

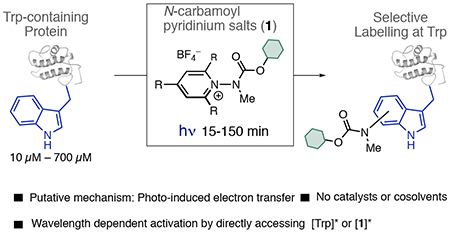
Abstract
We report here a photochemical process for the selective modification of tryptophan (Trp) residues in peptides and small proteins using electron-responsive N-carbamoylpyridinium salts and UV–B light. Preliminary mechanistic experiments suggest that the photoconjugation process proceeds through photoinduced electron transfer (PET) between Trp and the pyridinium salt, followed by fragmentation of the pyridinium N–N bond and concomitant transfer of this group to Trp. The reaction displays excellent site selectivity for Trp and is tolerant to other, redox-active amino-acid residues. Moreover, the reaction proceeds in pure aqueous conditions without the requirement of organic cosolvents or photocatalysts, is enhanced by glutathione, and operates efficiently over a wide range of peptide concentrations (10–700 μM). The scope of the process was explored through the labeling of 6-Trp-containing peptides and proteins ranging from 1 to 14 kDa. We demonstrate the versatility of the N-carbamoylpyridinium salt both by tuning the electrochemical and photochemical properties of the pyridinium scaffold to enable challenging photoconjugation reactions and by using the carbamoyl moiety to tether a plethora of productive functional groups, including reactive handles, purification tags, and removable protecting groups.
Graphical Abstract
The selective chemical manipulation of proteins has enabled the precision engineering of new properties and functions into biomolecular structure, in turn facilitating advancements in therapeutic development and chemical biology.1 Given the vast diversity of biomolecular structure as well as the demand for increasingly complex applications of bioconjugation across multiple disciplines, the development of a diverse array of chemical bioconjugation methods with complementary site selectivities and capabilities remains a necessity.2 In that regard, photochemically driven bioconjugation reactions, which are underexplored compared to thermal processes, can enable both spatiotemporal control of reactivity without the need for prefunctionalization with bio-orthogonal groups3 and access to site selectivity patterns that complement thermal processes.4a Efforts to achieve photochemical labeling of biomolecular structure have primarily focused upon the use of photocatalytic approaches toward protein cross-linking4b,c as well as the modification of cysteine,4d–f tyrosine,4g,h and C-termini.4i,j
We propose here that a catalyst-free approach to photobioconjugation, wherein the inherent photolability of native protein structure is leveraged to induce protein labeling, could offer a complementary approach to catalytic techniques and enable access to mild labeling conditions (pure aqueous buffer, low [protein], short reaction times) as well as circumvent the challenge of achieving site selectivity between amino acids possessing similar oxidation potentials. To that end, we noted tryptophan’s (Trp) propensity to participate in photoinduced electron transfer (PET).5 Trp possesses the largest molar absorptivity of the natural amino acids (ε ≈ 4500, 500 M−1 cm−1, 290, 300 nm respectively) as well as the highest propensity to undergo photoionization (Φe ≈ 0.008–0.08).5a,b As a result, Trp displays a rich varied profile of PET chemistry, participating in a wide variety of intraprotein PET events as both a reductive quencher and a photoreductant (Figure 1A).
Figure 1.
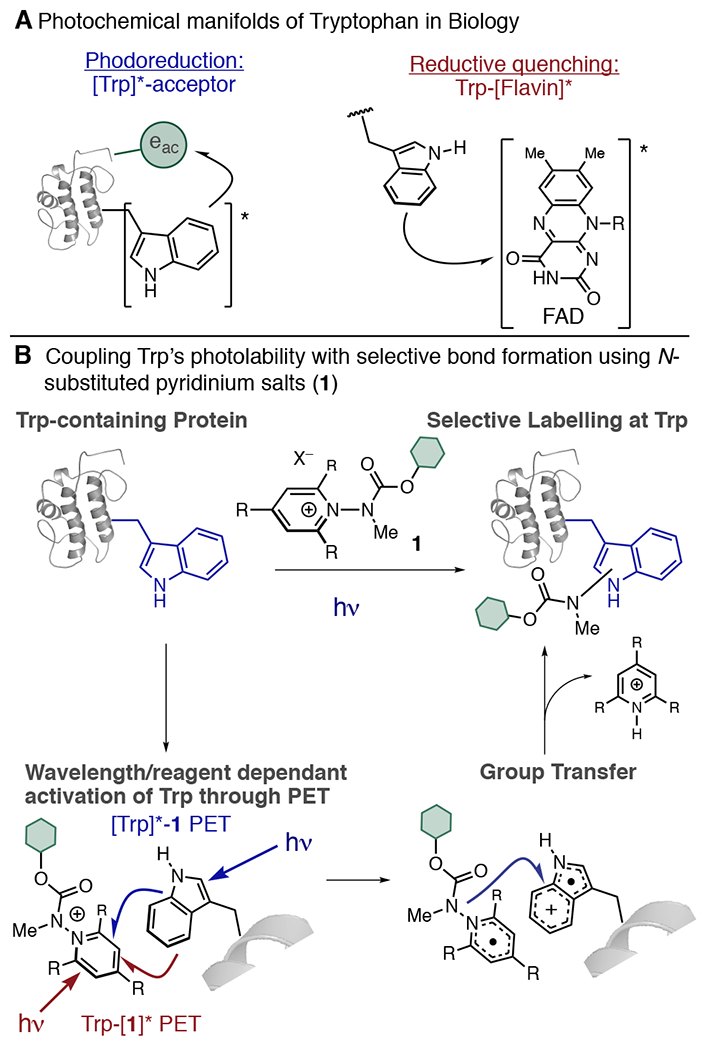
Pyridinium salt reagents for Trp-selective protein modification by mimicking biological PET.
