

Abstract
Aim
We previously performed the first trio‐based exome study for bipolar disorder and identified 71 de novo mutations. Among these mutations, the only mutation located at the splice donor site was in UNC13B. We focused on and analyzed the functions of the mutation.
Methods
In order to analyze the functional alterations, due to the mutation, we performed a minigene splicing assay.
Key results
We found that the mutation caused the loss of a wild‐type splicing variant, which was consistent with the computational splice prediction, and that an exon‐skipping variant increased significantly. The exon‐skipping variant also existed in the wild‐type minigene, although it was rare. Hence, we validated the expression of the exon‐skipping variant using total RNAs derived from the human cerebral cortex. We showed the possibility that the exon‐skipping variant was rare, but expressed even in those that do not carry the mutation.
Conclusions
Based on our results, we suggest that an abnormal splicing pattern of UNC13B occurred in the patient, which could be related to the pathophysiology of bipolar disorder.
Keywords: bipolar disorder, de novo mutation, splicing assay, UNC13B
We found that a de novo mutation of UNC13B found in a bipolar disorder patient altered splicing patterns and increased a rare exon‐skipping variant.

1. INTRODUCTION
Bipolar disorder is a common neuropsychiatric disorder with a lifetime prevalence of around 1%, and genetic factors are known to be important in this disorder.1 However, the pathogenesis of bipolar disorder remains unclear. Recently, the first trio‐based exome study for bipolar disorder was reported.2 The report suggested a possible relationship between de novo mutations and bipolar disorder and found 71 mutations in bipolar disorder patients. Among these mutations, UNC13B was reported as the only gene that possessed a mutation at a splice site.
UNC13B is thought to regulate a priming step in exocytosis, especially in presynaptic terminals,3, 4 and to possess a calcium ion‐binding domain. This is consistent with the fact that previous studies have shown the association of bipolar disorder with calcium ion channels, such as CACNA1C.5, 6 We have also reported a possible relationship between a calcium ion pump, ATP2A2, and psychosis, including bipolar disorder.7 Thus, the abnormality caused by the mutation of UNC13B might be related to the pathophysiology of bipolar disorder.
Herein, we analyzed the effects of the de novo UNC13B mutation found in the bipolar disorder patient using splice prediction Web sites and a minigene splicing assay.
2. METHODS
For the prediction of splicing patterns, NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/)8 and SPANR (http://tools.genes.toronto.edu/)9 were utilized.
To perform the minigene splicing assay, exons 37‐39 of UNC13B were amplified from DNA from the saliva of the patient carrying the heterozygous mutation who we previously reported.2 The amplified fragments were inserted into the pAcGFP‐C1 vector and transfected into HEK293 cells. Reverse transcription‐polymerase chain reaction (RT‐PCR) was performed using total RNAs, prepared from the transfected cells. PCR products were subcloned into the TOPO TA cloning kit (Thermo Fisher Scientific, Tokyo, Japan) and sequenced.
A cDNA fragment of exons 35‐39 of UNC13B was amplified using human total RNA derived from the cerebral cortex (#636561; Takara).
3. RESULTS
According to NetGene2, the mutation was predicted to disrupt the donor site of exon 38. We also used another splicing prediction software, SPANR, which showed that the mutation caused a decrease in the percent spliced in (PSI) and calculated the maximum difference between the reference and alternate allele in the PSI (dPSI) as −15.62. This dPSI was bigger than the expected value (|dPSI| = 5), which was predicted to influence the donor site function, indicating that the mutation might alter the splicing pattern of exon 38.
We performed a minigene splicing assay to analyze the functional alterations caused by the mutation. We generated two minigene constructs: UNC13B_WT and UNC13B_Mut (Figure 1A). Then, we transfected both minigenes into HEK293 cells and performed RT‐PCR, and we found different band patterns between UNC13B_WT and _Mut (Figure 1B). We subcloned and sequenced the PCR products. As a result, we detected that several splicing variants were generated in UNC13B_Mut and that the exon 38‐skipping variant was a major component of UNC13B_Mut (Figure 1C). The exon‐skipping variant caused a frameshift that resulted in a premature stop codon. We also found a significantly deviated distribution of variants between UNC13B_WT and _Mut (6 × 2 Fisher's exact test, P = 2.27 × 10−18).
Figure 1.
