantially reduced in their pathogenic potential in rodents, suggesting that the elimination of m6A residues by mutagenesis might represent a novel strategy for the development of attenuated viruses that could potentially serve as vaccines93. Although the addition of m6A residues to viral RNAs clearly increases their expression89,91, it has recently been reported that m6A addition to transcripts encoded by human HMPV also enables these viral RNAs to escape recognition by the host innate immune RNA sensor RIG-I, thus avoiding the host antiviral response and promoting virus replication95.
Although the majority of studies have concluded that m6A promotes viral gene expression, some exceptions have been reported. For example, in the case of KSHV, one group has reported that the addition of m6A to viral transcripts promotes lytic replication96, and another group has presented data arguing for the opposite conclusion: m6A can suppress lytic replication97. Importantly, a third group has reported that m6A can both promote and inhibit KSHV lytic replication, depending on the cell type being studied98. Thus it appears that, in the context of the complex, temporally regulated replication cycles characteristic of large DNA viruses such as KSHV, m6A may exert different phenotypic effects depending on the cellular context. In addition, several flaviviruses associated with acute human infections — including Zika virus, dengue virus, yellow fever virus and West Nile virus — have been reported to contain conserved clusters of m6A residues, yet it was also reported that knockdown of the m6A writer METTL3 increased Zika virus replication, while knockdown of the m6A eraser ALKBH5 or FTO exerted an inhibitory effect on viral replication99,100. This result, which is opposite to what was seen with the other RNA viruses discussed above, is difficult to understand, as it is not apparent why m6A sites would be evolutionarily conserved across several different flavivirus RNA genomes if they act in cis to inhibit viral gene expression.
Recently, it was reported that the human m6A reader YTHDF3 can inhibit HIV-1 replication, though the reported effect — a less than twofold inhibition in wild-type A3R5 T cells, when compared to YTHDF3 knockout cells over an ~4-day infection period — was very modest101. YTHDF3 was reported to be packaged into HIV-1 virions and to then reduce reverse transcription by ~30%101. The authors also reported that virion-associated YTHDF3 was efficiently degraded by the HIV-1 protease, which they propose serves as a viral countermeasure. By contrast, we previously reported that YTHDF2 overexpression in T cells increased HIV-1 replication, whereas YTHDF2 knockout reduced HIV-1 replication89. One possibility that was not considered is that YTHDF3 may act by competing with YTHDF2 for binding to m6A sites on HIV-1 RNA, thus reducing the positive effect on viral gene expression exerted by YTHDF2.
Box 2 Techniques used to map epitranscriptomic modifications.
Although high-performance liquid chromatography linked to tandem mass spectrometry (HPLC–MS/MS) can identify and precisely quantify RNA modifications, these methods do not provide location information. Methods to map the location of modifications usually involve RNA deep sequencing, which can be roughly separated into antibody-dependent methods, modification-interacting protein pulldowns and chemical methods. The figure depicts the core mechanisms of modification identification used in various mapping techniques, with immunoprecipitation-based techniques on the left and chemical methods on the right. The simplest method used to map N6-methyladenosine (m6A) sites on mRNA is m6A-seq (also known as methylated RNA immunoprecipitation sequencing (meRIP-seq)), in which RNA is typically fragmented by alkaline hydrolysis and then incubated with an m6A-specific antibody. The resulting RNA–antibody complexes are then recovered and sequenced132,133. However, the resolution of this mapping technique is limited by the average RNA fragment size, which is generally around 125 nucleotides, resulting in fairly broad peaks (that is, it identifies broad regions that contain one or more m6A residues). One variation of this method, called m6A individual-nucleotide-resolution crosslinking and immunoprecipitation (miCLIP), uses ultraviolet (UV) light crosslinking (wavelength of 254 nm) of the antibody to the motif characteristic of m6A modification sites (minimally 5ʹ-RAC-3ʹ, with R representing G or A), resulting in a cytidine-to-thymidine conversion of the cytidine immediately 3ʹ to the m6A modification in the reverse-transcribed cDNA library, allowing single-nucleotide resolution mapping134. Another variation is photo-crosslinking-assisted m6A-seq (PA-m6A-seq), in which cells are first pulsed with the highly photoactive uridine analogue 4-thiouridine (4SU). The 4SU-containing RNA is then isolated and UV-light-crosslinked (wavelength of 365 nm) in solution to an m6A antibody135. Because this method utilizes RNase to degrade any RNA not protected by the bound antibody, recovered bound RNA fragments reflect the sequence protected by the antibody, which is ~32 nucleotides. In addition, UV-light crosslinking of proteins to 4SU results in a thymidine-to-cytidine conversion during reverse transcription, which allows the removal of all background reads during final data analysis136. Antibody-based methods can easily be adapted to mapping different modifications, as we have previously demonstrated by performing PA-m6A-seq with m5C and ac4C antibodies80,106, whereas m6A-seq has also been adapted to ac4C80,104. Adapting miCLIP to other antibodies would require additional testing to discover the preferred mutation induced by UV-light crosslinking of the specific antibody.
Antibody-mapping results can be corroborated using the photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) technique136 to map the binding sites for modification-specific readers in living cells, if known. For example, the m6A reader YTHDF2 can be utilized in this way89. In a method for mapping 5-methylcytidine (m5C), confusingly also named miCLIP, mutation of the m5C writer NSUN2 at a key cysteine residue (C271A) causes NSUN2 to covalently crosslink to the target cytidine residue on the substrate RNA; thus, m5C modifications could be identified by immunoprecipitation of NSUN2–C271A-bound RNAs followed by RNase treatment, RNA fragment recovery and deep sequencing137.
