

Abstract
Introduction
Epigenetic information such as DNA methylation is a useful biomarker that reflects complex gene‐environmental interaction. Peripheral tissues such as blood and saliva are commonly collected as the source of genomic DNA in cohort studies. Epigenetic studies mainly use blood, while a few studies have addressed the epigenetic characteristics of saliva.
Methods
The effects of methods for DNA extraction and purification from saliva on DNA methylation were surveyed using Illumina Infinium HumanMethylation450 BeadChip. Using 386 661 probes, DNA methylation differences between blood and saliva from 22 healthy volunteers, and their functional and structural characteristics were examined. CpG sites with DNA methylation levels showing large interindividual variations in blood were evaluated using saliva DNA methylation profiles.
Results
Genomic DNA prepared by simplified protocol from saliva showed a similar quality DNA methylation profile to that derived from the manufacturer provided protocol. Consistent with previous studies, the DNA methylation profiles of blood and saliva showed high correlations. Blood showed 1,514 hypomethylated and 2099 hypermethylated probes, suggesting source‐dependent DNA methylation patterns. CpG sites with large methylation difference between the two sources were underrepresented in the promoter regions and enriched within gene bodies. CpG sites with large interindividual methylation variations in blood also showed considerable variations in saliva.
Conclusion
In addition to high correlation in DNA methylation profiles, CpG sites showing large interindividual DNA methylation differences were similar between blood and saliva, ensuring saliva could be a suitable alternative source for genomic DNA in cohort studies. Consideration of source‐dependent DNA methylation differences will, however, be necessary.
Keywords: biomarker, blood, epigenetics, interindividual variation
We compared quality of saliva methylome data collected by several DNA purification protocols and examined the characteristics of saliva methylome. Optimized protocol and identified characteristics such as common informative CpG sites to blood and unique epigenetic changes in saliva will contribute to promote the use of saliva for epigenetic studies in clinical settings and epidemiological cohort studies.

1. INTRODUCTION
DNA methylation is a reversible chemical modification of cytosine residue in the DNA sequence and is an important regulator of gene expression. It reflects the genetic background of individuals, as well as environmental factors.1, 2 DNA methylation is therefore closely associated with neuropsychiatric disorders3 and is an informative biological marker in cohort studies.4, 5
In epidemiological cohort studies, peripheral tissues such as blood and saliva are commonly used as the source of genomic DNA. Because cohort studies usually deal with hundreds to thousands of subjects in a longitudinal manner, saliva has clear advantages over blood because it is noninvasive, easy to use, and does not require trained medical professionals for sample collection.6 However, saliva has been used less often than blood in epigenetic studies, and only a few studies have addressed the epigenetic characteristics and uniqueness of saliva.6, 7, 8
When conducting large‐scale epigenome‐wide association studies (EWAS), a previous study has demonstrated that CpG sites whose DNA methylation levels show large interindividual variations are useful for identifying disease‐related epigenetic changes.9 That study used blood data to evaluate the CpG sites, and the validity of using saliva needs to be addressed.
In this study, we investigated the usefulness of saliva samples for providing epigenetic date in cohort studies. First, we compared the methods of DNA extraction and purification from saliva using the commercially available kit. Second, we identified differentially methylated CpG sites between blood and saliva, and revealed their characteristics. Third, we tested whether the CpG sites showing large interindividual methylation differences in blood9 also showed variations in saliva.
2. MATERIALS AND METHODS
2.1. Subjects
To evaluate DNA extraction and purification methods, saliva samples were collected from 3 Japanese females. To compare the DNA methylation profiles of blood and saliva, we collected blood and saliva from 22 age‐matched healthy volunteers (male: mean age 31.1 ± 4.9, N = 15; female: 29.7 ± 6.8, N = 7). This study conformed to the provisions of the Declaration of Helsinki. The ethics committees of the University of Tokyo Hospital and collaborative research organizations approved this study.
