

Abstract
Aims
As an extension of a phase 2/3 study evaluating the efficacy and safety of lisdexamfetamine dimesylate (LDX) 30, 50, or 70 mg/d for 4 weeks in Japanese patients aged 6‐17 years with attention‐deficit/hyperactivity disorder (ADHD), this study evaluated its long‐term safety and efficacy.
Methods
This was a multicenter, open‐label study of LDX for 53 weeks. Safety was assessed by regular medical examination for treatment‐emergent adverse events (TEAEs); regular recording of body weight, vital signs, and laboratory test values; and completion of dependence questionnaires. Efficacy was assessed using Japanese versions of the ADHD‐Rating Scale‐IV (ADHD‐RS‐IV) and Conners' 3rd edition Parent Rating Scale (Conners 3); plus Clinical Global Impression‐Improvement (CGI‐I), Clinical Global Impression‐Severity, and Parent Global Assessment (PGA) scales.
Results
Of 132 enrolled patients, 104 completed the trial. Most frequent treatment‐related TEAEs were decreased appetite (73.5%), initial insomnia (39.4%), and weight decrease (22.0%). Most TEAEs were mild (82.6% of patients). There were no serious or severe TEAEs or deaths. No treatment‐related TEAEs were associated with blood pressure or pulse rate, and no patient had a QTcF interval >500 ms. Statistically significant improvement from baseline to week 53 was observed in the mean ADHD‐Rating Scale‐IV total score and mean Conners 3 subscale scores. Most patients showed improvement on the CGI‐I (78%) and PGA (76.5%) scales.
Conclusions
No significant safety issues were observed with LDX 30, 50, or 70 mg/d administered for 1 year in Japanese children and adolescents with ADHD. LDX was associated with long‐term reductions in ADHD symptoms and severity.
Keywords: attention‐deficit/hyperactivity disorder, Japanese patients, lisdexamfetamine dimesylate, long‐term efficacy, long‐term safety
This multicenter, open‐label study evaluated the long‐term safety and efficacy of lisdexamfetamine dimesylate (LDX) in Japanese patients aged 6‐17 years with attention‐deficit/hyperactivity disorder. No significant safety issues were observed with LDX 30, 50, or 70 mg/d administered for 1 year. There were no serious or severe treatment‐emergent adverse events (TEAEs) or deaths. The most frequent treatment‐related TEAEs were decreased appetite, initial insomnia, and weight decrease. TEAEs were mild in 82.6% of patients.

1. INTRODUCTION
Attention‐deficit/hyperactivity disorder (ADHD) occurs in about 5%‐7% of children globally, although prevalence rates can vary by region and/or diagnostic criteria.1, 2 Large‐scale studies conducted in Japan have reported similar rates.3, 4 ADHD represents a significant burden to patients with regard to educational and vocational outcomes and to parents, caregivers, healthcare payors, and society as a whole.5
Currently, in Japan, three agents are licensed for treatment of ADHD in children. These are stimulant, osmotic‐release oral system (OROS) methylphenidate; and two nonstimulants, atomoxetine and extended‐release guanfacine. Pharmacologic treatment of ADHD is highly individualized taking into account a child's specific symptoms and comorbid conditions.6 As children can vary considerably in terms of their ability to respond to or tolerate ADHD medications, a wider range of options may be useful to facilitate personalized therapy.
Lisdexamfetamine dimesylate (LDX) is a pharmacologically inactive prodrug which, after oral administration, is rapidly absorbed from the gastrointestinal tract and enzymatically hydrolyzed mainly in the blood to release therapeutically active dexamphetamine and the amino acid l‐lysine.7, 8 As conversion of LDX to dexamphetamine is gradual, LDX is reported to have a clinical effect duration of up to 13 hours.7, 9 Slower release of dopamine in the central nervous system may translate to less euphoric activity and lower potential for abuse.11, 12
Lisdexamfetamine dimesylate has demonstrated efficacy in several large‐scale well‐designed studies in pediatric ADHD13 and is currently marketed for these indications in numerous countries. In long‐term open‐label safety studies of up to 104 weeks, the tolerability profile of LDX (at doses of 30‐70 mg/d) was similar to that reported in short‐term randomized trials and consistent with that of other stimulant medications.13 Treatment with LDX for 2 years was not associated with any clinically concerning trends in pubertal development,14 or with deterioration in cognitive function.15
An early phase I study of LDX revealed no differences between Japanese and Caucasian individuals in the safety and tolerability of 20, 50, and 70 mg doses (data on file, Shionogi). A subsequent phase 2, 4‐week, open‐label study in Japanese children and adolescents (aged 6‐17 years) with ADHD reported efficacy for LDX as early as 1 week after the start of treatment and a safety profile similar to that documented in other worldwide studies (data on file, Shionogi). Recently, a placebo‐controlled, randomized, double‐blind, fixed‐dose (30, 50, or 70 mg/d), phase 2/3 trial in Japanese pediatric patients with ADHD demonstrated marked efficacy for LDX over 4 weeks with no major safety or tolerability concerns.16 As an extension of the phase 2/3 trial, this open‐label study was conducted to evaluate the long‐term safety of LDX 30, 50, and 70 mg/d in Japanese children and adolescents with ADHD.
2. METHODS
2.1. Study design
This was a multicenter, open‐label, dose‐optimized, long‐term study of LDX comprising a 53‐week treatment period and a 1‐week follow‐up period for a total of 18 study visits (Figure 1). The study was conducted in accordance with the Ministerial Ordinance on Good Clinical Practice (GCP) of the Pharmaceutical Affairs Law of Japan. The study protocol was approved by each center's institutional review board. Study participants were children and adolescents with ADHD who had completed an earlier phase 2/3 study of LDX and who wished to continue LDX treatment, plus newly enrolled children and adolescents with ADHD. After receiving a thorough description of the study, written informed consent was obtained from the parent or legal guardian of all patients. Written informed assent was also obtained from patients aged ≥13 years.
Figure 1.