For sites of 2′O-methylation (Nm), no antibody is currently available, and mapping thus depends on chemical methods. RiboMethSeq exploits the fact that a methylation at the 2′O position can block alkaline hydrolysis at that position138,139. In 2′OMe-Seq, reverse transcription stops at an Nm site under low-dNTP conditions140. Nm-seq and RiboOxiSeq utilize the resistance of 3′-end Nm to periodate cleavage (IO4–)141,142. All three of these chemical Nm-mapping methods provide single-nucleotide resolution. However, because RiboMethSeq and 2′OMe-Seq depend on the absence of sequencing reads ending at the modification site, they require very high sequencing read depths. We note that the oxidation and elimination processes of Nm-seq and RiboOxiSeq lead to the loss of most of the RNA sample. Thus, all current Nm methods require large amounts of input RNA. Bisulfite sequencing has been used to map m5C residues on RNAs, exploiting the fact that m5C residues are resistant to the cytidine-to-uridine conversion induced by bisulfite treatment61. However, we have observed that cytidine residues located in RNA stems are also resistant to bisulfite treatment, potentially resulting in their incorrect identification as m5C residues.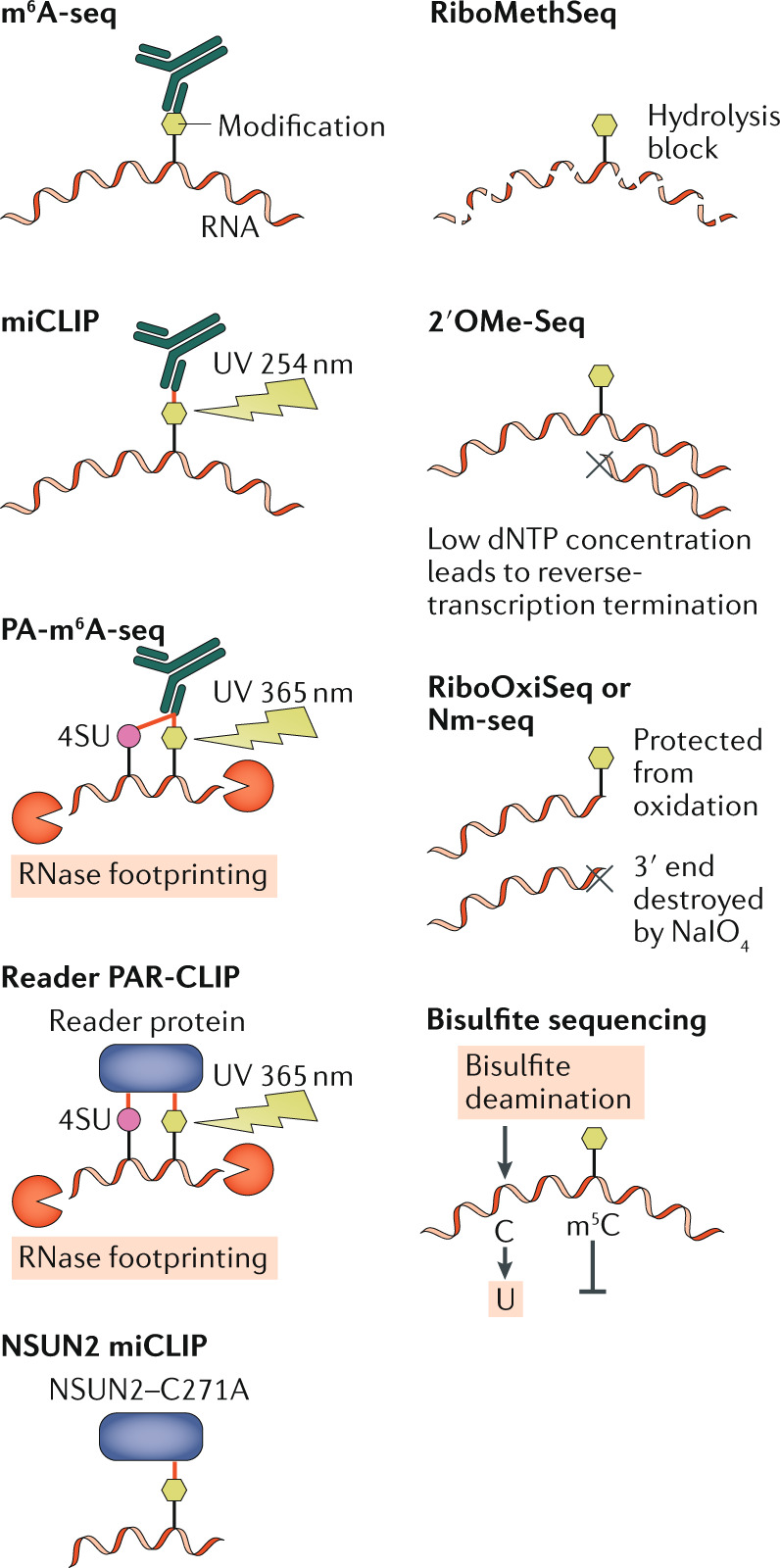
5-methylcytidine
Another relatively common epitranscriptomic mRNA modification is m5C, which represents ~0.05–0.1% of all cytidine residues found in cellular mRNAs, but up to ~1.4% of the cytidine residues in retroviral transcripts79,80,83. Although cells encode at least eight cytidine methyltransferases that act on RNA, all but one of which belong to the NSUN family of proteins, current data indicate that NSUN2 is the nuclear writer responsible for the large majority of, but not all, m5C residues added to mRNAs, including viral mRNAs80,102 (Table 1, Fig. 3). At present, no m5C readers are known. Analysis of the effect of loss of NSUN2 expression, and hence loss of m5C addition to mRNAs, in HIV-1-infected T cells revealed a specific loss of translation efficiency for HIV-1 and for cellular transcripts that are normally highly m5C-modified80. By contrast, cellular mRNAs that normally lack m5C, including several housekeeping genes, were unaffected by the loss of NSUN2 expression. Therefore, m5C seems to increase viral gene expression and replication by acting predominantly at the level of mRNA translation.
N4-acetylcytidine
Recently, ac4C residues were detected on cellular mRNAs at a level of ~1 ac4C residue per mRNA, and the ac4C writer was identified as nuclear N-acetyltransferase 10 (NAT10)103,104 (Fig. 3). However, no ac4C readers are currently known (Table 1). In the case of cellular mRNAs, ac4C was reported to enhance both the stability and the translation of cellular mRNAs, in the latter case by acting in open reading frames to improve ribosomal decoding, especially when ac4C was present in the wobble position of codons104. This may relate to the fact that ac4C can form more stable base pairs with guanosine residues105. Recently, we reported the mapping of ac4C residues in HIV-1 transcripts (Box 2) and reported that the loss of NAT10 expression results in a decline in HIV-1 gene expression in infected T cells106. However, unlike m5C, ac4C was found to increase HIV-1 gene expression by enhancing RNA stability, while viral mRNA translation was unaffected. In confirmation of this result, we also observed that the mutagenesis of ac4C clusters in the env gene of HIV-1, which forms part of the 3ʹ untranslated region of the viral gag mRNA, nevertheless reduced Gag mRNA and protein levels equivalently. Thus, in addition to RNA methylation, acetylations in the form of ac4C can also be utilized to enhance viral gene expression, through the stabilization of viral RNA transcripts.
