

Abstract
Background
Acute myeloid leukemia (AML) is a hematopoietic malignancy which is biologically, phenotypically and genetically very heterogeneous. Outcome of patients with AML remains dismal, highlighting the need for improved, less toxic therapies. Chimeric antigen receptor T-cell (CART) immunotherapies for patients with refractory or relapse (R/R) AML are challenging because of the absence of a universal pan-AML target antigen and the shared expression of target antigens with normal hematopoietic stem/progenitor cells (HSPCs), which may lead to life-threating on-target/off-tumor cytotoxicity. CD33-redirected and CD123-redirected CARTs for AML are in advanced preclinical and clinical development, and they exhibit robust antileukemic activity. However, preclinical and clinical controversy exists on whether such CARTs are myeloablative.
Methods
We set out to comparatively characterize in vitro and in vivo the efficacy and safety of 41BB-based and CD28-based CARCD123. We analyzed 97 diagnostic and relapse AML primary samples to investigate whether CD123 is a suitable immunotherapeutic target, and we used several xenograft models and in vitro assays to assess the myeloablative potential of our second-generation CD123 CARTs.
Results
Here, we show that CD123 represents a bona fide target for AML and show that both 41BB-based and CD28-based CD123 CARTs are very efficient in eliminating both AML cell lines and primary cells in vitro and in vivo. However, both 41BB-based and CD28-based CD123 CARTs ablate normal human hematopoiesis and prevent the establishment of de novo hematopoietic reconstitution by targeting both immature and myeloid HSPCs.
Conclusions
This study calls for caution when clinically implementing CD123 CARTs, encouraging its preferential use as a bridge to allo-HSCT in patients with R/R AML.
Keywords: cell engineering; immunotherapy, adoptive; T lymphocytes
Background
Acute myeloid leukemia (AML) is a biologically, phenotypically and genetically very heterogeneous malignant disease which results from the uncontrolled accumulation of differentiation-defective hematopoietic stem/progenitor cells (HSPCs) or immature myeloid cells.1 2 AML is one of the most common hematopoietic malignances, and its incidence increases with age.3 4 Intensive chemotherapy combos based on nucleoside analogs plus anthracyclines remain the standard front-line treatment of AML,5 followed by allogeneic HSPC transplant (allo-HSCT), based on patients’ eligibility, to consolidate complete remission (CR) and to prevent relapse.6 However, with the exception of a few molecular subgroups (the ‘so-called’ low-risk AMLs), relapses are common after consolidation therapy and/or allo-HSCT. Chemotherapy-related toxicity, refractoriness, and failure to eradicate leukemia-initiating cells are the major mechanisms underlying AML progression and relapse.7–10 Unfortunately, improved AML treatments have only experienced minor developments over the last four decades, and current 5-year event-free survival (EFS) remains ~20% in adults and <70% in children,11 12 highlighting the desperate need for safer and more efficient therapeutics.
Immunotherapy has generated unprecedented expectations in cancer treatment. In AML, both CD33- and CD123-specific antibody-drug conjugates have been used for combination therapy with standard chemotherapy with improved EFS,13 14 and Bispecific T-cell Engagers (BiTEs) for CD33 and Dual-Affinity Retargeting (DART) antibodies for CD123 are being clinically assayed.15 16 Adoptive cellular immunotherapy based on the engineering of human chimeric antigen receptor T-cells (CARTs) redirected against cell surface tumor antigens has shown robust clinical responses in patients with B-cell malignances, thanks to the high efficacy, specificity and persistence of CARTs.17–19 However, the clinical implementation of AML-specific and safe CARTs for patients with refractory or relapse (R/R) AML is still awaiting. Strategies targeting AML using CARTs have proven more challenging than in B-lineage malignancies because of two reasons: (1) the lack of a universal pan-AML target antigen due to the large disease heterogeneity, which hampers clinical implementation, since a wide range of CARs would be needed to cover the different leukemic phenotypes; and (2) the shared expression of immature target antigens between normal HSPCs and myeloid blasts, which compromises safety due to potential on-target/off-tumor cytotoxicity against HSPCs, leading to fatal aplasia.20 21
Adoptive immunotherapy for AML is in advanced preclinical and clinical development using CD33-redirected and CD123-redirected CARs (CD123 CARTs), and they exhibit robust antileukemic activity in vitro and in vivo.22–25 However, controversy exists on whether CD123-directed and CD33-directed CARTs are myeloablative. Some groups raised safety concerns leading to the development of complex target antigen knockout in HSPC or T-cell suicide strategies to circumvent such a toxicity.23 26 In contrast, other groups showed a safety profile with limited on-target/off-tumor toxicity of such CARs.22 27–31 Here, we set out to characterize in vitro and in vivo the efficacy and the safety of 41BB-costimulated and CD28-costimulated CD123CARs, based on a clinically relevant single chain variable fragment (scFv) from the CSL362 monoclonal antibody (MoAb). Analysis of a large cohort of diagnostic and relapse AML primary samples revealed that CD123 is a suitable target for AML, and CD123 CARTs were very efficient in vitro and in vivo in eliminating both AML cell lines and primary cells, regardless of the costimulation motif. However, clonogenic assays and several xenograft models revealed that both 41BB-costimulated and CD28-costimulated CD123 CARTs strongly ablate normal human hematopoiesis by targeting both HSCs and myeloid progenitors. This study calls for caution when clinically implementing CD123 CARTs and highlights its preferential use as a bridge to allo-HSCT in patients with R/R AML.
Materials and methods
Car design and vectors, lentiviral production and T-cell transduction
The anti-CD123 scFV derived from the clinically tested CSL362 MoAb was generated and cloned into the pCCL lentiviral-based second-generation CAR backbone containing a human CD8 transmembrane domain, a human costimulatory domain (either 41BB or CD28), CD3z endodomain, and a T2A–green fluorescence protein (GFP) cassette. The pCCL vector expressing GFP alone (MOCK vector) was used as a control. Chimeric antigen receptor (CAR)-expressing viral particles pseudotyped with vesicular stomatitis virus-G (VSV-G) were generated using HEK 293T cells with a standard polyethylenimine (PEI) transfection protocol. For each production, plasmid transfection was carried out using a 3:1 PEI:DNA ratio using 16 µg transfer vector, 16 µg of pSPAX2, and 8 µg VSV-G per plate, and viral particles were concentrated by ultracentrifugation as previously described.32 Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats from healthy volunteers by Ficoll-Hypaque gradient centrifugation. Buffy coats were obtained from the Barcelona Blood and Tissue Bank (BST) on institutional review board (IRB) approval (HCB/2018/0030). T-cells were activated by plate-bound OKT3 and anti-CD28 antibodies (BD Biosciences) for 2 days in the presence of interleukin (IL)-7 and IL-15 (10 ng/mL, Mitenyi Biotec).33 34 Surface expression of CAR123 was traced by fluorescence-activated cell sorting (FACS). CAR detection was confirmed by GFP expression and by using an AffiniPure F(ab')₂ Fragment Goat Anti-Human IgG (H+L) (Jackson ImmunoResearch). Activation and subsetting of lentivirally transduced T-cells were confirmed by surface staining with CD25/CD69 (data no shown) and CD3/CD4/CD8, respectively.
