

Summary
Centromeres are essential for accurate chromosome segregation and are marked by CENP-A nucleosomes. Mis-targeted CENP-A chromatin has been shown to seed centromeres at non-centromeric DNA. However, the requirements for such de novo centromere formation and transmission in vivo remain unknown. Here, we employ Drosophila melanogaster and the LacI/lacO system to investigate the ability of targeted de novo centromeres to assemble and be inherited through development. De novo centromeres form efficiently at six distinct genomic locations, which include actively transcribed chromatin and heterochromatin, and cause widespread chromosomal instability. During tethering, de novo centromeres sometimes prevail, causing the loss of the endogenous centromere via DNA breaks and HP1-dependent epigenetic inactivation. Transient induction of de novo centromeres and chromosome healing in early embryogenesis show that, once established, these centromeres can be maintained through development. Our results underpin the ability of CENP-A chromatin to establish and sustain mitotic centromere function in Drosophila.
Graphical Abstract
eTOC Blurb
Centromere identity is thought to be specified epigenetically by the centromeric histone CENP-A, but whether targeted CENP-A chromatin can mediate heritable de novo centromeres in vivo is unknown. Palladino et al. show that multiple genomic locations can acquire centromere activity and that these de novo centromeres can be transmitted mitotically.
Introduction
Centromeres are chromosomal structures that specify the site of kinetochore assembly and are thus essential for the accurate segregation of genetic material through mitosis and meiosis. In humans and Drosophila, centromeres are embedded within megabases of heterochromatin (McKinley and Cheeseman, 2016), are composed of satellite DNA and transposable elements (Miga et al., 2014; Chang et al., 2019), and are among the most rapidly evolving parts of genomes (Henikoff et al., 2001; Rosin and Mellone, 2017). While naive centromeric DNA (cenDNA) sequences can initiate the assembly of de novo centromeres (also known as ectopic centromeres) in human cells (Harrington et al., 1997; Ohzeki et al., 2012), centromeres can also form on non-centromeric DNA (Voullaire et al., 1993; Depinet et al., 1997; Tyler-Smith et al., 1999). Thus, the role of centromeric sequences in centromere identity and function remains elusive.
CenDNA is not conserved across species, yet the centromeric chromatin of nearly all eukaryotic organisms contains the histone H3 variant CENP-A, which is essential for the recruitment of kinetochore proteins (Régnier et al., 2005; Fachinetti et al., 2013). CENP-A is deposited at the centromere by specific assembly factors; the ones identified to date are called HJURP in humans (Dunleavy et al., 2009; Foltz et al., 2009; Barnhart et al., 2011), Scm3 in yeast (Camahort et al., 2007; Mizuguchi et al., 2007), and CAL1 in flies (Erhardt et al., 2008; Chen et al., 2014).
Neocentromeres (i.e. centromeres that assemble at a new location) have been shown to form during both meiosis (Rowe et al., 2000) and mitosis (Depinet et al., 1997; Voullaire et al., 2001), either as an attempt to rescue an acentric chromosomal fragment generated by a broken chromatid, or by replacing the endogenous centromere (Marshall et al., 2008). How neocentromeres form and become stable is still unclear.
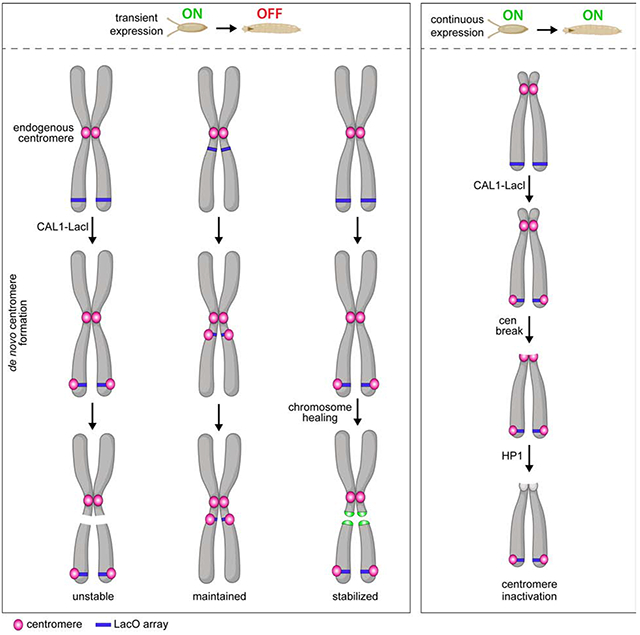
The identification of neocentromeres in humans and the central role played by CENP-A in centromere function lead to the proposal that centromeres are regulated epigenetically by CENP-A-containing chromatin (Karpen and Allshire, 1997). Consistent with this model, ectopic targeting of CENP-A in Drosophila tissue culture cells and overexpression of CENP-A Drosophila in tissue culture cells and animals results in de novo centromere formation (Heun et al., 2006; Mendiburo et al., 2011; Olszak et al., 2011), suggesting that CENP-A is sufficient to initiate the assembly of de novo centromeres. In Drosophila Schneider 2 (S2) cells, ectopic centromeres can be observed at a lacO array several cell divisions after the transient induction of LacI-tagged CAL1 or CENP-A and they are able to direct the deposition of newly synthesized CENP-A (Mendiburo et al., 2011; Chen et al., 2014), consistent with a self-propagation mechanism independent of cenDNA. However, we still know very little about where de novo centromeres can or cannot form, how they become stable, and how their formation is prevented in normal cells. Additionally, whether CENP-A-directed epigenetic propagation occurs in vivo is unknown. Even though the idea that centromeres are specified epigenetically is over two decades old (Karpen and Allshire, 1997), de novo centromere formation and transmission has not been systematically dissected in any animal model.
Dicentric chromosomes (i.e. chromosomes with two centromeres) are highly unstable and initiate breakage-fusion-bridge cycles (McClintock, 1939; Stimpson et al., 2012), therefore, it is critical that chromosomes contain only one centromere. In mouse 3T3 cells, assembly of ectopic kinetochores via CENP-T tethering induces genome instability and cellular transformation (Gascoigne and Cheeseman, 2013). In human tissue culture cells, engineering of dicentric chromosome through telomere fusion results in segregation defects, chromatin bridges, and chromothripsis (Maciejowski et al., 2015). Instability caused by dicentric chromosomes can be rescued by the inactivation of one of the two centromeres (Stimpson et al., 2012). In human cells and fission yeast, dicentric chromosomes can return to a monocentric state by either genetic or functional inactivation (Stimpson et al., 2010; Sato et al., 2012). Genetic inactivation involves loss of cenDNA along with CENP-A chromatin (Stimpson et al., 2010), while functional inactivation (also called epigenetic inactivation) occurs with no apparent loss of cenDNA via kinetochore disassembly, spreading of pericentric heterochromatin into centromeric chromatin and loss of CENP-A (Sato et al., 2012; Stimpson et al., 2012). Centromere inactivation also occurs during karyotype evolution (e.g. in humans (Chiatante et al., 2017) and stickleback fish (Cech and Peichel, 2016)). However, because these centromere inactivation events have occurred long before their identification, it remains unclear how they occurred and whether genetic and functional inactivation are mutually exclusive mechanisms.
Here, we employ genetics and chromosome engineering in Drosophila melanogaster to investigate the formation, epigenetic maintenance, and phenotypic effects of de novo centromeres using LacI/lacO-mediated tethering of CAL1. We find that CAL1 tethering efficiently initiates the formation of de novo centromeres at several distinct locations, irrespective of the surrounding chromatin environment. The resulting dicentric chromosomes cause chromosomal rearrangements and aneuploidy, leading to apoptosis, tissue defects, and inviability. We identify instances of inactivation of the endogenous centromere that are associated with intra-centromeric DNA breaks and are dependent on the heterochromatin component HP1. We further show that de novo centromeres have the potential to be maintained by the endogenous centromere-maintenance pathway. Not only do these findings further support the epigenetic model of centromere specification, but they also provide a framework for understanding the impact of dicentric chromosomes on genome stability, development, and the relationship between centromere dysfunction and disease.
Results
De novo centromeres can form at diverse genomic locations
To investigate the ability of de novo centromeres to form at different genomic locations, we took advantage of the well-established lac repressor/lac operator (GFP-LacI/lacO) protein-tethering system and of the existence of several Drosophila single lacO insertions (256 lacO repeats) spanning multiple chromosomal positions: subtelomeric on the X chromosome (Xsubtelo); telomeric and medial on chromosome 2 (2telo, 2medial); subtelomeric, medial, and pericentric on chromosome 3 (3subtelo, 3medial, 3peri) (Figure 1A; Vazquez et al., 2002; Li et al., 2003; Janssen et al., 2016). The precise genomic position of the lacO insertions were either known or were determined by us by inverse PCR. Using these coordinates and data from modENCODE (Kharchenko et al., 2011), we inferred the chromatin state of each of these positions, with the exception of 2telo for which no chromatin information is available (Figure 1A). To induce de novo centromere formation at these lacO insertions, we decided to tether the CENP-A assembly factor CAL1 because it efficiently assembles de novo centromeres in Drosophila S2 cells (Chen et al., 2014) and because tethering of CENP-A via LacI/lacO does not generate de novo centromeres in flies (Torras-Llort et al., 2010).
Figure 1: De novo centromeres can form at diverse genomic locations.
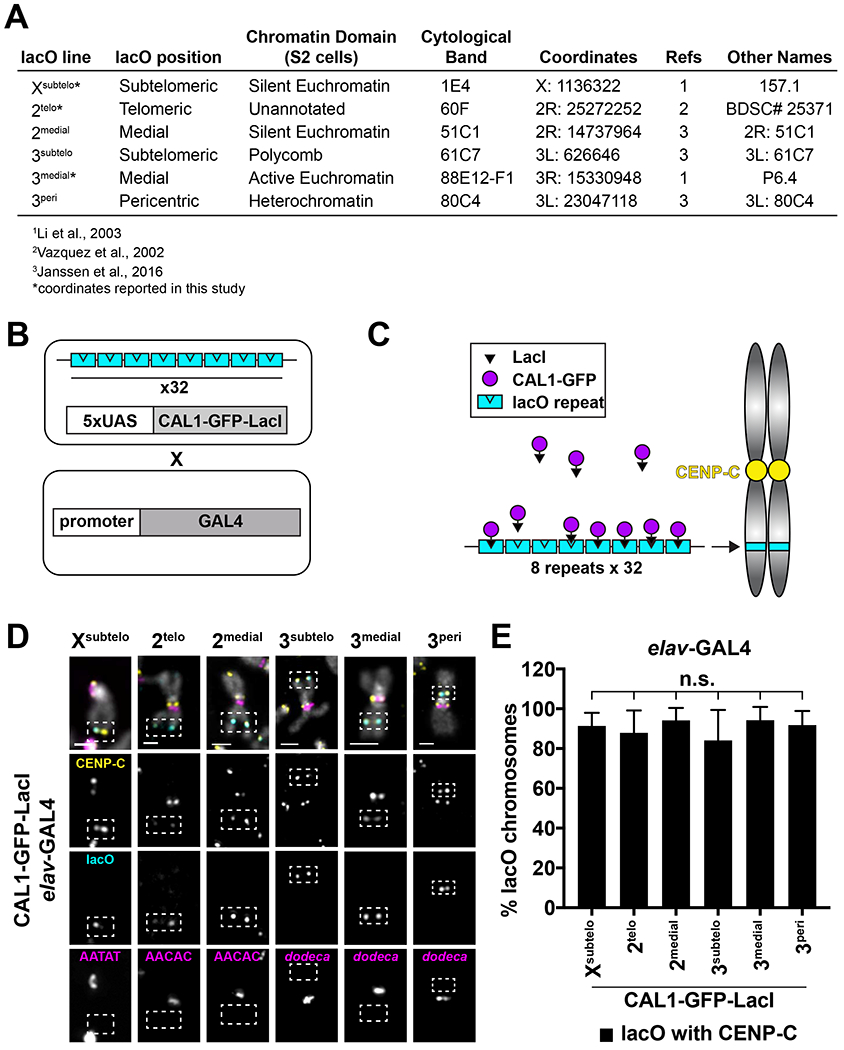
- Summary of lacO insertions and their positions (see also STAR methods).
- Schematic of crosses used to induce GAL4/UAS-mediated CAL1-GFP-LacI expression and lacO binding in Drosophila.
- Schematic of LacI/lacO tethering of CAL1.
- IF for CENP-C (yellow) and FISH for lacO (cyan) and satellites (magenta; AATAT, AACAC, dodeca) on Xsubtelo, 2telo, 2medial, 3subtelo, 3medial, and 3pen mitotic chromosome spreads from elav-GAL4/CAL1-GFP-LacI L3 brains. DAPI is shown in gray. Dashed white box indicates lacO. Scale bar = 1μm.
- De novo centromere formation efficiency on intact Xsubtelo, 2telo, 2medial, 3subtelo, 3medial, and 3peri chromosomes from (D). Shown is the mean ± SD for 3 brains (n=18–122 lacO chromosomes per brains). n.s. not significant (unpaired t-test). See also Figure S1.
We generated transgenic flies with CAL1-GFP-LacI or GFP-LacI (negative control) under the UAS promoter. These were crossed to each lacO line creating homozygous lines, with the exception of 3peri, which is homozygous lethal and had to be maintained as a heterozygous line. CAL1-GFP-LacI/lacO or GFP-LacI/lacO flies are then crossed to lines expressing the GAL4 transcriptional activator under the control of tissue-specific promoters to induce expression. As a result, the progeny is heterozygous for the lacO array (Figure 1B–C). Immunofluorescence (IF) with anti-GFP antibodies on polytene chromosomes from CAL1-GFP-LacI expressing salivary glands confirmed its specific binding to the lacO insertions (Figure S1A–F).
To assess if CAL1-GFP-LacI can assemble de novo centromeres at each lacO array, we analyzed metaphase spreads from 3rd instar larval (L3) brains expressing CAL1-GFP-LacI under the brain-specific elav-GAL4 driver (elav-GAL4/CAL1-GFP-LacI) using IF with anti-CENP-C antibodies (as a marker for active centromeres) combined with fluorescence in situ hybridization (FISH) with a lacO fluorescent LNA probe. Upon tethering CAL1-GFP-LacI to lacO arrays, CENP-C colocalizes with lacO in >80% of spreads irrespective of the lacO position (Figure 1D–E). Of note, de novo sister centromeres did not appear close together suggesting they lack cohesion, a finding consistent with a study that showed that, in flies, cohesin enrichment depends on heterochromatin and is independent of centromere proximity (Oliveira et al., 2014). IF on interphase brain cells with anti-CENP-A antibodies confirmed CAL1 tethering leads to de novo centromere formation via CENP-A deposition (Figure S1G). Control experiments in which GFP-LacI is tethered to lacO showed no localization of CENP-A or CENP-C at the lacO array (Figure S1H–I). CAL1-GFP-LacI expression alone (no lacO array) causes the formation of ectopic CENP-C foci in 63% of spreads (Figure S2A–B). These de novo centromeres form on the 2nd, 3rd and, less frequently, on the X chromosome with no overall preference for any sub-location (Figure S2C–D). The telomere appears to be a preferred region for de novo centromeres on the X (telomere: 26/45, arm: 11/45, pericentromere: 8/45, p<0.001 telomere versus arm and pericentromere, Fisher’s exact test) and on chromosome 2 (Figure S2E), while no preferential location was observed on the 3rd (Figure S2F). De novo centromeres were never observed on the highly repetitive and heterochromatic 4th and Y chromosomes. Collectively, these experiments show that de novo centromeres, whether targeted or spontaneous, can form successfully at several different chromatin contexts, including actively transcribed chromatin and heterochromatin, and at many genomic locations such as the middle of a chromosome and near the telomere.