2′O-methylation
The fourth and final internal epitranscriptomic modification that has, so far, been reported to affect viral replication is 2ʹO-methylation of the ribose moiety of all four ribonucleosides (Am, Cm, Gm and Um, collectively known as Nm). Each of the four Nm residues represents ~0.1% of the level of the relevant nucleoside found in cellular mRNAs, yet this level was found to be up to 20 times higher when HIV-1 or MLV genomic RNAs were analysed79,80. The Nm writer that acts on retroviral transcripts has been identified as the nucleolar protein FTSJ3 (ref.107), which was previously shown to function in pre-rRNA processing108 (Fig. 3). We note that FTSJ3 was reported to be incapable of adding 2′O-methyl groups to cytidine residues107, which appears inconsistent with the high levels of Cm detected on HIV-1 (1.02%) and MLV (0.74%) genomic RNAs79,80. Moreover, preliminary data suggest that the yeast FTSJ3 homologue (Spb1) is able to methylate cytidine residues109. Only one report has so far examined the phenotypic effect of Nm residues on HIV-1 replication, and these researchers did not report any effect of Nm residues on HIV-1 gene expression. Instead, they found that HIV-1 virions produced in cells in which FTSJ3 was knocked down by RNA interference were potent activators of the cytoplasmic viral RNA sensor MDA5, a key component of the host antiviral immune response, when the virions were used to infect dendritic cells107. Others have also reported that specific epitranscriptomic RNA modifications, including not only Nm but also pseudouridine, can attenuate cellular innate immune responses to transfected mRNA molecules110–113, whereas m6A was recently reported to prevent the activation of a second cytoplasmic RNA sensor, RIG-I95. Clearly, it will be important to examine whether other viruses also use epitranscriptomic RNA modifications, including but not limited to m6A and Nm, to escape viral RNA detection by host innate immunity factors.
In conclusion, the current evidence suggests that several different epitranscriptomic RNA modifications are able to promote viral replication, either directly, by increasing viral mRNA stability or translation, or indirectly, by allowing viruses to elude the recognition of their transcripts as foreign by cytoplasmic viral RNA sensors. It is therefore not surprising that viruses appear to have evolved so as to maximize the level of several epitranscriptomic modifications that are added to their mRNAs. By contrast, mammalian cells have evolved the capacity to recognize viral DNA as foreign and seek to silence that DNA epigenetically as one form of innate immune response. Viruses, in turn, have evolved mechanisms that allow them to avoid epigenetic silencing, either by the targeted destruction of relevant cellular factors, such as the components of PML-NBs, or by hiding in host-cell chromosomal DNA, as is seen for retroviruses.
Future perspectives
In this Review, we have argued that cells use epigenetic gene regulation as a potential mechanism to silence incoming viral DNA molecules, whereas viruses have evolved to recruit the cellular epitranscriptomic modification factors in order to heavily modify viral transcripts, as a means to boost viral gene expression and/or replication. Although the field of epigenetic gene regulation is fairly mature, the question of how cells distinguish between viral DNA and host-cell DNA remains unresolved. In particular, how unintegrated HIV-1 proviral DNA is recognized and silenced remains unknown. By contrast, the field of epitranscriptomic gene regulation is still in its infancy. For example, how writers select specific bases for modification remains largely unknown, and even in the case of m6A — for which the consensus editing sequence 5ʹ-RAC-3ʹ has been defined, where R is either G or A3,4 — only a minority of sites with that sequence are, in fact, modified. Moreover, although five m6A readers have been identified, the readers for all other RNA modifications remain unknown, and even for m6A, how the readers exert their phenotypic effects remains largely undefined. It will be important to understand why m6A clearly promotes viral replication in most published studies, yet has also been suggested in other reports to inhibit viral replication, as discussed above.
Both epigenetic and epitranscriptomic regulatory pathways could in principle be targeted for antiviral drug development. In the case of epigenetic viral gene regulation, two strategies have emerged. In an approach termed ‘shock-and-kill’, drugs that promote the formation of active chromatin, such as histone deacetylase (HDAC) inhibitors, have been proposed as tools to activate latent viruses, including EBV114–116 and HIV-1 (ref.117). In the case of HIV-1, this strategy envisions using these drugs to reactivate latent proviruses, in individuals who are also on antiretrovirals, as a means of selectively eliminating latently infected cells. However, this strategy has yet to prove clinically useful. An alternative approach envisions the use of drugs that instead keep viral DNA epigenetically suppressed, including drugs that inhibit the H3K9 demethylases LSD1 and JMJD2, which are recruited by HCF-1 to HSV-1 viral DNA in order to remove repressive marks. Inhibitors of LSD1 and JMJD2 have indeed been shown to suppress HSV-1 gene expression, replication and reactivation from latency, both in vitro and in vivo43,118,119. These drugs have also proved effective against other HCF-1-dependent herpesviruses, including HCMV and varicella zoster virus (VZV), and they might prove useful in the treatment of pathologies that result from the reactivation of latent herpesviruses, such as shingles. Although in principle LSD1 and JMJD2 inhibitors could be used to entirely repress the reactivation of latent DNA viruses, these drugs would then need to be taken long-term, and it seems unlikely that inhibition of host H3K9 demethylases for months or even years would be well-tolerated.