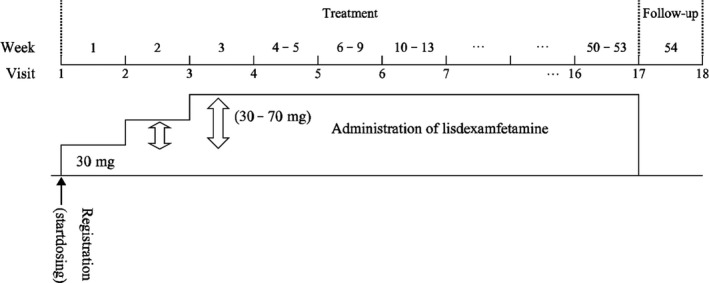
Study design
From study visit 1, patients began LDX treatment at a dosage of 30 mg once daily in the morning. During the treatment period, the dosage was increased or decreased in 20 mg increments or decrements, within the range 30‐70 mg/d, according to the following criteria: The dosage was increased if the Clinical Global Impression‐Improvement (CGI‐I) score was “minimally improved” (or less) and in the absence of safety concerns; the dosage was decreased if the patient had hypertension, or a pulse rate ≥160 beats/min (age <15 years) or ≥110 beats/min (age ≥15 years); the dosage was reduced (50 or 70 mg dosing), or LDX was discontinued (30 mg dosing) if a safety issue was identified.
Lisdexamfetamine dimesylate safety, the primary objective, was evaluated by regular medical examination for adverse events, by regular recording of bodyweight, blood pressure, electrocardiogram (ECG), pulse rate, and laboratory test values, and by completion of dependence questionnaires (D‐2‐A, D‐2‐B). LDX efficacy was assessed as a secondary objective using Japanese versions of the ADHD‐Rating Scale‐IV (ADHD‐RS‐IV) and Conners' 3rd edition Parent Rating Scale (Conners 3), plus the CGI‐I scale, Clinical Global Impression‐Severity (CGI‐S) scale, and Parent Global Assessment (PGA) scale.
2.2. Study participants
Study participants were male or female children or adolescents (aged 6‐17 years) meeting Diagnostic and Statistical Manual of Mental Disorders, 5th edition, criteria for the principal diagnosis of ADHD.17 To be eligible for inclusion, patients had to be functioning at an age‐appropriate intellectual level, able to swallow capsules, and at least “moderately ill” on the CGI‐S scale at the study registration visit. In addition, patients were required to have thyroid‐stimulating hormone and free thyroxine levels within normal ranges. Principal exclusion criteria were as follows: serious metabolic disease; serious disorders of the blood or bone marrow, heart, kidneys, liver, and lungs; psychiatric comorbidity (eg, bipolar disorder, schizophrenia); conduct disorder (excluding oppositional defiant disorder); current tics; history of seizures; low or high bodyweight; hypertension; QTc interval (Fridericia adjusted; QTcF) >430 ms; substance use disorder; and pregnancy or lactation.
2.3. Study objectives
The primary study objective was to evaluate the long‐term safety of LDX 30, 50, and 70 mg/d in Japanese pediatric patients with ADHD. Secondary study objectives were to assess the long‐term efficacy of LDX 30, 50, and 70 mg/d in this population.
2.4. Study assessments
All adverse events identified from provision of signed informed consent to the end of follow‐up (visit 18) were investigated, and all adverse events reported after the initial dose of study drug were considered to be treatment‐emergent adverse events (TEAEs). After trial commencement, an independent safety evaluation committee was established for impartial assessment of LDX dependence. Prior to database lock, the committee assessed dependence and withdrawal symptoms according to information recorded on case record forms including the D‐2‐A and D‐2‐B survey results.
A Japanese language translation of the ADHD‐RS‐IV encompassing 18 items of the DSM‐IV‐Text Revision was used to assess symptoms.18, 19, 20, 21 At each study visit, an investigator evaluated a patient's home and classroom behavior by grading each item of the ADHD‐RS‐IV on a 4‐point scale: “never or rarely” (0 points), “sometimes” (1), “often” (2), or “very often” (3). Behavior (over the past 2 weeks at visit 1, and from the previous to current visit at all other time points) was assessed by interviewing a parent or guardian of each patient.
From visit 1 onwards, parents or guardians used the Japanese version 110‐item Conners 322, 23 to assess ADHD‐related symptoms on a 4‐category scale: “Not true at all. It never (or seldom) happened” (0 points); “Just a little true. It happened occasionally” (1); “Pretty much true. It happened often (or quite a bit)” (2); or “Very much true. It happened very often (very frequently)” (3).
From visit 2 onwards, improvement in a patient's ADHD symptoms was assessed using the 7‐grade CGI‐I (investigators) and PGA (parents or guardians) scales: “very much improved,” “much improved,” “minimally improved,” “no change,” “minimally worse,” “much worse,” or “very much worse.” At baseline and throughout the trial, investigators assessed the severity of a patient's ADHD symptoms using the 7‐category CGI‐S scale: “normal, not at all ill,” “borderline ill,” “mildly ill,” “moderately ill,” “markedly ill,” “severely ill,” or “extremely ill.”
2.5. Statistical analyses
Safety analyses were performed in the safety analysis population, which was defined as all enrolled participants except those with major GCP noncompliance (eg, informed assent not obtained, attendance at a center without Institutional Review Board review); those not receiving study treatment; or those with no postbaseline safety data. Efficacy analyses were performed in the modified intent‐to‐treat (mITT) population, which was defined as all patients except those with major GCP noncompliance; those not receiving study treatment; or those with no observations (ie, ADHD‐RS‐IV total score not evaluated at baseline or from the start of study drug dosing to visit 17). Unless otherwise stated, continuous variables are summarized using mean and standard deviation (SD), and qualitative variables are described using number and percentage of patients by category. The Clopper‐Pearson method was used to calculate confidence intervals (CIs) of rates. CIs for continuous values were calculated based on the t‐distribution. Unless otherwise stated, two‐sided 95% CIs were calculated. A P value of <.05 indicates statistical significance.
3. RESULTS
3.1. Study population
A total of 132 patients were enrolled in the study and received LDX, 69 of whom had completed the preceding phase 2/3 trial, and 63 of whom were newly enrolled (Figure 2). Overall, 104 patients (78.8%) completed the trial. Of the 28 patients (21.2%) who discontinued the study, reasons were adverse events (12 patients), request for withdrawal by patient or guardian (7), inadequate efficacy (4), or “other” (5). Sixteen of these patients were continuing from the phase 2/3 trial, and 12 were newly enrolled; the reasons for discontinuation were similar in both groups (Table 1).