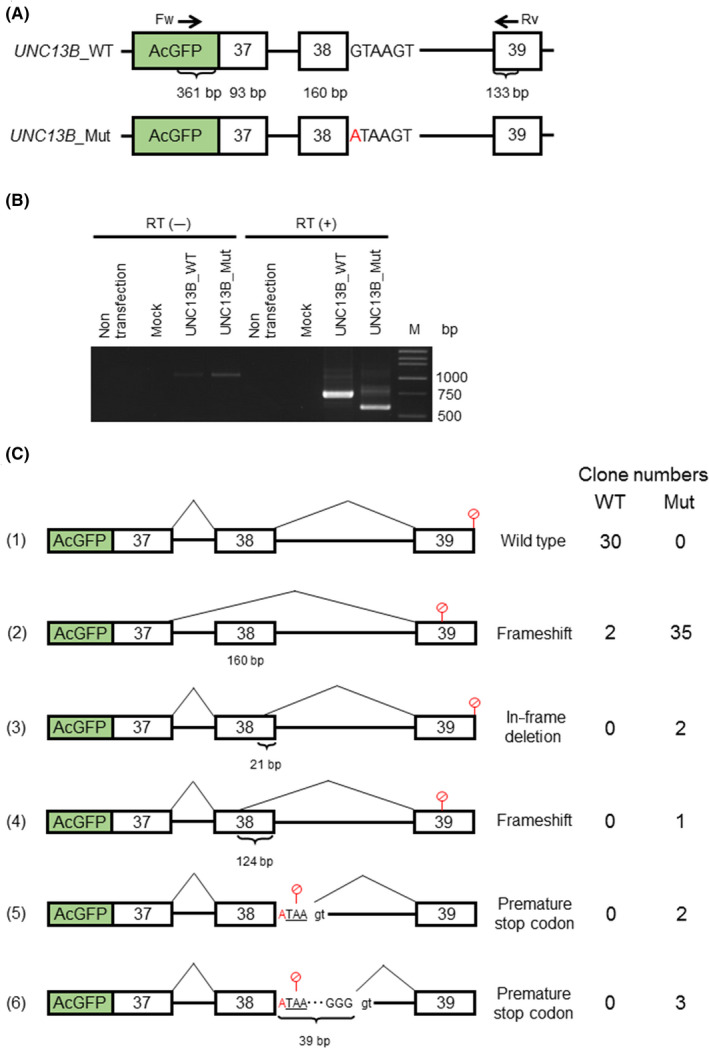
A, A schematic diagram of the minigenes; B, RT‐PCR of the transfected HEK293 cells; C, The exon‐skipping variant (2) increased significantly in UNC13B_Mut (Fisher's exact test, P = 2.27 × 10−18). M: marker
Unexpectedly, we also detected the exon‐skipping variant in UNC13B_WT, although the rate was less than 10%. To verify whether this variant in UNC13B_WT was an artifact, we performed RT‐PCR using human total RNA derived from the cerebral cortex. We designed a new primer set to amplify the other variant, including an in‐frame alternative exon (exon 36a). As expected, we validated two variants and also found a slight band at the expected height of the exon‐skipping variant (Figure 2A,B). We performed nested PCR using a primer set that specifically recognized the exon‐skipping variant and found the band for the exon‐skipping variant, although the wild‐type variant was also amplified nonspecifically (Figure 2C).
Figure 2.
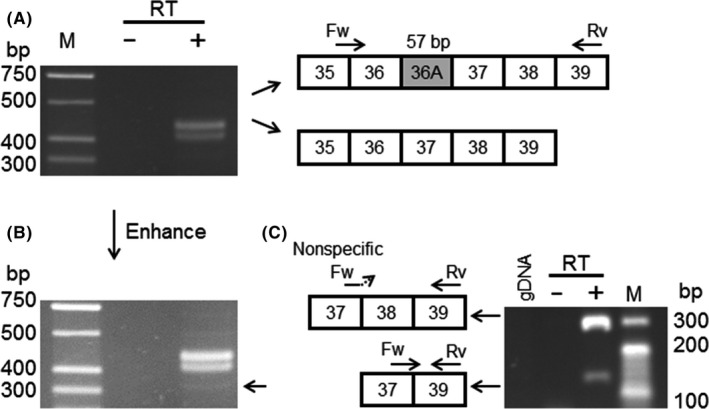
A, RT‐PCR of total RNA derived from the human cerebral cortex; B, An enhanced image of (A). A slight band was detected below the main band; C, Nested PCR using the forward primer that recognizes the junction of exons 37 and 39
4. DISCUSSION
In this study, we found that the de novo UNC13B mutation caused a significant increase in the exon‐skipping variant and resulted in a frameshift. We also showed that this exon‐skipping variant was expressed in the human cerebral cortex.