Immunophenotyping of healthy HSPCs, primary AML samples and cell lines
Diagnostic immunophenotyping data for the most commonly expressed antigens in AML (CD123, CD33, CD13, CD34, CD15, c-kit, and CD66) were obtained for 97 patients diagnosed at local hospitals: Germans Trias i Pujol (Barcelona, Spain), Hospital Clínico (Madrid, Spain), Hôpital Armand Trousseau (Paris, France) and Santa Creu i San Pau (Barcelona, Spain). CD123 expression was also compared in diagnostic-relapsed paired samples (n=68 patients) and in paired bulk leukemia–leukemia stem cells (LSCs) (n=37 patients).35 Cell lines were stained with CD123-APC, CD33-BV-421, CD14-PerCP-Cy5.5 and CD19-APC. The expression of CD123 and CD33 antigens was prospectively compared in CD34+ HSPCs derived from healthy cord blood (CB, n=22), mobilized peripheral blood (PB, n=10) and diagnostic primary AML samples (n=24). For HSPC subsetting, CD34+ cells were stained with CD34-PE or CD34-PE-Cy7, CD133-PE, CD19-FITC, CD90-APC, CD13-PE-Cy7 and CD71-APC-Cy7, which allow for the identification and quantification of immature HSCs (CD34++CD133+CD90+), myeloid progenitors (CD34+CD13++CD71−/low), erythroid progenitors (CD34+CD71++CD13 low), and B-cell progenitors (CD34+CD19+CD71−CD13−). Isotype-matched, non-reactive fluorochrome-conjugated MoAbs were always used as a fluorescence reference. All antibodies were purchased from Beckton Dickinson. Cells were incubated with MoAbs (30 min at 4°C in the darkness), then washed in phosphate-buffered saline (PBS) and analyzed in a FACSCanto-II flow cytometer equipped with FACSDiva software (Becton Dickinson).36–38 Determination of antigen density for CD33 and CD123 was performed using BDQuantibrite-PE (Becton Dickinson) according to the manufacturer’s instructions.
In vitro cytotoxicity assays and cytokine release determination
The cell lines THP-1, MOLM-13 and 697 were purchased from DSMZ (Germany) and expanded according to DSMZ recommendations. Primary AMLs and healthy CD34+ cells were obtained from the aforementioned hospitals and the BST, respectively (IRB approval: HCB/2018/0030). Target cells were incubated with CAR123 or MOCK T-cells at different effector:target (E:T) ratios for the indicated time periods. CART-mediated cytotoxicity was determined by analyzing the residual alive (7-AAD-) target cells at each time point and E:T ratio. For absolute cell counting, Trucount absolute count beads (Becton Dickinson) were used. Furthermore, FACS-sorted CD3+ mature T-cells from the bone marrow (BM) of CD123+ patients with AML were activated, transduced with CD123 CAR and tested against their autologous-matched CD123+ AML blasts. Target cells (1×105) were used for all cytotoxicity assays unless stated otherwise. Table 1 shows the clinical–biological features of the CD123+ AML samples used for in vitro experiments. The production of the proinflammatory cytokines IL-2, tumor necrosis factor alpha (TNF-α) and interferon gamma (IFN-γ) was assessed by ELISA (Human ELISA SET, BD Biosciences) using in vitro supernatants harvested at 16 hours post-T-cell exposure, and sera were collected from mice 10 days after CART infusion.
Table 1.
Biological and cytogenetic-molecular characteristics of blasts from diagnostic patients with AML
| Patient ID | Diagnostic | Cytogenetics | Molecular | Age (years) | Gender | Blasts (%) | Cd123 (%) | Use |
| 14 085 | AML | 46, XY | NPM1+, FLT3-ITD | 42 | M | 87 | 80 | AG density |
| 14 176 | AML-M1 | 46, XX | Normal | 44 | F | 83 | 75 | AG density ELISA |
| 14 093 | AML-M4 | 46, XX | NPM1MUT, FLT3-ITD | 52 | F | 81 | 86 | AG density |
| 14 184 | AML-M1 | 46, XX, t(8;21) | AML1-ETO | 14 | F | 90 | 84 | AG density |
| 14 268 | AML | 46, XY, inv(16) | NPM1MUT, FLT3-ITD | 69 | M | 95 | 92 | AG density Autologous |
| 14 269 | AML-M5 | 47, XX,+8 | Normal | 64 | F | 92 | 90 | AG density ELISA |
| 14 266 | AML | 46, XX, t(8;21) | AML1-ETO, FLT3-ITD+ | 48 | F | 90 | 92 | AG density ELISA |
| 14 123 | AML | 46, XY, t(3;3) | Normal | 28 | M | 42 | 77 | AG density |
| 14 185 | AML | 46, XY, inv(16) | Normal | 8 | M | 77 | 72 | AG density |
| 14 143 | AML-M1 | 46, XY | Normal | 43 | M | 90 | 90 | AG density |
| 14 156 | AML | 46, XY,11q23 | MLL-AF6 | 1 | M | 88 | 100 | AG density |
| 14 144 | AML | 46, XY, del(7)(q22) | Normal | 61 | M | 88 | 98 | AG density |
| 14 141 | AML-M5 | 46, XX, t(8;21) | AML1-ETO | 39 | F | 83 | 82 | AG density |
| 14 091 | AML-M4 | 47, XX,+8 | Normal | 61 | F | 95 | 80 | AG density |
| 14 272 | AML | 46, XX | NPM1MUT, FLT3-ITD | 44 | F | 73 | 88 | Autologous |
| 14 274 | AML | 46, XX, t(8;21) | AML1-ETO | 13 | F | 85 | 75 | Autologous |
| ABT3974 | AML | 46, XY,+9, inv(16)(p13;q22), der(17)t(11;17)(q13;q25) | CEBPA, FLT3-TKD, WT1 | 37 | M | 91 | 87 | AG density |
| ABT5270 | AML | 46, XY | NPM1, DNMT3A, IDH1 | 55 | M | 74 | 77 | AG density |
| ABT4435 | AML | 46, XX | IDH2, DNMT3A | 70 | F | 24 | 96 | AG density |
| ABT8326 | sAML | 44–45X-, Y, der(3), del(7)(q22), der(8), add(12)(p13),−18, add(21)(q28) | TET2, CALR | 61 | M | 6 | 56 | AG density |
| ABT7693 | AML | 46, XY | NPM1, IDH1, PTPN11 | 45 | M | 80 | 99 | AG density |
| ABT4470 | AML | 47, XX | NPM1, IDH1, BCOR | 72 | F | 71 | 76 | AG density |
| ABT5718 | AML | 46, XX | CEBPAbi, DNMT3A, TET2 | 52 | F | 34 | 65 | AG density |
| ABT8597 | AML | NA | NPM1, IDH1, NRAS | 69 | F | 71 | 100 | AG density |
| ABT3906 | AML | NA | NPM1, FLT3-ITDHIGH, IDH1 | 40 | M | 93 | 77 | AG density |
| ABT4685 | tAML | 46, XX | CEBPAbi, TET2, WT1 | 67 | F | 49 | 93 | AG density |
AG, antigen; AML, acute myeloid leukemia; F, female; M, male; sAML, secondary acute myeloid leukemia; tAML, therapy-related acute myeloid leukemia.