Formation of de novo centromeres causes genome instability and cell death
Dicentric chromosomes initiate cycles of chromosome breakages and fusions (Stimpson et al., 2012) and ectopic centromeres resulting from CENP-A overexpression in flies cause chromosome missegregation (Heun et al., 2006). Therefore, we anticipated that the formation of a single de novo centromere in our lacO lines would similarly cause genome instability. Interphase brain cells expressing elav-GAL4/CAL1-GFP-LacI displayed a significantly higher number of cells with γH2Av foci (a marker for double-strand DNA breaks (DSBs) (Madigan et al., 2002)) compared to GFP-LacI controls (Figure S3A–C).
DSBs induced by dicentric chromosomes in mitosis lead to chromosomal instability (CIN) (Stimpson et al., 2012; Gascoigne and Cheeseman, 2013). To characterize CIN associated with de novo centromere formation, we evaluated mitotic spreads from L3 brains for changes in chromosome number (aneuploidy), chromosome breakages, fusions, and number of satellite DNA foci (Figure 2A–C). Specific chromosomes were identified based on the presence of pericentric satellites with known localization, chromosome morphology, and pericentromeric DAPI staining. Expression of GFP-LacI alone or with a lacO array (Xsubtelo) is rarely associated with chromosome defects (Figure 2A, C), while expression of CAL1-GFP-LacI alone leads to a significant increase in aneuploidy (32%), chromosome breakages (16%), and fusions (10%) (Figure 2B–C and Tables S1–S5). Tethering CAL1-GFP-LacI to the Xsubtelo, 2medial, 2telo, 3medial, and 3subtelo lacO arrays results in significantly more defects in all the categories compared to GFP-LacI or CAL1-GFP-LacI controls (Figure 2C and Tables S1–S5). Even though lacO chromosomes showed more frequent missegregation (Figure S3D–E), CIN involved both lacO and non-lacO chromosomes (Figure S3D and S3F), suggesting global instability during mitosis. Interestingly, de novo centromeres at the 3peri lacO insertion give rise to fewer chromosomal aberrations than the 3medial and 3subtelo counterparts (Figure 2C), perhaps because the closer proximity to the endogenous centromere of 3peri allows for their co-orientation and less frequent breakage. Collectively, these analyses show that the presence of a single de novo centromere causes dramatic genome instability.
Figure 2: Formation of de novo centromeres causes genome instability and cell death.
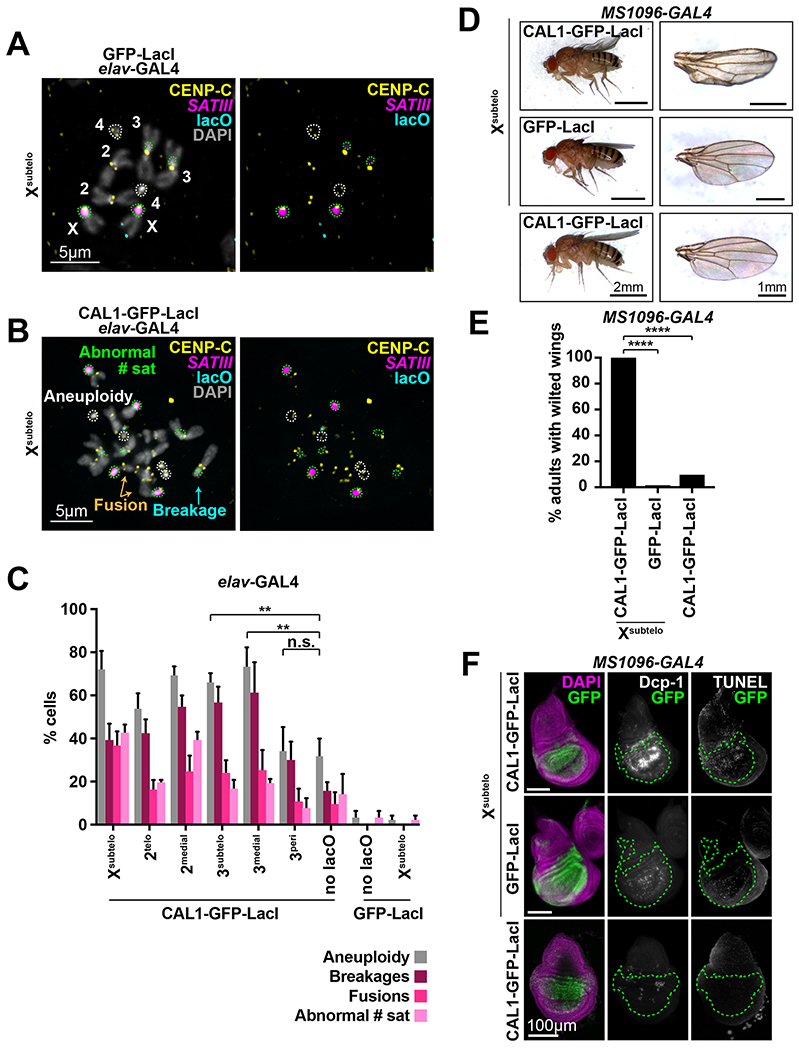
- IF for CENP-C (yellow) and FISH for the SATIII satellite (magenta) and lacO (cyan) on mitotic chromosomes from L3 brain cells expressing elav-GAL4/GFP-LacI/Xsubtelo. DAPI is shown in gray. Right panel excludes the DAPI channel. The identity of each chromosome is shown. The two chromosome 4’s and 4 SATIII foci are outlined in white and green, respectively.
- IF for CENP-C (yellow) and FISH for the SATIII satellite (magenta) and lacO (cyan) on mitotic chromosomes from L3 brain cells expressing elav-GAL4/CAL1-GFP-LacI/Xsubtelo. DAPI is shown in gray. Right panels exclude the DAPI channel. Chromosome 4’s are outlined in white, and SATIII foci in green. Note the spread shows aneuploidy for chromosome 4 (4 chromosomes instead of 2), an abnormal number of satellites (8 SATIII foci instead of 4), and the presence of broken (cyan arrows) and fused (orange arrows) chromosomes.
- Frequency of aneuploidy, chromosome breakages, fusions, and abnormal number of satellite foci in mitotic L3 elav-GAL4/CAL1-GFP-LacI/lacO brain cells. Controls are CAL1-GFP-LacI alone (“no lacO”), GFP-LacI alone (“GFP-LacI”) and GFP-Lac/Xsubtelo. Shown is the mean ± SD for 3 brains (n=38–60 spreads per brain). n.s. not significant, ** p<0.01 (unpaired t-test). See Tables S1–S4 for detailed statistical comparisons and Table S5 for details on the number of satellites per chromosome.
- Adult wing phenotypes in MS 1096-GAL4/CAL1-GFP-LacI and GFP-LacI/Xsubtelo, and CAL1-GFP-LacI alone animals. Left, whole adult progeny. Right, single wings.
- Incidence of wilted wing phenotype in MS1096-GAL4/CAL1-GFP-LacI/Xsubtelo (n=206), GFP-LacI/Xsubtelo (n=183), and CAL1-GFP-LacI alone (n=280) animals. **** p<0.0001, Fisher’s exact test.
- IF and TUNEL staining of wing imaginal discs from the crosses in (D). DAPI (purple), GFP (green), Dcp-1 (white), and TUNEL (white). The GAL4 expressing area is outlined in green. See also Figures S2–S3.
To establish if de novo centromere-induced CIN leads to cell death, as observed in spindle assembly checkpoint mutants (Dekanty et al., 2012; Silva et al., 2013), we analyzed the adult wings of flies expressing CAL1-GFP-LacI/Xsubtelo under a wing driver and observed a distinct wilted wing phenotype that is largely absent in controls (Figure 2D–E). IF for the apoptotic marker Dcp-1 and TUNEL staining on wing imaginal discs showed higher levels of both markers compared to controls (Figure 2F). These data indicate that the CIN resulting from de novo centromere formation causes cell death.
De novo centromere formation causes viability and developmental defects
The CIN caused by de novo centromeres varies between lacO insertions (Figure 2C), suggesting that their formation at a subset of genomic locations may be more detrimental to normal development than that at others. To investigate the effects of de novo centromeres in fly development and viability, we expressed CAL1-GFP-LacI under the control of the engrailed promoter (engrailed-GAL4), which is expressed in a striped pattern from embryogenesis through late pupation (Drees et al., 1987), and scored the percentage of pupae that hatched for each lacO line (Figure 3A). CAL1-GFP-LacI/Xsubtelo, 2medial, 3subtelo, or 3medial progeny display significantly higher hatching defects compared to GFP-LacI/lacO controls (Figure 3A). Furthermore, the flies that successfully hatch display severe developmental defects (e.g. deformed wings and thorax and motility defects) and die shortly after hatching. In contrast, CAL1-GFP-LacI/2telo or 3peri progeny show less severe viability defects (Figure 3A) and the flies that hatch appear morphologically normal and with normal motion. While 3peri shows lower levels of CIN relative to other insertions, 2telo might cause less lethality because of milder gene dosage imbalance associated with the chromosomal rearrangements occurring in this line.
Figure 3: De novo centromere formation causes viability and developmental defects.
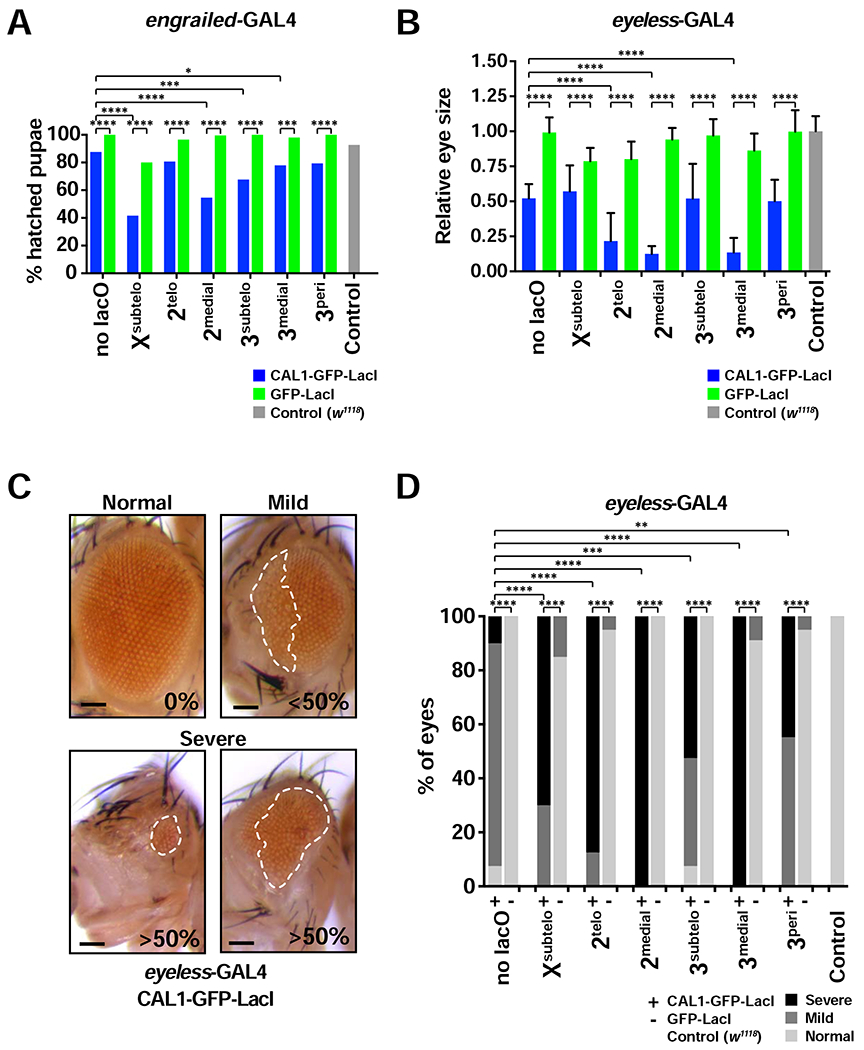
- Percent of hatched engrailed-GAL4/CAL1-GFP-LacI and GFP-LacI pupae with and without lacO inserted at the indicated positions. engrailed-GAL4/w1118 is used as a control. n=51–260 pupae per cross. * p<0.05, *** p<0.001, **** p<0.0001 (Fisher’s exact test).
- Adult eye size in eyeless-GAL4/CAL1-GFP-LacI or GFP-LacI with and without lacO inserted at the indicated locations. Eye sizes are shown relative to eyeless-GAL4/w1118 controls. Shown is the mean ± SD of average eye sizes per cross (n=17–20 flies per cross). **** p<0.0001 (unpaired t-test).
- Representative images depicting “Normal” (0% disorganization), “Mild” (<50% disorganized), and “Severe” (>50% disorganized and/or small in size) eye phenotypes resulting from eyeless-GAL4 driven de novo centromere assembly. A white outline designates the part of the eye that is disorganized. Scale bar = 100 μm.
- Adult eye phenotypes from flies shown in (B). Both eyes are quantified independently for each fly (n=17–20 flies per cross). ** p<0.01, *** p<0.001, **** p<0.000l (Chi-squared). See also Figure S4.
To investigate the effects of de novo centromeres on tissue development, we assessed eye morphology in adult lacO flies expressing CAL1-GFP-LacI or GFP-LacI under an eye-specific driver. Assembling de novo centromeres at all lacO insertions results in decreased eye size and increased defects in eye organization compared to GFP-LacI/lacO controls, with 2medial, 2telo, and 3medial displaying the strongest effects (Figures 3B–D and S4). Flies expressing CAL1-GFP-LacI (no lacO) also display a smaller eye size (Figure 3B). However, in this line, eye organizational defects are mild (Figures 3C–D and S4). Taken together, these data show that de novo centromeres lead to developmental and viability defects, consistent with the observed CIN and cell death (Figure 2C) and with previous CENP-A overexpression studies (Heun et al., 2006).
Inactivation of the endogenous centromere on dicentric Xsubtelo chromosomes
In our constitutive (elav-GAL4) CAL1-GFP-LacI/lacO expression system, over 80% of brain cells contain a dicentric chromosome, meaning that ~20% of cells do not contain a de novo centromere (Figure 1E). We reasoned that if these cells had previously contained a de novo centromere and inactivated it, they would exhibit some type of chromosomal aberration. Quantification of CIN among these relatively infrequent cells showed that the majority are euploid (14/19 for Xsubtelo; 12/14 for 2telo; 5/5 for 2medial; 20/22 for 3subtelo; 17/18 for 3medial; 9/9 for 3peri), suggesting that they never contained de novo centromeres. We conclude that inactivation of de novo centromeres is relatively rare.
To investigate if the endogenous centromere can be inactivated, we quantified the presence of CENP-C at the endogenous centromere of lacO chromosomes with de novo centromeres in elav-GAL4/CAL1-GFP-LacI/lacO brains. We focused our quantification on chromosomes that appeared intact based on morphology and positive hybridization with centromeric/pericentric FISH probes (chromosome X: AATAT; chromosome 2: AACAC; chromosome 3: dodeca). Control GFP-LacI/lacO chromosomes always display CENP-C signal at the endogenous centromere (i.e. no centromere loss; Figure 4A). In the 2medial, 3subtelo and 3peri lines, loss of centromeric CENP-C is very rare (~1% of lacO chromosomes with de novo centromeres; Figure 4A), while in the other two autosomal lines, 2telo and 3medial, loss of endogenous centromere is never observed (0%; Figure 4A), although cells with inactivation events may have died out and not be scored. In contrast, the Xsubtelo line loses endogenous CENP-C signal in 14% of Xsubtelo chromosomes with de novo centromeres (Figure 4A–B).
Figure 4: Inactivation of the endogenous centromere on dicentric Xsubtelo chromosomes.
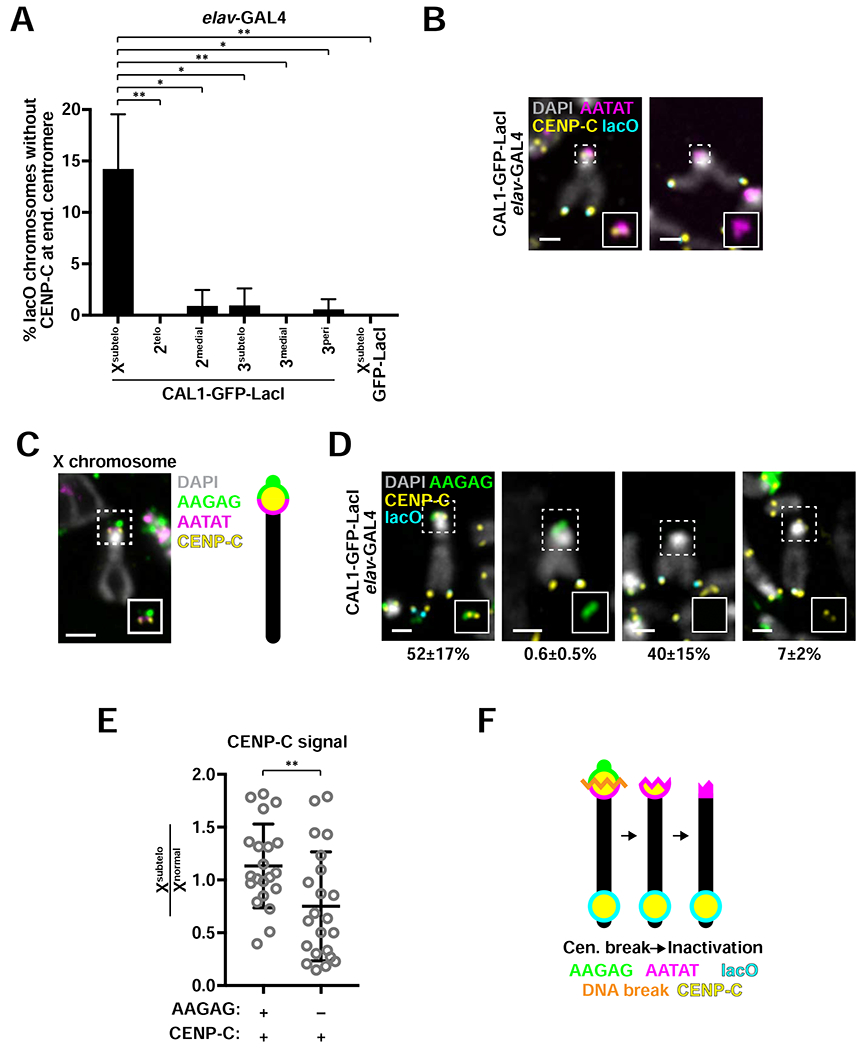
- Frequency of endogenous centromeres loss in elav-GAL4/CAL1-GFP-LacI/lacO flies. Shown is the mean ± SD for 3 brains (n=14—117 intact chromosomes with de novo centromeres per brain). * p<0.05, ** p<0.01 (unpaired t-test). Differences between 2telo, 2medial, 3subtelo, 3medial, 3peri and the GFP-LacI/Xsubtelo control were not statistically significant.
- Representative Xsubtelo chromosomes from L3 brains stained with CENP-C antibodies (yellow) and the lacO (cyan) and AATAT (magenta) FISH probes. DAPI is shown in gray. Insets show 2x zoom of CENP-C and AATAT without DAPI.
- Representative image of IF for CENP-C (yellow) and FISH for AAGAG (green) and AATAT (magenta) on the X chromosome from L3 brains of a laboratory strain (iso-1). DAPI is shown in gray. Inset omits DAPI. Diagram depicts the positions of AATAT (long arm side of the centromere) and AAGAG (short arm side of the centromere) in relation to CENP-C.
- IF for CENP-C (yellow) and FISH for lacO (cyan) and AAGAG (green) on L3 elav-GAL4/CAL1-GFP-LacI/Xsubtelo brain cells. DAPI is shown in gray. Insets exclude the DAPI channel. Mean ± SD for 3 brains are shown (n=98–180 Xsubtelo chromosomes with de novo centromeres per brain).
- Quantification of CENP-C intensity at Xsubtelo endogenous centromere with (+) and without (−) AAGAG relative to that of normal X chromosome centromeres. Shown is the mean ± SD for 3 brains (n=21–22 Xsubtelo chromosomes with de novo and endogenous centromeres). ** p<0.01 (unpaired t-test).
- Schematic showing a model for endogenous centromere loss in which a double-strand DNA break occurs within the endogenous centromere, which is subsequently inactivated epigenetically. See also Figure S5.
The X centromere cytologically localizes between a large block of pericentric AAGAG (on the short arm side of the primary constriction) and of AATAT satellite (on the long arm side; Figure 4C) and occupies a complex DNA island called Maupiti (Sun et al., 1997, 2003; Chang et al., 2019). Thus, the presence of the AAGAG satellite can be used to infer that centromeric DNA is still there. We found that Xsubtelo chromosomes displaying AAGAG signal at the endogenous centromere and no CENP-C at the endogenous centromere represents only 0.6% of Xsubtelo chromosomes with de novo centromeres (Figure 4D, middle left panel), suggesting that the majority of endogenous centromere loss events observed using AATAT FISH (Figure 4A–B) were in fact due to a DNA break resulting in removal of centromeric DNA.
Our experiments uncovered another category of chromosomes that have lost the AAGAG satellite yet still contain endogenous CENP-C (Figure 4D, far right panel). The endogenous CENP-C signal intensity on chromosomes in this category was approximately 50% lower than that of intact dicentrics (Figure 4E), suggesting that these are chromosomes in an intermediate inactivation state in which either a break removed the AAGAG satellite and a portion of centromeric chromatin with it, or partial epigenetic inactivation occurred following a break in pericentric AAGAG. Notably, we never observed chromosomes that lacked AATAT while displaying endogenous CENP-C signal. Therefore, it is possible that some of the Xsubtelo chromosomes that retain AATAT and have lost CENP-C might have started out with a break that removed pericentric AAGAG which was followed by epigenetic inactivation, leading to loss of CENP-C signal (Figure 4F). Taken together, our data suggest that while functional centromere inactivation is infrequent, it is facilitated by a DNA break within or near the centromere.
Functional endogenous centromere inactivation requires HP1
While inactivation of endogenous centromeres is infrequent, the existence of chromosomes lacking the pericentric AAGAG satellite block and displaying weak CENP-C staining, along with Xsubtelo chromosomes containing centromeric AATAT satellite but no CENP-C signal, suggest that breaks within or near the centromere may help initiate epigenetic centromere inactivation (Figure 4F). Heterochromatin has been implicated in centromere inactivation in dicentric chromosomes in S. pombe (Sato et al., 2012). Therefore, we investigated a possible involvement of the heterochromatin protein HP1 in centromere inactivation in the Xsubtelo line. We simultaneously induced de novo centromere formation at Xsubtelo and RNAi-mediated knock-down of HP1 (UAS-HP1RNAi) under the elav-GAL4 driver and analyzed metaphase spreads. IF with anti-HP1 antibodies confirmed that HP1 expression was strongly reduced when compared to the mCherryRNAi control (Figure 5A), and de novo centromere formation at Xsubtelo occurred with similar frequencies in HP1RNAi and in the mCherryRNAi control (Figure 5B). To identify epigenetic centromere inactivation events, we scored Xsubtelo chromosomes that contained the centromeric AATAT satellite but lacked the pericentromeric AAGAG satellite block and CENP-C (Figure 5C). While the incidence of chromosomes that lose AAGAG but still retain AATAT dropped upon HP1 RNAi (Figure 5D), suggesting fewer DNA breaks occurring in this region, there was a clear decrease in the incidence of chromosomes that lose both AAGAG and CENP-C (i.e. functional inactivation events; Figure 5E). These data indicate that following breaks within or near the centromeres, endogenous centromeres are inactivated in a manner requiring HP1.
Figure 5: Functional endogenous centromere inactivation requires HP1.
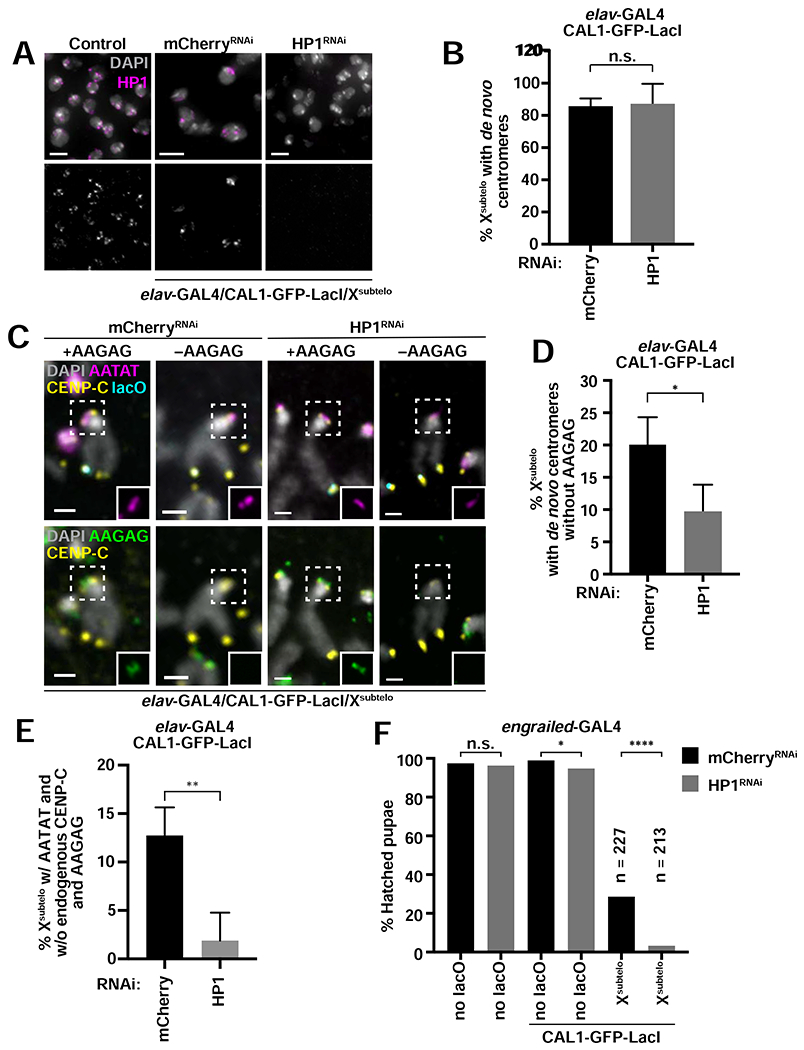
- IF for HP1 (magenta) on L3 brain tissue from control larvae (UAS-HP1RNAi without GAL4) and elav-GAL4/CAL1-GFP-LacI/Xsubtelo larvae with mCherryRNAi or HP1RNAi. DAPI is shown in gray.
- Percent of Xsubtelo chromosomes with de novo centromeres in elav-GAL4/CAL1-GFP-LacI/Xsubtelo with mCherryRNAi or HP1RNAi. Shown is the mean ± SD for 4 brains (n=25–72 Xsubtelo chromosomes with de novo centromeres per brain). n.s. not significant (unpaired t-test).
- IF for CENP-C (yellow) and FISH for lacO (cyan), AATAT (magenta) and AAGAG (green) on elav-GAL4/CAL1-GFP-LacI/Xsubtelo with either mCherryRNAi or HP1RNAi. DAPI is shown in gray. Insets show AATAT (top) and AAGAG (bottom) at the endogenous centromere.
- Percent of Xsubtelo chromosomes with de novo centromeres that have lost AAGAG at the endogenous centromere locus (as marked by AATAT). Shown is the mean ± SD for 4 brains. (n=26–66 Xsubtelo chromosomes with de novo centromeres per brain). * p<0.05 (unpaired t-test).
- Percent of Xsubtelo chromosomes that have lost AAGAG and CENP-C at the endogenous centromere in mCherryRNAi or HP1RNAi backgrounds. All chromosomes scored had AATAT at the endogenous centromere and CENP-C at lacO. Shown is the mean ± SD for 4 brains (n=26–66 Xsubteb chromosomes with de novo centromeres per brain). ** p<0.01 (unpaired t-test).
- Percent of pupae that hatched from engrailed-GAL4 crosses with mCherryRNAi alone, HP1RNAi alone, mCherryRNAi/CAL1-GFP-LacI, HP1RNAi/CAL1-GFP-LacI, mCherryRNAi/CAL1-GFP-LacI/Xsubtelo, and HP1RNAi/CAL1-GFP-LacI/Xsubtelo (n=141–271 pupae per cross). n.s. not significant, * p<0.05, **** p<0.0001 (Fisher’s exact test). See also Figure S5.
Even though de novo centromere inactivation events are rare, we tested if they are facilitated by HP1 by scoring Xsubtelo chromosomes that do not display CENP-C signal at lacO for the presence of CIN, as indicator of initial de novo centromere formation followed by inactivation. While in mCherryRNAi controls 10/34 spreads showed CIN, we observed significantly lower CIN in HP1RNAi (2/29, p=0.0279, Fisher’s exact test). These data suggest that HP1 is involved in the inactivation of de novo centromeres.
We next investigated whether endogenous centromere inactivation ameliorates or worsens the developmental defects resulting from de novo centromere formation. We assessed the viability of HP1RNAi/CAL1-GFP-LadI/Xsubtelo flies and compared it with a series of controls, including mCherryRNAi/CAL1-GFP-LacI. We found that HP1RNAi/CAL1-GFP-LacI/Xsubtelo results in a significant decrease in pupal viability compared to the control (3% of pupae hatched versus 29%; Figure 5F). These data suggest that epigenetic centromere inactivation alleviates at least in part the viability defects caused by de novo centromeres.
The strength of de novo centromeres correlates with the frequency of endogenous centromere loss
Our experiments identified instances in which de novo centromeres at Xsubtelo outcompeted the endogenous centromere (i.e. remained active while the endogenous centromere lost CENP-C). We hypothesized that such out-competition may be dependent upon the strength of the de novo centromere. To test this, we co-expressed GFP-LacI along with CAL1-GFP-LacI to create competition between these two fusion proteins for binding to Xsubtelo lacO (Figure S5A). Xsubtelo cells that co-express CAL1-GFP-LacI and GFP-LacI show lower incidence of de novo centromeres (22% versus 74%; Figure S5B). When we quantified CIN in cells without de novo centromeres, we found equal numbers in our competition compared to our control (27% versus 24%; Figure S5C), indicating that the decrease in the incidence of de novo centromeres is due to defective assembly rather than more frequent inactivation. Importantly, the Xsubtelo dicentrics from these flies display lower levels of CENP-C at the lacO compared to CAL1-GFP-LacI/Xsubtelo controls (Figure S5D–E), consistent with less CENP-A recruitment at the lacO array, resulting in weaker de novo centromeres.
We next inspected chromosomes with CENP-C at lacO for endogenous centromere loss and found that it is less frequent in the competition background (2% versus 23%; Figure S5F). Interestingly, the Xsubtelo chromosomes that lose their endogenous centromere display higher levels of CENP-C at the de novo centromere compared to dicentric Xsubtelo chromosomes, something that we did not observe in CAL1-GFP-LacI/Xsubtelo chromosomes with no endogenous CENP-C signal (Figure S5G–H). The CENP-C increase could be indicative of de novo centromeres that are not weak to begin with and successfully outcompete the endogenous centromere (as in Figure 4B and 4D–E) or de novo centromeres that start out as weak but become stronger after the endogenous centromere is lost. The observation that the largest majority of competition chromosomes display lower de novo/endogenous CENP-C average signal compared to controls (Figure S5E) supports the second possibility. Taken together, our data suggest a positive correlation between de novo centromere strength and endogenous centromere loss, and that weaker de novo centromeres are less efficient in outcompeting the endogenous centromere.
Epigenetic maintenance of de novo centromeres through development
The stable transmission of neocentromeres on the Y chromosome (Tyler-Smith et al., 1999) and on marker chromosomes (Voullaire et al., 1993; Depinet et al., 1997) in humans suggest that once non-centromeric DNA acquires CENP-A chromatin, it can be maintained through cell division and replication (Karpen and Allshire, 1997). However, these rare neocentromeres are analyzed long after their inception, leaving the mechanisms that led to their formation and stabilization difficult to assess. Due to the targeted nature of our de novo centromere assembly system, we have the opportunity to test if CENP-A chromatin at different chromatin and genomic contexts can be maintained in the fly.
In most of our experiments, we expressed CAL1-GFP-LacI under the elav-GAL4 driver, which is constitutively expressed in neurons and glioblasts from embryos to adults (Robinow and White, 1991; Berger et al., 2007). To address if de novo centromeres can be epigenetically maintained, we induced CAL1-GFP-LacI transiently using the nullo-GAL4 driver, which is expressed only during early embryogenesis (2–3 hours after egg laying; Rose and Wieschaus, 1992; Postner and Wieschaus, 1994). We assessed the presence of de novo centromeres in brain spreads from young larvae (2nd instar–L2; ~3 days after laying) and L3 larvae (~6 days after laying), long after the activity of the nullo promoter has ceased. Quantitative reverse-transcription PCR (qRT-PCR) confirmed that nullo-GAL4 larvae no longer express CAL1-GFP-LacI mRNA (Figure S6A–B).
Next, we assessed the viability of nullo-GAL4/CAL1-GFP-LacI or GFP-LacI flies. All CAL1-GFP-LacI/lacO lines, as well as the CAL1-GFP-LacI control, show viability defects (Figure S6C). Additionally, the viable adult flies display severe motion defects even though they are morphologically normal. Overall, the effect on viability observed with the nullo-GAL4 driver was more severe than that observed with engrailed-GAL4 (Figure 3A). This is likely because nullo is expressed in every cell (Rose and Wieschaus, 1992; Postner and Wieschaus, 1994), albeit transiently, whereas engrailed is not, leaving a large population of non-GAL4 expressing cells to compensate for cell death.
Quantification of the presence of CENP-C at the lacO site in nullo-GAL4/CAL1-GFP-LacI/lacO lines showed that, in L2 larvae, de novo centromeres are present in 36–61% of metaphase spreads (Figure 6A–B), indicative of centromere maintenance. In contrast, in L3 larvae, this percentage decreases to 7–16% for all insertions except for the 3peri, where the percentage remains around 55% at both stages (Figure 6B). The low frequencies of de novo centromeres in L3 larvae could be explained by either compensatory growth of cells that never contained de novo centromeres (Morata and Ripoll, 1975; Moreno et al., 2002) or to inefficient maintenance or inactivation of de novo centromeres over longer timescales. To distinguish between these possibilities, we again analyzed the karyotype of L3 brain cells, as cells without CIN (i.e. euploid) likely never formed de novo centromeres. We found that the majority of L3 brain cells (78–90%) from all lacO lines are euploid and by and large do not contain de novo centromeres (93–99%; Figure 6C–D), suggesting that cells with de novo centromeres die and are replaced by euploid cells. Of the very few spreads with CIN, a small percentage lacked de novo centromeres (Figure 6E), suggesting rare de novo centromeres inactivation events. Interestingly, 3peri displays significantly more euploid spreads with de novo centromeres compared to other insertions (45%; Figure 6D), consistent with the observation that the 3peri insertion causes less CIN than other lacO sites. (Figure 2C). The fact that 3peri de novo centromeres are maintained with good efficiency indicates that de novo centromeres have the potential to be maintained through development, so long as cells harboring them are not eliminated due to high levels of CIN and cell death. Importantly, we never observed instances in which the endogenous centromere is inactivated in these animals. We conclude that de novo centromeres cannot outcompete the endogenous when both centromeres are being epigenetically maintained.
Figure 6: Epigenetic maintenance of de novo centromeres through development.
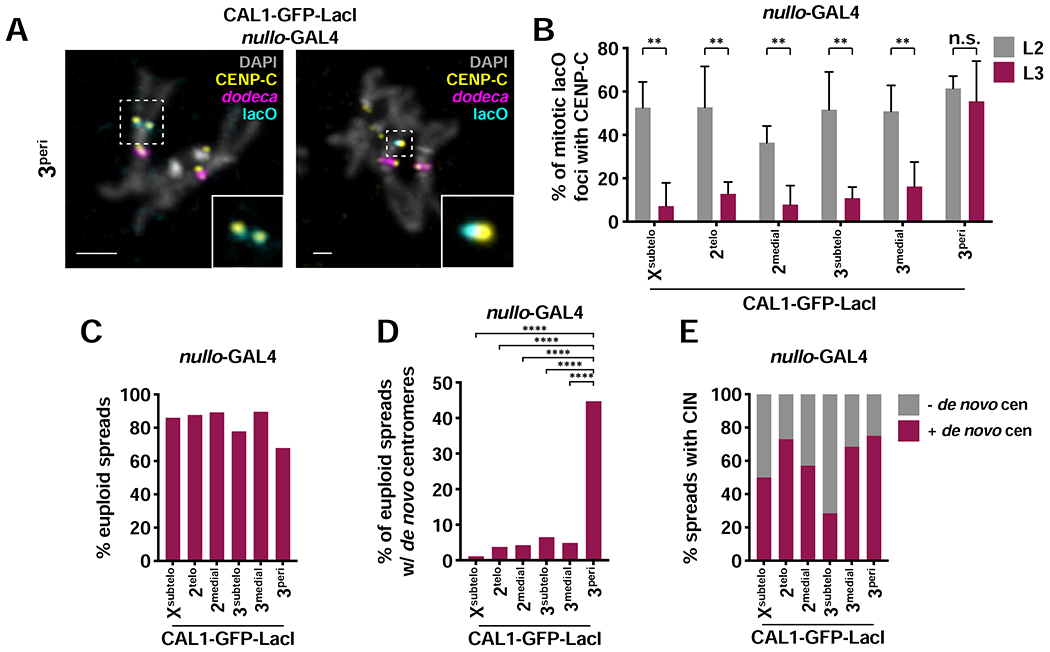
- IF for CENP-C (yellow) and FISH for lacO (cyan) and dodeca (magenta) on mitotic chromosomes from L2 and L3 nullo-GAL4/CAL1-GFP-LacI/lacO brains. DAPI is shown in gray. Insets show colocalization of lacO and CENP-C.
- Percent of co-localizing lacO and CENP-C on mitotic chromosomes from L2 and L3 nullo-GAL4/CAL1-GFP-LacI/lacO brains. Shown is the mean ± SD for 3–8 brains (L2: n=9–53 spreads per brain; L3: n=14–166 spreads per brain). n.s. not significant, ** p<0.01 (unpaired t-test).
- Percent of euploid mitotic spreads from L3 nullo-GAL4/CAL1-GFP-LacI/lacO brains (n=100–211 spreads from 3–8 brains).
- Percent of euploid mitotic spreads with de novo centromeres from L3 nullo-GAL4/CAL1-GFP-LacI/lacO brains (n=76–185 spreads from 3–8 brains). **** p<0.0001 (Fisher’s exact test).
- Percent of mitotic spreads displaying CIN that did (maroon) and did not (gray) contain de novo centromeres from L3 nullo-GAL4/CAL1-GFP-LacI/lacO brains (n=51–185 spreads from 3–8 brains. See also Figures S6–S7.
In Drosophila S2 cells, pulse-overexpression of CENP-A followed by chase results in the recovery of CENP-A islands preferentially located at euchromatin/heterochromatin boundaries (Olszak et al., 2011). To determine if spontaneous de novo centromeres resulting from transient CAL1-GFP-LacI expression are preferentially maintained at specific locations in vivo, we generated nullo-GAL4/CAL1-GFP-LacI progeny and followed ectopic centromeres at L2 and L3 stages. As with elav-GAL4/CAL1-GFP-LacI (Figure S2A), we did not observe de novo centromeres on the 4th or on the Y chromosomes. In L2 brain cells, 54% of spreads contained de novo centromeres (Figure S7A–B), more frequently on the 2 and 3 chromosomes than on the X (Figure S7C), but with no general preference for any sub-chromosomal position (Figure S7D). In L3 cells, only 8% of spreads still displayed de novo centromeres (Figure S7B), most often on chromosome 3, which was the only chromosome for which ectopic centromeres formed preferentially near the pericentromere (Figure S7C–D). We conclude that de novo centromeres are maintained irrespectively of their proximity to heterochromatin boundaries, as reported in a previous study in S2 cells (Olszak et al., 2011), suggesting that the requirements for de novo centromere maintenance are different in vivo.
Telomere capping facilitates the propagation of de novo centromeres
Our de novo centromere maintenance experiments show that, with the exception of 3peri, which is maintained through the L3 stage, de novo centromeres are lost with high frequency, likely due to cell death induced by CIN. Since CIN results from cycles of breaks and fusions initiated by dicentric chromosomes, we wondered if maintenance of de novo centromeres at the L3 stage would be facilitated by the stabilization of lacO-containing chromosome fragments. To test this, we implemented a system that protects broken chromosome ends by generating de novo telomeres (chromosome healing) (Kurzhals et al., 2017) through the expression of the telomere-capping protein HipHop (Gao et al., 2010). We hypothesized that HipHop and CAL1-GFP-LacI expression under nullo-GAL4 in the 3subtelo line, which shows low frequency of de novo centromeres in L3 (Figure 6B), would lead to the healing and maintenance through the L3 stage of lacO fragments generated from DSBs early in embryogenesis.
Although on average the percent of L3 spreads with CENP-C at the lacO is not significantly different between control (CAL1-GFP-LacI/3subtelo) and UAS-hiphop (8% versus 13%; p=0.1, unpaired t-test), we found that one of the five HipHop expressing brains displayed more de novo centromeres (HipHop 1,22%; Figure 7A). The same animal displayed more cells with lacO marker chromosomes (16%) compared to control and to other HipHop expressing larvae (1–7%; Figure 7B–C). The fact that only one in five animals shows a higher incidence of de novo centromeres and lacO chromosomes suggests that successful chromosome healing varies from organism to organism rather than from cell to cell. One possible explanation for this variability is that HipHop, like CAL1-GFP-LacI, is induced only transiently; therefore, breaks that occur after HipHop expression returns to endogenous levels cannot be healed, resulting in CIN and cell death. If an early embryonic cell experiences a break followed by healing on a chromatid, cell death may be delayed, but any remaining intact dicentric chromatid would continue to break during mitosis and fuse to other chromosomes once HipHop expression is back to endogenous levels (Figure 7D). A prediction of this model is that chromosome healing could delay cell death, resulting in more cells displaying CIN. Indeed, HipHop 1 shows significantly more cells with CIN (55%) compared to control and other HipHop larvae (12–27%; Figure 7E). These data indicate that de novo centromeres persist longer when cell death is delayed via chromosome healing.
Figure 7: Telomere capping facilitates the propagation of de novo centromeres.
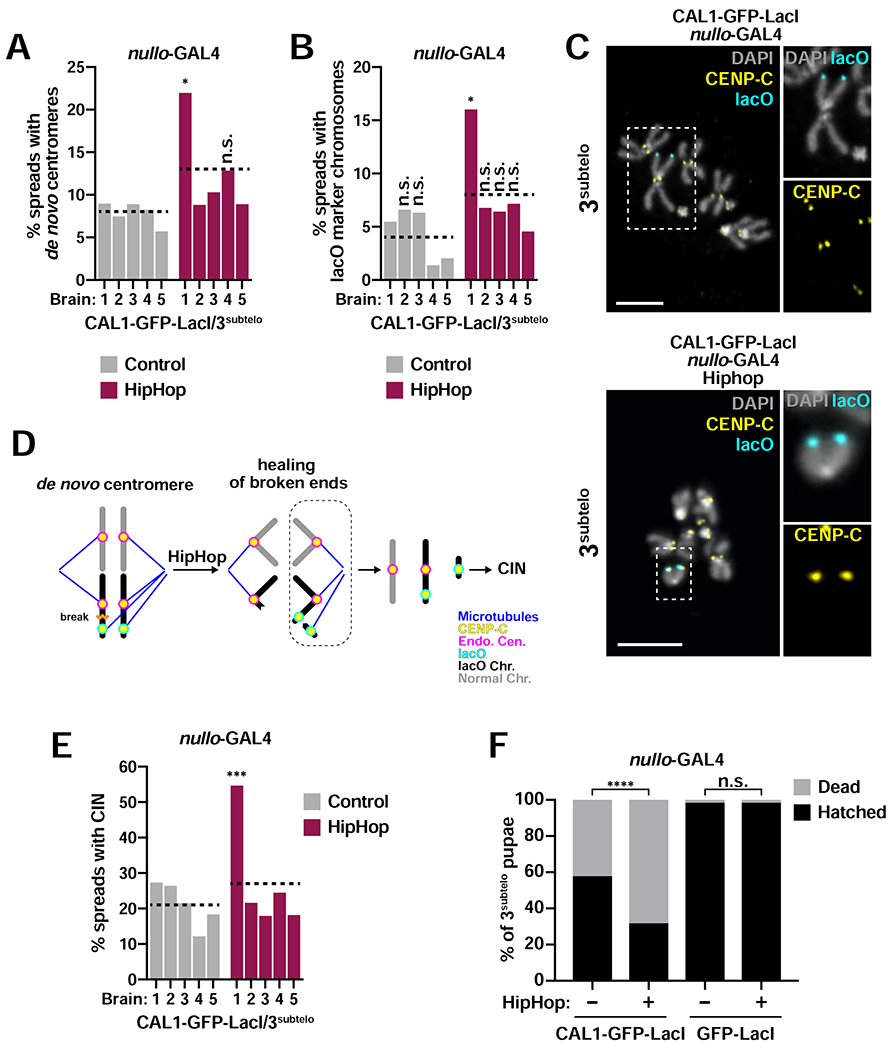
- Percent of mitotic spreads with de novo centromeres in L3 brains from control (nullo-GAL4/CAL1-GFP-LacI/3subtelo) and HipHop (nullo-GAL4/HipHop/CAL1-GFP-LacI/3subteb). Each bar represents an individual brain, and the dotted line represents the average of all five brains (n=91–145 spreads per brain). * p<0.05 (Fisher’s exact test) between Hiphop 1 and all other control and HipHop brains; (n.s. not significant).
- Percent of mitotic spreads with lacO marker chromosomes in L3 brains from control (nullo-GAL4/CAL1-GFP-LacI/3subtelo) and HipHop (nullo-GAL4/HipHop/CAL1-GFP-LacI/3subtelo). Each bar represents an individual brain, and the dotted line represents the average of all five brains (n=73–128 spreads per brain). * p<0.05 (Fisher’s exact test) between Hiphop 1 and all other control and HipHop brains; (n.s. not significant).
- IF for CENP-C (yellow) and FISH for lacO (cyan). DAPI is shown in gray. Top: example of spread from L3 stage nullo-GAL4/CAL1-GFP-Lad/3subtelo brain. Inset shows the 3subtelo chromosome without a de novo centromere (no CENP-C at lacO). Bottom: example of spread from L3 stage nullo-GAL4/HipHop/CAL1-GFP-LacI/3subtelo. Inset shows a lacO marker chromosome with a de novo centromere (CENP-C at lacO).
- Model of CIN resulting from chromosome healing. When a break occurs on one chromatid, the broken chromosome ends are capped by HipHop, preventing chromosome fusions and cell death. However, at later divisions, the remaining dicentric lacO chromatid can break and re-start the breakage-fusion-bridge cycle once HipHop expression returns to endogenous levels.
- Percent of mitotic spreads with CIN in L3 brains from control (nullo-GAL4/CAL1-GFP-LacI/3subtelo) and HipHop (nullo-GAL4/HipHop/CAL1-GFP-LacI/3subtelo). Each bar represents an individual brain, and the dotted line represents the average of all five brains (n=74–128 spreads per brain). *** p<0.001 (Fisher’s exact test) between Hiphop 1 and all other control and HipHop brains.
- Percent of hatched nullo-GAL4/CAL1-GFP-LacI/3subtelo and GFP-LacI/3subtelo pupae with and without HipHop (n=306–555 pupae per cross). n.s. not significant, **** p<0.0001 (Fisher’s exact test).
To establish if chromosome healing affects viability, we quantified pupal hatching in this genetic background. Expression of HipHop in CAL1-GFP-LacI/3subtelo animals results in a significant decrease in viability (32% of pupae hatched) compared to CAL1-GFP-LacI/3subtelo controls (68% of pupae hatched; Figure 7F). Even though chromosome fragments might be healed in these animals, they still missegregate (only 5/42 cells contain both fragments of the broken 3subtelo chromosome), causing gene dosage imbalance. Thus, the healing of broken chromosomes allows for the persistence of cells with chromosome fragments at later developmental stages. In contrast, the high degree of fusions and breaks in CAL1-GFP-LacI/3subtelo animals causes cell death earlier in development, leading to compensatory growth of non-dicentric cells. By counting hundreds of progeny, these viability assays reveal a clear genetic interaction between de novo centromere formation and HipHop expression, validating the efficacy of chromosome healing. Telomere capping therefore reveals that de novo centromeres can be epigenetically maintained at later stages of development.
Discussion
In this study, we have taken a systematic approach to induce de novo centromere formation at defined genomic locations with different characteristics—chromosomal location (pericentric, medial, telomeric and subtelomeric) and chromatin state (silent and active euchromatin, heterochromatin, polycomb chromatin)—by testing the ability of the Drosophila CENP-A assembly factor, CAL1, to deposit CENP-A at these sites. CAL1 tethering via LacI, successfully resulted in the recruitment of the active kinetochore mark CENP-C, showing that de novo centromere formation can occur in each of the genomic and chromatin contexts tested. The size of the lacO array insertion is only ~12kb, about ten times smaller than that of endogenous centromeres (101–171 kb; Chang et al., 2019). Given that CENP-A spreads beyond the lacO array following CENP-A-LacI and CAL1-LacI tethering in Drosophila S2 cells (Mendiburo et al., 2011; Chen et al., 2015), it is possible that CENP-A spreads to surrounding genomic sequences also upon tethering via CAL1-GFP-LacI in flies.
Since our CAL1-GFP-LacI/lacO system generates dicentric chromosomes containing an endogenous centromere and a de novo centromere, it is not surprising that chromosomal instability (CIN) is prevalent in the induced tissues. The dramatic, genome-wide karyotypic changes that ensue upon CAL1 tethering result in cell death, which in turn negatively affects proper tissue development and viability. In general, between 42% and 88% of pupae hatched, indicating that the animal can tolerate high CIN to complete development even though these flies were extremely unhealthy and died after eclosion. Early experiments in Drosophila showed that an imbalance of about one cytological band (i.e. ~1Mb; Adams et al., 2000) or more results in lethality (Lindsley et al., 1972), consistent with our findings. Interestingly, transient induction of de novo centromere formation during a short interval in early embryogenesis also had very strong effects on the viability of hatched flies. Together, these findings are consistent with Barbara McClintock’s model about the instability and effects on genome stability of dicentric chromosomes (McClintock, 1939, 1941).
Dicentric chromosomes are well-established players in the genomic instability and evolution of malignancies (MacKinnon and Campbell, 2011), yet some dicentric chromosomes are able to co-orient and segregate normally (Koshland et al., 1987; Sullivan and Willard, 1998). The severity of genome instability was not uniform between all lacO insertions, with 3peri causing less severe CIN, likely because it can co-orient with the endogenous centromere. While we did not have enough lacO insertions to test the correlation between inter-centromeric distance and dicentric stability, our observations with 3peri indicate that having two centromeres is not necessarily detrimental to genome stability.
Dicentric chromosomes can return to a monocentric state through functional inactivation or through physical removal of centromeric DNA (Koshland et al., 1987; Stimpson et al., 2010). In humans, a centromeric deletion typically removes the active centromere core (i.e. the DNA associated with CENP-A), rather than the entire centromere region (Chiatante et al., 2017). When CAL1-GFP-LacI is expressed constitutively, new CAL1-GFP-LacI is continuously tethered to the lacO, making the de novo centromere less susceptible to functional inactivation than the endogenous centromere. The endogenous centromere on the X is lost via a DSB within or near the centromere and, very rarely, by epigenetic inactivation alone. In some instances, DSB at the centromere or pericentromere leaves behind centromeric DNA with associated centromeric proteins, perhaps exposing this centromere to functional inactivation. The finding that HP1 knockdown decreases the frequency of these chromosomes suggests that epigenetic inactivation requires HP1. Interestingly, although these events are rare, HP1 seems to also affect the inactivation of de novo centromeres.
Our study provides a first step towards testing the epigenetic model for centromere specification in an animal model. By transiently inducing de novo centromere assembly early in embryogenesis, we can track the stability of the new centromeres through larval development. Interestingly, while all lacO insertions are able to assemble de novo centromeres that persist from embryogenesis to the L2 stage, only the 3peri lacO insertion maintains them with the same frequency through the L3 stage likely because it leads to less CIN and cell death. We think it is unlikely that the longer-term persistence of the 3peri centromere is due to its vicinity to heterochromatin, since 2telo, which is within an unannotated region within or near heterochromatin, does not persist as well. Instead, we propose that the loss of de novo centromeres during larval development is due to CIN and cell death rather than to defective maintenance. Consistent with this idea, we observe that most of the mitotic spreads at the L3 stage are euploid, and thus likely represent the population of cells that never assembled de novo centromeres. Additionally, ectopic centromeres resulting from both constitutive and transient expression of CAL1-GFP-LacI were found at many different positions within chromosomes, with no apparent preference for euchromatin/heterochromatin boundaries as reported for Drosophila S2 cells (Olszak et al., 2011). In our experiments, ectopic centromeres form upon overexpression of CAL1, not CENP-A, so they likely reflect CAL1-mediated CENP-A localization, which could be different from overexpression-induced CENP-A mislocalization, which in human cells has been shown to be mediated not by HJURP but by DAXX (Lacoste et al., 2014). Alternatively, our results could underscore differences between cell culture and in vivo studies.
CIN is a result of dicentric chromosomes that break during mitosis and initiate breakage-fusion-bridge cycles. Healing of the earliest chromosomal fragments that form from a broken dicentric prevents fusions and further breakages enabling longer-term de novo centromere maintenance. Co-expressing the telomere capping protein HipHop along with CAL1-GFP-LacI in a line in which de novo centromeres does not last through the L3 stage (3subtelo) shows that de novo centromeres can be maintained longer-term. However, these persisting chromosome fragments missegregate due to the lack of cohesion at the de novo centromere (Oliveira et al., 2014), worsening animal viability. Thus, proximity to cohesin-rich heterochromatin is likely a requirement for long-term maintenance of de novo centromeres on marker chromosomes, in agreement with previous studies (Murphy and Karpen, 1995). Collectively, our work is consistent with the epigenetic model of centromere maintenance in mitosis. Future experiments will be needed to determine if the same principles apply in meiosis and if endogenous centromeres are dispensable and can be replaced by an induced ectopic centromere.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Barbara Mellone (barbara.mellone@uconn.edu). Plasmids and available fly stocks will be shared upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila stocks and handling
All Drosophila melanogaster lines are listed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea pig polyclonal anti-CENP-C | Gary Karpen (Erhardt et al., 2008) | |
| Rabbit anti-CENP-A | Gary Karpen | |
| Alexa488 conjugated rabbit polyclonal anti-GFP | Thermo Fisher | Cat#A21311; RRID:AB_221477 |
| Mouse monoclonal anti-HP1 | DSHB | Cat#C1A9; RRID:AB_528276 |
| Rabbit polyclonal anti-Dcp-1 | Cell Signaling | Cat#9578S; RRID:AB_2721060 |
| Rabbit polyclonal anti-Histone H2AvD pS137 (γH2Av) | Rockland | Cat#600-401-914; RRID:AB_828383 |
| Alexa488 goat anti-rabbit | Thermo Fisher | Cat#A27034; RRID:AB_2536097 |
| Alexa546 goat anti-rabbit | Thermo Fisher | Cat#A11035; RRID:AB_2534093 |
| Alexa647 goat anti-rabbit | Thermo Fisher | Cat#A21244; RRID:AB_2535812 |
| Alexa488 goat anti-guinea pig | Thermo Fisher | Cat#A11073; RRID:AB_2534117 |
| Alexa546 goat anti-guinea pig | Thermo Fisher | Cat#A11074; RRID:AB_2534118 |
| Cy5 donkey anti-guinea pig | Fisher Scientific | Cat#NC0447750 |
| Alexa546 goat anti-mouse | Thermo Fisher | Cat#A11030; RRID:AB_2534089 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| KpnI | NEB | Cat#R0142S |
| BglII | NEB | Cat#R0144S |
| XhoI | NEB | Cat#R0146S |
| HhaI | NEB | Cat#R0139S |
| HpaII | NEB | Cat#R0171S |
| Taq DNA polymerase | NEB | Cat#M0273L |
| T4 Ligase | NEB | Cat#M0202S |
| Dispase/Collagenase | Roche | Cat#10269638001 |
| SlowFade Gold | Thermo Fisher Scientific | Cat#S36936 |
| iTaq Universal SYBR Green | BioRad | Cat#1725121 |
| Salmon sperm DNA | Invitrogen | Cat#15632011 |
| Dextran sulfate | Millipore | Cat#S4030 |
| DAPI | Invitrogen | Cat#D1306 |
| Formamide | Sigma-Aldrich | Cat#47670 |
| Triton-X-100 | Sigma-Aldrich | Cat#T9284 |
| Tween 20 | Sigma-Aldrich | Cat#P9416 |
| Formaldehyde | Sigma-Aldrich | Cat#252549 |
| Glacial acetic acid | Fisher Scientific | Cat#A38S-212 |
| TRI Reagent | Sigma-Aldrich | Cat#T9424 |
| Chloroform | Fisher Scientific | Cat#C298-500 |
| Isopropanol | Fisher Scientific | Cat#416-4 |
| Ethanol | Pharmco-Aaper | Cat#111000200 |
| Methanol | Fisher Scientific | Cat#A412-4 |
| RNaseZap RNase Decontamination Solution | Invitrogen | Cat#AM9780 |
| DEPC | Sigma-Aldrich | Cat#40718 |
| BSA | Sigma-Aldrich | Cat#A7906 |
| NGS | Sigma-Aldrich | Cat#G9023 |
| PIPES | Sigma-Aldrich | Cat#P6757 |
| Magnesium sulfate | Fisher Scientific | Cat#M65-500 |
| EGTA | Sigma-Aldrich | Cat#E0396 |
| Glycine | Fisher Scientific | Cat#BP381-5 |
| Sodium chloride | Sigma-Aldrich | Cat#S3014 |
| Sodium citrate | Sigma-Aldrich | Cat#W302600 |
| Sodium phosphate | Sigma-Aldrich | Cat#S9390 |
| Potassium chloride | Fisher Scientific | Cat#BP366-1 |
| Potassium phosphate | Sigma-Aldrich | Cat#60220 |
| Tris base | Fisher Scientific | Cat#BP152-5 |
| EDTA | Fisher Scientific | Cat#AC118432500 |
| Agarose | Fisher Scientific | Cat#BP160-500 |
| Critical Commercial Assays | ||
| In Situ Cell Death Detection Kit, TMR red | Roche | Cat#12156792910 |
| Expand high fidelity PCR system | Roche | Cat#EHIFI-RO |
| TURBO DNA-free Kit | Invitrogen | Cat#AM1907 |
| iScript cDNA Synthesis Kit | BioRad | Cat#1708891 |
| PureLink PCR Purification Kit | Invitrogen | Cat#K310001 |
| Nucleospin Plasmid | Macherey-Nagel | Cat#740588.250 |
| Plasmid Midi Kit | Qiagen | Cat#12144 |
| RNeasy Mini Kit | Qiagen | Cat#74106 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: Xsubtelo | Lori Wallrath (Li et al., 2003) | N/A |
| D. melanogaster: 2telo | (Vazquez et al., 2002) | BDSC: 26371; FlyBase: FBst0025371 |
| D. melanogaster: 2medial | Gary Karpen (Janssen et al., 2016) | N/A |
| D. melanogaster: 3subtelo | Gary Karpen (Janssen et al., 2016) | N/A |
| D. melanogaster: 3medial | Kristen Johansen (Li et al., 2003) | N/A |
| D. melanogaster: 3peri | Gary Karpen (Janssen et al., 2016) | N/A |
| D. melanogaster: MS1096-GAL4 | Bloomington Drosophila Stock Center | BDSC: 8860; FlyBase: FBst0008860 |
| D. melanogaster: Actin-GAL4/TM6B, Tb1 | Bloomington Drosophila Stock Center | BDSC: 3954; FlyBase: FBst0003954 |
| D. melanogaster: elav-GAL4/CyO | Bloomington Drosophila Stock Center | BDSC: 8765; FlyBase: FBst0008765 |
| D. melanogaster: engrailed-GAL4, UAS-GFP | Bloomington Drosophila Stock Center | BDSC: 25752; FlyBase: FBst0025752 |
| D. melanogaster: nullo-GAL4 | Bloomington Drosophila Stock Center | BDSC: 26875; FlyBase: FBst0026875 |
| D. melanogaster: eyeless-GAL4/CyO | Bloomington Drosophila Stock Center | BDSC: 5535; FlyBase: FBst0005535 |
| D. melanogaster: Sgs3-GAL4 | Bloomington Drosophila Stock Center | BDSC: 6870; FlyBase: FBst0006870 |
| D. melanogaster: UAS-CAL1-GFP-LacI | This paper | N/A |
| D. melanogaster: UAS-GFP-LacI | This paper | N/A |
| D. melanogaster: UAS-hiphop | Kent Golic (Kurzhals et al., 2017) | N/A |
| D. melanogaster: UAS-mCherryRNAi | Bloomington Drosophila Stock Center | BDSC: 35785; FlyBase: FBst0035785 |
| D. melanogaster: UAS-HP1RNAi | Bloomington Drosophila Stock Center | BDSC: 33400; FlyBase: FBst0033400 |
| D. melanogaster: iso-1 | Bloomington Drosophila Stock Center | BDSC: 2057; FlyBase: FBst0002057 |
| D. melanogaster: w1118 | Yih-Woei Fridell | N/A |
| D. melanogaster: w* | Bloomington Drosophila Stock Center | BDSC: 7198; FlyBase: FBst0007198 |
| D. melanogaster: T(2;3)TSTL | Bloomington Drosophila Stock Center | BDSC: 42713; FlyBase: FBst0042713 |
| D. melanogaster: CyO, Actin-GFP | Chris Doe | BDSC: 56554; FlyBase: FBst0056554 |
| Oligonucleotides | ||
| Cloning primer: BglII-CAL1 Forward (5’-CAGTAGATCTATGGCGAATGCGGTGGTG-3’) | This Paper | |
| Cloning primer: XhoI-GFP Forward (GCAGTCTCGAGATGAGTAAAGGAGAAGAACT-3’) | This paper | |
| Cloning primer: KpnI-LacI Reverse (5’-CAGTGGTACCTTAGGCAACCTTTCTCTTC-3’) | This paper | |
| Inverse PCR primer: inv-lacO Forward (5’-GCTTCGGCTATCGACGGGACCACCTTATGTTA-3’) | (Cryderman et al., 1998) | |
| Inverse PCR primer: inv-lacO Reverse (5’-GACGAAATGAACCACTCGGAACCATTTGAGCG-3’) | (Cryderman et al., 1998) | |
| qPCR primer: Rp49 Forward (5’-CCGCTTCAAGGGACAGTATC-3’) | (Chen et al., 2015) | |
| qPCR primer: Rp49 Reverse: (5’-GACAATCTCCTTGCGCTTCT-3’) | (Chen et al., 2015) | |
| qPCR primer: LacI Forward (5’-TATCCGCTGGATGACCAGGA-3’) | (Chen et al., 2015) | |
| qPCR primer: LacI Reverse (5’-CAGTCGCGTACCGTCTTCAT-3’) | (Chen et al., 2015) | |
| Recombinant DNA | ||
| pMT-CAL1-GFP-LacI | (Chen et al., 2014) | |
| pUASTattB | Konrad Basler (Bischof et al., 2007) | |
| Software and Algorithms | ||
| Prism | GraphPad | RRID:SCR_002798 |
| SoftWoRx 7.0.0 | GE Healthcare | http://incelldownload.gehealthcare.com/bin/download_data/SoftWoRx/7.0.0/SoftWoRx.htm |
| Illustrator | Adobe | RRID:SCR_010279 |
| Photoshop | Adobe | RRID:SCR_014199 |
| CellProfiler | Broad Institute | RRID:SCR_007358 |
| ZEN Digital Imaging for Light Microscopy | Zeiss | RRID:SCR_013672 |
| Other | ||
| FISH probe: lacOLNA, see Table S6. | This paper | |
| FISH probe: dodecaLNA, see Table S6. | (Bateman et al., 2012) | |
| FISH probe: AAGAG, see Table S6. | (Jagannathan et al., 2016) | |
| FISH probe: Sec6-AAGAG, see Table S6 | This paper | |
| FISH probe: AATAT, see Table S6. | (Jagannathan et al., 2016) | |
| FISH probe: SATIII, see Table S6. | (Bateman et al., 2012; Joyce et al., 2012) | |
| FISH probe: AACAC, see Table S6. | (Jagannathan et al., 2016) | |
| FISH probe: Sec6 secondary oligo, see Table S6. | (Beliveau et al., 2015) | |
Flies were reared on standard cornmeal, molasses, and yeast food (https://bdsc.indiana.edu) at 25°C, except for Xsubtelo and RNAi experiments (Figures 1,4–5 and S5) were reared at 29°C. All lacO; UAS-CAL1-GFP-LacI and lacO; UAS-GFP-LacI lines were maintained as homozygous lines, except 3peri which was maintained as a heterozygous line with the T(2;3)TSTL double balancer. For crosses with 3peri parents, only progeny with 3peri and UAS-trangenes was selected.
Crosses were set up using GAL4 males and lacO; UAS-transgene or UAS-transgene females, with the exception of Xsubtelo crosses in Figures 1D–E, 4A–E, 5B–E and S5, which were set up using Xsubtelo; elav-GAL4/CyO, Actin-GFP females and UAS-transgene males.
The following GAL4 drivers were used for mitotic and interphase cell preparations: elav-GAL4/CyO, Actin-GFP (brain) and nullo-GAL4 (early embryonic). For elav-GAL4/CyO, Actin-GFP crosses, progeny was counter selected for GFP.
For polytene chromosomes preparations the driver used was Sgs3-GAL4 (salivary glands). For expression in the wing, the driver used was MS1096-GAL4. For wing phenotypes only female progeny were scored as males do not inherit the MS1096-GAL4 containing X chromosome. For expression in the eye, the driver used was eyeless-GAL4/CyO. Adult progenies were aged for 72 hours following eclosion, frozen at −20°C, and imaged. For eyeless-GAL4 adult eye phenotypes, straight winged males and females were scored. For viability experiments the drivers used were nullo-GAL4 and engrailed-GAL4 (semi-ubiquitous) were used.
For RNA extraction for RT-qPCR, L2 larvae were collected from crosses set up on grape agar plates (Genesee Scientific), while L3 larvae were collected from crosses set up on standard food. UAS-CAL1-GFP-LacI females were crossed with either nullo-GAL4, Actin-GAL4/TM6B, Tb1, or w1118 males. Parents were allowed to lay for 6–18 hours before being passed to new food. For L2 stage, progeny were allowed to develop for 2–3 days before collection. For L3 stage, progeny were allowed to develop for 4–5 days before being collected.
METHOD DETAILS
Plasmid cloning and transgenic fly generation
Primers were used to amplify CAL1-GFP-LacI (BglII-CAL1 Forward: 5’-CAGTAGATCTATGGCGAATGCGGTGGTG-3’; KpnI-LacI Reverse: 5’-CAGTGGTACCTTAGGCAACCTTTCTCTTC-3’) and GFP-LacI (XhoI-GFP Forward: 5’-GCAGTCTCGAGATGAGTAAAGGAGAAGAACT-3’; KpnI-LacI Reverse: 5’-CAGTGGTACCTTAGGCAACCTTTCTCTT C-3’) from pMT-CAL1-GFP-LacI (Chen et al., 2014), including 5’ BglII and 3’ KpnI sites in CAL1-GFP-LacI amplicons and 5’ XhoI and 3’ KpnI sites in GFP-LacI amplicons (see underlined region of primer sequences). For GAL4/UAS mediated expression (Brand and Perrimon, 1993) and site-specific transgene integration, the vector pUASTattB (gift from Johannes Bischof (Bischof et al., 2007)), as well as the PCR amplicons, were digested using either BglII and KpnI for CAL1-GFP-LacI or XhoI and KpnI for GFP-LacI. CAL1-GFP-LacI and GFP-LacI were ligated into pUASTattB, placing them downstream of the UAS promoter, and upstream of attB, making UAS-CAL1-GFP-LacI-attB and UAS-GFP-LacI-attB. Transgenic flies were generated by Genetic Services, Inc. (Sudbury, MA) using phiC31 mediated integration with y, w; attP2 (Bloomington Drosophila Stock Center # 8862) and y, w; attP40 (N/A) flies. Transformants were selected by mini-white expression.
Inverse PCR
To identify the insertion site for lacO repeats, inverse PCR was performed according to Cryderman et al. (1998) and Huang et al. (2009). Genomic DNA was isolated from lacO containing stocks 3medial and 2telo. Three micrograms of DNA were then digested at 37°C for 2.5 hours with either HhaI (2telo) or HpaII (3medial), followed by heat inactivation at 65°C for 20 minutes. Ligation was performed by incubating 5μl of digested DNA with 2U of T4 ligase at 16°C overnight. To amplify circularized DNA containing the lacO integration site, PCR was performed using 5μl of circularized DNA and primers for p-element arms (inv-lacO Forward: 5’-GCTTCGGCTATCGACGGGACCACCTTATGTTA-3’; inv-lacO Reverse: 5’-GACGAAATGAACCACTCGGAACCATTTGAGCG-3’). Amplicons were reamplified and gel purified using the PureLink Quick Gel Extraction Kit (Invitrogen), and sequenced on an ABI sequencer (BioTech Center, University of Connecticut). Sequences for Xsubtelo flies was provided to us by Lori Wallrath. BLAST searches using these sequences on FlyBase were used to determine the genomic coordinates for the Xsubtelo, 2telo, and 3medial lacO insertions.
Characterization of lacO locations
lacO position was determined using a lacO FISH probe on mitotic chromosomes from cells expressing GFP-LacI (see Figure S1I). Cytological bands previously characterized (see Figure 1A, Refs column) were validated by tethering CAL1-GFP-LacI to lacO on polytene chromosomes (Figure S1A–F). lacO insertion coordinates for xsubtelo, 2telo, and 3medial were determined using inverse PCR combined with BLAST searches on FlyBase (see Inverse PCR). The lacO insertion coordinates for 2medial, 3subtelo, and 3peri were determined based on the position of MiMIC sites (Venken et al., 2011) used for integration by Janssen et al. (2016). Chromatin domains surrounding lacO insertion loci was determined using the “9-state model, S2 cells” track in the FlyBase genome browser.
Tissue monolayer immunofluorescence (IF)
L3 brains were dissected and immediately fixed in 3.7% formaldehyde, PBS for 5 minutes. Brains received four 5 minutes washes in PBS and were then incubated in 1mL of 0.4% Dispase/Collagenase (Roche), PBS for 10 minutes at 37°C. Individual brains were transferred to 2μL drops of PBS on a siliconized coverslip as squashed onto a Superfrost Plus (VWR) slide. Slides were frozen in liquid nitrogen, immediately fixed in 400μL of 3.7% formaldehyde, PBS-T for 5 minutes, and washed four times in PBS for 5 minutes. Slides were blocked in 1% milk, PBST for 1 hour. Slides were incubated with an anti-H2AvD-pS137 (referred to as γH2Av) antibody (rabbit, 1:250; Rockland) overnight at 4°C, washed three times in 1% milk, PBS-T for 5 minutes, then incubated with secondary antibodies either at 4°C overnight or at room temperature for 1 hour. Slides were washed three times in PBS-T for 5 minutes, then mounted in SlowFade with DAPI.
Whole mount IF and TUNEL staining
Imaginal discs were dissected from wandering L3 larvae in PBS and fixed in 4% formaldehyde, 100mM PIPES (pH 6.9), 0.3% Triton X-100, 2mM EGTA (pH 8), 1mM MgSO4 for 23 minutes, followed by two rinses and two 20 minute washes in PBS, 0.3% Triton X-100 (PBS-Tx3). Tissues were blocked in PBS-Tx3, 1% BSA, 0.1% NGS, 0.1mM Glycine for 30 minutes, rocking at room temperature. Tissues were incubated with anti-GFP (chicken, 1:500; Abcam) and anti-Dcp-1 (rabbit, 1:100; Cell Signaling) antibodies overnight at 4°C. Tissues were rinsed twice and washed twice for 15 minutes in blocking solution, and then incubated with secondary antibodies for 90 minutes at room temperature. Tissues were rinsed twice and washed twice for 30 minutes in PBS-Tx3. For TUNEL labelling, tissues were washed twice for 5 minutes in PBS, 0.5% Triton X-100 (PBS-Tx5) and then incubated in 100mM sodium citrate, 0.1% Triton X-100 at 65°C for 30 minutes. Tissues were washed twice for 5 minutes in PBS-T, twice for 10 minutes in 25μL Reaction Buffer (In Situ Cell Death DETC TMR Red; Roche), and incubated for 60 minutes in 36μL Reaction Buffer at 37°C. 4μL of TdT Enzyme Mix (In Situ Cell Death DETC TMR Red; Roche) was added to each reaction and incubated at 37°C for 3 hours. Tissues were washed twice for 5 minutes with PBS-Tx5 and mounted in SlowFade with DAPI.
Polytene chromosome preparation and IF
Polytene chromosomes were prepared according to Kennison (2008), with modifications. Larvae were dissected in PBS. Salivary glands were fixed with 45% acetic acid, 2% formaldehyde for 2 minutes, then placed in a 20μl drop of 60% acetic acid on a siliconized coverslip for 5 minutes and spread onto a Superfrost Plus (VWR) slide. Slides were frozen in liquid nitrogen and washed twice with PBS for 15 minutes, then washed once in PBS, 0.1% Triton X-100 (PBS-T) for 15 minutes. Slides were blocked with PBS-T, 1% BSA and incubated with an Alexa488 conjugated anti-GFP antibody (rabbit, 1:100; Invitrogen). Slides were mounted in SlowFade Gold containing 1μg/mL DAPI (SlowFade with DAPI).
Interphase cell preparation and IF
For HP1 staining, L3 brains were dissected in PBS and incubated in 0.5% sodium citrate for 2 minutes. Individual brains were fixed in a 5μL drop of methanol-acetic acid fixative (10 parts methanol, 2 parts acetic acid, 1 part H2O) on a siliconized coverslip for 2 minutes and squashed onto Shandon Polysine (Thermo) slides. Slides were frozen in liquid nitrogen and washed once in PBS for 5 minutes, once in 1% PBS, 1% Triton X-100 for 10 minutes and blocked in PBS, 1% milk for 30 minutes. Slides were incubated with an anti-HP1 antibody (mouse, 1:20; James and Elgin, 1986) overnight at 4°C, washed three times in PBS-T for 5 minutes, then incubated with secondary antibodies either at 4°C overnight or at room temperature for 1 hour. Slides washed three times in PBS-T for 5 minutes, and then mounted using SlowFade with DAPI.
For CENP-A staining, L3 brains were dissected in PBS and incubated in 0.5% sodium citrate for 8 minutes. Individual brains were fixed and teased apart in a 6μL drop of 3.7% formaldehyde, 0.45% sodium citrate on a siliconized coverslip for 4 minutes and squashed onto Shandon Polysine (Thermo) slides. Slides were frozen in liquid nitrogen and washed once in PBS for 5 minutes, once in PBS-T for 5 minutes and blocked in 3% BSA, PBS-T for 30 minutes. Slides were incubated with an anti-CENP-A antibody (rabbit serum, 1:500; gift of Gary Karpen) overnight at 4°C, washed three times in PBS-T for 5 minutes, then incubated with secondary antibodies either at 4°C overnight or at room temperature for 1 hour. Slides washed three times in PBS-T for 5 minutes, and then proceeded to FISH.
Mitotic chromosome IF
Larval mitotic chromosomes were prepared from L2 or L3 brains as previously described in Pimpinelli et al. (2011), with the following modifications. Brains from larvae were dissected and incubated in 0.5% (w/v) sodium citrate for 8 minutes. Individual brains were fixed in 6μL drops of 45% acetic acid, 2% formalin for 6 minutes on a siliconized coverslip and squashed onto Shandon Polysine™ (Thermo) slides. Slides were frozen in liquid nitrogen and washed once in PBS for 5 minutes, once in PBS-T for 5 minutes and blocked in 1% BSA, PBS for 30 minutes. As high concentrations of acetic acid in this protocol result in extraction of most if not all histones (Dick and Johns, 1968), we could not detect CENP-A with anti-CENP-A antibodies and chose to identify active centromeres using an anti-CENP-C antibody. Slides were incubated with an anti-CENP-C antibody (guinea pig, 1:500; Erhardt et al., 2008) overnight at 4°C, washed three times in PBS-T for 5 minutes, then incubated with secondary antibodies either at 4°C overnight or at room temperature for 1 hour. Slides washed three times in PBS-T for 5 minutes, and then proceeded to FISH. In nullo-GAL4 experiments, L2 brains contained fewer mitotic spreads than L3 (approximately 20-90 versus 80-210).
Fluorescence in situ hybridization (FISH)
Slides were post-fixed in 4xSSC, 3.7% formaldehyde for 6 minutes, followed by three 3 minute washes in 4xSSC. Slides were washed once in 2xSSC, 0.1% Tween-20 (2xSSC-T) for 5 minutes, and once in 50% formamide, 2xSSC-T for 5 minutes. 4–40pmol of each probe was resuspended in hybridization buffer [50% formamide, 2xSSC-T, 10% dextran sulfate, 0.8μg/mL salmon sperm DNA]. Probes and slides were denatured at 95°C for 5 minutes and allowed to hybridize at 37°C overnight. Slides were then washed once in 0.1xSSC at 37°C for 10 minutes, followed by two 10 minute washes in 0.1xSSC at room temperature. Slides were mounted using SlowFade with DAPI. See Table S6 for FISH probe information.
Sequential FISH
Following FISH and imaging for Figure 5C–E, coverslips were removed and slides were washed three times for 5 minutes in PBS-T, three times for 3 minutes in 4xSSC, once for 5 minutes in 2xSSC-T, and once for 5 minutes in 50% formamide, 2xSSC-T. 40pmol of Sec6-AAGAG probe was resuspended in hybridization buffer (50% formamide, 2xSSC-T, 10% dextran sulfate, 0.8μg/mL salmon sperm DNA). Probes and slides were denatured at 95°C for 5 minutes and allowed to hybridize at 37°C overnight. Slides were then washed once in 2xSSC-T at 60°C for 15 minutes, once in 2xSSC-T at room temperature for 10 minutes, and once in 0.2xSSC for 5 minutes. Slides were incubated with 40pmol of Sec6 secondary oligo in 30% formamide, 2xSSC for 30 minutes at room temperature. Slides were then washed once in 2xSSC-T at 60°C for 15 minutes, once in 2xSSC-T at room temperature for 10 minutes, and once in 0.2xSSC for 5 minutes. Slides were mounted using SlowFade with DAPI.
Pupal viability assay
Ten female flies containing lacO and either UAS-CAL1-GFP-LacI or UAS-GFP-LacI were crossed with 5–8 males of either the genotype w*/Y; engrailed-GAL4, UAS-GFP or nullo-GAL4 on normal food at 25°C. Flies were allowed to mate for 24 hours before being transferred to a new vial. Vials were incubated at 25°C, with eclosed flies being dumped and counted twice a day. After 20 days, pupal viability was scored based on the percent of pupae that were empty, signifying successful eclosion.
RT-qPCR for CAL1-GFP-LacI expression
Total RNA was extracted from 27–30 L2 progeny or 10 L3 progeny resulting from females containing UAS-CAL1-GFP-LacI crossed with either nullo-GAL4, Actin-GAL4/TM6B, Tb1 (positive control; progeny that inherited TM6B, Tb1 were excluded), or w1118 (negative control) males were collected, flash frozen, and homogenized in 500μL of TRI Reagent (Sigma Aldrich). Samples were spun at 12,000 × g for 10 minutes at 4°C and supernatant was transferred to a new tube and then incubated for 5 minutes at room temperature. After the incubation, 100μL of chloroform was added and samples were shaken by hand for 15 seconds or until solution became opaque. Samples were incubated for 3 minutes at room temperature and spun at 10,000 × g for 15 minutes at 4°C. The RNA (aqueous phase) was transferred to an RNase free tube with 250μL of isopropanol, incubated at room temperature for 10 minutes, and spun at 12,000 × g for 10 minutes at 4°C. Pellets were washed with 75% ethanol and spun at 7,500 × g for 5 minutes at 4°C. Pellets were resuspended in 50μL of DEPC-treated H2O and quantified using a NanoDrop 2000c (Thermo). Two Turbo DNase (Invitrogen) treatments were performed on 10μg of RNA with RNeasy (Qiagen) RNA cleanups following each DNase treatment.
Reverse-transcription (RT) was performed using iScript cDNA synthesis kit (Bio-Rad) and 500ng of total RNA. Quantitative PCR (qPCR) was performed using SYBR-green (Bio-Rad) and primers for LacI (LacI Forward: 5’-TATCCGCTGGATGACCAGGA-3’; LacI Reverse: 5’-CAGTCGCGTACCGTCTTCAT-3’) and Rp49 (Rp49 Forward 5’-CCGCTTCAAGGGACAGTATC-3’; Rp49 Reverse: 5’-GACAATCTCCTTGCGCTTCT-3’) and 12.5ng of cDNA on a CFX96 Real-Time System (Bio-Rad).
Imaging
Images of cells and tissues were acquired at 25°C using an Inverted Deltavision RT restoration Imaging System (Applied Precision) equipped with a Cool Snap HQ2 camera (Photometrics) and either 100x/1.40 NA, 60x/1.42 NA oil objectives or 20x/0.75 NA air objective (Olympus). Image acquisition and processing was performed using softWoRx software (Applied Precision). Images taken with the 60x/1.42 NA or 100x/1.40 NA oil objectives were deconvolved for 5 cycles using the conservative method. For mitotic chromosomes, cells were imaged using the 100x objective. For interphase cells and tissue monolayers, cells were imaged using either 60x or 100x objectives. For polytene chromosomes, whole polytenes were imaged using the 60x objective and stitched together without deconvolution. Insets were taken using the 100x objective. For whole mount tissues, images were taken using the 20x objective and stitched together. Images are presented as maximum intensity projections. Images were saved as Photoshop files.
Images of adult flies were acquired using a SteREO Discovery V12 stereomicroscope equipped with an Axiocam 506 color camera (Zeiss). Single z’s were taken of individual wings, while whole flies and eyes were imaged as z-stacks. Stack alignment, projections, and image processing was performed using ZEN pro and the Extended Focus module (Zeiss). Images were saved as TIFF files.
Images were scaled using Photoshop (Adobe) and assembled into figures using Illustrator (Adobe).
QUANTIFICATION AND STATISTICAL ANALYSIS
Scoring pupal viability
Quantification pupal viability was based on the percent of pupae that were empty, indicative of complete development to adulthood. For the pupal viability of progeny resulting from crosses with engrailed-GAL4, UAS-GFP (Figure 3A). For pupal viability of progeny resulting from crosses with nullo-GAL4 (Figure S6C). For experiments comparing pupal viability between progeny resulting from crosses with nullo-GAL4 with and without UAS-hiphop (Figure 7F).
Scoring centromere inactivation
For quantification of loss of CENP-C at endogenous centromeres (Figure 4A) we reanalyzed lacO chromosomes quantified from Figure 1E that showed CENP-C colocalizing with lacO. Quantification of loss of CENP-C at the endogenous centromere of the Xsubtelo chromosome (Figures 4A, 4D and 5D–E) was performed using FISH probes for AAGAG and/or AATAT to mark the endogenous centromere locus. We inferred the presence of X centromere sequences indirectly through the presence of intact AAGAG satellite. We could not use Oligopaint FISH for Maupiti, because of variability of FISH signal (i.e. the absence of signal would not necessarily imply that the centromere island is missing).
Quantification of centromere signal intensity
For quantification of endogenous and de novo centromere’s CENP-C signal, we used SoftWoRx (GE Healthcare) to acquire total signal intensities of endogenous and de novo centromeres on Xsubtelo chromosomes, as well as total signal intensities of endogenous centromeres on normal X chromosomes from maximum intensity quick projected images. For Figure 4E, plotted data points represent the total signal intensity of de novo centromeres divided by that of the endogenous centromere of the same chromosome. For Figure S5E, plotted data points represent the total signal intensity of the endogenous centromere of Xsubtelo chromosomes with CENP-C at lacO divided by the total signal intensity of the endogenous centromere of a normal X chromosome within the same spread. For Figure S5H, plotted data points represent the total signal intensity of the de novo centromere of Xsubtelo chromosomes without CENP-C at the endogenous centromere (marked by SATIII FISH) divided by the total signal intensity of the endogenous centromere of a normal X chromosome within the same spread.
Karyotype analysis and genome instability
For quantification of genome instability in crosses with elav-GAL4 (Figure 2C), IF for CENP-C and FISH using a lacO probe and a satellite probe (Xsubtelo: SATIII; 2telo and 2medial: AACAC; 3subtelo, 3medial and 3peri; dodeca; “no lacO”: AATAT) was performed on mitotic chromosome spreads from the L3 brains. Chromosomal instability (CIN) was scored based on chromosomal abnormalities separated into four categories: 1) aneuploidy as indicated by a non-diploid (2n≠8) number of chromosomes, having marker chromosomes, and/or having greater than or less than 2 copies of any chromosome; 2) an abnormal number of satellite loci per cell, scored based on any change in the number of chromosome specific satellite foci (satellite loci on the Y chromosome were excluded from analysis; see Table S5); 3) broken chromosomes; 4) chromosome fusions.
For quantification of CIN in crosses with nullo-GAL4 (Figures 6C–E), mitotic chromosome spreads from L3 brains were scored based on 1) whether or not the spread contained a de novo centromere, and 2) whether or not the spread displayed markers of CIN (i.e. marker chromosomes, chromosome breaks, chromosome fusions, and/or an abnormal number of chromosomes). CIN is sometimes rare in L3 larvae (i.e. n<5 aneuploid spreads), therefore, to avoid misrepresentation of CIN and de novo centromere maintenance events caused by brains with a low number of spreads showing CIN, these data are represented as percentages per genotype, and statistical significance is determined by Fisher’s exact test. We excluded L2 cells from CIN quantification as L2 brains have a low number of mitotic cells, and the mitotic chromosomes often overlap and are ambiguous in regard to chromosomal stability.
For quantification of chromosome-specific aneuploidy in crosses with elav-GAL4 (Figure S3D), mitotic chromosome spreads from L3 brains were scored based on whether they contained abnormal numbers of non-lacO chromosomes (≠2), lacO chromosomes (≠1) and their normal, homologous chromosomes (≠1).
For quantification of lacO chromosome nondisjunction in crosses with elav-GAL4 (Figure S3E), mitotic chromosomes spreads from L3 brains were scored based on whether they contained a number of lacO chromosome greater than 1.
For quantification of lacO marker chromosomes in crosses with elav-GAL4 (Figure S3F), marker chromosomes from L3 brains were scored based on whether they contained lacO foci.
Calculating expression of CAL1-GFP-LacI
Fold change in expression of CAL1-GFP-LacI was determined using the ΔCT method (i.e. Fold change = 2−ΔCT = 2−(gene of Interest-Internal control)) (Schmittgen and Livak, 2008) with Rp49 as a reference gene.
Automated DSB quantification
Quantification of cells containing DSBs was performed using the open source software CellProfiler (cellprofiler.org). Images were acquired using the same exposure parameters for all samples. Images were converted into two grayscale TIFF files for nuclei (DAPI) and DSBs (γH2Av). Nuclei were identified as objects ranging from 25–125 pixels. DSBs were identified as objects contained within nuclei that range from 2–10 pixels. All objects outside of these ranges were discarded. Automated quantification was compared against manually counted γH2Av foci for validation purposes. Data was exported as Excel files for further analysis. See Table S6 for detailed experimental parameters.
Statistical analyses and representation
Descriptions for the representation of quantification data and the statistical significance can be found in the associated figure legend.
Percentages, averages, and standard deviation were calculated using Excel (Microsoft). Statistical analyses were performed using Prism (GraphPad). Quantification involving mitotic and interphase cells from larval brains is generally represented as either mean ± SD or scatter plots using unpaired t-test to determine statistical significance, except in the case of a brain that had less than 9 spreads, in which case counts between brains were added together, represented as either percentages (in graphs) or fractions (in text) using Fisher’s exact test to determine statistical significance. Quantification of eye sizes is represented as mean ± SD using unpaired t-test to determine statistical significance. Quantification of wing phenotypes and viability are represented as percentages using Fisher’s exact test to determine statistical significance. Quantification of eye organization is represented as percentages in a stack graph using Chi-squared to determine statistical significance. Graphs were constructed using Prism (GraphPad) and assembled into figures using Illustrator (Adobe).
DATA AND CODE AVAILABILITY
Not applicable.
ADDITIONAL RESOURCES
Not applicable.
Supplementary Material
Highlights.
De novo centromeres can be targeted to several genomic locations in vivo
De novo centromeres cause widespread genome instability and cell and organism death
Continuously induced de novo centromeres lead to endogenous centromere loss on the X
De novo centromeres are maintained epigenetically in mitotic tissues
Acknowledgements
We are grateful to Lori Wallrath, Kristen Johansen, Andrew Belmont, and Gary Karpen for providing the lacO lines. We also thank Kent Golic for suggesting the HipHop experiment and providing the UAS-hiphop line, Johannes Bischof for the pUASTattB plasmid, Ran-Der Hwang for early help with eye imaging, Pariksheet Nanda for help with automated image quantification in CellProfiler, and Rebecca Marrero and members of the Mellone lab for helpful input. This work was supported by NIH grants R01GM108829 and R35GM131868 to B.G.M. Stocks obtained from the Bloomington Drosophila Stock Center (BDSC; NIH P40OD018537) were used in this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. (2000). The Genome Sequence of Drosophila melanogaster. Science 287, 2185–2195. [DOI] [PubMed] [Google Scholar]
- Barnhart MC, Kuich PHJL, Stellfox ME, Ward JA, Bassett EA, Black BE, and Foltz DR (2011). HJURP is a CENP-A chromatin assembly factor sufficient to form a functional de novo kinetochore. J. Cell Biol 194, 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman JR, Larschan E, D’Souza R, Marshall LS, Dempsey KE, Johnson JE, Mellone BG, and Kuroda MI (2012). A Genome-Wide Screen Identifies Genes That Affect Somatic Homolog Pairing in Drosophila. G3 GenesGenomesGenetics 2, 731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliveau BJ, Boettiger AN, Avendaño MS, Jungmann R, McCole RB, Joyce EF, Kim-Kiselak C, Bantignies F, Fonseka CY, Erceg J, et al. (2015). Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat. Commun 6, 7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger C, Renner S, Lüer K, and Technau GM (2007). The commonly used marker ELAV is transiently expressed in neuroblasts and glial cells in the Drosophila embryonic CNS. Dev. Dyn 236, 3562–3568. [DOI] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, and Basler K (2007). An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. U. S. A 104, 3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, and Perrimon N (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Dev. Camb. Engl 118, 401–415. [DOI] [PubMed] [Google Scholar]
- Camahort R, Li B, Florens L, Swanson SK, Washburn MP, and Gerton JL (2007). Scm3 is essential to recruit the histone h3 variant cse4 to centromeres and to maintain a functional kinetochore. Mol. Cell 26, 853–865. [DOI] [PubMed] [Google Scholar]
- Cech JN, and Peichel CL (2016). Centromere inactivation on a neo-Y fusion chromosome in threespine stickleback fish. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol 24, 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-H, Chavan A, Palladino J, Wei X, Martins NMC, Santinello B, Chen C-C, Erceg J, Beliveau BJ, Wu C-T, et al. (2019). Islands of retroelements are major components of Drosophila centromeres. PLoS Biol. 17, e3000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-C, Dechassa ML, Bettini E, Ledoux MB, Belisario C, Heun P, Luger K, and Mellone BG (2014). CAL1 is the Drosophila CENP-A assembly factor. J. Cell Biol 204, 313–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C-C, Bowers S, Lipinszki Z, Palladino J, Trusiak S, Bettini E, Rosin L, Przewloka MR, Glover DM, O’Neill RJ, et al. (2015). Establishment of Centromeric Chromatin by the CENP-A Assembly Factor CAL1 Requires FACT-Mediated Transcription. Dev. Cell 34, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiatante G, Giannuzzi G, Calabrese FM, Eichler EE, and Ventura M (2017). Centromere Destiny in Dicentric Chromosomes: New Insights from the Evolution of Human Chromosome 2 Ancestral Centromeric Region. Mol. Biol. Evol 34, 1669–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryderman DE, Cuaycong MH, Elgin SC, and Wallrath LL (1998). Characterization of sequences associated with position-effect variegation at pericentric sites in Drosophila heterochromatin. Chromosoma 107, 277–285. [DOI] [PubMed] [Google Scholar]
- Dekanty A, Barrio L, Muzzopappa M, Auer H, and Milán M (2012). Aneuploidy-induced delaminating cells drive tumorigenesis in Drosophila epithelia. Proc. Natl. Acad. Sci 109, 20549–20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depinet TW, Zackowski JL, Earnshaw WC, Kaffe S, Sekhon GS, Stallard R, Sullivan BA, Vance GH, Van Dyke DL, Willard HF, et al. (1997). Characterization of Neo-Centromeres in Marker Chromosomes Lacking Detectable Alpha-satellite DNA. Hum. Mol. Genet 6, 1195–1204. [DOI] [PubMed] [Google Scholar]
- Dick C, and Johns EW (1968). The effect of two acetic acid containing fixatives on the histone content of calf thymus deoxyribonucleoprotein and calf thymus tissue. Exp. Cell Res 51, 626–632. [DOI] [PubMed] [Google Scholar]
- Drees B, Ali Z, Soeller WC, Coleman KG, Poole SJ, and Kornberg T (1987). The transcription unit of the Drosophila engrailed locus: an unusually small portion of a 70,000 bp gene. EMBO J. 6, 2803–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, and Almouzni-Pettinotti G (2009). HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell 137, 485–497. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Mellone BG, Betts CM, Zhang W, Karpen GH, and Straight AF (2008). Genome-wide analysis reveals a cell cycle–dependent mechanism controlling centromere propagation. J. Cell Biol 183, 805–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinetti D, Folco HD, Nechemia-Arbely Y, Valente LP, Nguyen K, Wong AJ, Zhu Q, Holland AJ, Desai A, Jansen LET, et al. (2013). A two-step mechanism for epigenetic specification of centromere identity and function. Nat. Cell Biol 15, 1056–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LET, Bailey AO, Yates JR, Bassett EA, Wood S, Black BE, and Cleveland DW (2009). Centromere specific assembly of CENP-A nucleosomes is mediated by HJURP. Cell 137, 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Walser J-C, Beaucher ML, Morciano P, Wesolowska N, Chen J, and Rong YS (2010). HipHop interacts with HOAP and HP1 to protect Drosophila telomeres in a sequence-independent manner. EMBO J. 29, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, and Cheeseman IM (2013). Induced dicentric chromosome formation promotes genomic rearrangements and tumorigenesis. Chromosome Res. 21, 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington JJ, Van Bokkelen G, Mays RW, Gustashaw K, and Willard HF (1997). Formation of de novo centromeres and construction of first-generation human artificial microchromosomes. Nat. Genet 15, 345–355. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K, and Malik HS (2001). The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293, 1098–1102. [DOI] [PubMed] [Google Scholar]
- Heun P, Erhardt S, Blower MD, Weiss S, Skora AD, and Karpen GH (2006). Mislocalization of the Drosophila centromere-specific histone CID promotes formation of functional ectopic kinetochores. Dev. Cell 10, 303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AM, Rehm EJ, and Rubin GM (2009). Recovery of DNA sequences flanking P-element insertions in Drosophila: inverse PCR and plasmid rescue. Cold Spring Harb. Protoc. 2009, pdb.prot5199. [DOI] [PubMed] [Google Scholar]
- Jagannathan M, Warsinger-Pepe N, Watase GJ, and Yamashita YM (2016). Comparative Analysis of Satellite DNA in the Drosophila melanogaster Species Complex. G3 GenesGenomesGenetics 7, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James TC, and Elgin SC (1986). Identification of a nonhistone chromosomal protein associated with heterochromatin in Drosophila melanogaster and its gene. Mol. Cell. Biol 6, 3862–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, Breuer GA, Brinkman EK, van der Meulen AI, Borden SV, van Steensel B, Bindra RS, LaRocque JR, and Karpen GH (2016). A single double-strand break system reveals repair dynamics and mechanisms in heterochromatin and euchromatin. Genes Dev. 30, 1645–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce EF, Williams BR, Xie T, and Wu C-T (2012). Identification of genes that promote or antagonize somatic homolog pairing using a high-throughput FISH-based screen. PLoS Genet. 8, e1002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpen GH, and Allshire RC (1997). The case for epigenetic effects on centromere identity and function. Trends Genet. 13, 489–496. [DOI] [PubMed] [Google Scholar]
- Kennison JA (2008). Dissection of Larval Salivary Glands and Polytene Chromosome Preparation. Cold Spring Harb. Protoc. 2008, pdb.prot4708. [DOI] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. (2011). Comprehensive analysis of the chromatin landscape in Drosophila. Nature 471, 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland D, Rutledge L, Fitzgerald-Hayes M, and Hartwell LH (1987). A genetic analysis of dicentric minichromosomes in saccharomyces cerevisiae. Cell 48, 801–812. [DOI] [PubMed] [Google Scholar]
- Kurzhals RL, Fanti L, Ebsen ACG, Rong YS, Pimpinelli S, and Golic KG (2017). Chromosome Healing Is Promoted by the Telomere Cap Component Hiphop in Drosophila. Genetics 207, 949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Danzer JR, Alvarez P, Belmont AS, and Wallrath LL (2003). Effects of tethering HP1 to euchromatic regions of the Drosophila genome. Dev. Camb. Engl 130, 1817–1824. [DOI] [PubMed] [Google Scholar]
- Lindsley DL, Sandler L, Baker BS, Carpenter AT, Denell RE, Hall JC, Jacobs PA, Miklos GL, Davis BK, Gethmann RC, et al. (1972). Segmental aneuploidy and the genetic gross structure of the Drosophila genome. Genetics 71, 157–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, and de Lange T (2015). Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 163, 1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon RN, and Campbell LJ (2011). The Role of Dicentric Chromosome Formation and Secondary Centromere Deletion in the Evolution of Myeloid Malignancy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madigan JP, Chotkowski HL, and Glaser RL (2002). DNA double-strand break-induced phosphorylation of Drosophila histone variant H2Av helps prevent radiation-induced apoptosis. Nucleic Acids Res. 30, 3698–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall OJ, Chueh AC, Wong LH, and Choo KHA (2008). Neocentromeres: new insights into centromere structure, disease development, and karyotype evolution. Am. J. Hum. Genet 82, 261–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B (1939). The Behavior in Successive Nuclear Divisions of a Chromosome Broken at Meiosis. Proc. Natl. Acad. Sci. U. S. A 25, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B (1941). The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 26, 234–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, and Cheeseman IM (2016). The molecular basis for centromere identity and function. Nat. Rev. Mol. Cell Biol 17, 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendiburo MJ, Padeken J, Fülöp S, Schepers A, and Heun P (2011). Drosophila CENH3 is sufficient for centromere formation. Science 334, 686–690. [DOI] [PubMed] [Google Scholar]
- Miga KH, Newton Y, Jain M, Altemose N, Willard HF, and Kent WJ (2014). Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res. 24, 697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi G, Xiao H, Wisniewski J, Smith MM, and Wu C (2007). Nonhistone Scm3 and histones CenH3-H4 assemble the core of centromere-specific nucleosomes. Cell 129, 1153–1164. [DOI] [PubMed] [Google Scholar]
- Morata G, and Ripoll P (1975). Minutes: Mutants of Drosophila autonomously affecting cell division rate. Dev. Biol 42, 211–221. [DOI] [PubMed] [Google Scholar]
- Moreno E, Basler K, and Morata G (2002). Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature 416, 755–759. [DOI] [PubMed] [Google Scholar]
- Murphy TD, and Karpen GH (1995). Localization of Centromere Function in a Drosophila Minichromosome. Cell 82, 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohzeki J, Bergmann JH, Kouprina N, Noskov VN, Nakano M, Kimura H, Earnshaw WC, Larionov V, and Masumoto H (2012). Breaking the HAC Barrier: Histone H3K9 acetyl/methyl balance regulates CENP-A assembly. EMBO J. 31, 2391–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira RA, Kotadia S, Tavares A, Mirkovic M, Bowlin K, Eichinger CS, Nasmyth K, and Sullivan W (2014). Centromere-Independent Accumulation of Cohesin at Ectopic Heterochromatin Sites Induces Chromosome Stretching during Anaphase. PLOS Biol. 12, e1001962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszak AM, van Essen D, Pereira AJ, Diehl S, Manke T, Maiato H, Saccani S, and Heun P (2011). Heterochromatin boundaries are hotspots for de novo kinetochore formation. Nat. Cell Biol 13, 799–808. [DOI] [PubMed] [Google Scholar]
- Pimpinelli S, Bonaccorsi S, Fanti L, and Gatti M (2011). Immunostaining of Mitotic Chromosomes from Drosophila Larval Brain. Cold Spring Harb. Protoc. 2011, pdb.prot065524. [DOI] [PubMed] [Google Scholar]
- Postner MA, and Wieschaus EF (1994). The nullo protein is a component of the actin-myosin network that mediates cellularization in Drosophila melanogaster embryos. J. Cell Sci 107 ( Pt 7), 1863–1873. [DOI] [PubMed] [Google Scholar]
- Régnier V, Vagnarelli P, Fukagawa T, Zerjal T, Burns E, Trouche D, Earnshaw W, and Brown W (2005). CENP-A Is Required for Accurate Chromosome Segregation and Sustained Kinetochore Association of BubR1. Mol. Cell. Biol 25, 3967–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinow S, and White K (1991). Characterization and spatial distribution of the ELAV protein during Drosophila melanogaster development. J. Neurobiol 22, 443–461. [DOI] [PubMed] [Google Scholar]
- Rose LS, and Wieschaus E (1992). The Drosophila cellularization gene nullo produces a blastoderm-specific transcript whose levels respond to the nucleocytoplasmic ratio. Genes Dev. 6, 1255–1268. [DOI] [PubMed] [Google Scholar]
- Rosin LF, and Mellone BG (2017). Centromeres Drive a Hard Bargain. Trends Genet. TIG 33, 101–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe AG, Abrams L, Qu Y, Chen E, and Cotter PD (2000). Tetrasomy 15q25→qter: Cytogenetic and molecular characterization of an analphoid supernumerary marker chromosome. Am. J. Med. Genet 93, 393–398. [PubMed] [Google Scholar]
- Sato H, Masuda F, Takayama Y, Takahashi K, and Saitoh S (2012). Epigenetic inactivation and subsequent heterochromatinization of a centromere stabilize dicentric chromosomes. Curr. Biol. CB 22, 658–667. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, and Livak KJ (2008). Analyzing real-time PCR data by the comparative CT method. Nat. Protoc 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Silva S.M. da, Moutinho-Santos T, and Sunkel CE (2013). A tumor suppressor role of the Bub3 spindle checkpoint protein after apoptosis inhibition. J Cell Biol 201, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stimpson KM, Song IY, Jauch A, Holtgreve-Grez H, Hayden KE, Bridger JM, and Sullivan BA (2010). Telomere Disruption Results in Non-Random Formation of De Novo Dicentric Chromosomes Involving Acrocentric Human Chromosomes. PLOS Genet. 6, e1001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stimpson KM, Matheny JE, and Sullivan BA (2012). Dicentric chromosomes: unique models to study centromere function and inactivation. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol 20, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan BA, and Willard HF (1998). Stable dicentric X chromosomes with two functional centromeres. Nat. Genet 20, 227–228. [DOI] [PubMed] [Google Scholar]
- Sun X, Wahlstrom J, and Karpen G (1997). Molecular Structure of a Functional Drosophila Centromere. Cell 91, 1007–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Le HD, Wahlstrom JM, and Karpen GH (2003). Sequence analysis of a functional Drosophila centromere. Genome Res. 13, 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torras-Llort M, Medina-Giró S, Moreno-Moreno O, and Azorín F (2010). A Conserved Arginine-Rich Motif within the Hypervariable N-Domain of Drosophila Centromeric Histone H3 (CenH3CID) Mediates BubR1 Recruitment. PLoS ONE 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler-Smith C, Gimelli G, Giglio S, Floridia G, Pandya A, Terzoli G, Warburton PE, Earnshaw WC, and Zuffardi O (1999). Transmission of a Fully Functional Human Neocentromere through Three Generations. Am. J. Hum. Genet 64, 1440–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez J, Belmont AS, and Sedat JW (2002). The dynamics of homologous chromosome pairing during male Drosophila meiosis. Curr. Biol. CB 12, 1473–1483. [DOI] [PubMed] [Google Scholar]
- Venken KJT, Schulze KL, Haelterman NA, Pan H, He Y, Evans-Holm M, Carlson JW, Levis RW, Spradling AC, Hoskins RA, et al. (2011). MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat. Methods 8, 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voullaire L, Saffery R, Earle E, Irvine DV, Slater H, Dale S, Sart D. du, Fleming T, and Choo KHA (2001). Mosaic inv dup(8p) marker chromosome with stable neocentromere suggests neocentromerization is a post-zygotic event. Am. J. Med. Genet 102, 86–94. [DOI] [PubMed] [Google Scholar]
- Voullaire LE, Slater HR, Petrovic V, and Choo KH (1993). A functional marker centromere with no detectable alpha-satellite, satellite III, or CENP-B protein: activation of a latent centromere? Am. J. Hum. Genet 52, 1153–1163. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.