Inhibitors that block the epitranscriptomic modification of viral RNAs clearly would be potentially even more interesting, as this should inhibit viral gene expression and/or promote antiviral immune responses. These drugs would presumably be targeted to the cellular writers that add epitranscriptomic marks to mRNAs (Table 1), and the emergence of drug-resistant viral mutants would therefore seem to be unlikely. Conversely, as such drugs would also prevent the epitranscriptomic modification of cellular mRNAs, toxicity might be an issue, and we therefore envision that such drugs would be used only briefly, during the 5–7-day acute phase of infection by viruses such as IAV, RSV and possibly severe acute respiratory syndrome coronavirus 2, in order to reduce the peak viral load and limit viral pathogenicity until the adaptive immune system becomes effective.
Because the addition of m6A to mRNAs has been implicated as a driver in some forms of cancer120–122, several biotech companies are already attempting to identify effective inhibitors of METTL3 function, and it would clearly be of interest to test these, once they have been identified, in animal models that support pathogenic infections by human viruses. In the interim, some data in fact already suggest that drugs that inhibit mRNA methylation could prove to be effective pan-viral inhibitors. Specifically, several drugs, including 3-deaza-adenosine (DAA), are known to act as inhibitors of S-adenosylhomocysteine (SAH) hydrolase and, as a result, to deplete cells of SAM, the methyl donor used not only by METTL3 but also by NSUN2, and likely also by the enzymes that add Nm residues to mRNAs. Importantly, DAA was shown to inhibit m6A addition to mRNAs, while the formation of 7-methylguanosine, which forms part of the mRNA 5ʹ cap, was largely unaffected123. DAA acts as a potent pan-viral inhibitor in culture and is able to effectively inhibit a wide range of DNA and RNA viruses at doses that are >100-fold lower than the level found to exert a toxic effect on cultured primary cells124. In vivo, DAA is also a remarkably effective broad-spectrum antiviral. For example, a single dose of DAA given one or two days after infection reduced peak viraemia in Ebolavirus-infected mice by >1,000 fold and resulted in the survival of almost all the treated animals, whereas untreated animals showed a 0% survival rate125. Similarly, DAA drastically reduced viral titers in cotton rats infected with RSV at doses of DAA that were not detectably toxic126. Although DAA may not itself be a potentially useful drug, these studies do make the point that the epitranscriptomic modification of viral mRNAs may represent a potential viral Achilles heel, and that the identification of inhibitors of this process could therefore lead to the development of a novel class of potent, broad-spectrum antivirals.
Acknowledgements
This research was funded in part by NIH grants R01-DA046111 and U54-AI150470 to B.R.C., and by a Duke University Center for AIDS Research (CFAR, P30-AI064518) pilot award to K.T.
Glossary
- Chromatin
A DNA–protein complex consisting of genomic DNA wrapped around organizing proteins called histones (see Box 1).
- Methylation
The addition of a small chemical group called a methyl group (CH3), found as a chemical modification on DNA, RNA and proteins (in particular, histones).
- Latent infections
A viral life-cycle stage in which the viral genomic material persists long-term in the host cell with minimal viral gene expression and replication.
- Acetylation
The addition of a small chemical group called an acetyl group (CH3CO), found on RNA and proteins including histones.
- Histones
A family of highly basic proteins used to organize genomic DNA (see Box 1).
- Heterochromatic marks
Epigenetic markers that mark a segment of chromatin for transcriptional repression, which usually coincide with ‘closed chromatin’ that is less accessible to transcription factors.
- Immunofluorescence microscopy
Microscopy assays in which subjects of interest are visualized though specific binding of antibodies labelled with fluorescent dyes.
- Interferon
A family of proteins that are expressed by eukaryotic cells upon invasion by viruses, used to signal neighbouring cells to mount an antiviral response through the expression of a variety of interferon-stimulated genes.
- Tegument
The space in viral particles between the outer membrane and the inner protein capsid shell; the term is most commonly used in the herpesvirus family, where proteins packaged in this space are termed tegument proteins.
- Ubiquitin
A small peptide that can be ligated onto other proteins, best known to mark proteins for degradation through the proteasome.
- Covalently closed circular DNA
The HBV genomic DNA is packaged in the viral particle as a gapped, partially double-stranded DNA with open DNA ends. The ends are closed up upon infection of a host cell, resulting in a fully double-stranded circular DNA genome, termed the covalently closed circular DNA, which acts as a functional template for gene expression and replication.
- Integrase
An essential retrovirus enzyme that catalyses the integration of reverse-transcribed viral DNA into the host genomic DNA.
- Lytic replication
A viral life-cycle stage in which viruses undergo active replication in the host cell, in most cases resulting in the lysis of the host cell.
- tRNAs
Transfer RNAs; a short (76–90 nt), highly structured family of RNA that, during protein translation, recognize the coding sequence on the template mRNA in order to bring in the appropriate amino acid.
- Non-coding RNAs
Cellular RNAs that do not encode proteins.
- Nucleolar protein
A protein that normally localizes to the subnuclear structure called the nucleolus, which is where the RNA components of the ribosome are produced.