Trp is a desirable bioconjugation target, as it is a rare (~1% natural abundance) amino acid. Thus, any method selective for Trp would allow for access to bespoke protein constructs with high precision, structural homogeneity, and reproducibility. Current strategies for Trp modification generally rely upon the generation of Trp-selective electrophiles such as metallocarbenoids,6a–c nitroxides,6d sulfenyl chlorides,6e trifluoromethyl radicals,6f transition metal complexes,6g and oxidative strategies.6h While elegant, indoles are only moderately nucleophilic7 and must compete with other biological nucleophiles. Exploiting Trp’s inherent photolability could offer a complementary means of engaging Trp functionalization under more biologically compatible conditions. Recently, Shi reported a method for photochemical modification of peptides that modified Trp, His, and C-termini through a unique, photoredox-mediated alkenylation process.8
Here, we propose to use a Katritsky-type9 N-substituted pyridinium salt (1) to couple PET and Trp labeling into a single process (Figure 1B). 1 features a cationic π-electron system that is readily reduced10 by SET. Variation of the substituents on the pyridinium ring, as well as the N-substituent, can lead to homolytic cleavage of the labile N–N bond, restoring pyridinium aromaticity and generating a reactive new radical species that can be harnessed for a bond forming reaction.11 Using Trp as the single electron reductant to activate 1, we propose to simultaneously generate a Trp•+ and a reactive N-centered radical that could recombine to result in a labeled protein with selective modification at Trp (Figure 1B), thereby enabling a labeling process that is kinetically viable and avoids photodegradation pathways typically associated with Trp photoionization.5c
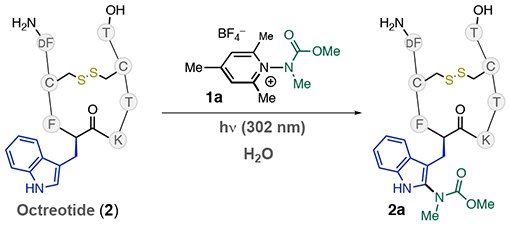
We sought to test this hypothesis in the context of a photolabile substrate, thereby necessitating the development of a robust process to achieve labeling. We chose Octreotide·OAc (2), a 1 kDa Somatostatin analogue that possesses a d-Trp residue and an established record of photolability,12 as a substrate for our preliminary studies (Table 1). We commenced by irradiating a solution of 2 (100 μM) and pyridinium salt 1a (10 mM) in NH4OAc buffer with UV-B light (UVP lamp, 302 nm, 8 W) for 30 min (Table 1, entry 1). We observed high conversion levels of 2, but with only moderate conversion to the labeled peptide 2a (34% by LC-MS, entry 1). Further optimization initially failed to improve upon this, with 7 mM of 1a only showing formation of 2a in 27% (entry 2). In these instances, we noted the formation of undefinable byproducts, which were interpreted as photodegradation of 2. When the reaction was performed in the presence of 1 mM of glutathione (GSH), we observed complete conversion (>95%) to 2a with a mono/di label ratio of >20/1 (entry 3). Given that GSH-disulfide was observed postreaction by LC/MS, we propose that GSH enhances the reaction by acting in its key biological function as a reactive oxygen species (ROS) scavenger.13 Notably, we did not observe any adducts of GSH with 2, 2a, or 1a under these conditions. Tandem MS revealed that Trp is the exclusive site of modification in the reaction. These conditions also allowed the labeling of 2 at lower concentrations, with complete conversion when [2] = 10 μM (entry 4). We next explored the role of buffer in the reaction, finding that the labeling reaction proceeded smoothly in 20 mM pH 7.4 phosphate (entry 5) and 20 mM NaOAc (entry 6), although the former condition required a slightly longer reaction time and resulted in a smaller mono/di labeling ratio (3:1). Tandem MS suggests the second label is also located on the Trp residue. Finally, no reaction took place in the absence of light (entry 7). 1a displays both excellent aqueous solubility (>100 mM) and stability, with <10% decomposition over 48 h in pH 6.9 NH4OAc buffer, millimolar M concentrations of buffered (pH 7.4) GSH, and human serum. We attribute this stability to the methylated 2, 4, and 6 positions of 1a, which likely suppresses nucleophilic addition.
Table 1.
Selected Optimization
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | [2] μM | [1] mM | Buffera | Additive | t (min) | Conversion of 2b | %2a (mono/di)b |
| 1 | 100 | 10 | NH4OAc | 30 | >95% | 34% (>20:1) | |
| 2 | 100 | 7 | NH4OAc | 30 | >95% | 27% (>20:1) | |
| 3 | 100 | 7 | NH4OAc | 1 mM GSH | 30 | >95% | 95% (>20:1) |
| 4 | 10 | 7 | NH4OAc | 1 mM GSH | 30 | >95% | 95% (>20:1) |
| 5 | 100 | 7 | Na2HPO4 | 1 mM GSH | 45 | >95% | <95% (3:1) |
| 6 | 100 | 7 | NaOAc | 1 mM GSH | 30 | >95% | 95% (>20:1) |
| 7c | 100 | 7 | NH4OAc | 1 mM GSH | 30 | 0% | 0% |
[Buffer] = 20 mM. NH4OAc: pH = 6.9. Na2HPO4: pH = 7.4. NaOAc: pH = 8.9.