Figure 2.
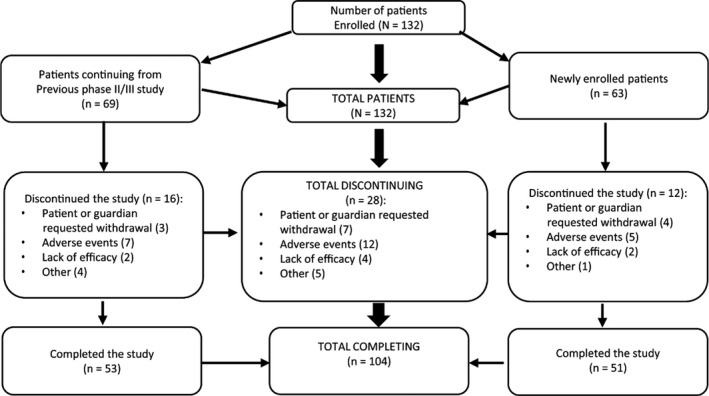
Patient disposition
Table 1.
Patient dispositiona
| Continuing ptsb | New ptsc (n = 63) | All pts (n = 132) | ||||
|---|---|---|---|---|---|---|
| LDX 30 mg (n = 18) | LDX 50 mg (n = 16) | LDX 70 mg (n = 16) | Placebo (n = 19) | |||
| Completed | 12 (66.7) | 14 (87.5) | 14 (87.5) | 13 (68.4) | 51 (81.0) | 104 (78.8) |
| Discontinued | 6 (33.3) | 2 (12.5) | 2 (12.5) | 6 (31.6) | 12 (19.0) | 28 (21.2) |
| Reasons for discontinuation | ||||||
| Ineligible | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Lost to follow‐up | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Withdrawal by patient or representative | 2 (11.1) | 0 (0.0) | 0 (0.0) | 1 (5.3) | 4 (6.3) | 7 (5.3) |
| Adverse events | 3 (16.7) | 0 (0.0) | 2 (12.5) | 2 (10.5) | 5 (7.9) | 12 (9.1) |
| Lack of efficacy | 0 (0.0) | 1 (6.3) | 0 (0.0) | 1 (5.3) | 2 (3.2) | 4 (3.0) |
| Other | 1 (5.6) | 1 (6.3) | 0 (0.0) | 2 (10.5) | 1 (1.6) | 5 (3.8) |
Abbreviations: LDX, lisdexamfetamine; pts, patients.
Data shown are n (%) of pts.
Pts from previous phase 2/3 study of LDX.
Pts newly enrolled for the current study.
Mean treatment compliance was 95.4%, with most patients (96.2%) being ≥80% compliant with treatment.
3.2. Patient demographics
Baseline characteristics of the study population are shown in Table 2. The population was majority male (84.1%), and mean age (±SD) was 10.5 (±2.9) years. Approximately three‐quarters of patients (72.7%) were aged <13 years. Most patients had ADHD of the combined (58.3%) or predominantly inattentive (38.6%) subtype. The mean baseline ADHD‐RS‐IV total score was 34.7 (±8.1). Most patients (73.5%) had an ADHD‐RS‐IV total score <40 at baseline.
Table 2.
Baseline characteristics of the study population
| Characteristic | Continuing pts (n = 69) | Newly enrolled pts (n = 63) | Total pts (n = 132) |
|---|---|---|---|
| Male gender, n (%) | 58 (84.1) | 53 (84.1) | 111 (84.1) |
| Age, y; mean (SD) | 9.9 (2.7) | 11.1 (3.0) | 10.5 (2.9) |
| Height, cm; mean (SD)a | 137.5 (15.4) | 144.8 (17.6) | 141.0 (16.8) |
| Bodyweight, kg; mean (SD)a | 35.1 (12.6) | 41.1 (15.4) | 38.0 (14.3) |
| Previous medical conditions; n (% pts)a | 14 (20.3) | 21 (33.3) | 35 (26.5) |
| Concurrent medical conditions; n (% pts)a | 53 (76.8) | 48 (76.2) | 101 (76.5) |
| Previous drug treatment; n (% pts)a | 22 (31.9) | 28 (44.4) | 50 (37.9) |
| ADHD subtype; n (% pts) | |||
| Combined | 45 (65.2) | 32 (50.8) | 77 (58.3) |
| Predominantly inattentive | 22 (31.9) | 29 (46.0) | 51 (38.6) |
| Predominantly hyperactive‐impulsive | 2 (2.9) | 2 (3.2) | 4 (3.0) |
| ADHD‐RS‐IV total score <40 at baseline; n (% pts)a | 44 (63.8) | 53 (84.1) | 97 (73.5) |
| ADHD‐RS‐IV total score ≥40 at baseline; n (% pts)a | 25 (36.2) | 10 (15.9) | 35 (26.5) |
| Baseline ADHD‐RS‐IV total score; mean (SD)a | 37.8 (7.0) | 31.3 (8.0) | 34.7 (8.1) |
| Baseline ADHD‐RS‐IV inattention subscale score; mean (SD)a | 22.3 (4.0) | 20.3 (3.7) | 21.3 (4.0) |
| Baseline ADHD‐RS‐IV hyperactivity‐impulsivity subscale score; mean (SD)a | 15.5 (6.3) | 11.1 (6.1) | 13.4 (6.6) |
Abbreviations: ADHD, attention‐deficit/hyperactivity disorder; ADHD‐RS‐IV, ADHD‐Rating Scale‐IV; pts, patients; SD, standard deviation.
Values from an antecedent phase 2/3 study15 were used for continuing pts, and values measured in the current study were used for newly enrolled pts.
3.3. Safety
The mean duration of treatment was 324 days, and 104 patients (78.8%) received LDX for ≥52 weeks. The final LDX dosage was 30 mg/d (22.0% of patients), 50 mg/d (37.9%), or 70 mg/d (40.2%).
In the safety population (n = 132), 12 patients (9.1%) had 18 TEAEs that led to treatment discontinuation. These were as follows: decreased appetite (6 events), initial insomnia (2), weight decrease (2), insomnia (1), circadian rhythm sleep disorder (1), somnolence (1), nausea (1), vomiting (1), increased heart rate (1), QT prolongation (1), and decreased blood pressure (1). Of these events, four episodes of decreased appetite, two of initial insomnia, and one each of vomiting, somnolence, nausea, and insomnia were classified as moderate; all other events were mild in intensity. All patients who discontinued LDX due to TEAEs recovered or the adverse event resolved. Except for decreased appetite (2 events) and insomnia (1), no other TEAE required treatment with medication. No death, serious TEAE, or severe TEAE was reported during the study.
Of 130 patients in the safety population who experienced TEAEs, 112 (84.8%) recovered, six (4.5%) were recovering at the time of last assessment, and 12 (9.1%) had not yet achieved resolution. Unresolved TEAEs were weight decrease (6 events), initial insomnia (3), anemia (1), decreased appetite (1), tachycardia (1), dental caries (1), acne (1), atopic dermatitis (1), prurigo (1), maculopapular rash (1), osteochondrosis (1), menorrhagia (1), irregular menstruation (1), and skin abrasion (1). In the majority of patients (119/132 patients; 90.2%), TEAEs occurred during the first 9 weeks of LDX administration. The most frequent treatment‐related TEAEs reported within this time frame were decreased appetite (87 events), initial insomnia (45), weight decrease (14), influenza (1), and headache (11).
Treatment‐emergent adverse events with an incidence of ≥5% are summarized in Table 3. Events of nasopharyngitis, influenza, contusion, and gastroenteritis were considered unrelated to study drug. Most TEAEs were classified as mild (82.6% of patients) or moderate (15.9%) in intensity. Moderate TEAEs with an incidence of >1% were decreased appetite (7.6% of patients), initial insomnia (2.3%), influenza (1.5%), and insomnia (1.5%).
Table 3.
Treatment‐emergent adverse events with an incidence of ≥5%a
| Preferred term | All LDX dosages (n = 132) |
|---|---|
| Patients with any TEAE | 130 (98.5) |
| Decreased appetite | 97 (73.5) |
| Initial insomnia | 52 (39.4) |
| Nasopharyngitis | 50 (37.9) |
| Weight decrease | 29 (22.0) |
| Influenza | 19 (14.4) |
| Headache | 17 (12.9) |
| Abdominal pain | 11 (8.3) |
| Diarrhea | 10 (7.6) |
| Gastroenteritis | 10 (7.6) |
| Constipation | 9 (6.8) |
| Contusion | 9 (6.8) |
| Stomatitis | 8 (6.1) |
| Nausea | 7 (5.3) |
| Tachycardia | 7 (5.3) |
Abbreviations: LDX, lisdexamfetamine; TEAEs, treatment‐emergent adverse events.
Number (%) of patients from the safety population with TEAEs.
From baseline to last observation, mean increases of 1.58 mm Hg for systolic blood pressure, 3.72 mm Hg for diastolic blood pressure, and 8.95 beats/min for pulse rate were recorded in the safety population. TEAEs related to blood pressure or pulse rate included tachycardia (7 patients), increased heart rate (5), palpitations (3), postural hypotension (2), increased blood pressure (2), hypertension (1), and decreased blood pressure (1). One event each of decreased blood pressure and increased heart rate led to study drug discontinuation, but both events resolved without any specific intervention. There was a mean change of −5.06 ms in the QTcF interval from baseline to last observation in the safety population. Nine patients had a QTcF interval of >430 ms at any time point, but only one of these patients discontinued study treatment; no patient had a QTcF interval >500 ms at any time point.
Mean change in bodyweight from baseline to the last evaluation point in the safety population was −0.72 (±3.44) kg. Mild “bodyweight decreased” events were recorded in 29 patients (22.0%) and were assessed as TEAEs. Two of these patients discontinued study treatment, and the TEAE resolved without additional measures. Higher final LDX dosages did not lead to greater bodyweight changes: Mean bodyweight changes with LDX 30, 50, and 70 mg final doses were −0.81 kg (n = 29), −1.46 kg (n = 50), and +0.02 kg (n = 53), respectively. The mean height change from baseline to last observation in the safety population was an increase of 3.43 (±2.36) cm. Mean height increases with LDX 30, 50, and 70 mg final doses were 2.57 cm (n = 29), 3.48 cm (n = 50), and 3.85 cm (n = 53), respectively.
There were no substantial changes in laboratory parameters during the treatment period.
After reviewing data for all patients, the independent safety evaluation committee found no suspected cases of drug dependence development or withdrawal symptoms.
A total of 28 events of special situations were reported in 20 patients, including 17 medication errors in 14 patients, nine events of misuse in eight patients, and two overdoses in two patients. All medication errors were violations of the up‐down titration criteria, while most instances of misuse resulted from dose omissions. There were no safety concerns in patients with violations of the up‐down titration criteria, or in patients with dose omissions; all patients, except for two with reported misuse, continued the study. Two patients with reported overdose took two LDX 70 mg capsules on the same day by mistake; no adverse events occurred, and the patients continued the study.
3.4. Efficacy
The time course of mean change from baseline in the ADHD‐RS‐IV total score is shown in Figure 3A. At week 53, mean improvement was −20.05 (95% CI: −22.02, −18.07); the corresponding value at last observation was −17.93 (95% CI: −19.75, −16.11). At all time points from week 1 to week 53 and last observation, the upper limit of the 95% CI for mean change vs baseline in the ADHD‐RS‐IV total score was below zero, indicating statistical significance (P < .05). Similar findings were noted for the ADHD‐RS‐IV inattention subscale score (mean changes −11.57 [week 53], and −10.33 [last observation]; Figure 3B) and for the ADHD‐RS‐IV hyperactivity‐impulsivity subscale score (mean changes −8.48 [week 53], and −7.60 [last observation]; Figure 3C).
Figure 3.
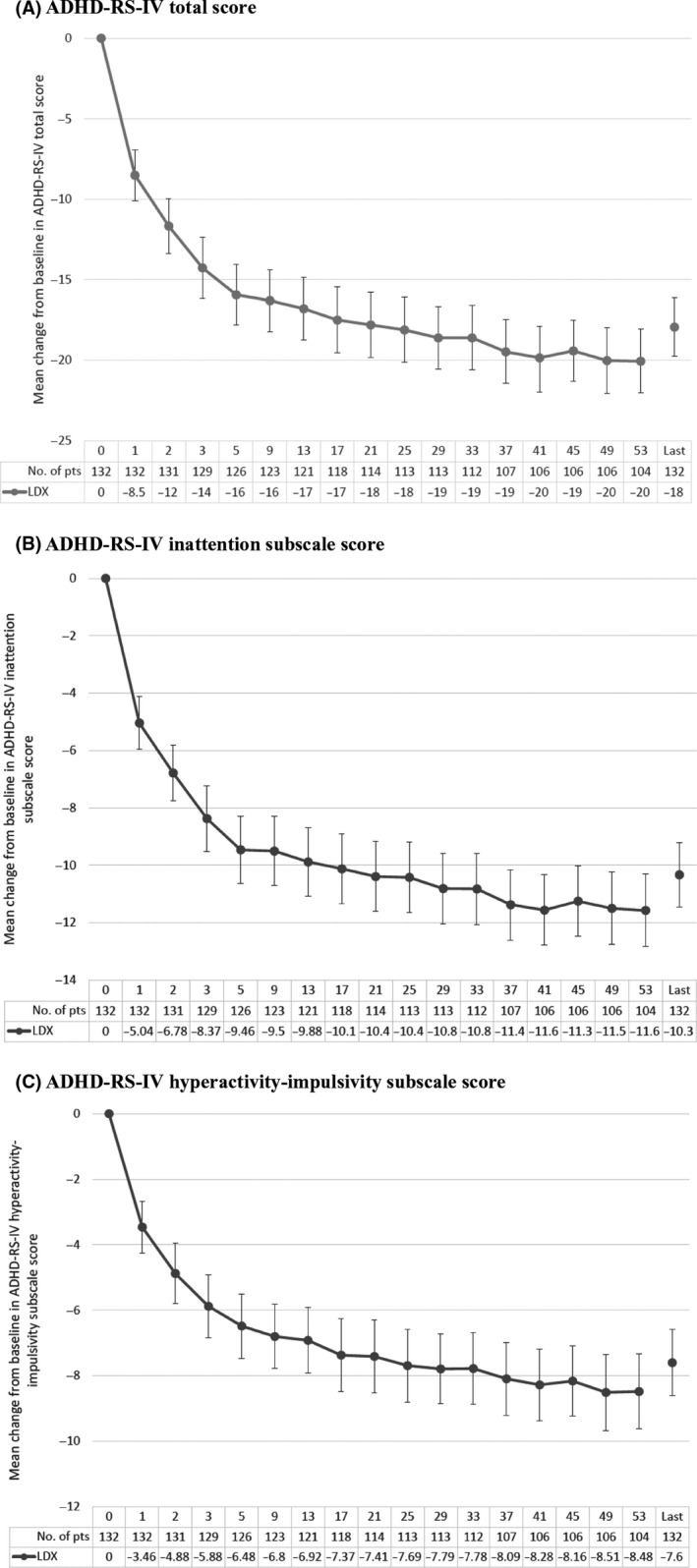
Changes from baseline in ADHD‐RS‐IV scores (mean ± 95% confidence intervals). A, Total score; B, Inattention subscale score; C, Hyperactivity‐impulsivity subscale score. Data tabulated beneath the figures are study week number, then numbers of patients (pts) at each time point, and then mean decrease from baseline in ADHD‐RS‐IV total score with lisdexamfetamine (LDX)
All Conners 3 subscale scores were significantly improved (P < .05) from baseline at each evaluation time point. Mean changes (95% CIs) from baseline to last observation in the inattention, hyperactivity‐impulsivity, conduct disorder, oppositional defiant disorder, and inattention plus hyperactivity‐impulsivity subscale scores were −6.19 (−7.47, −4.90), −7.36 (−8.75, −5.96), −1.38 (−2.08, −0.68), −2.59 (−3.55, −1.63), and −13.55 (−15.91, −11.18), respectively. Improvement was most marked in the Conners 3 inattention plus hyperactivity‐impulsivity subscale (Table 4).
Table 4.
Changes from baseline to last observation in Conners 3 inattention plus hyperactivity‐impulsivity subscale scores
| Time point (wk) | Number of pts | Actual value; mean (SD) | Mean change from baseline (SD) | 95% CI |
|---|---|---|---|---|
| Baseline | 132 | 34.14 (13.28) | — | — |
| Week 13 | 122 | 22.29 (11.34) | −12.18 (11.99) | −14.33, −10.03 |
| Week 25 | 113 | 21.43 (11.59) | −13.37 (12.42) | −15.69, −11.06 |
| Week 37 | 107 | 21.02 (12.00) | −14.35 (12.88) | −16.81, −11.88 |
| Week 53 | 104 | 19.83 (11.57) | −15.48 (13.28) | −18.06, −12.90 |
| Last observation | 132 | 20.60 (11.66) | −13.55 (13.75) | −15.91, −11.18 |
Abbreviations: CI, confidence interval; Conners 3, Japanese version Conners' 3rd edition Parent Rating Scale; pts, patients; SD, standard deviation.
The CGI‐I rate (ie, the proportion of patients “much improved” or “very much improved” at each assessment point) increased gradually from week 1 (36.4%) to week 5 (76.2%), then stabilized at around 75%. The CGI‐I rate (95% CI) was 86.5% (95% CI: 78.4%, 92.4%) at week 53 and 78.0% (95% CI: 70.0%, 84.8%) at last observation. Similar findings were evident for the PGA. The proportion of patients who were “much improved” or “very much improved” at each assessment point increased gradually from week 1 (34.8%) to week 5 (73.8%), then stabilized at around 70%. The PGA improvement rate was 86.5% (95% CI: 78.4%, 92.4%) at week 53 and 76.5% (95% CI: 68.4%, 83.5%) at last observation. At baseline, 1 patient (0.8%) had a CGI‐S rating of “normal, not at all ill” or “borderline ill”; this increased to 37 patients (28.0%) at last observation.