Author contributions
The authors contributed equally to all aspects of the article.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holtzman L, Gersbach CA. Editing the epigenome: reshaping the genomic landscape. Annu. Rev. Genomics Hum. Genet. 2018;19:43–71. doi: 10.1146/annurev-genom-083117-021632. [DOI] [PubMed] [Google Scholar]
- 3.Li S, Mason CE. The pivotal regulatory landscape of RNA modifications. Annu. Rev. Genomics Hum. Genet. 2014;15:127–150. doi: 10.1146/annurev-genom-090413-025405. [DOI] [PubMed] [Google Scholar]
- 4.Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169:1187–1200. doi: 10.1016/j.cell.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J, Harada BT, He C. Regulation of gene expression by N6-methyladenosine in cancer. Trends Cell Biol. 2019;29:487–499. doi: 10.1016/j.tcb.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delaunay S, Frye M. RNA modifications regulating cell fate in cancer. Nat. Cell Biol. 2019;21:552–559. doi: 10.1038/s41556-019-0319-0. [DOI] [PubMed] [Google Scholar]
- 7.Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: moving forward. PLoS Genet. 2018;14:e1007362. doi: 10.1371/journal.pgen.1007362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knipe DM. Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity. Virology. 2015;479–480:153–159. doi: 10.1016/j.virol.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knipe DM, Raja P, Lee J. Viral gene products actively promote latent infection by epigenetic silencing mechanisms. Curr. Opin. Virol. 2017;23:68–74. doi: 10.1016/j.coviro.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diner BA, Lum KK, Toettcher JE, Cristea IM. Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. mBio. 2016;7:e01553-16. doi: 10.1128/mBio.01553-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orzalli MH, Conwell SE, Berrios C, DeCaprio JA, Knipe DM. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc. Natl Acad. Sci. USA. 2013;110:E4492–E4501. doi: 10.1073/pnas.1316194110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boutell C, Everett RD. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J. Gen. Virol. 2013;94:465–481. doi: 10.1099/vir.0.048900-0. [DOI] [PubMed] [Google Scholar]
- 13.Everett RD, Chelbi-Alix MK. PML and PML nuclear bodies: implications in antiviral defence. Biochimie. 2007;89:819–830. doi: 10.1016/j.biochi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 14.Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007;8:1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- 15.Luciani JJ, et al. PML nuclear bodies are highly organised DNA-protein structures with a function in heterochromatin remodelling at the G2 phase. J. Cell Sci. 2006;119:2518–2531. doi: 10.1242/jcs.02965. [DOI] [PubMed] [Google Scholar]
- 16.Maul GG. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays. 1998;20:660–667. doi: 10.1002/(SICI)1521-1878(199808)20:8<660::AID-BIES9>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 17.Guldner HH, Szostecki C, Grotzinger T, Will H. IFN enhance expression of Sp100, an autoantigen in primary biliary cirrhosis. J. Immunol. 1992;149:4067–4073. [PubMed] [Google Scholar]
- 18.Lavau C, et al. The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene. 1995;11:871–876. [PubMed] [Google Scholar]
- 19.Stadler M, et al. Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene. 1995;11:2565–2573. [PubMed] [Google Scholar]
- 20.Tsai K, Messick TE, Lieberman PM. Disruption of host antiviral resistances by gammaherpesvirus tegument proteins with homology to the FGARAT purine biosynthesis enzyme. Curr. Opin. Virol. 2015;14:30–40. doi: 10.1016/j.coviro.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isaac A, Wilcox KW, Taylor JL. SP100B, a repressor of gene expression preferentially binds to DNA with unmethylated CpGs. J. Cell Biochem. 2006;98:1106–1122. doi: 10.1002/jcb.20841. [DOI] [PubMed] [Google Scholar]
- 22.Seeler JS, Marchio A, Sitterlin D, Transy C, Dejean A. Interaction of SP100 with HP1 proteins: a link between the promyelocytic leukemia-associated nuclear bodies and the chromatin compartment. Proc. Natl Acad. Sci. USA. 1998;95:7316–7321. doi: 10.1073/pnas.95.13.7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michaelson JS, Leder P. RNAi reveals anti-apoptotic and transcriptionally repressive activities of DAXX. J. Cell Sci. 2003;116:345–352. doi: 10.1242/jcs.00234. [DOI] [PubMed] [Google Scholar]
- 24.Hollenbach AD, McPherson CJ, Mientjes EJ, Iyengar R, Grosveld G. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J. Cell Sci. 2002;115:3319–3330. doi: 10.1242/jcs.115.16.3319. [DOI] [PubMed] [Google Scholar]
- 25.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl Acad. Sci. USA. 2010;107:14075–14080. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue Y, et al. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc. Natl Acad. Sci. USA. 2003;100:10635–10640. doi: 10.1073/pnas.1937626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stratmann SA, Morrone SR, van Oijen AM, Sohn J. The innate immune sensor IFI16 recognizes foreign DNA in the nucleus by scanning along the duplex. eLife. 2015;4:e11721. doi: 10.7554/eLife.11721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Unterholzner L, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson KE, et al. IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog. 2014;10:e1004503. doi: 10.1371/journal.ppat.1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gariano GR, et al. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog. 2012;8:e1002498. doi: 10.1371/journal.ppat.1002498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lo Cigno I, et al. The nuclear DNA sensor IFI16 acts as a restriction factor for human papillomavirus replication through epigenetic modifications of the viral promoters. J. Virol. 2015;89:7506–7520. doi: 10.1128/JVI.00013-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Y, et al. Nuclear sensor interferon-inducible protein 16 inhibits the function of hepatitis B virus covalently closed circular DNA by integrating innate immune activation and epigenetic suppression. Hepatology. 2020;71:1154–1169. doi: 10.1002/hep.30897. [DOI] [PubMed] [Google Scholar]
- 33.Roy A, Ghosh A, Kumar B, Chandran B. IFI16, a nuclear innate immune DNA sensor, mediates epigenetic silencing of herpesvirus genomes by its association with H3K9 methyltransferases SUV39H1 and GLP. eLife. 2019;8:e49500. doi: 10.7554/eLife.49500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Diner BA, Lum KK, Javitt A, Cristea IM. Interactions of the antiviral factor interferon gamma-inducible protein 16 (IFI16) mediate immune signaling and herpes simplex virus-1 immunosuppression. Mol. Cell Proteom. 2015;14:2341–2356. doi: 10.1074/mcp.M114.047068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bechtel JT, Winant RC, Ganem D. Host and viral proteins in the virion of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005;79:4952–4964. doi: 10.1128/JVI.79.8.4952-4964.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johannsen E, et al. Proteins of purified Epstein–Barr virus. Proc. Natl Acad. Sci. USA. 2004;101:16286–16291. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varnum SM, et al. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 2004;78:10960–10966. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cliffe AR, Knipe DM. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 2008;82:12030–12038. doi: 10.1128/JVI.01575-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen C, et al. Promyelocytic leukemia (PML) nuclear bodies (NBs) induce latent/quiescent HSV-1 genomes chromatinization through a PML NB/histone H3.3/H3.3 chaperone axis. PLoS Pathog. 2018;14:e1007313. doi: 10.1371/journal.ppat.1007313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oh J, Fraser NW. Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J. Virol. 2008;82:3530–3537. doi: 10.1128/JVI.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Placek BJ, et al. The histone variant H3.3 regulates gene expression during lytic infection with herpes simplex virus type 1. J. Virol. 2009;83:1416–1421. doi: 10.1128/JVI.01276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 2009;15:1312–1317. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang Y, et al. Targeting the JMJD2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency. Sci. Transl. Med. 2013;5:167ra5. doi: 10.1126/scitranslmed.3005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narayanan A, Ruyechan WT, Kristie TM. The coactivator host cell factor-1 mediates Set1 and MLL1 H3K4 trimethylation at herpesvirus immediate early promoters for initiation of infection. Proc. Natl Acad. Sci. USA. 2007;104:10835–10840. doi: 10.1073/pnas.0704351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JS, Raja P, Knipe DM. Herpesviral ICP0 protein promotes two waves of heterochromatin removal on an early viral promoter during lytic infection. mBio. 2016;7:e02007–e02015. doi: 10.1128/mBio.02007-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orzalli MH, Broekema NM, Knipe DM. Relative contributions of herpes simplex virus 1 ICP0 and vhs to loss of cellular IFI16 vary in different human cell types. J. Virol. 2016;90:8351–8359. doi: 10.1128/JVI.00939-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diner BA, Lum KK, Cristea IM. The emerging role of nuclear viral DNA sensors. J. Biol. Chem. 2015;290:26412–26421. doi: 10.1074/jbc.R115.652289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cantrell SR, Bresnahan WA. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 2006;80:6188–6191. doi: 10.1128/JVI.02676-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lukashchuk V, McFarlane S, Everett RD, Preston CM. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 2008;82:12543–12554. doi: 10.1128/JVI.01215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saffert RT, Kalejta RF. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate–early gene expression. J. Virol. 2006;80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsai K, Thikmyanova N, Wojcechowskyj JA, Delecluse HJ, Lieberman PM. EBV tegument protein BNRF1 disrupts DAXX–ATRX to activate viral early gene transcription. PLoS Pathog. 2011;7:e1002376. doi: 10.1371/journal.ppat.1002376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang H, et al. Structural basis underlying viral hijacking of a histone chaperone complex. Nat. Commun. 2016;7:12707. doi: 10.1038/ncomms12707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsai K, et al. Viral reprogramming of the Daxx histone H3.3 chaperone during early Epstein–Barr virus infection. J. Virol. 2014;88:14350–14363. doi: 10.1128/JVI.01895-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cristea IM, et al. Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J. Virol. 2010;84:7803–7814. doi: 10.1128/JVI.00139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li T, Chen J, Cristea IM. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe. 2013;14:591–599. doi: 10.1016/j.chom.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dell’Oste V, et al. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J. Virol. 2014;88:6970–6982. doi: 10.1128/JVI.00384-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hong X, Kim ES, Guo H. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: implications for epigenetic therapy against chronic hepatitis B. Hepatology. 2017;66:2066–2077. doi: 10.1002/hep.29479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murphy CM, et al. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep. 2016;16:2846–2854. doi: 10.1016/j.celrep.2016.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niu C, et al. The Smc5/6 complex restricts HBV when localized to ND10 without inducing an innate immune response and is counteracted by the HBV X protein shortly after infection. PLoS One. 2017;12:e0169648. doi: 10.1371/journal.pone.0169648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Decorsiere A, et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. 2016;531:386–389. doi: 10.1038/nature17170. [DOI] [PubMed] [Google Scholar]
- 61.Sakai H, et al. Integration is essential for efficient gene expression of human immunodeficiency virus type 1. J. Virol. 1993;67:1169–1174. doi: 10.1128/jvi.67.3.1169-1174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schwartzberg P, Colicelli J, Goff SP. Construction and analysis of deletion mutations in the pol gene of Moloney murine leukemia virus: a new viral function required for productive infection. Cell. 1984;37:1043–1052. doi: 10.1016/0092-8674(84)90439-2. [DOI] [PubMed] [Google Scholar]
- 63.Geis FK, Goff SP. Unintegrated HIV-1 DNAs are loaded with core and linker histones and transcriptionally silenced. Proc. Natl Acad. Sci. USA. 2019;116:23735–23742. doi: 10.1073/pnas.1912638116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang GZ, Wang Y, Goff SP. Histones are rapidly loaded onto unintegrated retroviral DNAs soon after nuclear entry. Cell Host Microbe. 2016;20:798–809. doi: 10.1016/j.chom.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu Y, Wang GZ, Cingoz O, Goff SP. NP220 mediates silencing of unintegrated retroviral DNA. Nature. 2018;564:278–282. doi: 10.1038/s41586-018-0750-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chougui G, Margottin-Goguet F. HUSH, a link between intrinsic immunity and HIV latency. Front. Microbiol. 2019;10:224. doi: 10.3389/fmicb.2019.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schroder AR, et al. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 68.Lusic M, Siliciano RF. Nuclear landscape of HIV-1 infection and integration. Nat. Rev. Microbiol. 2017;15:69–82. doi: 10.1038/nrmicro.2016.162. [DOI] [PubMed] [Google Scholar]
- 69.Irwan ID, Karnowski HL, Bogerd HP, Tsai K, Cullen BR. Reversal of epigenetic silencing allows robust HIV-1 replication in the absence of integrase function. mBio. 2020;11:e01038-20. doi: 10.1128/mBio.01038-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lieberman PM. Epigenetics and genetics of viral latency. Cell Host Microbe. 2016;19:619–628. doi: 10.1016/j.chom.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kristie TM, Vogel JL, Sears AE. Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc. Natl Acad. Sci. USA. 1999;96:1229–1233. doi: 10.1073/pnas.96.4.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tempera I, Wiedmer A, Dheekollu J, Lieberman PM. CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog. 2010;6:e1001048. doi: 10.1371/journal.ppat.1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Arvey A, et al. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe. 2012;12:233–245. doi: 10.1016/j.chom.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pich D, et al. First days in the life of naive human B lymphocytes infected with Epstein–Barr virus. mBio. 2019;10:e01723-19. doi: 10.1128/mBio.01723-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tempera I, Lieberman PM. Epigenetic regulation of EBV persistence and oncogenesis. Semin. Cancer Biol. 2014;26:22–29. doi: 10.1016/j.semcancer.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Everett RD. Dynamic response of IFI16 and promyelocytic leukemia nuclear body components to Herpes simplex virus 1 infection. J. Virol. 2016;90:167–179. doi: 10.1128/JVI.02249-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Merkl PE, Orzalli MH, Knipe DM. Mechanisms of Host IFI16, PML, and Daxx protein restriction of herpes simplex virus 1 replication. J Virol. 2018;92:e00057-18. doi: 10.1128/JVI.00057-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cuchet-Lourenco D, Anderson G, Sloan E, Orr A, Everett RD. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J. Virol. 2013;87:13422–13432. doi: 10.1128/JVI.02474-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Courtney DG, et al. Extensive epitranscriptomic methylation of A and C residues on murine leukemia virus transcripts enhances viral gene expression. mBio. 2019;10:e01209-19. doi: 10.1128/mBio.01209-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Courtney DG, et al. Epitranscriptomic Addition of m5C to HIV-1 transcripts regulates viral gene expression. Cell Host Microbe. 2019;26:217–227. doi: 10.1016/j.chom.2019.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McIntyre W, et al. Positive-sense RNA viruses reveal the complexity and dynamics of the cellular and viral epitranscriptomes during infection. Nucleic Acids Res. 2018;46:5776–5791. doi: 10.1093/nar/gky029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014;15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li X, Xiong X, Yi C. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat. Methods. 2016;14:23–31. doi: 10.1038/nmeth.4110. [DOI] [PubMed] [Google Scholar]
- 84.Ke S, et al. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017;31:990–1006. doi: 10.1101/gad.301036.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhao BS, Nachtergaele S, Roundtree IA, He C. Our views of dynamic N6-methyladenosine RNA methylation. RNA. 2018;24:268–272. doi: 10.1261/rna.064295.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Darnell RB, Ke S, Darnell JE., Jr Pre-mRNA processing includes N6 methylation of adenosine residues that are retained in mRNA exons and the fallacy of ‘RNA epigenetics’. RNA. 2018;24:262–267. doi: 10.1261/rna.065219.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Krug RM, Morgan MA, Shatkin AJ. Influenza viral mRNA contains internal N6-methyladenosine and 5′-terminal 7-methylguanosine in cap structures. J. Virol. 1976;20:45–53. doi: 10.1128/jvi.20.1.45-53.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lavi S, Shatkin AJ. Methylated simian virus 40-specific RNA from nuclei and cytoplasm of infected BSC-1 cells. Proc. Natl Acad. Sci. USA. 1975;72:2012–2016. doi: 10.1073/pnas.72.6.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kennedy EM, et al. Posttranscriptional m6A editing of HIV-1 mRNAs enhances viral gene expression. Cell Host Microbe. 2016;19:675–685. doi: 10.1016/j.chom.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lichinchi G, et al. Dynamics of the human and viral m6A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol. 2016;1:16011. doi: 10.1038/nmicrobiol.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Courtney DG, et al. Epitranscriptomic enhancement of influenza A virus gene expression and replication. Cell Host Microbe. 2017;22:377–386. doi: 10.1016/j.chom.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsai K, Courtney DG, Cullen BR. Addition of m6A to SV40 late mRNAs enhances viral structural gene expression and replication. PLoS Pathog. 2018;14:e1006919. doi: 10.1371/journal.ppat.1006919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xue M, et al. Viral N6-methyladenosine upregulates replication and pathogenesis of human respiratory syncytial virus. Nat. Commun. 2019;10:4595. doi: 10.1038/s41467-019-12504-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hao H, et al. N6-methyladenosine modification and METTL3 modulate enterovirus 71 replication. Nucleic Acids Res. 2019;47:362–374. doi: 10.1093/nar/gky1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lu M, et al. N6-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat. Microbiol. 2020;5:584–598. doi: 10.1038/s41564-019-0653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ye F, Chen ER, Nilsen TW. Kaposi’s sarcoma-associated herpesvirus utilizes and manipulates RNA N6-adenosine methylation to promote lytic replication. J. Virol. 2017;91:e00466-17. doi: 10.1128/JVI.00466-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tan B, et al. Viral and cellular N6-methyladenosine and N6,2′-O-dimethyladenosine epitranscriptomes in the KSHV life cycle. Nat. Microbiol. 2018;3:108–120. doi: 10.1038/s41564-017-0056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hesser CR, Karijolich J, Dominissini D, He C, Glaunsinger BA. N6-methyladenosine modification and the YTHDF2 reader protein play cell type specific roles in lytic viral gene expression during Kaposi’s sarcoma-associated herpesvirus infection. PLoS Pathog. 2018;14:e1006995. doi: 10.1371/journal.ppat.1006995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lichinchi G, et al. Dynamics of human and viral RNA methylation during Zika virus infection. Cell Host Microbe. 2016;20:666–673. doi: 10.1016/j.chom.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gokhale NS, et al. N6-methyladenosine in Flaviviridae viral RNA genomes regulates infection. Cell Host Microbe. 2016;20:654–665. doi: 10.1016/j.chom.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jurczyszak D, et al. HIV protease cleaves the antiviral m6A reader protein YTHDF3 in the viral particle. PLoS Pathog. 2020;16:e1008305. doi: 10.1371/journal.ppat.1008305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang X, et al. 5-methylcytosine promotes mRNA export—NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res. 2017;27:606–625. doi: 10.1038/cr.2017.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Castello A, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 104.Arango D, et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell. 2018;175:1872–1886. doi: 10.1016/j.cell.2018.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kamble AS, Kumbhar BV, Sambhare SB, Bavi RS, Sonawane KD. Conformational preferences of modified nucleoside 5-taurinomethyluridine, taum5U occur at ‘wobble’ 34th position in the anticodon loop of tRNA. Cell Biochem. Biophys. 2015;71:1589–1603. doi: 10.1007/s12013-014-0382-x. [DOI] [PubMed] [Google Scholar]
- 106.Tsai K, et al. Acetylation of cytidine residues boosts HIV-1 gene expression by increasing viral RNA stability. Cell Host. 2020 doi: 10.1016/j.chom.2020.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ringeard M, Marchand V, Decroly E, Motorin Y, Bennasser Y. FTSJ3 is an RNA 2′-O-methyltransferase recruited by HIV to avoid innate immune sensing. Nature. 2019;565:500–504. doi: 10.1038/s41586-018-0841-4. [DOI] [PubMed] [Google Scholar]
- 108.Morello LG, et al. The human nucleolar protein FTSJ3 associates with NIP7 and functions in pre-rRNA processing. PLoS One. 2011;6:e29174. doi: 10.1371/journal.pone.0029174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bartoli, K. M., Schaening, C., Carlile, T. M. & Gilbert, W. V. Conserved methyltransferase Spb1 targets mRNAs for regulated modification with 2′-O-methyl ribose. Preprint at https://www.biorxiv.org/content/10.1101/271916v2 (2018).
- 110.Robbins M, et al. 2′-O-methyl-modified RNAs act as TLR7 antagonists. Mol. Ther. 2007;15:1663–1669. doi: 10.1038/sj.mt.6300240. [DOI] [PubMed] [Google Scholar]
- 111.Kariko K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 112.Anderson BR, et al. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010;38:5884–5892. doi: 10.1093/nar/gkq347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Durbin AF, Wang C, Marcotrigiano J, Gehrke L. RNAs containing modified nucleotides fail to trigger RIG-I conformational changes for innate immune signaling. mBio. 2016;7:e00833–16. doi: 10.1128/mBio.00833-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Israel BF, Kenney SC. Virally targeted therapies for EBV-associated malignancies. Oncogene. 2003;22:5122–5130. doi: 10.1038/sj.onc.1206548. [DOI] [PubMed] [Google Scholar]
- 115.Mentzer SJ, Perrine SP, Faller DV. Epstein–Barr virus post-transplant lymphoproliferative disease and virus-specific therapy: pharmacological re-activation of viral target genes with arginine butyrate. Transpl. Infect. Dis. 2001;3:177–185. doi: 10.1034/j.1399-3062.2001.003003177.x. [DOI] [PubMed] [Google Scholar]
- 116.Archin NM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Archin NM, Margolis DM. Emerging strategies to deplete the HIV reservoir. Curr. Opin. Infect. Dis. 2014;27:29–35. doi: 10.1097/QCO.0000000000000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liang Y, et al. A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency. mBio. 2013;4:e00558-12. doi: 10.1128/mBio.00558-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hill JM, et al. Inhibition of LSD1 reduces herpesvirus infection, shedding, and recurrence by promoting epigenetic suppression of viral genomes. Sci. Transl. Med. 2014;6:265ra169. doi: 10.1126/scitranslmed.3010643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ianniello Z, Paiardini A, Fatica A. N6-methyladenosine (m6A): a promising new molecular target in acute myeloid leukemia. Front. Oncol. 2019;9:251. doi: 10.3389/fonc.2019.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lan Q, et al. The critical role of RNA m6A methylation in cancer. Cancer Res. 2019;79:1285–1292. doi: 10.1158/0008-5472.CAN-18-2965. [DOI] [PubMed] [Google Scholar]
- 122.Jaffrey SR, Kharas MG. Emerging links between m6A and misregulated mRNA methylation in cancer. Genome Med. 2017;9:2. doi: 10.1186/s13073-016-0395-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Backlund PS, Jr, Carotti D, Cantoni GL. Effects of the S-adenosylhomocysteine hydrolase inhibitors 3-deazaadenosine and 3-deazaaristeromycin on RNA methylation and synthesis. Eur. J. Biochem. 1986;160:245–251. doi: 10.1111/j.1432-1033.1986.tb09963.x. [DOI] [PubMed] [Google Scholar]
- 124.de Clercq E, Montgomery JA. Broad-spectrum antiviral activity of the carbocyclic analog of 3-deazaadenosine. Antivir. Res. 1983;3:17–24. doi: 10.1016/0166-3542(83)90011-6. [DOI] [PubMed] [Google Scholar]
- 125.Bray M, Driscoll J, Huggins JW. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-L-homocysteine hydrolase inhibitor. Antivir. Res. 2000;45:135–147. doi: 10.1016/s0166-3542(00)00066-8. [DOI] [PubMed] [Google Scholar]
- 126.Wyde PR, Ambrose MW, Meyer HL, Zolinski CL, Gilbert BE. Evaluation of the toxicity and antiviral activity of carbocyclic 3-deazaadenosine against respiratory syncytial and parainfluenza type 3 viruses in tissue culture and in cotton rats. Antivir. Res. 1990;14:215–225. doi: 10.1016/0166-3542(90)90003-p. [DOI] [PubMed] [Google Scholar]
- 127.Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 2014;157:95–109. doi: 10.1016/j.cell.2014.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. 2011;123:2145–2156. doi: 10.1161/CIRCULATIONAHA.110.956839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res. 2011;21:421–434. doi: 10.1038/cr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253–1265. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Goldberg AD, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 133.Meyer KD, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Linder B, et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods. 2015;12:767–772. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chen K, et al. High-resolution N6-methyladenosine (m6A) map using photo-crosslinking-assisted m6A sequencing. Angew. Chem. Int. Ed. Engl. 2015;54:1587–1590. doi: 10.1002/anie.201410647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hafner M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hussain S, et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 2013;4:255–261. doi: 10.1016/j.celrep.2013.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Marchand V, Blanloeil-Oillo F, Helm M, Motorin Y. Illumina-based RiboMethSeq approach for mapping of 2′-O-Me residues in RNA. Nucleic Acids Res. 2016;44:e135. doi: 10.1093/nar/gkw547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Birkedal U, et al. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew. Chem. Int. Ed. Engl. 2015;54:451–455. doi: 10.1002/anie.201408362. [DOI] [PubMed] [Google Scholar]
- 140.Incarnato D, et al. High-throughput single-base resolution mapping of RNA 2-O-methylated residues. Nucleic Acids Res. 2017;45:1433–1441. doi: 10.1093/nar/gkw810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dai Q, et al. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat. Methods. 2017;14:695–698. doi: 10.1038/nmeth.4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhu Y, Pirnie SP, Carmichael GG. High-throughput and site-specific identification of 2′-O-methylation sites using ribose oxidation sequencing (RibOxi-seq) RNA. 2017;23:1303–1314. doi: 10.1261/rna.061549.117. [DOI] [PMC free article] [PubMed] [Google Scholar]