Estimated by LC-MS TIC, average of 2 runs.
Reaction was performed in the dark.
With these conditions in hand, we next explored the ability of 1 to deliver reporter handles to 2 (Figure 2). The carbamate group of 1a provides a simple and convenient handle for derivatization, and we were pleased to observe that removable protecting groups (2b), purification tags (2c), and linking groups (2d) can easily be transferred using this chemistry (Figure 2A). Labeling of 2 could also be performed at higher concentrations to enable milligram scale preparation; a 3.5 mg scale labeling of 2 ([2] = 700 μM) with 1a yielded 2a in an average of 68% isolated yield after HPLC purification. We additionally synthesized salt 1c, which displays an amine group that can be used for further derivatization. Linking 1c with an alkyne-containing NHS ester followed by photochemical labeling enabled us to transfer an alkyne to 2 in excellent conversion (Figure 2B). LC/MS traces of crude reaction mixtures show that the labeling process produces clean reaction profiles (Figure 2C).
Figure 2.
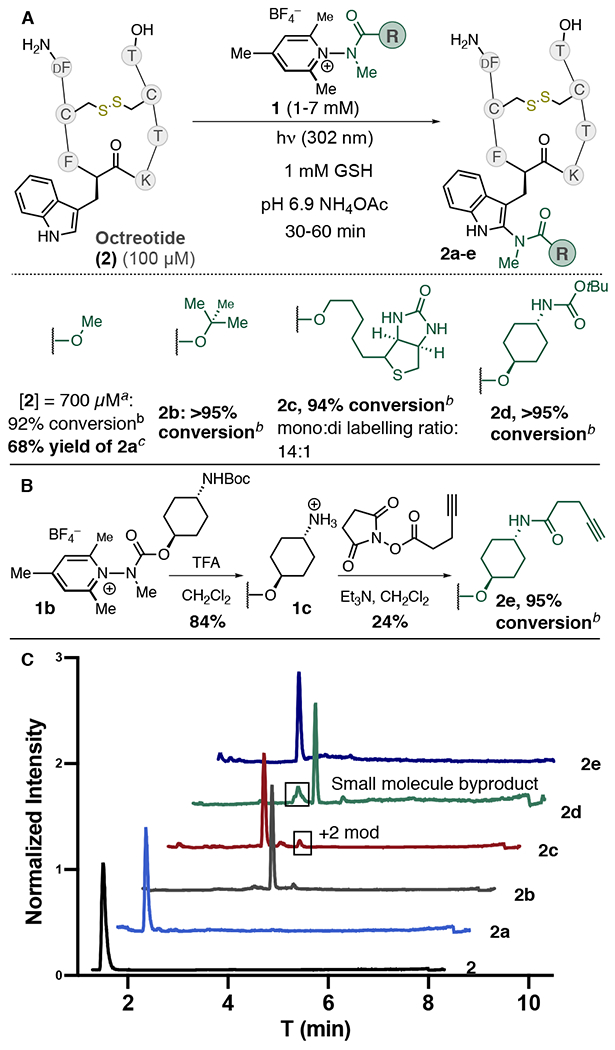
(A) Transferring group scope. (B) Synthesis of alkyne-labeled 2 via derivatizable pyridinium salt 1c. (C) LC/MS TIC traces of crude reaction mixtures. a 120 min reaction time. [GSH] = 5 mM, [1a] = 20 mM. b % conversion estimated by TIC, average of 2–3 experiments. Ratio of mono/di label >20:1 unless stated otherwise. c Isolated after HPLC purification, average of three experiments.
We next explored the labeling of Trp residues on peptides and proteins possessing both differing topologies and redox/photolabile residues (Figure 3). Leuprorelin (3), a 1.2 kDa peptide that possesses a single Trp, Tyr, and His residue, furnished singly modified conjugate 3a in high conversion when irradiated with 1a. MS-MS experiments confirmed Trp as the exclusive site of modification. 3 was also amenable to labeling on a 3 mg scale ([3] = 500 μM), furnishing 3a with a single modification at Trp in a 73% average isolated yield. Additionally, antibiotic daptomycin (4) (1.6 kDa, 1 Trp, 1 kynurenine), Melittin (5) (2.7 kDa 1, Trp), and glucagon (6) (3.4 kDa 1 Trp, 1 His, 2 Tyr, 1 Met, carboxylate terminus) each underwent selective Trp labeling in good conversion. The concentrations of peptide used in each instance were varied in accordance with the solubility properties of each specific peptide, wherein 4–6 aggregate at concentrations ≥100 μM.14
Figure 3.
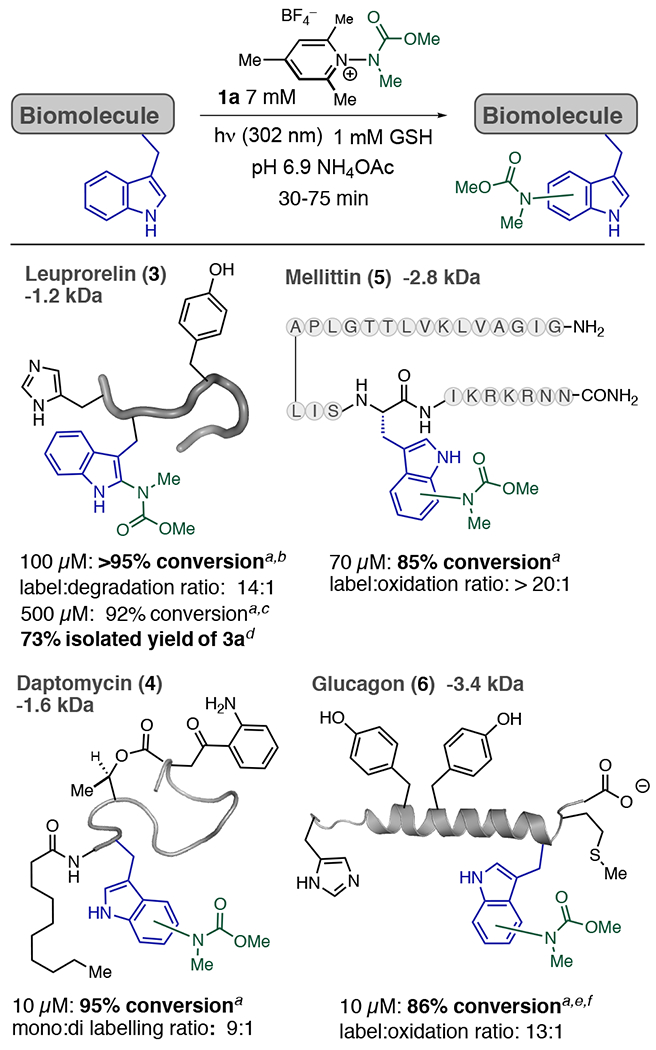
Scope of the Trp photobioconjugation process. a % conversion was measured by TIC, average of 2 runs. b [1a] = 2 mM. c 150 min reaction, [GSH] = 5 mM, [1a] = 10 mM. d Isolated after HPLC purification, average of two experiments. e 20 mM 1a. f Tris(2-carboxyethyl)phosphine (TCEP) (1 mM) used as an additive.
We next sought to gain a preliminary mechanistic understanding of the labeling process that could help explain the selectivity of this process for Trp (Figure 4). Temporal control experiments, consisting of successive exposures of the reaction mixture to UV-B light, clearly demonstrated that formation of 2a only occurs in the presence of UV-B light (Figure 4A). Reactions performed either in the dark or with 365 nm UV-A irradiation showed no conversion. Spin-trap TEMPO and NaI, which is both an efficient quencher of Trp fluorescence15 and known to form charge transfer species with pyridinium species,16 also inhibited the reaction. A large excess of prenyl alcohol, which could serve to trap free N-centered radicals, had no effect on the reaction (Figure 4B). Stern–Volmer analysis of 2 with 1a showed nonlinear (280 nm) and linear (295 nm) quenching (see Supporting Information); but the analysis of these curves is complicated by the overlap of absorption of Trp and 1c (Figure 4C). Given that both 2 and 1a have overlapping absorbance, it is possible that the labeling process could be initiated by photoexcitation of either/both Trp and 1a. We confirmed the photoexcitation of Trp by observation of complete deuteration of the C4-position of d,l-Trp in D2O under 302 nm irradiation5a (Figure 4D). Given that 1a does not absorb beyond 295 nm, we irradiated 2 and 1a with light that was filtered using a 305 nm long-pass filter, and the reaction proceeded efficiently to completion, albeit at a slower rate (Figure 4B). We also estimated the quantum yield of the labeling of 2 under conditions that solely excite Trp, observing Φlabeling = 0.08 ± 0.02. This value is in good agreement with the quantum yields for Trp-photionization,5d suggesting electron transfer from [Trp]* is a mechanistic component of the labeling process. NMR analysis of peptide conjugates 2a and 3a revealed that labeling occurs at the indole C2 position through C–N bond formation; no coupling at the methyl group was observed (Figure 4D).17 Taken together, we propose that PET is the key mechanistic process that drives the reaction (Figure 1B), though a clear understanding of the nature of the group transfer remains obscure. Trp selectivity can be partially rationalized by its large ε values compared to Tyr (~50 times larger) as well as potentially by preorganization of Trp and 1 through precomplexation.18 A number of alternative mechanisms, including amination via a free N-centered radical11a or a nitrenium19 species, are possible. However, the propensity of these two species to either rapidly rearrange17,19b or aminate aromatic structures analogous to Phe and Tyr led us to disfavor these two possibilities.
Figure 4.
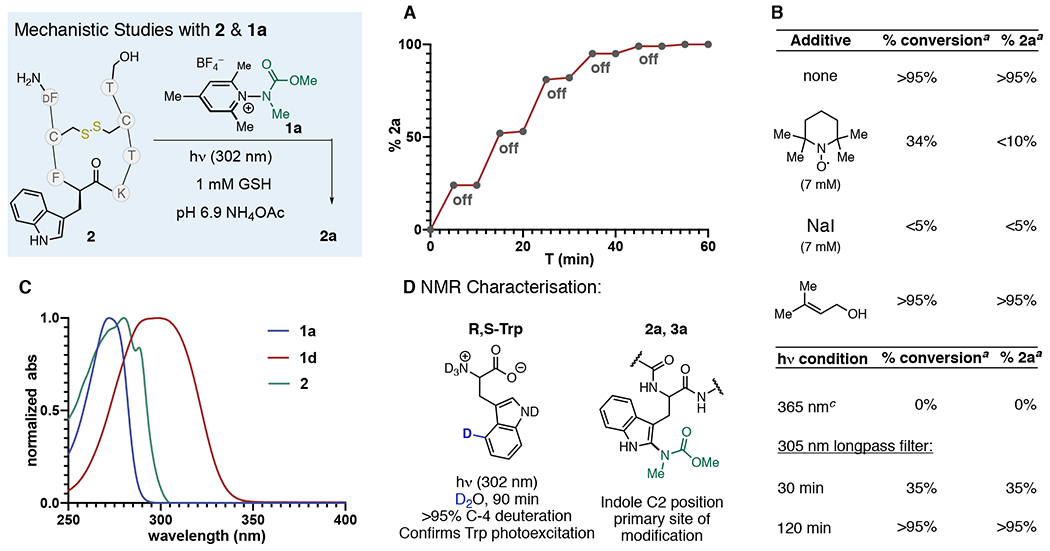
Mechanistic studies. (A) Temporal control experiments with 2 and 1a. (B) Additive and light-perturbation experiments. (C) Absorption spectra of 2, 1a, and 1d. (D) Mechanistic and NMR experiments performed on Trp analogs. a % conjugate observed by TIC. b Performed in a foiled reaction vessel that was irradiated. c UVP 365 nm lamp (8 W).
Finally, we investigated the modification of lysozyme (7), a photolabile,20 14.3 kDa protein displaying 6Trp residues, 3Tyr residues, and a myriad of other redox active residues (Figure 5). While we observed labeling of lysozyme using 1a, we also observed cys-glutathionylation (verified via TCEP reduction), which likely arises either through reduction of proximal disulfides by [Trp]*20a or through thiyl radical exchange21 (Figure 5C). Seeking to avoid this undesired side reaction, we hypothesized that by altering the mechanism of the labeling process such that Trp → [1]* became the dominant pathway, then degradation by intraprotein PET could be minimized. We therefore synthesized phenyl-substituted salt 1d. 1d red-shifts absorbance (λmax = 299 nm, 1a λmax = 272 nm) (Figure 4C) and displays an irreversible reduction potential that is anodically shifted by nearly 200 mV compared to the irreversible reduction potential of 1a. Using 1d, labeling of Lysozyme could be achieved in >90% conversion in 15 min with a 6:1 mono/di labeled conjugate, and with glutathionylation being reduced to trace levels (Figure 5D). In these instances, we found it beneficial to use higher concentrations of GSH in order to fully suppress glutathionylation. Since 1d possesses significant absorption beyond 320 nm, we irradiated 7 with 1d using a 320 nm long-pass filter to suppress Trp excitation, leading to smooth conversion of labeled lysozyme of 94% conversion in 45 min (Figure 5E). In this instance, we observed that the labeling reaction proceeded to completion more rapidly using lower concentrations of GSH (3 mM). Digestion/MS2 revealed Trp-62 to be the site of carbamylation. The doubly labeled conjugate was formed in insufficient quantities to establish the site of second label. While modification of 7 occurs in the active site, modified 7 still displays bacteriolytic activity, indicating that both the carbamate label and the reaction conditions do not denature the protein.
Figure 5.
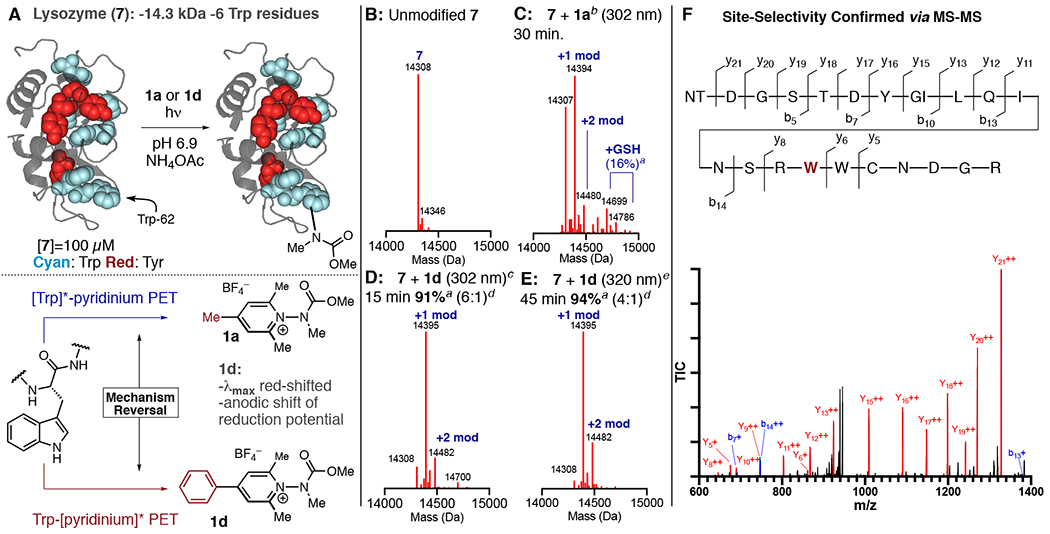
(A) Photochemical labeling of 7 and mechanistic reversal of the labeling process using 1d. Trp residues in 7 are highlighted in cyan. (B) Unmodified 7. (C) 7 + 1a. (D) 7 + 1d, 15 min irradiation time at 302 nm. (E) 7 + 1d, 45 min irradiation time with 320 nmlong pass filter. (F) Confirmation of site selectivity by MS-MS. a Conversion estimated by TIC, average of 2 runs. b 7 mM 1a, 1 mM GSH, 20 mM pH 6.9 NH4OAc buffer. c 3 mM 1d, 6 mM GSH, 20 mM pH 6.9 NH4OAc buffer. d Ratio of +1 to +2 modifications. e 3 mM 1d, 3 mM GSH, 20 mM pH 6.9 NH4OAc buffer, 320 nm long-pass filter used.