4. DISCUSSION
This multicenter, open‐label, dose‐optimized, long‐term study indicated that LDX offers a safe and effective therapeutic alternative for treatment of Japanese children and adolescents with ADHD. Consistent with findings in other long‐term (up to 2 years) studies of LDX in non‐Japanese children and adolescents with ADHD,24, 25 we observed a low discontinuation rate (9.1%) of LDX due to TEAEs despite nearly 80% of patients having been titrated to optimized final doses of 50 or 70 mg/d. TEAEs were predominantly mild and, similar to that observed in other long‐term studies, occurred mainly during the first several weeks of treatment (within 9 weeks in 90.2% of patients), suggesting that patients accustomize to LDX with continued use. The most frequent treatment‐related TEAEs of decreased appetite, initial insomnia, and weight decrease are expected with the stimulant class of ADHD medication and are consistent with the safety profile of LDX reported in other long‐term studies.24, 25 There were no TEAEs related to height, no treatment‐related TEAEs associated with blood pressure or pulse rate, and no patient had a QTcF interval >500 ms at any time point. There were no suspected cases of drug dependence development. Improvements from baseline were observed in all efficacy measures which included the ADHD‐RS‐IV, Conners 3, CGI‐I, CGI‐S, and PGA scales.
The favorable long‐term safety and efficacy we observed with LDX in Japanese patients with ADHD concur with results of similar long‐term studies in non‐Japanese populations. For instance, among 272 children aged 6‐12 years with ADHD in the United States (US), no clinically meaningful changes were observed in blood pressure or ECG parameters with LDX 30‐70 mg/d, and the mean ADHD‐RS total score was significantly improved (−27.2 points; P < .0001). Most common TEAEs were decreased appetite, headache, weight decrease, and insomnia.24 Likewise, in a study of 269 US adolescents with ADHD treated with LDX, no clinically meaningful changes were observed in vital signs or ECG parameters, and the mean ADHD‐RS‐IV total score was significantly improved (−26.2 points; P < .001). Most common TEAEs were upper respiratory infection, decreased appetite, headache, and weight decrease.25 Finally, a phase IV study from Europe which evaluated open‐label, dose‐optimized LDX 30‐70 mg/d over 2 years in 314 children aged 6‐17 years reported no new safety signals across an extensive range of assessments, and a mean improvement of −25.8 points (P < .001) in the ADHD‐RS‐IV total score. Most common TEAEs considered related to study drug were decreased appetite, weight decrease, and insomnia.26
Much contention exists as to whether stimulants influence growth in prepubertal children.27, 28 A follow‐up of the Multimodal Treatment Study in the United States reported a small (1.29 cm) adult height decrement associated with extended use of stimulant medication in patients with ADHD relative to controls.29 However, other longitudinal studies have not identified any links between stimulant treatment in ADHD and the magnitude of peak height velocity or final adult height.27, 28 In our study, mean height increased by 3.43 cm during the study period. In the absence of a control group, further investigation is warranted to confirm the impact of long‐term LDX treatment on growth in Japanese children and adolescents with ADHD.
In the current study, the independent safety evaluation committee found no suggestion of development of drug dependence. Studies in non‐Japanese populations have also reported low rates of nonmedical use of prescription stimulants. For instance, in almost 150 000 assessments from the US National Addictions Vigilance Intervention and Prevention Program system, previous 30 day nonmedical use of prescription stimulants (1.3%) was significantly less than that of prescription opioids (19.8%) or sedatives (10.6%).30 Among stimulants, nonmedical use of LDX and OROS methylphenidate appeared to be considerably lower than that of short‐acting and long‐acting mixed amphetamine salts, and of short‐acting methylphenidate. The principal source of diversion was family or friends, and the main route of administration was oral. A smaller‐scale survey (n = 10 000) reported similar findings.31
Our study demonstrated that LDX in the indicated dosage range of 30‐70 mg/d significantly reduced the mean ADHD‐RS‐IV total score and ADHD‐RS‐IV inattention and hyperactivity‐impulsivity subscale scores from baseline over 53 weeks in Japanese children and adolescents aged 6‐17 years with ADHD; reductions were evident as early as 1 week after treatment start and were maintained throughout the trial. All Conners 3 subscale scores (particularly inattention plus hyperactivity‐impulsivity subscale scores) were significantly reduced from baseline at each evaluation time point and, at last observation, more than three‐quarters of patients were either "much improved" or “very much improved” on the CGI‐I and PGA scales.
Study limitations include the open‐label design, modest sample size, comparatively limited observation period of 53 weeks, and “mixed” population of patients either continuing from a previous phase 2/3 trial (52.3%) or newly enrolled (47.7%). Ideally, all patients would have been newly enrolled, and hence without potentially confounding conditioning from previous clinical study experience. Notwithstanding, our study provides important evidence of the safety and efficacy of LDX in Japanese pediatric patients with ADHD. Additional studies, or pharmacovigilance assessments, would be appropriate in this population to confirm that LDX lacks major potential to cause growth retardation or adverse cardiovascular effects (eg, QTcF prolongation),32 or to be linked with abuse liability. Further studies specifically in Japanese patients with ADHD would also be useful to clearly define the overall place in therapy of LDX.
Our study highlighted the absence of significant safety concerns associated with long‐term administration of LDX 30, 50, or 70 mg/d in Japanese children and adolescents (aged 6‐17 years) with ADHD. The efficacy results confirmed that treatment with LDX at these dosages for 1 year was associated with sustained reductions in ADHD symptoms and severity.
The prevalence of ADHD in Japan has been estimated at 7.7% in children aged 4‐12 years living in the greater Tokyo area.4 However, only three medicines are approved in Japan for treatment of ADHD: OROS methylphenidate, atomoxetine, and extended‐release guanfacine. Alternative options are needed for patients who may not respond adequately to these medications in order to reduce the burden of disease. The major clinical significance of this multicenter, open‐label, dose‐optimized study is that it demonstrates that LDX has favorable long‐term safety and tolerability profiles in Japanese children and adolescents with ADHD. No significant safety issues were associated with administration of LDX 30, 50, or 70 mg/d for more than 1 year, and these treatment schedules led to long‐term reductions in ADHD symptoms and severity. The results signify that LDX may be a valuable therapeutic alternative to enhance ADHD management in Japanese children and adolescents.
CONFLICT OF INTEREST
HI received research and funding support from Shionogi & Co. Ltd. and Shire Development LLC during conduct of the study; receives or has received lecture, manuscript and other fees from AbbVie GK, Eli Lilly Japan KK, Hisamitsu Pharmaceutical Co. Inc, Janssen Pharmaceutical KK, Meiji Seika Pharma Co. Ltd., Otsuka Pharmaceutical Co. Ltd., Shionogi & Co. Ltd., Shire Development LLC, and Taisho Pharmaceutical Co. Ltd. outside the submitted work. TM received research and funding support from Shionogi & Co. Ltd. and Shire Development LLC during conduct of the study; receives or has received lecture, consulting and other fees from Astellas Pharma Inc, Eli Lilly Japan KK, Otsuka Pharmaceutical Co. Ltd., Shionogi & Co. Ltd. and Shire Development LLC outside the submitted work. YY received research and funding support from Shionogi & Co. Ltd. and Shire Development LLC during conduct of the study; receives or has received lecture, editorial supervising and other fees from Eli Lilly Japan KK, Janssen Pharmaceutical KK, Otsuka Pharmaceutical Co. Ltd., Shionogi & Co. Ltd., Shire Development LLC, and Taisho Pharmaceutical Co. Ltd. outside the submitted work. MF and AF are employees of Shionogi & Co. Ltd and own Shionogi shares. KS received research and funding support from Shionogi & Co. Ltd. and Shire Development LLC during conduct of the study; receives or has received lecture, editorial supervising and other fees from Eli Lilly Japan KK, Hisamitsu Pharmaceutical Co. Inc, Janssen Pharmaceutical KK, Meiji Seika Pharma Co. Ltd., Mitsubishi Tanabe Pharma Corporation, Otsuka Pharmaceutical Co. Ltd., Shionogi & Co. Ltd., Shire Development LLC, Shire Japan KK, Sumitomo Dainippon Pharma Co. Ltd., Taisho Pharmaceutical Co. Ltd., and Yoshitomiyakuhin Corporation outside the submitted work.
AUTHOR CONTRIBUTIONS
HI, TM, YY, MF, AF, and KS contributed to the conception and design of the study, analysis and interpretation of the data, drafting of the manuscript, critical review, and final approval of the version to be published. YY contributed to acquisition of the data. The authors alone are accountable for all aspects of the work.
DATA REPOSITORY
Researchers can request access to detailed information about Shionogi's clinical trials, including trial protocols and individual patient data, on the portal site https://clinicalstudydatarequest.com. Sharable information includes data about Shionogi's clinical trials conducted in patients in Japan. The information will become sharable after the medicinal products for which the trials are performed have been approved in Japan. Note that all documents will be provided in Japanese language as they have been prepared in Japanese.
APPROVAL OF THE RESEARCH PROTOCOL BY AN INSTITUTIONAL REVIEWER BOARD
The study protocol was approved by each center's institutional review board.
INFORMED CONSENT
After receiving a thorough description of the study, written informed consent was obtained from the parent or legal guardian of all patients. Written informed assent was also obtained from patients aged ≥13 years.
REGISTRY AND THE REGISTRATION NO. OF THE STUDY/TRIAL
This clinical trial is registered at the Japan Pharmaceutical Information Center (JapicCTI‐152771).
DISCLAIMER
This information or content and conclusions are those of the authors.
ACKNOWLEDGMENTS
The authors wish to thank the patients and investigators who took part in the study. The authors thank Content Ed Net for their writing and editorial assistance in the preparation of this manuscript, with funding from Shionogi & Co. Ltd, Osaka, Japan and Shire Development LLC, Lexington, Massachusetts, USA.
Ichikawa H, Miyajima T, Yamashita Y, Fujiwara M, Fukushi A, Saito K. Long‐term study of lisdexamfetamine dimesylate in Japanese children and adolescents with attention‐deficit/hyperactivity disorder. Neuropsychopharmacol Rep. 2020;40:52–62. 10.1002/npr2.12091
Clinical Trial Registry: This clinical trial is registered at the Japan Pharmaceutical Information Center (JapicCTI‐152771).
REFERENCES
- 1. Thomas R, Sanders S, Doust J, Beller E, Glasziou P. Prevalence of attention‐deficit/hyperactivity disorder: a systematic review and meta‐analysis. Pediatrics. 2015;135:e994–1001. [DOI] [PubMed] [Google Scholar]
- 2. Taylor E. Attention deficit hyperactivity disorder: overdiagnosed or diagnoses missed? Arch Dis Child. 2017;102:376–9. [DOI] [PubMed] [Google Scholar]
- 3. Special Needs Education Division, the Elementary and Secondary Education Bureau, Japanese Ministry of Education, Culture, Sports, Science, and Technology . Results of research about children/students with probable developmental disabilities who need special educational support in the regular class: December 5, 2012 [cited 2018 May 31]. Available from ; (Report) http://www.mext.go.jp/a_menu/shotou/tokubetu/material/1328729.htmhttp://www.mext.go.jp/a_menu/shotou/tokubetu/material/__icsFiles/afieldfile/2012/12/10/1328729_01.pdf
- 4. Yamashita Y. Prevalance of ADHD. Japan J Clin Psychopharmacol. 2005;8:871–4 (in Japanese). [Google Scholar]
- 5. Zimovetz EA, Beard SM, Hodgkins P, Bischof M, Mauskopf JA, Setyawan J. A cost‐utility analysis of lisdexamfetamine versus atomoxetine in the treatment of children and adolescents with attention‐deficit/hyperactivity disorder and inadequate response to methylphenidate. CNS Drugs. 2016;30:985–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Felt BT, Biermann B, Christner JG, Kochhar P, Harrison RV. Diagnosis and management of ADHD in children. Am Fam Physician. 2014;90:456–64. [PubMed] [Google Scholar]
- 7. Steer C, Froelich J, Soutullo CA, Johnson M, Shaw M. Lisdexamfetamine dimesylate: a new therapeutic option for attention‐deficit hyperactivity disorder. CNS Drugs. 2012;26:691–705. [DOI] [PubMed] [Google Scholar]
- 8. Hutson PH, Pennick M, Secker R. Preclinical pharmacokinetics, pharmacology and toxicology of lisdexamfetamine: a novel d‐amphetamine pro‐drug. Neuropharmacology. 2014;87:41–50. [DOI] [PubMed] [Google Scholar]
- 9. Adler LA, Alperin S, Leon T, Faraone SV. Pharmacokinetic and pharmacodynamic properties of lisdexamfetamine in adults with attention‐deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2017;27:196–9. [DOI] [PubMed] [Google Scholar]
- 10. Dolder PC, Strajhar P, Vizeli P, Hammann F, Odermatt A, Liechti ME. Pharmacokinetics and pharmacodynamics of lisdexamfetamine compared with d‐amphetamine in healthy subjects. Front Pharmacol. 2017;8:617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jasinski DR, Krishnan S. Abuse liability and safety of oral lisdexamfetamine dimesylate in individuals with a history of stimulant abuse. J Psychopharmacol. 2009;23:419–27. [DOI] [PubMed] [Google Scholar]
- 12. Heal DJ, Buckley NW, Gosden J, Slater N, France CP, Hackett D. A preclinical evaluation of the discriminative and reinforcing properties of lisdexamfetamine in comparison to D‐amfetamine, methylphenidate and modafinil. Neuropharmacology. 2013;73:348–58. [DOI] [PubMed] [Google Scholar]
- 13. Frampton JE. Lisdexamfetamine dimesylate: a review in paediatric ADHD. Drugs. 2018;78:1025–36. [DOI] [PubMed] [Google Scholar]
- 14. Banaschewski T, Johnson M, Nagy P, Otero IH, Soutullo CA, Yan B, et al. Growth and puberty in a 2‐year open‐label study of lisdexamfetamine dimesylate in children and adolescents with attention‐deficit/hyperactivity disorder. CNS Drugs. 2018;32:455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coghill DR, Banaschewski T, Bliss C, Robertson B, Zuddas A. Cognitive function of children and adolescents with attention‐deficit/hyperactivity disorder in a 2‐year open‐label study of lisdexamfetamine dimesylate. CNS Drugs. 2018;32:85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ichikawa H, Miyajima T, Yamashita Y, Fujiwara M, Fukushi A, Saito K. Phase II/III study of lisdexamfetamine dimesylate in japanese pediatric patients with attention‐deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2019; 10.1089/cap.2019.0076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. American Psychiatric Association . Diagnostic and statistical manual of mental disorders, 5th ed Arlington, VA: American Psychiatric Publishing, 2013. [Google Scholar]
- 18. Dupaul GJ, Power TJ, Anastopoulos AD, Reid R. ADHD rating scale‐IV: checklists, norms, and clinical interpretation. New York, NY: Guildford Press, 1998. [Google Scholar]
- 19. Ichikawa H, Tanaka Y. ADHD rating scale‐IV: checklists, norms, and clinical interpretation. Tokyo: AkashiShoten, 2008. (in Japanese). [Google Scholar]
- 20. Ohnishi M, Okada R, Tani I, Nakajima S, Tsujii M. Japanese version of school form of the ADHD‐RS: an evaluation of its reliability and validity. Res Dev Disabil. 2010;31:1305–12. [DOI] [PubMed] [Google Scholar]
- 21. Tani I, Okada R, Ohnishi M, Nakajima S, Tsujii M. Japanese version of home form of the ADHD‐RS: an evaluation of its reliability and validity. Res Dev Disabil. 2010;31:1426–33. [DOI] [PubMed] [Google Scholar]
- 22. Conners KC. Conners 3. Toronto, ON: Multi‐Health Systems Inc, 2008. [Google Scholar]
- 23. Conners KC (translated by Tanaka Y, Sakamoto R). Conners 3 Japanese version manual. Tokyo: KanekoShobo, 2011. (in Japanese). [Google Scholar]
- 24. Findling RL, Childress AC, Krishnan S, McGough JJ. Long‐term effectiveness and safety of lisdexamfetamine dimesylate in schoolaged children with attention‐deficit/hyperactivity disorder. CNS Spectr. 2008;13:614–20. [DOI] [PubMed] [Google Scholar]
- 25. Findling RL, Cutler AJ, Saylor K, Gasior M, Hamdani M, Ferreira‐Cornwell MC, et al. A long‐term open‐label safety and effectiveness trial of lisdexamfetamine dimesylate in adolescents with attention‐deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2013;23:11–21. [DOI] [PubMed] [Google Scholar]
- 26. Coghill DR, Banaschewski T, Nagy P, Otero IH, Soutullo C, Yan B, et al. Long‐term safety and efficacy of lisdexamfetamine dimesylate in children and adolescents with ADHD: a phase IV, 2‐year, open‐label study in Europe. CNS Drugs. 2017;31:625–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Biederman J, Spencer TJ, Monuteaux MC, Faraone SV. A naturalistic 10‐year prospective study of height and weight in children with attention‐deficit hyperactivity disorder grown up: sex and treatment effects. J Pediatr. 2010;157:635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harstad EB, Weaver AL, Katusic SK, Colligan RC, Kumar S, Chan E, et al. ADHD, stimulant treatment, and growth: a longitudinal study. Pediatrics. 2014;134:e935–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Swanson JM, Arnold LE, Molina BSG, Sibley MH, Hechtman LT, Hinshaw SP, et al. Young adult outcomes in the follow‐up of the multimodal treatment study of attention‐deficit/hyperactivity disorder: symptom persistence, source discrepancy, and height suppression. J Child Psychol Psychiatry. 2017;58:663–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cassidy TA, McNaughton EC, Varughese S, Russo L, Zulueta M, Butler SF. Nonmedical use of prescription ADHD stimulant medications among adults in a substance abuse treatment population: early findings from the NAVIPRPO surveillance system. J Atten Disord. 2015;19:275–83. [DOI] [PubMed] [Google Scholar]
- 31. Cassidy TA, Varughese S, Russo L, Budman SH, Eaton TA, Butler SF. Nonmedical use and diversion of ADHD stimulants among U.S. adults ages 18–49: a national internet survey. J Atten Disord. 2015;19:630–40. [DOI] [PubMed] [Google Scholar]
- 32. Hennissen L, Bakker MJ, Banaschewski T,Carucci S, Coghill D, Danckaerts M, et al. Cardiovascular effects of stimulant and non‐stimulant medication for children and adolescents with ADHD: a systematic review and meta‐analysis of trials of methylphenidate, amphetamines and atomoxetine. CNS Drugs. 2017;31:199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]