2.2. Genomic DNA extraction
From each participant, we obtained 2 mL of saliva using the Oragene‐DNA collection kit10 (DNAgenotek Inc., Ontario, Canada). Genomic DNA was extracted according to the protocol of the prepIT‐C2D Genomic DNA MiniPrep kit (DNAgenotek Inc). The protocol contained a step to purify DNA using the MiniPrep column and another step of RNA degradation using RNase A. To evaluate the DNA extraction and purification methods, we compared four protocols: (1) employing the MiniPrep column for DNA purification and RNase A (the manufacturer's protocol without modification); (2) employing the MiniPrep column without using RNase A; (3) employing ethanol precipitation instead of the MiniPrep column and using RNase A; and (4) employing ethanol precipitation without using RNase A. Genomic DNA extraction from the saliva of 22 subjects was performed using protocol (1). Genomic DNA extraction from blood was performed using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA).
2.3. DNA methylation analysis and data analysis
We analyzed DNA methylation using Infinium HumanMethylation450 BeadChip (Illumina, CA, USA) according to the manufacturer's instructions. The assay contained >485 000 CpG targets and covered 99% NCBI Reference Sequence genes. DNA methylation data were processed under R environment.11 Color normalization was performed using the background correction and internal control probes included in each chip. We excluded probes that (1) showed detection P value ≥.05; (2) were located on X or Y chromosomes; (3) had potential SNPs; (4) might cross‐hybridize with unspecific genomic regions;12 and (5) lacked data for at least one sample. Differentially methylated probes were defined as P value <.05 by the Wilcoxon signed‐rank test with a difference of mean β value between blood and saliva larger than 0.2. Gene ontology (GO) analysis was performed using PANTHER,13 applying Bonferroni correction. Principal component analysis (PCA) and hierarchical clustering analysis were conducted using the maptools package in R. The CpG sites showing large interindividual methylation variation in blood were retrieved from a previous report.9 According to the definition,9 we used CpG sites whose reference interval (RI) was larger than 30. Correlation between standard deviation (SD) of DNA methylation values in this study and RI in the previously identified CpGs was calculated using Spearman's rank correlation. Preference for genomic region of the differentially methylated probes was assessed using Fisher's exact test.
3. RESULTS AND DISCUSSION
We performed Illumina Infinium HumanMethylation450 BeadChip assay using genomic DNA derived from the saliva of three subjects. For each subject, we performed 4 patterns of DNA extraction and purification, including the protocol provided by the manufacturer. The protocol included column purification followed by RNase A treatment, and we performed either column purification or ethanol precipitation with or without RNase A treatment. Evaluation of data quality was made by comparing the (1) total number of detected probes at the levels of detection P value; (2) average intensities of the array probes; and (3) PCA. The comparisons revealed that there were no statistically significant differences in the total number of detected probes or the intensities of the probe signals (ANOVA, P > .05) (Figure 1A). In addition, PCA showed sample‐dependent, rather than protocol‐dependent, separation (Figure 1B). Therefore, we concluded that the simplest protocol (ie, ethanol precipitation without RNase A treatment) was also effective in DNA methylation analysis. Although our modifications were relatively minor, they will significantly improve the time and cost for epidemiological cohort studies dealing with hundreds to thousands of subjects.
Figure 1.
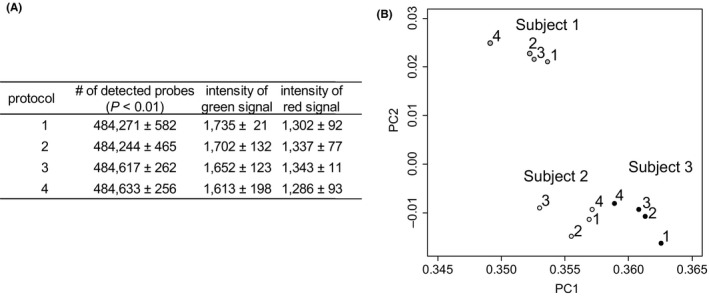
Comparison of the protocols for DNA extraction and purification. A, Comparison of summary data from Infinium HumanMethylation450 BeadChip assay. Data are given in mean and standard deviations. Protocol 1; Column + RNase A, 2; Column – RNase A, 3; Ethanol precipitation + RNase A, 4; Ethanol precipitation – RNase A. B, Result of PCA. The number for each subject represents the employed protocol for DNA extraction and purification
We then compared the DNA methylation profiles of saliva and blood taken from 22 subjects. After filtering, the remaining 386 661 probes were further analyzed. Both clustering analysis and PCA showed that the DNA methylation profile of saliva was clearly separated from that of blood (Figure S1 A). However, as expected, the average DNA methylation profiles showed a high correlation (R = 0.977) between the two sample sources, indicating a close relationship between them (Supplementary Figure S1 B). Age and sex would be important factors for DNA methylation status at the specific genomic regions. However, our PCA and clustering analysis suggested that the overall DNA methylation profiles were not affected by age or sex. Therefore, we did not consider the effect of these factors in detail in this study. We then attempted to estimate what constituted the main cell population of our saliva samples. We calculated the correlation of the overall DNA methylation level with the publicly available data from samples separated into several blood cell lineages.14 Our saliva data showed best correlation with granulocytes (R = 0.947). However, our saliva samples did not separate according to specific blood cell lineages by clustering analysis and PCA, likely due to differences in the races, ages, and experimental batches (data not shown).