Colony-forming unit (CFU) assays
CB-derived CD34+ cells were exposed for 24 hours to either CD123 CARTs or MOCK T-cells (E:T 1:1) and then plated (2×103) onto serum-free methylcellulose H4435 (Stem Cell Technologies). CFUs were then counted and scored after 12–14 days following standard procedures.
In vivo xenograft models for AML, HSPCs and CARTs
Non-obese diabetic-Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (8–12 weeks old, Jackson Laboratory) were bred and housed under pathogen-free conditions in the animal facility of the Barcelona Biomedical Research Park. In experiments addressing CAR123 efficacy, mice were intravenously transplanted with 0.25×106 Luc-mCherry-expressing patient-derived xenograft AML cells (PDX-579)39 5 days before intravenous infusion of 3×106 of either 41BB-CD123 or CD28-CD123 CARTs derived from healthy PBMCs. Tumor burden was monitored at the indicated time points by bioluminescence (BLI) using the Xenogen in vivo imaging system (IVIS) 50 Imaging System (Perkin Elmer).32 In experiments addressing the myeloablative effect of CAR123, CD34+ HSPCs (0.1×106) were intra-BM transplanted in sublethally irradiated (2 Gy) NSG mice, followed by intravenous infusion of 3×106 of 41BB-CD123, CD28-CD123 CARTs or MOCK T-cells either 1 day or 6 weeks after CD34+ transplantation. BM and PB were FACS-analyzed for human chimerism at sacrifice. Cells were stained with anti-HLA.ABC-PE and CD45-BV450. Engrafted mice were assessed for multilineage engraftment using anti-CD123-APC for myeloid cells, anti-CD19-BV421 for lymphoid cells, and anti-CD34-PE.Cy7 for immature cells. Human absolute engraftment in PB and BM was quantified using BD Trucount tubes according to manufacturer’s instructions.
Statistical analysis
For comparison of CD123 expression between paired ‘diagnostic-relapse’ and ‘bulk leukemic cells-LSC’, the Mann-Whitney U test was used. For differences in antigen density and engraftment among groups, a one-way analysis of variance test was used. For the remaining comparisons, Student’s t-test was used. All p values were considered statistically significant at <0.05 (*).
Results
CD123 represents a bona fide immunotarget for AML
We first analyzed by FACS the expression levels of the most common diagnostic myeloid markers in a cohort of 97 patients with AML at presentation. We found that CD123 was the most common and homogeneously expressed antigen (86.4%±26.8 of AML blasts) followed by CD33 (77.4%±32.1) (figure 1A). Important, in 82% of the patients with AML analyzed, >80% of the blasts were CD123+, while only 66% of the patients showed positivity for CD33 in >80% of the blasts (figure 1A). A target antigen for immunotherapy in AML should ideally be absent in HSPCs. CD123 and CD33 are both partially expressed in healthy CD34+ HSPCs,22 23 so we next quantified the density (molecules/cell) of both antigens in fresh primary AML blasts (n=24), healthy CB-derived (n=22) and healthy mobilized PB-derived CD34+ HSPCs (n=10). Of note, 67% (16/24) of patients with AML displayed levels of CD123 significantly higher than those found in both CB-derived and PB-derived CD34+ HSPCs, while only 41% (10/24) of patients with AML displayed levels of CD33 that segregate them from CB-derived and PB-derived CD34+ HSPCs (figure 1B). This suggests that CD123 represents, a priori, a less myeloablative target than CD33. Of note, analysis of paired diagnostic (DX)-relapse (RX) AML samples revealed that CD123 expression is maintained at relapse, and in AML-LSC (identified as CD34+CD38−)35 (figure 1C), reinforcing CD123 as a bona fide immunotarget for R/R AML.
Figure 1.

Expression of CD123 in AML and design, detection and expansion CD123 CARTs. (A) Immunophenotyping of the indicated diagnostic myeloid markers in a cohort of 97 patients with AML at presentation. Each dot denotes an individual patient. Red circles identify patients with >80% of blasts positive for the indicated marker. (B) Comparative antigen density (measured as antigen molecules/cell) for CD123 and CD33 in primary AML samples (n=24), CB-derived (n=22) and PB-derived (n=10) CD34+ cells from healthy donors. AML blasts were identified as 7AAD−CD3−CD45+/lowCD123+CD33+. One-way analysis of variance; *p<0.05, **p<0.01, ***p<0.001. (C) Comparison of CD123 expression in 68 paired diagnostic-relapse AML samples (left panel) and in bulk tumor versus AML-LSC (n=37, right panel).33 (D) Scheme of the CD123 CAR structure. (E) CAR detection in primary T-cells using an antihuman IgG F(ab')2 antibody and GFP. (F) Successful CAR123 transduction and detection in CD4+ and CD8+ T-cells (n=3). (G) Robust expansion of activated T-cells transduced with either MOCK (black line) or CAR123 (red line) (n=3). AML, acute myeloid leukemia; CAR, chimeric antigen receptor; CART, chimeric antigen receptor T-cell; CB, cord blood; DX, diagnostic; GFP, green fluorescence protein; LSC, leukemia stem cell; PB, peripheral blood; RX, relapse.