In summary, we have developed a photobioconjugation process that operates directly on native biological structure using N-carbamoyl pyridinium salts. The reaction proceeds with excellent selectivity for tryptophan, features short reaction times, and does not require catalysts or organic solvent. We demonstrate the versatility of the N-carbamoyl pyridinium salt scaffold by transferring relevant reporter handles to Trp residues and by adjusting the properties of the reagent to enable photochemical transformations on photolabile substrates.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully thank the Wyoming SBC COBRE program (5P20GM121310-02) and the Wyoming NASA space grant consortium (NNX15AI08H) for funding. Partial instrumentation support was provided by the NSF (CHE-1429615). S.J.T. and W.J.H. acknowledge P20GM103432 for studentships. T.E.M. acknowledges the Wyoming Research Scholars program for support. The University of Wyoming is acknowledged for start-up funds. We thank Profs. J. M. Fox and M. J. Gaunt as well as E. L. Clennan for comments on the manuscript and insightful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications Web site. The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c03039.
Experimental details and procedures; mechanistic studies; NMR spectra (PDF)
The authors declare no competing financial interest.
Contributor Information
Samantha J. Tower, University of Wyoming, Department of Chemistry, Laramie, Wyoming 82071, United States
Wesley J. Hetcher, University of Wyoming, Department of Chemistry, Laramie, Wyoming 82071, United States
Tyler E. Myers, University of Wyoming, Department of Chemistry, Laramie, Wyoming 82071, United States
Nicholas J. Kuehl, University of Wyoming, Department of Chemistry, Laramie, Wyoming 82071, United States
Michael T. Taylor, University of Wyoming, Department of Chemistry, Laramie, Wyoming 82071, United States.
REFERENCES
- (1).(a) Hermanson G Bioconjugate Techniques, 3rd ed.; Elsevier: London, 2013. [Google Scholar]; (b) Sletten EM; Bertozzi CR Bioorthogonal Chemistry: Fishing for Selectivity in a sea of Functionality. Angew. Chem., Int. Ed 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Koniev O; Wagner A Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem. Soc. Rev 2015, 44, 5495–5551. [DOI] [PubMed] [Google Scholar]; (d) Boutureira O; Bernardes GJL Advances in Chemical Protein Modification. Chem. Rev 2015, 115, 2174–2195. [DOI] [PubMed] [Google Scholar]; (e) deGruyter JN; Malins LR; Baran PS Residue-Specific Modification: A Chemist’s Guide. Biochemistry 2017, 56, 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Takaoka Y; Ojida A; Hamachi I Protein Organic Chemistry and Applications for Labeling and Engineering in Live-Cell Systems. Angew. Chem., Int. Ed 2013, 52, 4088–4106. [DOI] [PubMed] [Google Scholar]; (b) Hu Q-Y; Berti F; Adamo R Towards the next generation of biomedicines by site-selective conjugation. Chem. Soc. Rev 2016, 45, 1691–1719. [DOI] [PubMed] [Google Scholar]; (c) Rudra A; Li J; Shakur R; Bhagchandani; Langer R Trends in Therapeutic Conjugates: Bench to Clinic. Bioconjugate Chem. 2020, 31, 462. [DOI] [PubMed] [Google Scholar]; (d) Spicer CD; Pashuck ET; Stevens MM Achieving Controlled Biomolecule-Biomaterial Conjugation. Chem. Rev 2018, 118, 7702–7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Herner A; Lin Q Photo-Triggered Click Chemistry for Biological Applications. Top. Curr. Chem 2016, 374, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ankenbruck N; Courtney T; Naro Y; Deiters A Optochemical Control of Biological Processes in Cells and Animals. Angew. Chem., Int. Ed 2018, 57, 2768–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Bottecchia C; Noël T Photocatalytic Modification of Amino Acids, Peptides, and Proteins. Chem. - Eur. J 2019, 25, 26–42. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fancy DA; Kodadek T Chemistry for the analysis of protein-protein interactions: Rapid and efficient cross-linking triggered by long wavelength light. Proc. Natl. Acad. Sci U. S. A 1999, 96, 6020–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim K; Fancy DA; Carney D; Kodadek T Photoinduced Protein Cross-Linking Mediated by palladium Porphyrins. J. Am. Chem. Soc 1999, 121, 11896–11897. [Google Scholar]; (d) Tyson EL; Niemeyer ZL; Yoon TP Redox Mediators in Visible Light Photocatalysis: Photocalytic Radical Thiol-Ene Additions. J. Org. Chem 2014, 79, 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Vara BA; Li X; Berritt S; Walters CR; Petersson EJ; Molander GA Scalable thioarylation of unprotected peptides and biomolecules under Ni/photoredox catalysis. Chem. Sci 2018, 9, 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Bottecchia C; Rubens M; Gunnoo SB; Hessel V; Madder A; Noël T Visible-Light-Mediated Selective Arylation of Cysteine in Batch and Flow. Angew. Chem., Int. Ed 2017, 56, 12702–12707. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Sato S; Nakamura H Angew. Chem., Int. Ed 2013, 52, 8681–8684. [DOI] [PubMed] [Google Scholar]; (h) Ichiishi N; Caldwell JP; Lin M; Zhong W; Zhu X; Streckfuss E; Kim H; Parish CA; Krska SW Protecting group free radical C-H trifluoromethylation of peptides. Chem. Sci 2018, 9, 4168–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Bloom S; Liu C; Kölmel DK; Qiao JX; Zhang Y; Poss MA; Ewing WR; MacMillan DWC Decarboxylative alkylation for site-selective bioconjugation of native proteins via oxidation potentials. Nat. Chem 2018, 10, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Garreau M; Le Vaillant F; Waser J C-Terminal bioconjugation of Peptides through Photoredox Catalyzyed Decarboxylative Alkynylation. Angew. Chem., Int. Ed 2019, 58, 8182–8186. [DOI] [PubMed] [Google Scholar]
- (5).(a) Bent DV; Hayon E Excited State Chemistry of Aromatic Amino Acids and Related Peptides. III. Tryptophan. J. Am. Chem. Soc 1975, 97, 2612–2619. [DOI] [PubMed] [Google Scholar]; (b) Creed D The Photophysics and Photochemistry of the Near-UV Absorbing Amino Acids-I. Tryptophan and its Simple Derivatives. Photochem. Photobiol 1984, 39, 577–583. [Google Scholar]; (c) Pattison DI; Rahmanto AS; Davies MJ Photo-oxidation of proteins. Photochem. Photobiol. Sci 2012, 11, 38–53. [DOI] [PubMed] [Google Scholar]; (d) Tsentalovich YP; Snytnikova OA; Sagdeev RZ Properties of Excited States of aquous Tryptophan. J. Photochem. Photobiol., A 2004, 162, 371–379. [Google Scholar]
- (6).(a) Antos JM; Francis MB Selective Tryptophan Modification with Rhodium Carbenoids in Aqueous Solution. J. Am. Chem. Soc 2004, 126, 10256–10257. [DOI] [PubMed] [Google Scholar]; (b) Antos JM; McFarland JM; Iavarone AT; Francis MB Chemoselective Tryptophan Labeling with Rhodium Carbenoids at Mild pH. J. Am. Chem. Soc 2009, 131, 6301–6308. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ball ZT Designing Enzyme-like Catalysts: A Rhodium(II) Metallopeptide Case Study. Acc. Chem. Res 2013, 46, 560–570. [DOI] [PubMed] [Google Scholar]; (d) Seki Y; Ishiyama T; Sasaki D; Abe J; Sohma Y; Oisaki K; Kanai M Transition Metal-Free Tryptophan-Selective Bioconjugation of Proteins. J. Am. Chem. Soc 2016, 138, 10798–10801. [DOI] [PubMed] [Google Scholar]; (e) Scoffone E; Fontana A; Rocchi R Sulfenyl halides as Modifying Reagents for Polypeptides and Protein I. Modification of Tryptophan Residues. Biochemistry 1968, 7, 971–979. [DOI] [PubMed] [Google Scholar]; (f) Imiolek M; Karunanithy G; Ng W-L; Baldwin AJ; Gouverneur VE; Davis BG Selective radical trifluoromethylation of native residues in proteins. J. Am. Chem. Soc 2018, 140, 1568–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Wang W; Lorion MM; Shah J; Kapdi AR; Ackermann L Late-Stage Peptide Diversification by Position-Selective C-H activation. Angew. Chem., Int. Ed 2018, 57, 14700–14717. [DOI] [PubMed] [Google Scholar]; (h) Petersen J; Christensen KE; Nielsen MT; Mortensen KT; Komnatnyy VV; Nielsen TE; Qvortrup K Oxidative Modification of Tryptophan-Containing Peptides. ACS Comb. Sci 2018, 20, 344–349. [DOI] [PubMed] [Google Scholar]
- (7).Lakhdar S; Westermaier M; Terrier R; Boubaker T; Ofial AR; Mayr H Nucleophilic Reactivites of Indoles. J. Org. Chem 2006, 71 (24), 9088–9095. [DOI] [PubMed] [Google Scholar]
- (8).Yu Y; Zhang LK; Buevich AV; Li G; Tang H; Vachal P; Colletti SL; Shi ZC Chemoselective peptide Modification via Photocataliyic Tryptophan 1-position Conjugation. J. Am. Chem. Soc 2018, 140, 6797–6800. [DOI] [PubMed] [Google Scholar]
- (9).Bapat JB; Blade RJ; Boulton AJ; Epsztajn J; Katritzky AR; Lewis J; Molina-Buendia P; Nie P-L; Ramsden CA Pyridines as Leaving Groups in Synthetic Transformation: Nucleophilic Displacements of Amino Group, and Novel Preparations of Nitriles and Isocyanates. Tetrahedron Lett. 1976, 17, 2691–2694. [Google Scholar]
- (10).Wardman P Reduction Potentials of One-Electron Couples Involving Free Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar]
- (11).(a) Greulich TW; Daniliuc CG; Studer AN Aminopyridinium Salts as Precursors for N Centered Radicals - Direct Amidation of Arenes and Heteroarenes. Org. Lett 2015, 17, 254–257. [DOI] [PubMed] [Google Scholar]; For applications of this reactivity profile using similar pyridinium salts, see:; (b) Marquet J; Moreno-Mañas M; Pacheco P; Prat M C-alkylation of -diketones with Benzylpyridnium Salts. Evidence for Chain Radical Mechanisms. Tetrahedron 1990, 46, 5333–5346. [Google Scholar]; (c) Klauck FJR; James MJ; Glorius F Deaminative Strategy for the Visible-Light-Mediated Generation f Alkyl Radicals. Angew. Chem. Int. Ed 2017, 56, 12336–12339. [DOI] [PubMed] [Google Scholar]; (d) Basch CH; Liao J; Xu J; Piane JJ; Watson MP Harnessing Alkyl Amines as Electrophiles for Nickel-Catalyzed Cross Couplings via C-N Bond Activation. J. Am. Chem. Soc 2017, 139, 5313–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wu J; He L; Noble A; Aggarwal VK Photoinduced Deaminative Borylation of Alkylamines. J. Am. Chem. Soc 2018, 140 (34), 10700–10704. [DOI] [PubMed] [Google Scholar]
- (12).Mozziconacci O; Schöneich C Effect of Conformation on the Photodegradation of Trp- and Cystine-Containing Cyclic Peptides: Octreotide and Somatostatin. Mol. Pharmaceutics 2014, 11, 3537–3546. [DOI] [PubMed] [Google Scholar]
- (13).Forman HJ; Zhang H; Rinna A Glutathione: Overview of its protective roles, measurement, biosynthesis. Mol. Aspects Med 2009, 30, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Jackson MA; Caputo N; Castle JR; David LL; Roberts CT; Ward WK Stable Liquid Glucagon Formulations for Rescue Treatment of Bi-Hormonal Closed-Loop Pancreas. Curr. Diabetes Rep 2012, 12, 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wilcox W; Eisenberg D Thermodynamics of melittin tetramerization determined by circular dichroism and implications for protein folding. Protein Sci. 1992, 1, 641–653. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Qiu J; Kirsch LE Evaluation of lipopeptide (daptomycin) aggregation using fluoresecnece, light scattering, and nuclear magnetic resonance spectroscopy. J. Pharm. Sci 2014, 103, 853–861. [DOI] [PubMed] [Google Scholar]
- (15).Lehrer SS Solute Perturbation of Protein Fluorescence. The Quenching of the Tryptophyl Fluorescence of Model Compounds and of Lysozyme by Iodide Ion. Biochemistry 1971, 10, 3254–3263. [DOI] [PubMed] [Google Scholar]
- (16).Kosower EM The Effect of Solvent on Spectra. I. A New Empirical Measure of Solvent Polarity: Z-Values. J. Am. Chem. Soc 1958, 80, 3253–3260. [Google Scholar]
- (17).McManus JB; Onuska NPR; Nicewicz DA Generation and Alkylation of alpha-carbamyl radicals via organic-photoredox catalysis. J. Am. Chem. Soc 2018, 140, 9056–9060. [DOI] [PubMed] [Google Scholar]
- (18).Lee KY; Kochi JK Charge-transfer Structures of Aromatic EDA Complexes with N-Heteroatom-substituted Pyridinium Cations. J. Chem. Soc., Perkin Trans 2 1992, 1011–1017. [Google Scholar]
- (19).(a) Takeuchi H; Hayakawa S; Tanahashi T; Kobayashi A; Adachi T; Higuchi D Novel Generation of Parent, Alkyl, Dialkyl and Alicyclic Nitrenium ions in Photolyses of Pyridinium, Quinolonium, Bipyridinium and Phenanthrolium Salts and Aromatic N-substitution by Nitrenium Ions. J. Chem. Soc., Perkin Trans 2 1991, 847–855. [Google Scholar]; (b) Falvey DE Discrete Existence of Singlet Nitrenium Ions Revisited: Computational Studies of Non-Aryl Nitrenium Ions and Their Rearrangements. ACS Omega. 2018, 3, 10418–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Wu L-Z; Sheng YB; Xie J-B; Wang W Photo-excitation of tryptophan groups induced reduction of disulfide bonds in hen egg white lysozyme. J. Mol. Struct 2008, 882, 101–106. [Google Scholar]; (b) Silva CO; Petersen SB; Reis CP; Rijo P; Molpeceres J; Vorum H; Neves-Petersen MT Lysozyme Photochemistry as a Function of Temperature. The protective Effect of nanoparticles on Lysozyme Photostability. PLoS One 2015, 10, No. e0144454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fava A; Reichenbach G; Peron U Kinetics of the Thiol-Disulfide Exchange. II. Oxygen-Promoted Free-Radical Exchange between Aromatic Thiols and Disulfides. J. Am. Chem. Soc 1967, 89, 6696–6700. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.