Based on the statistical analysis and the extent of DNA methylation difference, we identified 1514 hypomethylated probes in blood, associated with 574 genes, and 2099 hypomethylated probes in saliva, associated with 1117 genes. GO analyses revealed that GO terms such as cell periphery, immune system, and plasma membrane commonly appeared in both sources (Figure 2A). Notably, GO terms related to leukocytes were found in the top list of hypomethylated genes in blood, and those related to enzyme binding and vesicle‐mediated transport were found in the top list of hypomethylated genes in saliva, likely reflecting that upregulated genes in each source were hypomethylated in that source. Compared with the previous study, which examined differential DNA methylation profiles between peripheral whole blood and saliva using HumanMethylation27 BeadChip,8 GO terms such as plasma membrane and immune system process are commonly appeared. However, those related to regulation of signaling and cell communication were not detected in the previous report. This difference may be due to the differences in the age and race of subjects and type of array platform. We next examined the genomic context of the differentially methylated probes. We found that differentially methylated probes were underrepresented in the CpG island and promoter regions (TSS1500 and TSS200) and were enriched in the 5’‐UTR and gene body (Figure 2B). Overall, these observations were generally consistent with those of previous studies.7, 8
Figure 2.
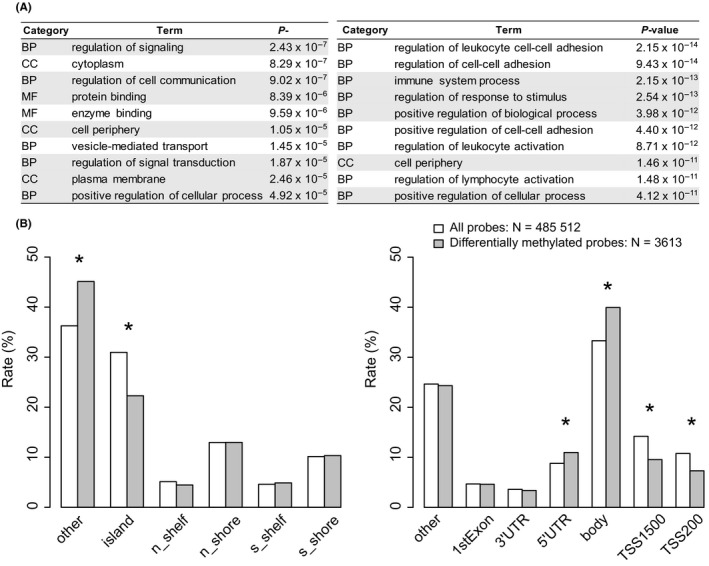
Characteristics of differentially methylated CpG sites. A, Gene Ontology (GO) analysis of the genes associated with differentially methylated probes. GO analysis in hypomethylated genes in saliva (2099 probes for 1117 genes, left) and blood (1514 probes for 574 genes, right). GO terms shaded represent commonly appearing terms in saliva and blood. Note that some common terms are not found, because only top‐ranked terms are listed here. BP, biological process; MF, molecular function; CC, cellular component. B, Classification of the genomic locations of the differentially methylated CpG sites. CpG sites were classified according to the positions related to either CpG island (left) or gene structure (right). * indicates P < .05 in Fisher's exact test. N shelf, 2–4 kb upstream to the CpG island; N shore, 0–2 kb upstream to the CpG island; S shelf, 2–4 kb downstream to the CpG island; S shore, 0–2 kb downstream to the CpG island; TSS 1500, 201–1500 bp upstream of the transcription start site; TSS 200, 1–200 bp upstream to the transcription start site
We also inspected in detail those genes showing DNA methylation differences in blood and saliva (Table S1). Genes such as TBX1, S1PR4, and SPEG were hypomethylated in blood, unlike saliva. TBX1 encodes a transcription factor involved in development and reported to be related to heart disease and DiGeorge syndrome.15 S1PR4 encodes a G‐protein‐coupled receptor important for immune response.16, 17 SPEG encodes a protein similar to the myosin light chain kinase family, which is important for cytoskeletal and cardiovascular development.18 On the other hand, RIN2, DOT1L, and BZRAP1 showed hypomethylation in saliva. RIN2 encodes a protein that works as a guanine nucleotide exchange factor for RAB5, and its mutations are known to cause several syndromes related to defects in connective tissue.19 DOT1L encodes a histone methyltransferase with important functions in cartilage and blood vessel homeostasis.20, 21 BZRAP1 encodes a protein forming benzodiazepine receptor complex in mitochondria.22
Hachiya and colleagues previously identified CpG sites where DNA methylation levels showed large interindividual differences in blood cells from the whole genome bisulfite sequencing data.9 They also showed that such CpG sites increased the efficacy in detecting differential DNA methylation in EWAS. To evaluate the usefulness for saliva of the CpG sites previously identified in blood, we compared the variance of DNA methylation levels in our data with that of the previous study. We used 23 076 probes that were available in both our data set and previous data set. As expected, we observed a positive correlation between our blood data and that of the previous study (R = 0.362, P < 2.2 × 10–16). We also found a significant correlation between our saliva data and the previous blood data (R = 0.323, P < 2.2 × 10–16), suggesting that saliva does reflect the interindividual DNA methylation profiles identified in blood.
In conclusion, saliva can be used as an alternative to blood in DNA methylation analysis for cohort studies. The informative CpG sites to be examined in the cohort study are generally common between the two sources. However, a group of CpG sites showed saliva‐dependent DNA methylation profiles, so target CpG sites should be carefully designed.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTION
M.B., K.K., and K.I. designed the research. S.K., S.F., and M.B. collected samples and performed experiment. Y.M., A.F., S.K., T.I., Z.Z., and S.J. analyzed the data. Y.N. managed the data. Y.M., A.F., and K.I. prepared the manuscript.
Supporting information
ACKNOWLEDGMENTS
This study was supported in part by JSPS KAKENHI Grant Number 16H06395, 16H06399, and 16K21720. This research was also partly supported by AMED under Grant Number JP19dm0107123, JP19dm0207074, JP19dm0307001, JP19dm0307004, and JP19dm0207069. This work was also supported in part by UTokyo Center for Integrative Science of Human Behavior (CiSHuB) and by the International Research Center for Neurointelligence (WPI‐IRCN) at The University of Tokyo Institutes for Advanced Study (UTIAS).
Murata Y, Fujii A, Kanata S, et al. Evaluation of the usefulness of saliva for DNA methylation analysis in cohort studies. Neuropsychopharmacol Rep. 2019;39:301–305. 10.1002/npr2.12075
Murata and Fujii are contributed equally.
Data Availability Statement: Raw data have been deposited in Gene Expression Omnibus and are accessible through GSE130153.
Contributor Information
Miki Bundo, Email: bundo@kumamoto-u.ac.jp.
Kazuya Iwamoto, Email: iwamotok@kumamoto-u.ac.jp.
DATA ACCESSIBILITY
Raw data have been deposited in Gene Expression Omnibus and are accessible through GSE130153.
REFERENCES
- 1. Graff J, Kim D, Dobbin MM, Tsai LH. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev. 2011;91(2):603–49. [DOI] [PubMed] [Google Scholar]
- 2. Kular L, Kular S. Epigenetics applied to psychiatry: clinical opportunities and future challenges. Psychiatry Clin Neurosci. 2018;72(4):195–11. [DOI] [PubMed] [Google Scholar]
- 3. Nishioka M, Bundo M, Kasai K, Iwamoto K. DNA methylation in schizophrenia: progress and challenges of epigenetic studies. Genome Med. 2012;4(12):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Terry MB, Delgado‐Cruzata L, Vin‐Raviv N, Wu HC, Santella RM. DNA methylation in white blood cells: association with risk factors in epidemiologic studies. Epigenetics. 2011;6(7):828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Okada N, Ando S, Sanada M, Hirata‐Mogi S, Iijima Y, Sugiyama H, et al. Population‐neuroscience study of the Tokyo TEEN Cohort (pn‐TTC): Cohort longitudinal study to explore the neurobiological substrates of adolescent psychological and behavioral development. Psychiatry Clin Neurosci. 2019;73(5):231–42. [DOI] [PubMed] [Google Scholar]
- 6. Langie S, Moisse M, Declerck K, Koppen G, Godderis L, Vanden Berghe W, et al. Salivary DNA methylation profiling: aspects to consider for biomarker identification. Basic Clin Pharmacol Toxicol. 2017;121(Suppl 3):93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith AK, Kilaru V, Klengel T, Mercer KB, Bradley B, Conneely KN, et al. DNA extracted from saliva for methylation studies of psychiatric traits: evidence tissue specificity and relatedness to brain. Am J Med Genet B Neuropsychiatr Genet. 2015;168B(1):36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thompson TM, Sharfi D, Lee M, Yrigollen CM, Naumova OY, Grigorenko EL. Comparison of whole‐genome DNA methylation patterns in whole blood, saliva, and lymphoblastoid cell lines. Behav Genet. 2013;43(2):168–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hachiya T, Furukawa R, Shiwa Y, Ohmomo H, Ono K, Katsuoka F, et al. Genome‐wide identification of inter‐individually variable DNA methylation sites improves the efficacy of epigenetic association studies. NPJ Genom Med. 2017;2:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nunes AP, Oliveira IO, Santos BR, et al. Quality of DNA extracted from saliva samples collected with the Oragene DNA self‐collection kit. BMC Med Res Methodol. 2012;12:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. R Core Team . R: A language and environment for statistical computing. Vienna: Austria: R Foundation for Statistical Computing; 2018. https://www.R-project.org [Google Scholar]
- 12. Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross‐reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13(9):2129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén SE, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS ONE. 2012;7(7):e41361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baldini A, Fulcoli FG, Illingworth E. Tbx1: transcriptional and developmental functions. Curr Top Dev Biol. 2017;122:223–43. [DOI] [PubMed] [Google Scholar]
- 16. Olesch C, Ringel C, Brune B, Weigert A. Beyond immune cell migration: the emerging role of the sphingosine‐1‐phosphate receptor S1PR4 as a modulator of innate immune cell activation. Mediators Inflamm. 2017;2017:6059203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dillmann C, Ringel C, Ringleb J, Mora J, Olesch C, Fink AF, et al. S1PR4 signaling attenuates ILT 7 internalization to limit IFN‐alpha production by human plasmacytoid dendritic cells. J Immunol. 2016;196(4):1579–90. [DOI] [PubMed] [Google Scholar]
- 18. Quick AP, Wang Q, Philippen LE, Barreto‐Torres G, Chiang DY, Beavers D, et al. SPEG (striated muscle preferentially expressed protein kinase) is essential for cardiac function by regulating junctional membrane complex activity. Circ Res. 2017;120(1):110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rosato S, Syx D, Ivanovski I, Pollazzon M, Santodirocco D, DeMarco L, et al. RIN2 syndrome: expanding the clinical phenotype. Am J Med Genet A. 2016;170(9):2408–15. [DOI] [PubMed] [Google Scholar]
- 20. Monteagudo S, Cornelis F, Aznar‐Lopez C, Yibmantasiri P, Guns L‐A, Carmeliet P, et al. DOT1L safeguards cartilage homeostasis and protects against osteoarthritis. Nat Commun. 2017;8:15889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dafflon C, Craig VJ, Mereau H, Gräsel J, Schacher Engstler B, Hoffman G, et al. Complementary activities of DOT1L and Menin inhibitors in MLL‐rearranged leukemia. Leukemia. 2017;31(6):1269–77. [DOI] [PubMed] [Google Scholar]
- 22. Galiegue S, Jbilo O, Combes T, Bribes E, Carayon P, LeFur G, et al. Cloning and characterization of PRAX‐1. A new protein that specifically interacts with the peripheral benzodiazepine receptor. J Biol Chem. 1999;274(5), 2938–52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data have been deposited in Gene Expression Omnibus and are accessible through GSE130153.