41BB-based and CD28-based CD123 CARTs efficiently eliminate AML primary cells in vitro and in vivo
We next designed second-generation 41BB-based and CD28-based CD123CARs coupled in-frame with GFP through a T2A sequence (figure 1D and online supplementary figure S1A). The expression of both 41BB-CD123 and CD28-CD123 CAR in T-cells was confirmed through codetection of scFv and GFP (figure 1E and online supplementary figure S1B) and did not affect the CD4:CD8 ratio (figure 1F). Importantly, activated (CD69+CD25+) T-cells continuously expanded ~50-fold over a 10-day period, similar to MOCK T-cells (figure 1G), demonstrating that redirecting T-cells against CD123 does not hamper T-cell expansion.
jitc-2020-000845supp001.pdf (288.4KB, pdf)
We then tested the functionality of our 41BB-CD123 and CD28-CD123 CARs in vitro and in vivo (figure 2 and online supplementary figure S1, S2). In vitro, both 41BB-CD123 (figure 2A) and CD28-CD123 (online supplementary figure S1C) CARTs, but not MOCK T-cells, specifically eliminated the CD123+ AML cell lines THP1 and MOLM13 in an E:T ratio-dependent manner (online supplementary figure S2) while sparing the CD123− B-ALL cell line 697. In fact, CD123+ AML cells barely survived exposure to CD123 CARTs in a 48-hour absolute number assay at a 1:1 E:T ratio (figure 2B and online supplementary figure S1C). We then examined in an autologous setting whether CD3+ T-cells deriving from patients with AML can be isolated, modified to express CD123 CAR, expanded and used as cytotoxic effector cells (figure 2C). Patient-derived CD123 CARTs were successfully generated from magnetic-activated cell sorting (MACS)-sorted CD3+ T-cells (>95% purity) and specifically eliminated autologous patient-matched CD123+ AML blasts (figure 2D). Important, both CD123 CARTs produced high levels of the proinflammatory cytokines IL-2, TNF-α, and IFN-γ on coculture with both AML cell lines (figure 2E and online supplementary figure S1D) and primary blasts (figure 2F), confirming their robust cytotoxicity.
Figure 2.

41BB-CD123 CARTs specifically target and eliminate CD123+ AML cells in vitro and in vivo. (A) Surface expression of CD123 (red) in THP-1, MOLM-13 and 697 cell lines. (B) Absolute counts of alive residual target cells measured by FACS in 48-hour cytotoxicity assays at 1:1 E:T ratio (n=3). Data are presented as mean±SEM; *p<0.05, **p<0.01, ***p<0.001. (C) Graphical cartoon of the experimental design for autologous cytotoxic assays. Normal CD3+ T-cells were FACS-purified from the BM of patients with AML (n=3), infected with CD123 CAR, expanded, and exposed to autologous total PBMCs (1:1 E:T). Residual CD123+ blasts were quantified 48 hours post-41BB-CD123 CART exposure. (D) Left: representative FACS analysis of the cytotoxicity assay. T-cells are shown in black and CD123+ blasts in blue. Right: absolute counts of alive AML blasts in 48-hour cytotoxicity assays at 1:1 E:T ratio (n=3). (E, F) ELISA showing robust secretion of proinflammatory cytokines by 41BB-CD123 CARTs after exposure to CD123+ cell lines (E) and AML primary blasts (F) for 16 hours at 1:2 E:T ratio (n=3). (G) Experimental design to assess in vivo the efficacy of both 41BB-based and CD28-based CD123 CAR. NSG mice were intravenously injected with 2.5×105 Luc-expressing xenograft AML cells (PDX-579) followed 5 days after by a single intravenous injection of 3×106 CD123 CARTs (either 41BB or CD28) generated from healthy PBMCs. Tumor burden was monitored every 7–10 days by BLI using IVIS imaging. (H) IVIS imaging of tumor burden monitored by BLI at the indicated time points. (I) Left: total radiance quantification (p/s/cm2/sr) at the indicated time points for 41BB-CD123 CARTs, CD28-CD123 CARTs and untreated mice. *P<0.05. Right: absolute counts of residual AML cells in PB and BM at endpoint. (J) T-cell persistence in PB and BM at endpoint. (K) In vivo quantification by ELISA of IFN-γ in PB sera collected in the acute phase (10 days post-CART infusion). *p<0.05. AML, acute myeloid leukemia; BLI, bioluminescence; BM, bone marrow; CART, chimeric antigen receptor Tcell; E:T, effector:target; FACS, fluorescence-activated cell sorting; IFN-γ, interferon gamma; IVIS, in vivo imaging system; ns, non-significant; NSG, non-obese diabetic-Cg-Prkdcscid Il2rgtm1Wjl/SzJ; PB, peripheral blood; PBMC, peripheral blood mononuclear cell.
jitc-2020-000845supp002.pdf (63.1KB, pdf)
We next compared the cytotoxic activity of 41BB-CD123 and CD28-CD123 CARTs in vivo using Luc-expressing CD123+ AML xenograft (figure 2G). NSG mice were transplanted with 0.25×106 Luc-expressing AML PDX cells 5 days prior to intravenous infusion of 3×106 41BB-CD123 or CD28-CD123 CARTs, and leukemia establishment was followed up weekly by BLI until disease signs were evident (figure 2H). While control mice increasingly showed aggressive disease and disseminated leukemia, CD123 CART-treated mice showed extensive disease control across the experiment, regardless of the costimulation domain used (figure 2H1). Of note, T-cells persisted in PB and BM at sacrifice, although at higher levels in 41BB-CD123 CART-treated mice, in line with the reported longer persistence/effector function of 41BB-costimulated CARTs (figure 2J).40 Similarly, both CD123 CARTs, especially 41BB-costimulated CD123 CARTs produced high levels of the IFN-γ in vivo (figure 2K). Collectively, both 41BB-CD123 and CD28-CD123 CARTs have similarly potent and specific antileukemic activity against AML cells in vitro and in vivo.
On-target/off-tumor targeting of immature HSPCs and myeloid progenitors render both 41BB-CD123 and CD28-CD123 CARTS severely myeloablative in vitro and in vivo
There is controversy on whether CD123-redirected T-cells are myeloablative. To prospectively assess the potential myelotoxicity of CD123 CARTs, we first addressed in vitro whether exposure to CD123 CARTs hampers the viability and clonogenic capacity of CD34+ HSPCs (figure 3A). As compared with MOCK T-cells, both CD123 CARTs induced a massive reduction in CD34+ cell counts in a 72 hours (E:T ratio 2:1) assay (figure 3B). Similarly, CD34+ HSPCs pre-exposed to either CD123 CARTs (E:T ratio 1:1) for only 24 hours showed 50%–80% reduction in their clonogenic capacity (figure 3D).
Figure 3.

Both 41BB-CD123 and CD28-CD123 CARTs eliminate healthy CD34+ HSPCs in vitro and in vivo. (A) Experimental scheme for in vitro assessment of CAR123 cytotoxicity on healthy CD34+ cells. (B) Representative FACS showing the residual CD34+ HSPCs (red) after exposure to CAR123 CARTs or MOCK T- cells for 72 hours at an E:T ratio of 2:1. (C) Absolute quantification of remaining alive CD34+ cells after exposure to either 41BB-CD123 or CD28-CD123 CARTS (72 hours, E:T 2:1). (D) Clonogenic assays performed with residual alive CD34+ HSPCs after 24 hours of coincubation with either 41BB-CD123 CARTs, CD28-CD123 CARTs or MOCK T-cells (E:T 1:1) (n=3 donors). (E) Schematic representation of the in vivo experimental plan. CD34+ cells were intra-BM transplanted into NSG mice, and 6 weeks later, the level of human engraftment was assessed by FACS analysis in PB and BM. Mice then received 3×106 of either CD123 CARTs (41BB or CD28) or MOCK T-cells. PB bleedings were performed biweekly and PB/BM were analyzed at sacrifice (6 weeks after CART infusion). (F, G) Analysis of murine PB (F) and BM (G) multilineage reconstitution (CD19+ B lymphoid, CD123+ myeloid and CD34+ immature) at the indicated weeks post-CART infusion. Final engraftment (POST) of myeloid, B lymphoid and immature HSPCs is presented as fold change in comparison to pre-CART/MOCK infusion (PRE). (H) Schematic representation of the in vivo experimental plan. CD34+ cells were intra-BM transplanted into NSG mice, followed the day after by infusion of either 3×106 CD123 CARTs (41BB or CD28) or MOCK T-cells. Mice were sacrificed 6 weeks after and PB and BM were analyzed. (I, J) Analysis of murine PB (I) and BM (J) multilineage reconstitution (CD19+ B lymphoid, CD123+ myeloid and CD34+ immature) 6 weeks after CART infusion. *P<0.05, **P<0.01, ***P<0.001. BM, bone marrow; CART, chimeric antigen receptor T-cell; E, erythroid colony-forming unit; E:T, effector:target; FACS, fluorescence-activated cell sorting; G, granulocytic colony-forming unit; GEMM, granulocytic, erythroid, myelomonocytic colony-forming unit; GM, granulomonocytic colony-forming unit; HSPC, hematopoietic stem/progenitor cell; M, monocytic colony-forming unit; NSG, non-obese diabetic-Cg-Prkdcscid Il2rgtm1Wjl/SzJ; PB, peripheral blood.
We next assessed the myeloablative potential of both 41BB-CD123 and CD28-CD123 CARTs in vivo using xenograft models of human hematopoietic reconstitution. In an initial set of experiments, sublethally irradiated NSG mice were reconstituted with 0.1×106 CD34+ HSPCs, and 6 weeks later, when human multilineage engraftment was established, mice received 3×106 41BB-CD123, CD28-CD123 CARTs or MOCK T-cells (figure 3E). Human engraftment was biweekly analyzed in PB (figure 3F) and BM (figure 3G) over 6 weeks. MOCK T-cell-treated mice consistently showed increased myeloid (HLA-ABC +CD45+CD123+CD33+), B-lymphoid (HLA-ABC+CD45+CD123 CD19+) and immature (HLA-ABC+CD45+CD34+) hematopoietic engraftment than that observed on the day of CART infusion. In contrast, both 41BB-CD123 and CD28-CD123 CART-treated mice showed an impaired multilineage engraftment in both PB and BM (figure 3F). However, in this xenograft model of existing hematopoiesis, CD28-based CD123 CARTs proved less myeloablative than 41BB-CD123 CARTs.
Next, we assessed the capacity of both CD123 CARTs in preventing de novo establishment of normal hematopoiesis by transplanting sublethally irradiated NSG mice with CD34+ HSPC and either 41BB-CD123, CD28-CD123 CARTs or MOCK T-cells 1 day after (figure 3H). Long-term multilineage human engraftment was found in both PB and BM in MOCK T-cell-treated mice; however, human hematopoiesis was barely reconstituted in both 41BB-CD123 and CD28-CD123 CART-treated mice (figure 3I), indicating that both 41BB-CD123 and CD28-CD123 CARTs prevent healthy hematopoietic reconstitution.
Finally, to further characterize the myeloablative effects of CD123 CARTs, we exposed total CD34+ HSPCs to either CD123 CARTs or MOCK T-cells for 48 hours at 1:1 E:T ratio, and quantified afterwards whether the myeloablative effects were CD34+ subset-specific (figures 3A, 4A). We found a significant loss of both immature/early HSPCs (CD34 ++CD133+CD90+) and myeloid progenitors (CD34 +CD13+CD71 low), while B-cell progenitors (CD34 +CD19+CD13-CD71−) and erythroid progenitors (CD34+CD71+CD13 low) were unaffected by CD123 CARTs exposure (figure 4A). Of note, CD123 CART-mediated cytotoxicity correlated well with the expression levels of CD123 in the different CD34+ subsets (figure 4C). Collectively, our results suggest that CD123 CARTs ablate human hematopoiesis by targeting both early/immature HSPCs and myeloid progenitors.
Figure 4.
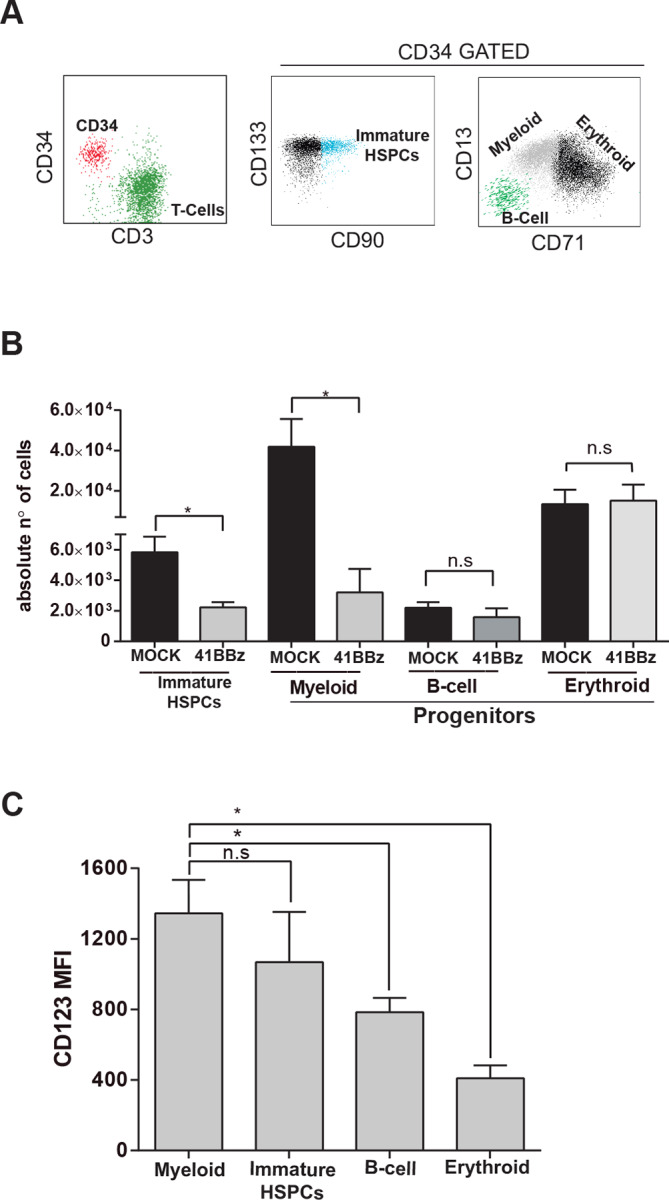
CAR123-mediated cytotoxicity is CD34 subset specific. (A) Flow cytometry characterization of different subsets of CD34+ HSPCs post CD123 CART exposure. Left: identification of CD34+ HSPCs and CD123 CARTS. Middle right: identification of CD90+ CD133+ early-immature CD34+ HSPCs (turquoise dots), CD13−CD71−CD19+ B-lymphoid CD34+ HSPCs (green dots), CD13++CD71 dim myeloid CD34+ HSPCs (gray dots) and CD13lowCD71++ erythroid CD34+ HSPCs (black dots). (B) Absolute quantification by FACS of the different CD34+ subsets (as identified in A) on exposure to CD123 CARTS or MOCK T-cells (48 hours, E:T 1:1) (n=3). (C) MFI levels of CD123 in the different CD34+ cell subsets (n=3). *P<0.05. CART, chimeric antigen receptor T-cell; FACS, fluorescence-activated cell sorting; MFI, mean fluorescence intensity.
Discussion
AML is a very heterogeneous stem cell malignant disease characterized by the progressive acquisition of (epi)genetic alterations resulting in a clonal rapid expansion of differentiation-defective HSPC in BM and PB.1 Unfortunately, the prognosis of AML remains unfavorable, especially in patients >60 years old, due to common relapses, disease refractoriness and treatment-related toxicities.41 Unfortunately, improved AML treatments have only experienced minor developments over the last four decades, reinforcing the high-demand for new therapeutics with improved efficacy and reduced toxicity.20 23 In this context, the undisputable clinical improvements of cancer immunotherapy have not gone unnoticed in AML and undoubtedly represent the great hope of the next decade in the treatment of AML. In fact, immunotherapeutic targeting in AML is already well advanced in clinical trials using MoAb, antibody–drug conjugates, BiTEs, DARTs and CAR T-cell therapies against CD33 and CD123.13–16 20 22 29 However, clinical progress and regulatory approval of such immunotherapies have been hampered by the challenge to find a specific and safe targetable surface antigen.41 42
CD33 and CD123 are the most extensively explored antigens for AML and blastic plasmocytoid dendritic cell neoplasm immunotherapy.42 43 In this study, we aimed to better characterize the suitability of CD33 and CD123 in a large cohort of diagnostic and relapse AML primary samples. We show that CD123 represents a bona fide target for AML with a potentially safer profile than CD33. Not only is the most common and homogeneously expressed antigen in AML, but also its expression is also fully retained at relapse and in AML-LSC. This lack of antigen plasticity, a phenomenon widely observed during the progression and relapse of acute leukemias,44 further strengthens the potential of CD123 as immunotarget for AML. More importantly, a target antigen for immunotherapy in AML should ideally spare HSPCs. Previous studies about the expression of CD123 in CD34+ HSPCs have provided conflicting results based on the source of CD34+ cells and the MoAb used.20 22 29 Here, we demonstrate that CD123 is expressed in both CB-derived and PB-derived CD34+ HSPCs; however, in contrast to CD33, which discriminates more poorly AML from either CB-derived or PB-derived CD34+ HSPCs, two-thirds of patients with AML express CD123 at levels significantly higher than those in healthy CD34+ cells, suggesting CD123 as a safer target than CD33 for AML.
Extensive evidence supports CD123 targeting as a therapeutic approach for AML. First, major phenotypical (immature, granulocytic and monocytic) and cytogenetic (FLT3- and NPM1-mutated) AML subgroups express CD123.35 45 46 Second, CD123+ AML cells are capable of initiating leukemogenesis when transplanted in immunodeficient mice, thus marking AML-LSC.47–49 Third, the presence of CD34+ CD38 CD123+cells in AML at presentation is associated with lower disease-free and overall survival and failure to achieve complete remission.50 51 Fourth, CD123 expression enhance AML cell proliferation and induces downregulation of CXCR4, favoring the egress of BM AML-LSCs into the circulation.52 Based on this background, we prompted to characterize and compare in vitro and in vivo the efficacy and safety profile of the second-generation 41BB-based and CD28-based CD123CARs derived from the clinically tested CSL362 humanized MoAb.14 53 Regardless of the costimulation motif, CD123 CARTs were very efficient in vitro and in vivo in eliminating both AML cell lines and primary cells, even at relatively low E:T ratios. Importantly, however, CD123 CARTs ablated existing normal human hematopoiesis and prevented the establishment of de novo hematopoietic reconstitution by directly targeting both myeloid progenitors and early/immature HSPCs, with subsequent functional consequences in all downstream normal hematopoietic progenitors, rendering severe impairment of multilineage hematopoiesis in BM and PB. This study adds information to the existing controversy about the myeloablative potential of CD123 CARTs. Despite several reports showing a limited cytotoxic effect on CD34+ HSPCs,22 25 29 our data support the work by Gill and coworkers who reported a myeloablative in vivo potential of 41BB-based CD123 CARTs on CD34+ HSPCs.
The myeloablative effects observed here were not limited to the 41BB-based CD123 CARTs but were similarly observed with the CD28-costimulated CD123CARTs. Previous studies used different sources of CD34+ cells, different vector designs, and distinct CD123 scFvs. Therefore, current conflicting data may be attributed to distinct vector architectures, CAR-binding affinity, target density, source of healthy CD34+ cells, or even experimental designs.31 The robust myeloablative effects reported in this study calls for caution when clinically implementing CD123 CARTs. Unfortunately, however, immunotherapies for AML different from CARTs, such as DARTs or CD123-directed MoAbs, resulted in limited clinical efficacy unable to control the disease in the medium–long term.14 54 55 A potentially safer clinical approach to circumvent myeloablation would be the use of potent CD123 CARTs to achieve CR followed by allo-HSCT as a rescue therapy. Finally, alternative sources of effector cells, such as natural killer (NK) cells, cytokine-induced killer (CIK) cells or Vδ1 γδ T-cells (DOTs) are being explored in order to better control the in vivo persistence of CD123 CAR bearing cells.56 57
Acknowledgments
We deeply thank Marixtell Vinyoles and Paolo Petazzi for their technical support and Juan Jose Rodríguez-Sevilla for critically reviewing the manuscript.
Footnotes
Correction notice: Since the online publication of this article, it was noticed that 'Samanta Romina Zanetti' was incorrectly spelt as 'Samanta Zanetti'. This error has been corrected.
Authors’ contribution: MLB designed the experiments, analyzed/interpreted the data, and wrote the manuscript. DSM, FGA, HRH, MC, SZ, TVH, JC, and RDdlG performed experiments. EA, SV, JN, HL, AEB, VHJvdV, JJ, PMa, AB, JE, AL, BV, IJ and MS provided clinical samples and biological data. CB designed the experiments, interpreted the data, and financially supported the work. PMe conceived the study, designed the experiments, wrote the manuscript, and financially supported the work.
Funding: We thank CERCA/Generalitat de Catalunya and Fundació Josep Carreras‐Obra Social la Caixa for their institutional support. PM acknowledges financial support from the European Research Council (CoG-2014–6 46 903, PoC-2018–8 11 220), the Spanish Ministry of Economy and Competitiveness (MINECO, SAF2016-80481-R), the Spanish Cancer Research Association (AECC-Semilla19), the Fundación Uno entre Cienmil, the Obra Social La Caixa (LCF/PR/HR19/52160011), the Leo Messi Foundation, the Banco Santander Foundation and the “Heroes hasta la médula” initiative. CB was supported by the AECC and the Health Institute Carlos III/FEDER (PI17/01028). MLB is supported by an FPI scholarship from MINECO. DSM is supported by a Sara Borrell fellowship from ISCIII. SZ and TVH are supported by Marie Sklodowska Curie Fellowships. PMa is an investigator of the Spanish Cell Therapy Network or TERCEL.
Competing interests: None declared.
Patient consent for publication: Not required.
Ethics approval: This study was approved by the institutional review board, Barcelona Clinic Hospital Ethics Committee (HCB/2017/1056). All in vivo procedures were approved by the Animal Care Committee of The Barcelona Biomedical Research Park (HRH-17-0029-P1).
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: All data relevant to the study are included in the article and uploaded as supplementary information. The datasets and materials generated in this study are available from the corresponding author on reasonable request.
References
- 1. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730–7. 10.1038/nm0797-730 [DOI] [PubMed] [Google Scholar]
- 2. Li S, Mason CE, Melnick A. Genetic and epigenetic heterogeneity in acute myeloid leukemia. Curr Opin Genet Dev 2016;36:100–6. 10.1016/j.gde.2016.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Young AL, Challen GA, Birmann BM, et al. . Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun 2016;7:12484. 10.1038/ncomms12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bowman RL, Busque L, Levine RL. Clonal hematopoiesis and evolution to hematopoietic malignancies. Cell Stem Cell 2018;22:157–70. 10.1016/j.stem.2018.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Robak T. Purine nucleoside analogues in the treatment of myleoid leukemias. Leuk Lymphoma 2003;44:391–409. 10.1080/1042819021000035608 [DOI] [PubMed] [Google Scholar]
- 6. Cornelissen JJ, Blaise D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood 2016;127:62–70. 10.1182/blood-2015-07-604546 [DOI] [PubMed] [Google Scholar]
- 7. Thol F, Schlenk RF, Heuser M, et al. . How I treat refractory and early relapsed acute myeloid leukemia. Blood 2015;126:319–27. 10.1182/blood-2014-10-551911 [DOI] [PubMed] [Google Scholar]
- 8. Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol 2017;18:17. 10.1007/s11864-017-0456-2 [DOI] [PubMed] [Google Scholar]
- 9. De Grandis M, Mancini SJ, Aurrand-Lions M. In quest for leukemia initiating cells in AML. Oncoscience 2018;5:9–10. 10.18632/oncoscience.394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guardia RDdela, González-Silva L, López-Millán B, et al. . Bone marrow clonogenic myeloid progenitors from NPM1-Mutated AML patients do not harbor the NPM1 mutation: implication for the Cell-Of-Origin of NPM1+ AML. Genes 2020;11:73. 10.3390/genes11010073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Creutzig U, van den Heuvel-Eibrink MM, Gibson B, et al. . Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood 2012;120:3187–205. 10.1182/blood-2012-03-362608 [DOI] [PubMed] [Google Scholar]
- 12. Thein MS, Ershler WB, Jemal A, et al. . Outcome of older patients with acute myeloid leukemia: an analysis of SEER data over 3 decades. Cancer 2013;119:2720–7. 10.1002/cncr.28129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hills RK, Castaigne S, Appelbaum FR, et al. . Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol 2014;15:986–96. 10.1016/S1470-2045(14)70281-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee EM, Yee D, Busfield SJ, et al. . Efficacy of an Fc-modified anti-CD123 antibody (CSL362) combined with chemotherapy in xenograft models of acute myelogenous leukemia in immunodeficient mice. Haematologica 2015;100:914–26. 10.3324/haematol.2014.113092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jitschin R, Saul D, Braun M, et al. . CD33/CD3-bispecific T-cell engaging (BiTE®) antibody construct targets monocytic AML myeloid-derived suppressor cells. J Immunother Cancer 2018;6:116. 10.1186/s40425-018-0432-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Al-Hussaini M, Rettig MP, Ritchey JK, et al. . Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood 2016;127:122–31. 10.1182/blood-2014-05-575704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brentjens RJ, Rivière I, Park JH, et al. . Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011;118:4817–28. 10.1182/blood-2011-04-348540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. . T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015;385:517–28. 10.1016/S0140-6736(14)61403-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kochenderfer JN, Wilson WH, Janik JE, et al. . Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010;116:4099–102. 10.1182/blood-2010-04-281931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gill S, Tasian SK, Ruella M, et al. . Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014;123:2343–54. 10.1182/blood-2013-09-529537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jetani H, García-Cadenas I, Nerreter T, et al. . Car T cells targeting FLT3 on AML confer potent anti-leukemic activity and act synergistically with the FLT3 inhibitor midostaurin. Blood 2017;130:1351. [DOI] [PubMed] [Google Scholar]
- 22. Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, et al. . Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia 2014;28:1596–605. 10.1038/leu.2014.62 [DOI] [PubMed] [Google Scholar]
- 23. Tasian SK, Kenderian SS, Shen F, et al. . Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood 2017;129:2395–407. 10.1182/blood-2016-08-736041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kenderian SS, Ruella M, Shestova O, et al. . CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015;29:1637–47. 10.1038/leu.2015.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tettamanti S, Marin V, Pizzitola I, et al. . Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol 2013;161:389–401. 10.1111/bjh.12282 [DOI] [PubMed] [Google Scholar]
- 26. Kim MY, Yu K-R, Kenderian SS, et al. . Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell 2018;173:1439–53. 10.1016/j.cell.2018.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stevens BM, Zhang W, Pollyea DA, et al. . Cd123 CAR T cells for the treatment of myelodysplastic syndrome. Exp Hematol 2019;74:52–63. 10.1016/j.exphem.2019.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luo Y, Chang L-J, Hu Y, et al. . First-In-Man CD123-Specific chimeric antigen receptor-modified T cells for the treatment of refractory acute myeloid leukemia. Blood 2015;126:3778 10.1182/blood.V126.23.3778.3778 [DOI] [Google Scholar]
- 29. Mardiros A, Dos Santos C, McDonald T, et al. . T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood 2013;122:3138–48. 10.1182/blood-2012-12-474056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Budde L, Song JY, Kim Y, et al. . Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-Specific CAR T cells: a first-in-human clinical trial. Blood 2017;130:811. [Google Scholar]
- 31. Bôle-Richard E, Fredon M, Biichlé S, et al. . CD28/4-1BB CD123 CAR T cells in blastic plasmacytoid dendritic cell neoplasm. Leukemia 2020. 10.1038/s41375-020-0777-1. [Epub ahead of print: 28 Feb 2020]. [DOI] [PubMed] [Google Scholar]
- 32. Sánchez-Martínez D, Baroni ML, Gutierrez-Agüera F, et al. . Fratricide-resistant CD1a-specific CAR T cells for the treatment of cortical T-cell acute lymphoblastic leukemia. Blood 2019;133:2291–304. 10.1182/blood-2018-10-882944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gomes-Silva D, Srinivasan M, Sharma S, et al. . CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 2017;130:285–96. 10.1182/blood-2017-01-761320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mamonkin M, Rouce RH, Tashiro H, et al. . A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood 2015;126:983–92. 10.1182/blood-2015-02-629527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bras AE, de Haas V, van Stigt A, et al. . Cd123 expression levels in 846 acute leukemia patients based on standardized immunophenotyping. Cytometry B Clin Cytom 2019;96:134–42. 10.1002/cyto.b.21745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bueno C, Roldan M, Anguita E, et al. . Bone marrow mesenchymal stem cells from patients with aplastic anemia maintain functional and immune properties and do not contribute to the pathogenesis of the disease. Haematologica 2014;99:1168–75. 10.3324/haematol.2014.103580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Menendez P, Catalina P, Rodríguez R, et al. . Bone marrow mesenchymal stem cells from infants with MLL-AF4+ acute leukemia harbor and express the MLL-AF4 fusion gene. J Exp Med 2009;206:3131–41. 10.1084/jem.20091050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rodriguez R, Tornin J, Suarez C, et al. . Expression of FUS-CHOP fusion protein in immortalized/transformed human mesenchymal stem cells drives mixoid liposarcoma formation. Stem Cells 2013;31:2061–72. 10.1002/stem.1472 [DOI] [PubMed] [Google Scholar]
- 39. Ebinger S, Zeller C, Carlet M, et al. . Plasticity in growth behavior of patients' acute myeloid leukemia stem cells growing in mice. Haematologica 2020. 10.3324/haematol.2019.226282. [Epub ahead of print: 06 Feb 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guedan S, Posey AD, Shaw C, et al. . Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018;3 10.1172/jci.insight.96976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ossenkoppele G, Löwenberg B. How I treat the older patient with acute myeloid leukemia. Blood 2015;125:767–74. 10.1182/blood-2014-08-551499 [DOI] [PubMed] [Google Scholar]
- 42. Tasian SK. Acute myeloid leukemia chimeric antigen receptor T-cell immunotherapy: how far up the road have we traveled? Ther Adv Hematol 2018;9:135–48. 10.1177/2040620718774268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ugo Testa, Elvira Pelosi, Germana Castelli Cd123 as a therapeutic target in the treatment of hematological malignancies. Cancers 2019;11:1358 10.3390/cancers11091358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dorantes-Acosta E, Pelayo R. Lineage switching in acute leukemias: a consequence of stem cell plasticity? Bone Marrow Res 2012;2012:1–18. 10.1155/2012/406796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Testa U, Riccioni R, Diverio D, et al. . Interleukin-3 receptor in acute leukemia. Leukemia 2004;18:219–26. 10.1038/sj.leu.2403224 [DOI] [PubMed] [Google Scholar]
- 46. Riccioni R, Diverio D, Riti V, et al. . Interleukin (IL)-3/granulocyte macrophage-colony stimulating factor/IL-5 receptor alpha and beta chains are preferentially expressed in acute myeloid leukaemias with mutated FMS-related tyrosine kinase 3 receptor. Br J Haematol 2009;144:376–87. 10.1111/j.1365-2141.2008.07491.x [DOI] [PubMed] [Google Scholar]
- 47. Al-Mawali A, Gillis D, Lewis I. Immunoprofiling of leukemic stem cells CD34+/CD38-/CD123+ delineate FLT3/ITD-positive clones. J Hematol Oncol 2016;9:61. 10.1186/s13045-016-0292-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Angelini DF, Ottone T, Guerrera G, et al. . A leukemia-associated CD34/CD123/CD25/CD99+ immunophenotype identifies FLT3-Mutated clones in acute myeloid leukemia. Clin Cancer Res 2015;21:3977–85. 10.1158/1078-0432.CCR-14-3186 [DOI] [PubMed] [Google Scholar]
- 49. Guzman ML, Neering SJ, Upchurch D, et al. . Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 2001;98:2301–7. 10.1182/blood.V98.8.2301 [DOI] [PubMed] [Google Scholar]
- 50. Zahran AM, Aly SS, Rayan A, et al. . Survival outcomes of CD34+CD38-LSCs and their expression of CD123 in adult AML patients. Oncotarget 2018;9:34056–65. 10.18632/oncotarget.26118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Arai NH M, Abe M, Baba Y, et al. . Int J Hematol 2019. [Google Scholar]
- 52. Wittwer NL, Brumatti G, Marchant C, et al. . High CD123 levels enhance proliferation in response to IL-3, but reduce chemotaxis by downregulating CXCR4 expression. Blood Adv 2017;1:1067–79. 10.1182/bloodadvances.2016002931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nievergall E, Ramshaw HS, Yong ASM, et al. . Monoclonal antibody targeting of IL-3 receptor α with CSL362 effectively depletes CML progenitor and stem cells. Blood 2014;123:1218–28. 10.1182/blood-2012-12-475194 [DOI] [PubMed] [Google Scholar]
- 54. He SZ, Busfield S, Ritchie DS, et al. . A phase 1 study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leuk Lymphoma 2015;56:1406–15. 10.3109/10428194.2014.956316 [DOI] [PubMed] [Google Scholar]
- 55. Barrett AJ. Antibody darts on target for acute myelogenous leukemia. Ann Transl Med 2017;5:80. 10.21037/atm.2017.01.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Correia DV, Fogli M, Hudspeth K, et al. . Differentiation of human peripheral blood Vδ1+ T cells expressing the natural cytotoxicity receptor NKp30 for recognition of lymphoid leukemia cells. Blood 2011;118:992–1001. 10.1182/blood-2011-02-339135 [DOI] [PubMed] [Google Scholar]
- 57. Rotolo R, Leuci V, Donini C, et al. . CAR-Based strategies beyond T lymphocytes: integrative opportunities for cancer adoptive immunotherapy. Int J Mol Sci 2019;20. 10.3390/ijms20112839. [Epub ahead of print: 11 Jun 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2020-000845supp001.pdf (288.4KB, pdf)
jitc-2020-000845supp002.pdf (63.1KB, pdf)