The exon‐skipping variant generates a premature stop codon in the last exon of UNC13B. This abnormal mRNA is predicted to escape nonsense‐mediated mRNA decay (NMD) and generate a truncated UNC13B protein in people that carry the mutation. The truncated protein loses part of the lipid‐binding domain, the C2C domain,10 such that the exocytosis function that is regulated by UNC13B might be disrupted. The ExAC database shows that the possibility for a loss‐of‐function mutation (pLi) in UNC13B is zero, which suggests that a loss‐of‐function mutation of UNC13B is not rare in the general population. However, it is possible that the truncated protein causes a dominant‐negative effect and has a larger effect on the proper functions of UNC13B in a patient.
We also showed the possibility that the exon‐skipping variant, which was not registered in University of California Santa Cruz (UCSC) Genome Browser, is expressed in the human cerebral cortex. Although the expression level of this variant is much lower than that of the wild‐type protein, it might play a role in the function of UNC13B. Further research is necessary to clarify the role of this variant.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest related to this study.
DATA REPOSITORY
All relevant data are included in Supporting Information.
APPROVAL OF THE RESEARCH PROTOCOL BY AN INSTITUTIONAL REVIEWER BOARD
The study was approved by the Wako first ethics committee of RIKEN and conforms to the provisions of the Declaration of Helsinki.
INFORMED CONSENT
Informed consent was obtained from the subject.
REGISTRY AND THE REGISTRATION NO. OF THE STUDY/TRIAL
n/a.
ANIMAL STUDIES
n/a.
AUTHOR CONTRIBUTIONS
T.N. conceived all experiments. K.J. performed the minigene splicing assay. T.N. conducted the validation of the rare splice variant of UNC13B. K.N. and T.T. supervised the experiments. T.K. conceived and organized the project. All authors wrote the manuscript.
Supporting information
ACKNOWLEDGMENT
We thank the Research Resources Division at the RIKEN Center for Brain Science for providing technical support.
Nakamura T, Jimbo K, Nakajima K, Tsuboi T, Kato T. De novo UNC13B mutation identified in a bipolar disorder patient increases a rare exon‐skipping variant. Neuropsychopharmacol Rep. 2018;38:210–213. 10.1002/npr2.12027
Funding information
The work was supported in part by the Advanced Genome Research and Bioinformatics Study to Facilitate Medical Innovation (GRIFIN) from the Japan Agency for Medical Research and Development (AMED) (16815678) to T.K.
Takumi Nakamura and Kotori Jimbo contributed equally to this work.
REFERENCES
- 1. Craddock N, Sklar P. Genetics of bipolar disorder. Lancet. 2013;381:1654–62. [DOI] [PubMed] [Google Scholar]
- 2. Kataoka M, Matoba N, Sawada T, et al. Exome sequencing for bipolar disorder points to roles of de novo loss‐of‐function and protein‐altering mutations. Mol Psychiatry. 2016;21:885–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Michelassi F, Liu H, Hu Z, Dittman JS. A C1‐C2 module in Munc13 inhibits calcium‐dependent neurotransmitter release. Neuron. 2017;95:577–590.e575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lai Y, Choi UB, Leitz J, et al. Molecular mechanisms of synaptic vesicle priming by Munc13 and Munc18. Neuron. 2017;95:591–607.e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sklar P, Smoller JW, Fan J, et al. Whole‐genome association study of bipolar disorder. Mol Psychiatry. 2008;13:558–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moskvina V, Craddock N, Holmans P, et al. Gene‐wide analyses of genome‐wide association data sets: evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Mol Psychiatry. 2009;14:252–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakamura T, Kazuno AA, Nakajima K, Kusumi I, Tsuboi T, Kato T. Loss of function mutations in ATP2A2 and psychoses: a case report and literature survey. Psychiatry Clin Neurosci. 2016;70:342–50. [DOI] [PubMed] [Google Scholar]
- 8. Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouze P, Brunak S. Splice site prediction in Arabidopsis thaliana pre‐mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24:3439–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015;347:1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pinheiro PS, Houy S, Sorensen JB. C2‐domain containing calcium sensors in neuroendocrine secretion. J Neurochem. 2016;139:943–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials