

Abstract
DNA transposases are enzymes that catalyze the movement of discrete pieces of DNA from one location in the genome to another. Transposition occurs through a series of controlled DNA strand cleavage and subsequent integration reactions that are carried out by nucleoprotein complexes known as transpososomes. Transpososomes are dynamic assemblies which must undergo conformational changes that control DNA breaks and ensure that, once started, the transposition reaction goes to completion. They provide a precise architecture within which the chemical reactions involved in transposon movement occur, but adopt different conformational states as transposition progresses. Their components also vary as they must, at some stage, include target DNA and sometimes even host-encoded proteins. A very limited number of transpososome states have been crystallographically captured, and here we provide an overview of the various structures determined to date. These structures include examples of DNA transposases that catalyze transposition by a cut-and-paste mechanism using an RNaseH-like nuclease catalytic domain, those that transpose using only single-stranded DNA substrates and targets, and the retroviral integrases that carry out an integration reaction very similar to DNA transposition. Given that there are a number of common functional requirements for transposition, it is remarkable how these are satisfied by complex assemblies that are so architecturally different.
1. Introduction to DNA transposition
1.1. What is DNA transposition?
Transposition is the movement of discrete segments of genetic material (transposons or transposable elements (TEs)) from one site (the donor site) to another (the target site) within a genome. There are two major transposon types (Fig. 1): those that use RNA intermediates (often called Class I elements) and those that use only DNA intermediates (often called Class II elements; Finnegan, 1989; Wicker et al. 2007). Among the Class I elements, both the long-terminal repeat (LTR)- and non-LTR retrotransposons use a self-encoded reverse transcriptase enzyme to convert RNA to a nucleic acid form that can be inserted into a new site. This review will be restricted to DNA transposition including that carried out by retro-viruses. Interested readers are referred to another recent review of this field (Montaño & Rice, 2011).
Fig. 1.
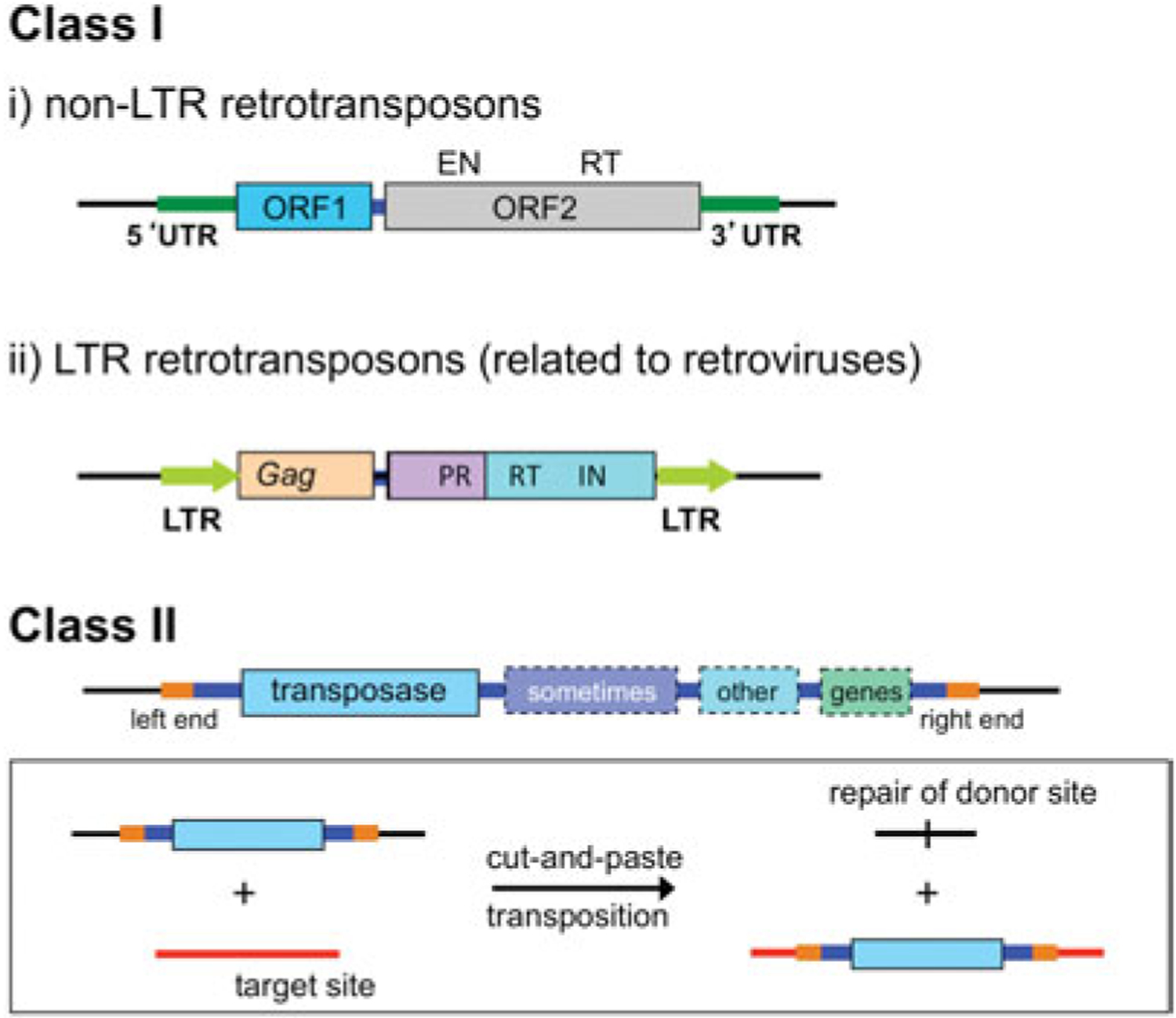
Major transposon types. Among the Class I elements, non-LTR retrotransposons encode two proteins required for retrotransposition: ORF1, a non-specific nucleic acid binding protein, and ORF2 which has endonuclease (EN) and reverse transcriptase (RT) activities. In their simplest form, LTR retrotransposons have a gag gene that encodes structural proteins, and a pol gene that encodes a protease (PR), integrase (IN), and an RT, bounded by LTRs. Class II elements use only DNA intermediates, and although eukaryotic DNA transposons usually encode only the transposase enzyme required for movement (depicted in light blue, and shown in the inset for cut-and-paste DNA transposons), prokaryotic DNA transposons often carry other genes such as those encoding antibiotic resistance proteins (shown in varying shades). Specific binding sites for the transposase (in orange) are located at each end of the transposon.
In contrast to homologous recombination, DNA transposition does not require significant homology between the TE and the target sequence. It is catalyzed by element-specific enzymes, transposases, usually encoded by the TE. These ensure specific DNA strand cleavages at the TE ends and their subsequent transfer into a DNA target. Although many DNA TEs transpose via double-stranded DNA (dsDNA) intermediates, families of TEs have now been identified which use single-stranded DNA (ssDNA) intermediates (Guynet et al. 2008).
Only a very limited number of structurally different protein domains that can cleave DNA have been discovered at the core of DNA transposase enzymes (reviewed in Curcio & Derbyshire, 2003). The vast majority of DNA transposases possess an RNaseH-like catalytic domain characterized by the convergence of three acidic residues (DDE or DDD) which coordinate two divalent metal ions in the active site (Hickman et al. 2010a; Yuan & Wessler, 2011). Retroviral integration also relies on enzymes that possess RNaseH-like catalytic domains (Dyda et al. 1994). Reactions catalyzed by RNaseH-like enzymes do not involve the formation of covalently linked protein–DNA intermediates, and for reasons that will become clear in Sections 1.3.4 and 1.3.5, DNA transposases with RNaseH-like nuclease domains are often called cut-and-paste transposases. The two-metal ion mechanism for DNA cleavage was first described for the 3′–5′ exonuclease domain of Escherichia coli polymerase I (Beese & Steitz, 1991), and it has been well-studied in the context of other nucleic acid processing enzymes (e.g., see Castro et al. 2009; Rosta et al. 2011).
A second class of DNA transposases has a completely different type of nuclease domain, referred to as an ‘HUH’ domain for the motif consisting of two histidine residues separated by a generic hydrophobic residue (Koonin & Ilyina, 1993). HUH nuclease domains are widespread. They are found in enzymes that catalyze the initiation of rolling circle replication, conjugative DNA transfer, and initiate the replication of certain ssDNA viruses. Their structure and catalytic mechanism was first revealed in the context of the endonuclease domains of the Adeno-associated virus Rep protein (Hickman et al. 2002), the tomato yellow leaf curl virus (Campos-Olivas et al. 2002), the TraI relaxase encoded by the conjugative F plasmid (Datta et al. 2003) and TrwC encoded by plasmid R388 (Guasch et al. 2003). The two histidine residues, together with a third polar side chain, create a binding site for a single divalent metal ion that is the essential cofactor for the DNA cleavage reaction. HUH nucleases always use ssDNA as their substrates and strand cleavage is always coupled to the formation of a covalent 5′-phosphotyrosine intermediate at the site of cleavage, features that have been cleverly exploited by ssDNA transposases (see Section 2.5).
A third type of enzyme associated with TE mobility are the tyrosine and serine site-specific recombinases. Little is known about these in the context of transposition although they are related to the well-characterized phage lambda Int and Tn3 family resolvase proteins (Azaro & Landy, 2002; Churchward, 2002).
1.2. Why is transposition important?
TEs were first identified more than half-a-century ago as ‘factors’ that influence the color of corn (maize) kernels (McClintock, 1950). They have since been observed in almost all organisms examined where they are often present in high copy number and sometimes represent an astounding percentage of their host genome (Biémont, 2010). A recent study concluded that ‘transposases’ are by far the most frequently occurring functional group of proteins in the annotated public sequence databases (Aziz et al. 2010) and, in view of the present quality of TE annotation, their numbers are almost certainly underestimated.
Their properties render TEs important actors in generating genetic diversity. These properties include generally low or no sequence specificity in DNA target choice, the ability to generate mutation by insertion, to invert and delete DNA segments, to sequester and mobilize genes and to influence neighboring gene expression. Since they are often present in more than a single copy, they also provide substrates for homologous recombination. It has recently been shown that the only demonstrably active TEs in humans, the L1 retrotransposons, are active in neuronal stem cells (Coufal et al. 2009) and that mobilization of retroelements in brain tissue leads to somatic genome mosaicism (Baillie et al. 2011). Thus, these TEs may have an important role in nervous system development and diversity.
An increasing number of eukaryotic TEs are being identified which have been ‘domesticated’ and now perform specific cellular functions (reviewed in Sinzelle et al. 2009). For some, the ability to move discrete pieces of DNA has been retained, as in the cases of the RAG1 protein of the V(D)J system responsible for generating antibody diversity (Jones & Gellert, 2004), which is related to the hAT-like transposases of the Transib family (Kapitonov & Jurka, 2005), and the transposases that generates the somatic nucleus (macronucleus) from the germline nucleus (micronucleus) in certain ciliates (Baudry et al. 2009; Nowacki et al. 2009; Schoeberl & Mochizuki, 2011). A transposase-like protein has also recently been described that plays a role in mating-type switching in the yeast, Kluyveromyces lactis (Barsoum et al. 2010). For other domesticated transposases, the crucial function that appears to have been retained is sequence-specific DNA binding. For example, DAYSLEEPER, a hAT-like transposase in Arabidopsis, is essential for plant growth and binds to a specific motif upstream of the Ku70 transcription start site (Bundock & Hooykaas, 2005). The SETMAR protein in primates (also known as Metnase), a chimera between a histone methylase and a mariner transposase (Cordaux et al. 2006), is widely expressed in human tissues and has been reported to possess many of the activities of a transposase although its exact function is not yet clear (Liu et al. 2007). Indeed, it has been suggested that the remnants of transposases can be identified in many present day genes (Britten, 2004).
In addition to their role in the natural world, there is an enormous potential for using TEs as tools for in vitro and in vivo genetic engineering and for gene therapy (reviewed in Copeland & Jenkins, 2010; Ivics et al. 2009). Several DNA transposons are currently in active use. These include piggyBac, Tol2 and Sleeping Beauty (see e.g., Grabundzija et al. 2010; Mátés et al. 2009; Yusa et al. 2011). Flourishing areas of interest include devising ways to make TEs more active in eukaryotes or more active in specific cell types, to render them site- or sequence-specific, to increase their gene-carrying capacity, and to apply them with minimal unintended genomic consequences.
1.3. How does transposition work and what is a transpososome?
Successful DNA transposition requires precise recognition of the TE ends, their endonucleolytic cleavage, and their insertion into a suitable target DNA site. These reactions must be coordinated both spatially and temporally to avoid non-productive transposition resulting in potentially fatal physical damage to genome structure e.g., the introduction of chromosome breaks. Another reason for tight control of the reactions is that incomplete transposition could trigger DNA repair events that in turn might damage or even eliminate the mobile element itself. Moreover, the frequency of transposition must be sufficiently low to prevent excessive genetic rearrangement which would lead to severe and potentially lethal changes in genome organization.
1.3.1. The transpososome
The key to productive transposition is the transpososome, a nucleoprotein complex involving both transposon ends and the transposase. Transpososomes are dynamic assemblies which must undergo conformational changes that presumably control DNA breaks and ensure that, once started, the reaction goes to completion. They provide a precise architecture within which the chemical reactions involved in transposon movement occur, but adopt different conformational states as transposition progresses. They also vary in their components at various stages as they must eventually also include target DNA and may recruit host-encoded proteins. For prokaryotic systems, these may include DNA-shaping proteins such as IHF, HU and HNS (reviewed in Haniford, 2006; Mizuuchi, 1992). Protein chaperones may also be involved (e.g. GroEL in the case of IS1; Ton-Hoang et al. 2004). There are a number of prokaryotic transposases whose transpososomes have been well-characterized, including those of Mu (Mizuuchi, 1992), transposon Tn7 (Skelding et al. 2002), Tn10 (Sakai et al. 1995), Tn5 (Bhasin et al. 2000), and IS911 (Normand et al. 2001). However, step-wise transpososome conformational changes through transposition have received little attention from a structural perspective except for one or two systems.
Only a handful of transpososome states have been crystallographically captured (Fig. 2) and hence have provided structural information about their organization to better than about 4Å resolution. These include those from the prokaryotic elements Tn5 (Davies et al. 2000) and the ssDNA transpososomes of IS608 and ISDra2 (Barabas et al. 2008; Hickman et al. 2010b; Ronning et al. 2005), together with the eukaryotic element Mos1 (Richardson et al. 2009) and the retroviral intasome of the prototype foamy virus (PFV) (Hare et al. 2010; Maertens et al. 2010). These form the basis of our current understanding of transpososome architecture and are the subject of this review. Structural information has also been obtained using electron microscopy for the phage Mu transpososome (Yuan et al. 2005) and the V(D)J signal end complex (Grundy et al. 2009), but in light of their lower resolution will not be discussed here in detail.
Fig. 2.
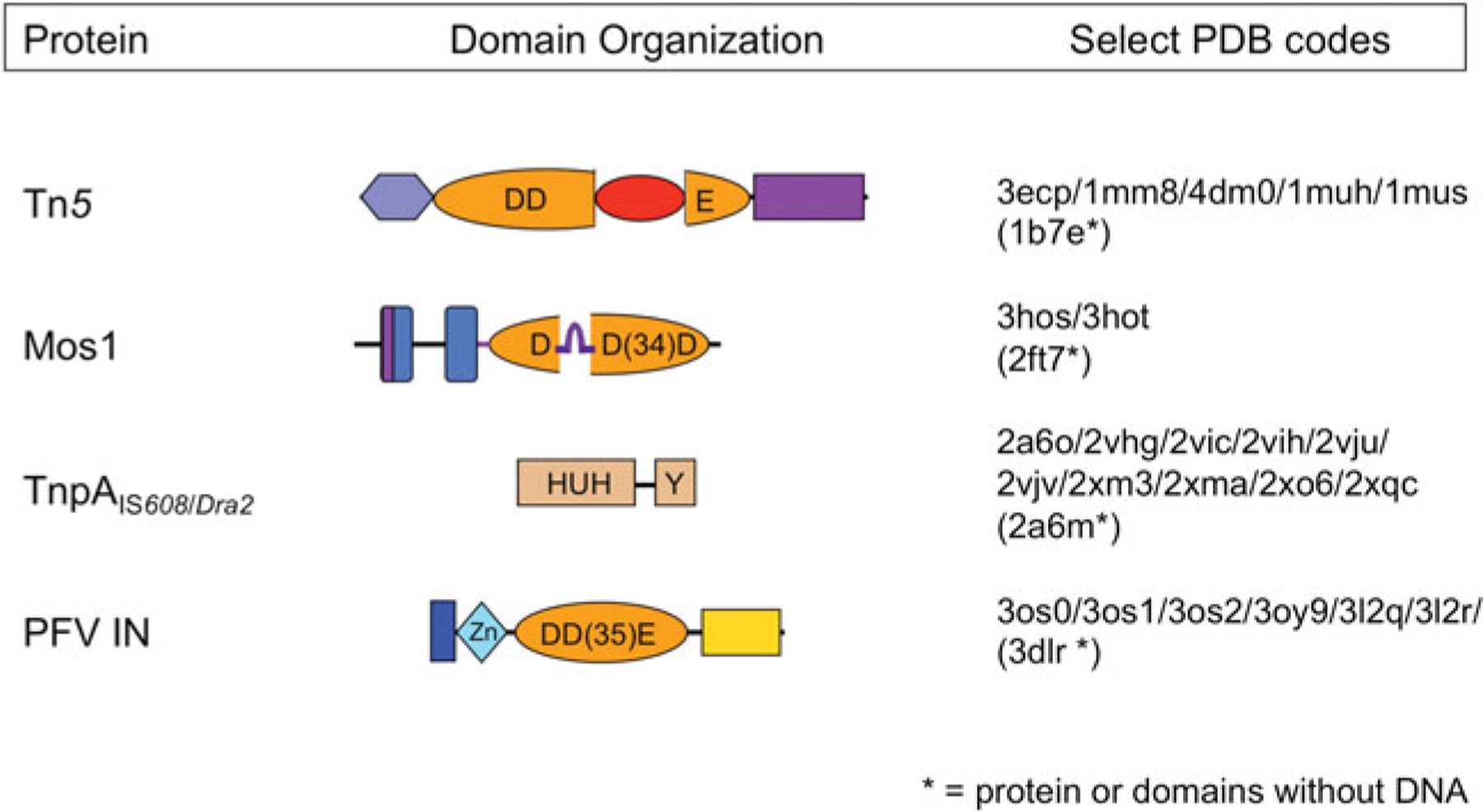
Domain organization of select transposases. The four transposases for which there are high resolution transpososome structures are shown schematically, with DNA-binding domains in shades of blue and nuclease catalytic domains in shades of orange. Regions important for multimerization are shown in purple. The Tn5 and Mos1 transposases, and PFV integrase (PFV IN) have RNaseH-like catalytic domains which use three acidic residues (DDD or DDE) to coordinate two divalent metal ions required for catalysis. The TnpA ssDNA transposases use an HUH nuclease domain to coordinate a single metal ion which, in conjunction with a catalytic tyrosine, comprises the enzyme active site. The four beta-strand insertion into the Tn5 catalytic domain is shown in red, and the ‘clamp-loop’ of Mos1, inserted relative to the standard RNaseH topology between beta strands 1 and 2, is shown in purple.
1.3.2. Transposase recognition of TE ends
Transpososome assembly is initiated by recognition of the TE ends by the transposase. Although, theoretically, a transposition system could use one protein to recognize the left end (LE) and another to deal with the right end (RE), in practice, this is not observed. Many TEs carry a single subterminal binding site that is the same or very similar on each end (Mahillon & Chandler, 1998). If there is a regulatory need to distinguish LE from RE, this may be achieved by slight sequence differences between the sequences at the ends; these binding sites are variously referred to as inverted terminal repeats (TIRs), ITRs, or simply inverted repeats (IRs). The ssDNA transposons also have a subterminal binding site on each end, in this case a conserved inverted palindromic sequence that folds into a hairpin (Ronning et al. 2005).
Other TEs, for example, those of bacteriophage Mu (Mizuuchi, 1992), transposon Tn7 (Arciszewska et al. 1989, 1991) and the eukaryotic element Ac of maize (Kunze & Starlinger, 1989), distinguish LE from RE by carrying multiple transposase-binding sites organized differently at each end. Multiple binding sites in these systems serve to sequester transposase and can generate intricately complex transpososome architectures (Holder & Craig, 2010; Yin et al. 2007). They impose an additional level of regulation as, for some systems, a copy of each end is essential for formation of an active transpososome. For others, it is not clear why artificial elements with one set of ends is active while the other is not – for example, Tn7 will transpose if it has two RE sequences but not with two LEs (Arciszewska et al. 1989).
Transposase binding to its cognate ends is itself highly regulated. Little is known for eukaryote systems but in prokaryotes in which transcription and translation are physically coupled, it may be coordinated with transposase expression. Early studies revealed that RNaseH-like transposases act preferentially on the element from which they are expressed (cis activity, reviewed in Duval-Valentin & Chandler, 2011). This prevents activation of other copies of the same TE in the host genome. Cis activity can arise in several ways. Transposase instability is partly responsible for cis activity of IS903 (and probably Tn5): enhanced trans activity is observed in a Lon protease-deficient host and hyperactive transposase mutants refractive to proteolysis have been isolated (Derbyshire et al. 1990). Cis activity can also be overcome by increased translation levels or increased transposase mRNA half-life, transcript release from its template or increased translation efficiency (e.g., IS10; Jain & Kleckner, 1993a, b). Another type of control is co-translational binding of the transposase, facilitated by the location of the sequence-specific DNA-binding domain at the N-terminal end of most bacterial transposases. This is therefore the first domain to be translated thus favoring transposase binding to sites close to its point of synthesis (see, e.g., IS903; Grindley & Joyce, 1980).
It has been observed that several full-length prokaryotic transposases bind poorly to their cognate ends when compared with their isolated N-terminal-binding domains (Haren et al. 1998; Zerbib et al. 1987). The C-terminal domains (CTDs) thus appear to sterically mask the N-terminal domain (NTD). This would favor co-translational binding of the nascent peptide after synthesis of the DNA-binding domain but before synthesis of more CTDs. This has recently been shown to occur in vitro for IS911 (Duval-Valentin & Chandler, 2011).
1.3.3. Bringing the ends together
For productive transposition, cleavage and strand transfer reactions occurring at one transposon end must be coordinated with those at the other end, and pairing of the TE ends before initiating transposition chemistry seems to be the principal way in which this is achieved. A priori there are several ways to ensure that transposon ends are paired. One is to pre-assemble a transposase multimer in which two monomers each bind one end; this seems to be the case for the Hermes hAT transposase (Hickman et al. 2005) and the TnpA proteins of ssDNA transposition (Ronning et al. 2005). Purified RAG1 of the V(D)J recombination system also forms dimers before binding to recombination signal sequences (Rodgers et al. 1999). Alternatively, the ends could be recognized by a transposase monomer, and subsequent protein multimerization would necessarily pair the ends. A combination of these pathways could also be imagined, in which the binding of one transposon end triggers multimerization, followed by the binding of the second end. An additional level of complexity (or control, depending on one’s perspective) could be imposed if a transposase monomer has multiple DNA-binding domains able to recognize distinct regions of each end.
Formation of the so-called paired-end complex (PEC), in which synapsed ends are anchored by multiple transposase monomers, is often another key regulatory checkpoint. In the few cases where it has been analyzed, no chemistry occurs before this step (e.g., Namgoong & Harshey, 1998; Naumann & Reznikoff, 2000; Williams et al. 1999). As will be seen for the structurally characterized RNaseH-like transposases, this is because the architecture of the transpososome dictates that a transposase monomer bound to one end through its sequence-specific DNA-binding domain cleaves the other end within the PEC. This is referred to as cleaving ‘in trans’, and may well prove to be a general principle.
For prokaryotic ISs, coupling between transposase expression and binding activity provides a possible scenario to ensure PEC formation since it restricts binding activity to immediately neighboring TE ends. However, it remains to be explored how this might also be coupled to transposase multimerization.
Moreover, some transposon derivatives are extremely short and approach the in vitro DNA persistence length (Duret et al. 2008; Gratias et al. 2008). In principle, this would disfavor bringing both ends together. In these cases, PEC formation might be facilitated in vivo by modification of the persistence length by host-encoded DNA-binding proteins. The fact that several such short TE derivatives clearly transpose efficiently in vivo and in vitro should provide experimental systems to analyze how transpososome assembly occurs with such substrates.
1.3.4. DNA cleavage at the transposon ends
RNaseH-like and HUH nuclease domains have active sites that accomplish the same task of introducing breaks into the sugar-phosphate backbone of DNA in different ways. As shown in Fig. 3a for the structurally characterized transposon systems, RNaseH-like transposons are liberated from their donor sites by a choreographed sequence of nucleophilic attacks by either activated water (depicted as OH–) or free 3′-OH groups that is characteristic of the different transposase types. The transposon strand carrying a terminal 3′-OH group is subsequently transferred into target DNA and is therefore known as the ‘transferred strand’ (TS), whereas the complementary strand is known as the ‘non-transferred strand’ (NTS). For Tn5 (reviewed in Reznikoff, 2008), the first nucleophilic attack is on the TS; this generates a 3′-OH group that subsequently attacks the NTS to form a hairpin on the transposon end and liberates the TE from its flanking DNA. The hairpin is then opened by a second nucleophilic attack by a water molecule to regenerate the 3′-OH at the end of the TS. In contrast, for Mos1 (Dawson & Finnegan, 2003), the initial nucleophilic attack is on the NTS, three base pairs into the transposon end. A second nucleophilic attack by an activated water molecule then occurs on the TS. Retroviral integrases (reviewed in Craigie, 2001) do not require two sequential strand cleavage reactions because their substrate is a dsDNA product of reverse transcription and has no flanking DNA. In preparation for strand transfer, integrases catalyze the removal of two bases from the TS in a process known as 3′-processing, creating a new 3′-OH at the end of each TS. In all the three of these systems, the result of the DNA cleavage reactions is a double-stranded TE which has been cut out of its donor site and which has a 3′-OH at each end.
Fig. 3.
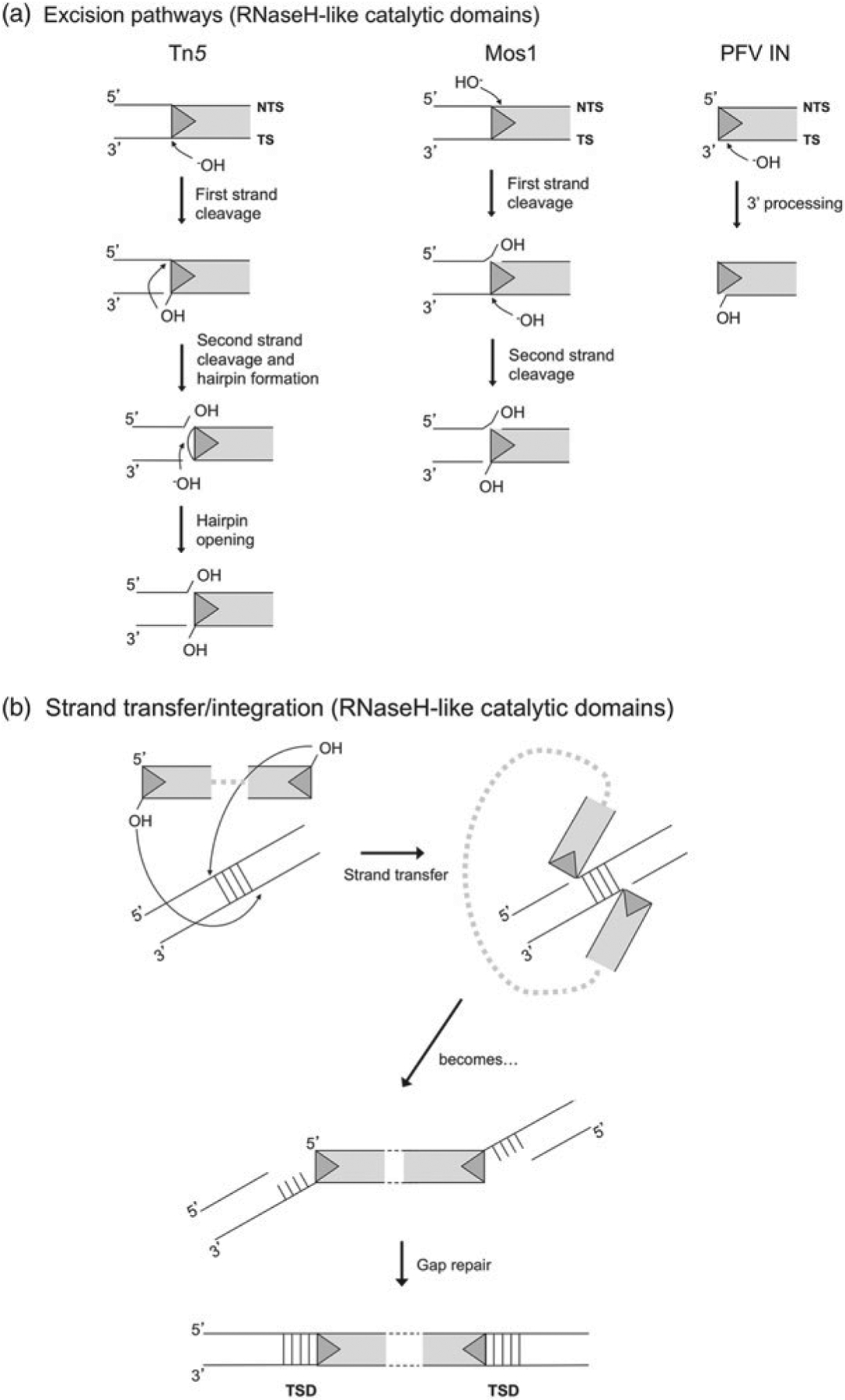
(a) Excision pathways for Tn5, Mos1, and PFV integrase. Only reactions occurring on the TE LEs are shown. For Tn5, cleavage on both the NTS and the TS occur precisely at the transposon end. For Mos1, NTS cleavage occurs three bases within the transposon end and TS cleavage is precisely at the transposon end. For retroviral integrases such as that of PFV, the product of reverse transcription is a blunt-ended provirus from which integrase removes two 3′-OH nucleotides from the TS in a step known as 3′-processing. (b) Strand transfer pathway for DDE transposases. Shown is the specific pathway for PFV integrase, which catalyzes concerted integration into target DNA with a 4 bp stagger. As a result, after gap repair, the TE is flanked on both sides by a 4 bp TSD. For Tn5, staggered sites for strand transfer are offset by 9 bp and for Mos1 by 2 bp.
There are other major pathways for DNA cleavage catalyzed by RNaseH-like transposases which are not discussed here in detail as representative transpososomes have not yet been captured in high-resolution crystal structures. One pathway is mechanistically different from the Tn5 pathway, in which the order of NTS and TS cleavage is reversed and hairpins are formed on the flanking DNA rather than the transposon end. This pathway is used by hAT transposases such as Hermes (Zhou et al. 2004) and the RAG1/2 proteins of the V(D)J recombination system (Roth et al. 1992). Another pathway used by phage Mu and the Tn3 family of transposons, involves replicative transposition, and in this case, only one strand is cleaved before the transposon – and its accompanying flanking DNA – is joined to a new site. Replicative transposition involving transposases with an RNaseH-like catalytic domain has not yet been reported in eukaryotes. The third major alternative pathway is commonly known as copy-and-paste. This is used by a large number of bacterial IS families including IS3, IS30, IS256 and IS110 (see Rousseau et al. 2002). It is an asymmetrical mechanism and occurs by cleavage at one end of the IS, transfer of the resulting 3′-OH to attack the same strand at the opposite end to generate a single-branched strand bridge and subsequent replication to generate a dsDNA closed circular intermediate which can then undergo integration. The process occurs without loss of the IS from its original site (Rousseau et al. 2002).
Single-stranded DNA transposases catalyze ssDNA cleavage using an active site that employs a tyrosine residue whose OH group acts as the nucleophile to attack the DNA backbone. The precise positioning and polarization of the scissile phosphate is achieved by a divalent metal ion which is coordinated by the histidine residues of the HUH motif and another polar protein side chain whose exact identity is less conserved. As shown in Fig. 4b, the product of cleavage at each transposon end is a free 3′-OH group on one side of the DNA break and a covalent 5′-phosphotyrosine intermediate on the other. After cleavage at both ends, the free 3′-OH groups then serve as nucleophiles to attack the opposite 5′-phosphotyrosine linkages to generate a sealed donor backbone and an excised single-stranded circular transposon intermediate. Integration is essentially a replay of these same steps (Fig. 4c). The details of ssDNA transposition will be discussed in Section 2.5.
Fig. 4.
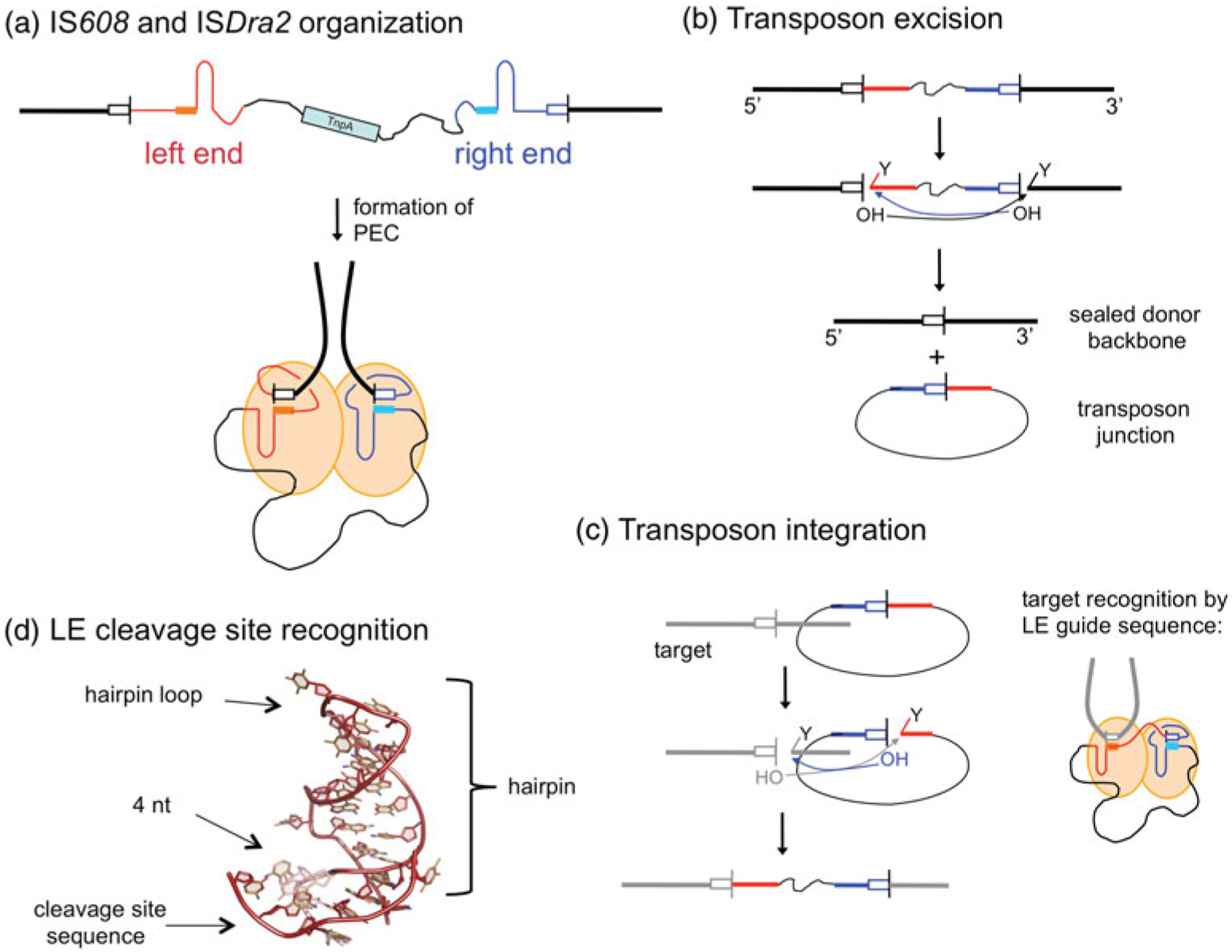
ssDNA transposition. (a) One strand of the transposon is shown, with the LE in red and the RE in blue. Both ends have imperfect palindromic sequences located close to the ends which form hairpins (as shown) that are recognized by the TnpA transposase. In the PEC, dimeric TnpA binds one LE and one RE. The cleavage sites (tetra- or pentanucleotides represented by white boxes) are recognized through non-linear base pairing with DNA (shown as solid boxes) at the base of the hairpins; cleavage occurs at the 3′ ends of the cleavage sites. Flanking DNA is represented as a thick black line, and transposon DNA as a thin black line. (b) Transposon excision. Each active site within the dimer cleaves one end. At the LE, this results in a free 3′-OH on flanking DNA and a covalent 5′-phosphotyrosine intermediate on the transposon end (represented by the ‘Y’). At the RE, the reaction with the same polarity results in a free 3′-OH on the transposon end and a covalent 5′-phosphotyrosine intermediate on flanking DNA. When the 3′-OH of one end attacks the phosphotyrosine intermediate of the other, the resulting products are an excised circular transposon junction and a precisely sealed donor backbone. (c) Transposon integration into a target site proceeds through two cleavage steps of the same polarity as for excision. The subsequent attacks of the 3′-OH groups on the 5′-phosphotyrosine intermediates result in an integrated transposon. The target cleavage site is recognized by non-linear base pairing with the DNA at the base of the LE hairpin, as shown for LE cleavage in (a) and (d).
1.3.5. Target binding and integration
Just as transposases with RNaseH-like and HUH nuclease domains carry out transposon end cleavage in different ways, how they bind target DNA and then catalyze integration also differ. For the RNaseH-like transposases, once the TS has been processed and the newly created 3′-OH is available (and, where needed, the NTS has also been cleaved), the transpososome is ready to find its target into which DNA integration will occur (Fig. 3b). One notable exception to this is the Tn7 transpososome that requires target binding before strand cleavage (Bainton et al. 1993). The Tn7 transpososome is substantially more complicated than others discussed here. It has two alternative transposition strategies (reviewed in Peters & Craig, 2001): one involves insertion into a specific site, a safe haven highly conserved in many bacteria, whereas in the other it recognizes distortions in the DNA such as Okazaki fragment ends.
As shown in Fig. 3b, for the RNaseH-like transposases, the integration or strand-transfer step is accomplished by attack on the two target DNA strands by the two 3′-OH transposon ends, at backbone phosphodiester positions separated by few nucleotides. The consequences of joining the 3′-OH transposon ends to the two target strands in this staggered manner are gaps of a few nucleotides on one strand at each end, flanking the integrated transposon. These short gaps appear to be readily repaired by host repair and replication machineries. This repair process generates duplicated sequences directly flanking the ends of the integrated transposon, known as target site duplications (TSDs). As their exact length is a characteristic of the transposon variety, these TSDs are genomic signatures indicating integrated elements.
It seems likely that transpososomes go through a series of conformational changes as they proceed through the steps of target binding and integration. One state, referred to as a strand transfer complex (STC) in which the two TS have carried out their nucleophilic attacks on the target DNA (Fig. 3b), is often the most stable form of the various complexes that exist during transposition. As none of the characterized transposition systems use high-energy cofactors (with the exception of the P element from Drosophila which requires GTP – although not its hydrolysis – for transposition; Tang et al. 2005), transition through the consecutive chemical states which occur during transposition must be an energetically ‘downhill’ process. Transposase binding to the DNA, transpososome assembly, and target binding to the transpososome results in complexes of increasing stability (e.g., Holder & Craig, 2010; Yanagihara & Mizuuchi, 2003). In some cases, the final assembly is so stable that an external energy source in the form of the ATP is needed to disassemble the complex (Burton & Baker, 2003; Levchenko et al. 1995; North & Nakai, 2005).
The situation is very different for the ssDNA transposases, which have specific tetra- or pentanucleotide target sites (Kersulyte et al. 2002). For both the IS608 and ISDra2 systems (Barabas et al. 2008; Guynet et al. 2009; Hickman et al. 2010b), the single-stranded target site is recognized in precisely the same manner as the LE cleavage site (Fig. 4a). This is achieved through DNA–DNA interactions between the target site and a short stretch of DNA within the transposon LE.
2. State of the union: transpososome architectures
2.1. So few transpososome structures
Considering the limited number of transpososome structures available to date, and the immense differences that exist between them (Fig. 5), it has been difficult to discern any grand unifying themes or overarching paradigms. Rather, the field is at the stage of possessing only a few really informative structural data points and eagerly awaiting the unveiling of the next transpososome structure to see what it might reveal. It has also been tempting to use the limited information on only one step of a transposition reaction to imagine or model the entire pathway, a process that is obviously littered with caveats.
Fig. 5.
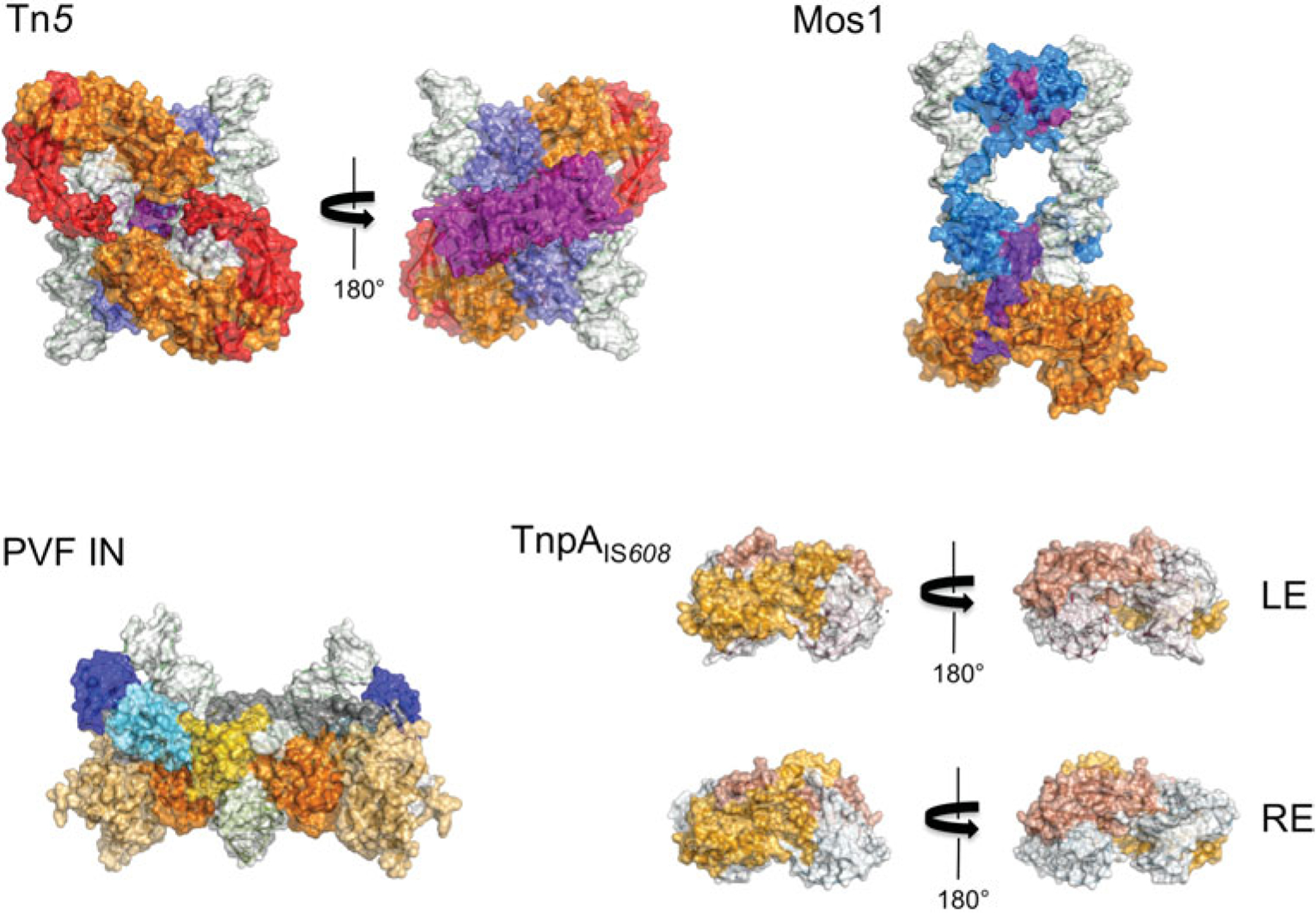
Space-filling representation of transpososomes (to scale). The domain colors correspond to those in Fig. 2. DNA is shown with a white surface. In the case of PVF integrase, a target capture complex is shown.
It is unfortunate that results are emerging at a slow pace, no doubt, in large part, due to the immense experimental difficulties involved. It is thought that there are two main limitations to progress. The first is that DNA transposases are often poorly soluble, and the second is that they are flexible. Although this latter property is a natural consequence of their need to adopt multiple conformational states as they go through the transposition event from start to finish, it also means that the configurational free energy landscape has a number of shallow minimums and the system can move between them. This movement makes crystallization attempts most perilous.
Why is flexibility a problem for structure determination? Current high-resolution experimental methods such as X-ray crystallography, cryo-electron microscopy, or nuclear magnetic resonance (NMR) spectroscopy are, in principle, capable of revealing structural details at better than (or at least around) 4Å resolution, a resolution that is required to understand mechanism. As most transposomes are large assemblies, typically larger than about 50 kDa, direct determination of their structure with NMR alone is usually not feasible. Both X-ray crystallography and cryo-electron microscopy rely on averaging the structures of a large number of individual molecules. This is because the experimental signal measured from a single molecule is very weak and a large assembly of molecules is needed to amplify it. When molecules are flexible and able to adopt many configurations, the average signal will be ‘smeared’, as the individual molecules in the population will assume different conformations; indeed, in the case of crystallography, crystals may not even form. For molecular assemblies as complex as transpososomes, a large number of iterations of buffer conditions, protein constructs, and DNA sequences must be tried in the attempt to find a magical combination where most of molecules ‘sit tight’ in an energy minimum which will allow determination of their structure.
2.2. Our first view: Tn5
The first transpososome structure elucidated was that of IS50, a component of the prokaryotic compound transposon Tn5 (Davies et al. 2000). Tn5 consists of two IS50 insertion sequences flanking antibiotic resistance genes (reviewed in Reznikoff, 2008). This initial view of a complete transpososome was enormously informative as it provided the first illustration of how protein and DNA interlace to pair two transposon ends, and it provided an elegant explanation for the mutual dependency of DNA binding and dimerization in the Tn5 system.
The Tn5 (IS50) transpososome can be viewed as the simplest possible system in that the active assembly consists of two transposase monomers and two transposon ends (Fig. 6). Since the uncomplexed Tn5 transposase is a monomer (Braam et al. 1999), the crystal structure shows how DNA binding and multimerization are inextricably linked. The N-terminal site-specific DNA-binding domain (shown in blue in Figs 5 and 6) of one transposase monomer specifically recognizes the 19-mer transposon end between nts 5 and 16 of each DNA molecule (counting inward from the transposon end). It is believed that formation of these initial subterminal ‘cis’ DNA contacts in the context of a monomer induces a conformational change affecting the CTD (shown in purple), thereby creating an interface for transposase dimerization (Braam et al. 1999). Upon dimerization, the very end of the transposon (and hence the cleavage site) is inserted into the active site of the other monomer forming a set of ‘trans’ contacts. The dimer is further stabilized by these trans DNA interactions with the nucleotides at the very end of the transposon.
Fig. 6.
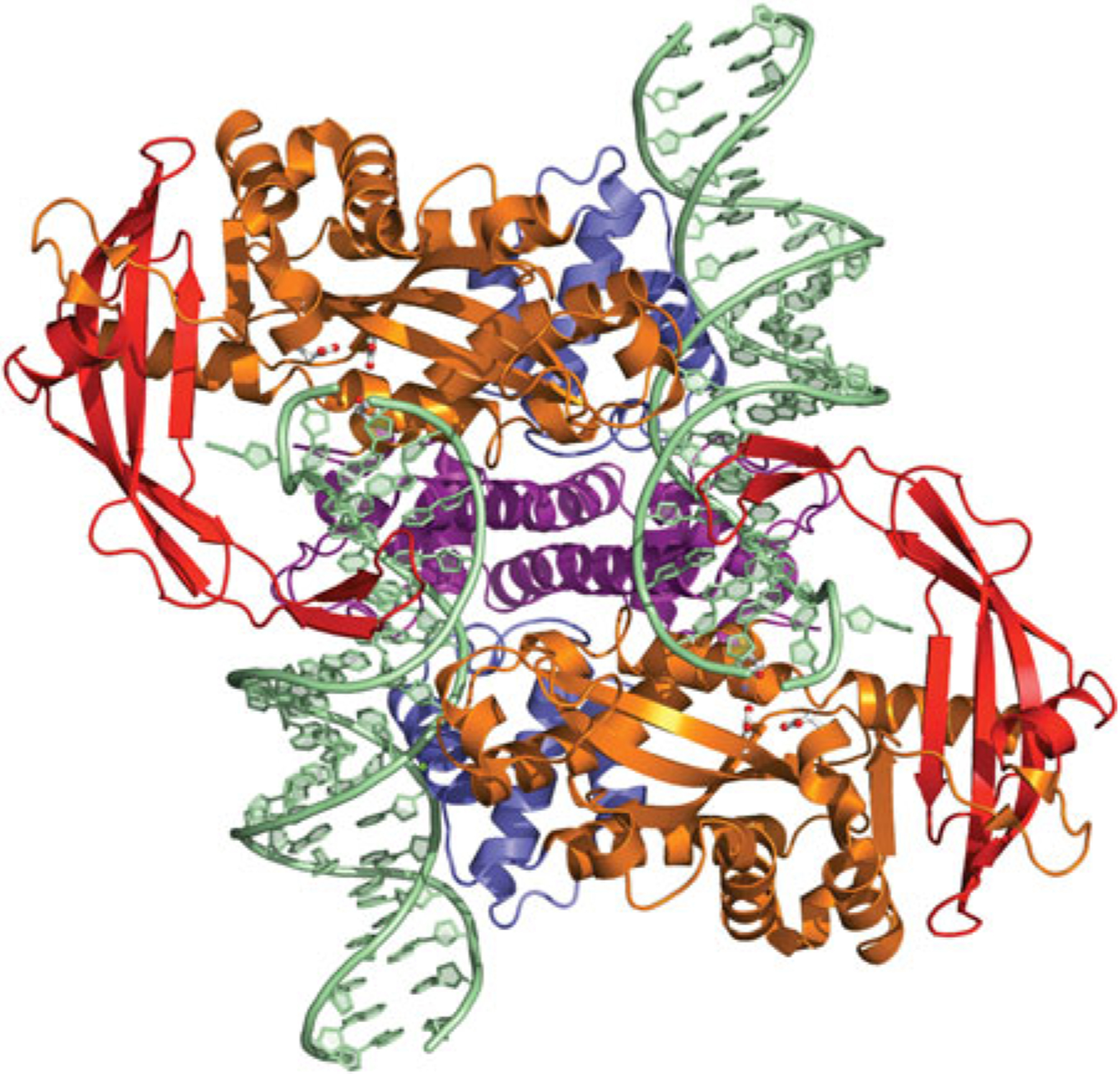
The dimeric Tn5 transpososome. The domain colors correspond to those shown in Fig. 2, where the NTD is in blue, the catalytic core is in orange, and the C-terminal dimerization domain is in purple. The four-stranded insertion into the RNaseH-like catalytic domain is shown in red. The residues comprising the DDE motif (D97, D188, and E326) are shown in ball-and-stick representation. PDB code used to generate figure: 1MUH.
Several subsequent crystal structures of the Tn5 transpososome have used slightly different DNA transposon end sequences in an attempt to understand the conformational changes that occur as the ends are processed in preparation for strand transfer (Lovell et al. 2002; Klenchin et al. 2008; Steiniger-White et al. 2002; PDB depositions 4DM0 and 1MUS). Although the transpososome state in which the transposase is bound to intact hairpin intermediates remains elusive, it is clear that the transposase engages in a number of interactions with the transposon ends that presumably would stabilize a tight hairpin. The observed differences between the various crystal structures most likely reflect the need for flexibility by a single active site that must catalyze three different nucleophilic attack reactions.
There are clearly a number of conformational states of the Tn5 transpososome, and the currently available structures capture only one stage of the transposition process. An approximate model for the target capture complex can be made, as the positions of the two terminal 3′-OH groups of the TS are clear and there is an obvious target DNA-binding surface. Although one might suspect that a Tn5 STC should be stable and suitable for crystallization, to date, such a structure has not been reported.
One of the most intriguing remaining questions is how the N-terminal DNA-binding domain and the C-terminal dimerization domain of the transposase inhibit each others’ activities at the early stages of the reaction. For example, full-length Tn5 transposase has not been observed to bind to its transposon end as a monomer, yet when the C-terminal dimerization domain is deleted, the truncated protein does so readily (York & Reznikoff, 1996). Conversely, when the N-terminal DNA-binding domain is removed, the remaining protein can dimerize (Braam et al. 1999), whereas the full-length transposase does not. These observations suggest that a number of important conformational rearrangements must occur during transpososome assembly to relieve these reciprocal inhibitions, and it would be very exciting to understand the structural dynamics of this process.
2.3. The first eukaryotic transpososome, Mos1
The second crystal structure describing a transpososome containing an RNaseH-like catalytic domain is that of the eukaryotic mariner element, Mos1, from Drosophila mauritiana (Richardson et al. 2009). This was a particularly important step forward as it illustrated a cut-and-paste system that, in contrast to the Tn5 transpososome, generates double-strand breaks (DSB) at both ends of the transposon without forming hairpins either on the transposon ends or on flanking DNA at the cleavage site.
2.3.1. Mos1 biochemistry and mechanism
Biochemical studies have established that the Mos1 transposase first catalyzes cleavage of the NTS and only then does it cleave the TS (Dawson & Finnegan, 2003). It accomplishes this with a transpososome that contains only a transposase dimer. In contrast to the Tn5 transpososome, evidence suggests that Mos1 transposase is a dimer before recognizing and binding its ITRs (Augé-Gouillou et al. 2005a). For Mos1, the left and right ITRs differ only by four base pairs, and it is believed that the initial DNA binding is to the right ITR, as the binding affinity of the transposase to the left ITR is substantially weaker (Augé-Gouillou et al. 2001). It is thought that a transposase dimer bound to the RE (called the single-end complex SEC2 where ‘2’ indicates that the Mos1 transposase is dimeric) then captures the left ITR forming a PEC. Although it has been suggested that the active form of the Mos1 transposome is tetrameric (Augé-Gouillou et al. 2005b), more recent data including the PEC crystal structure argue convincingly that this is not the case (Augé-Gouillou et al. 2005a; Richardson et al. 2009).
One important outstanding mechanistic question is whether the first strand cleavage of the NTS occurs in the context of the SEC2, or if PEC assembly is first required. The literature is inconsistent in this regard. Using a mutated right ITR, a near-normal level of NTS cleavage was observed in the absence of PEC formation (Dawson & Finnegan, 2003). However, a point mutant of the Mos1 transposase, L124S, that is probably defective for PEC formation also failed to support efficient NTS cleavage (Augé-Gouillou et al. 2005a). The idea that a transposase may introduce damaging DNA nicks at one ITR before finding the other, a search that might not be successful, is inherently unappealing. Nevertheless, current data have not yet formally excluded this possibility. These issues have also been investigated in the context of other mariner DNA transposases, such as the Hsmar1 transposase that was re-constructed as an ancestral consensus sequence from about 200 defective copies scattered in the human genome (Miskey et al. 2007). For this transposase, the NTS is cleaved only after PEC formation (Bouuaert et al. 2011).
Once both NTS are cleaved, the products – the newly created 3′-OH group on each NTS and the 5′-phosphate group on the other side of the DNA strand break – have to vacate the active sites to make way for the TS. With a dimeric transposase, we cannot invoke alternative active sites from other transposase monomers to catalyse TS cleavage. Furthermore, it is clear that no other proteins are involved. (This is contrasted with the Tn7 system that indeed uses a second nuclease, unrelated to RNaseH-like enzymes, to carry out NTS cleavage; Hickman et al. 2000; Sarnovsky et al. 1996.) This leaves the obvious implication that a major reconfiguration of the PEC must take place to make room for the TS after both NTS have been cleaved. This is consistent with the result reported for the Hsmar1 transposase that TS cleavage is the rate-limiting step in the entire reaction sequence (Bouuaert & Chalmers, 2010).
2.3.2. The Mos1 transpososome
The structure of the Mos1 transpososome (Fig. 7) captures the point of the reaction pathway immediately following TS cleavage. In this configuration, two cleaved transposon ends are bound by the transposase dimer in which each active site is occupied by the 3′-OH end of the TS. As shown in Fig. 3a, Mos1 catalyses a staggered cut at each transposon end in which the NTS cleavage point lies 3 nts into the transposon, and the structural work faithfully recapitulates this with two identical RE 28-mer transposon ends in which the NTS has been recessed by 3 nts. The TS is, therefore, longer than the NTS as it leads into the active site, and the structure reveals that this short ssDNA stretch of the TS is specifically recognized by the transposase. The NTS, which would already have been cleaved by this stage of the reaction, has moved away from the active site.
Fig. 7.
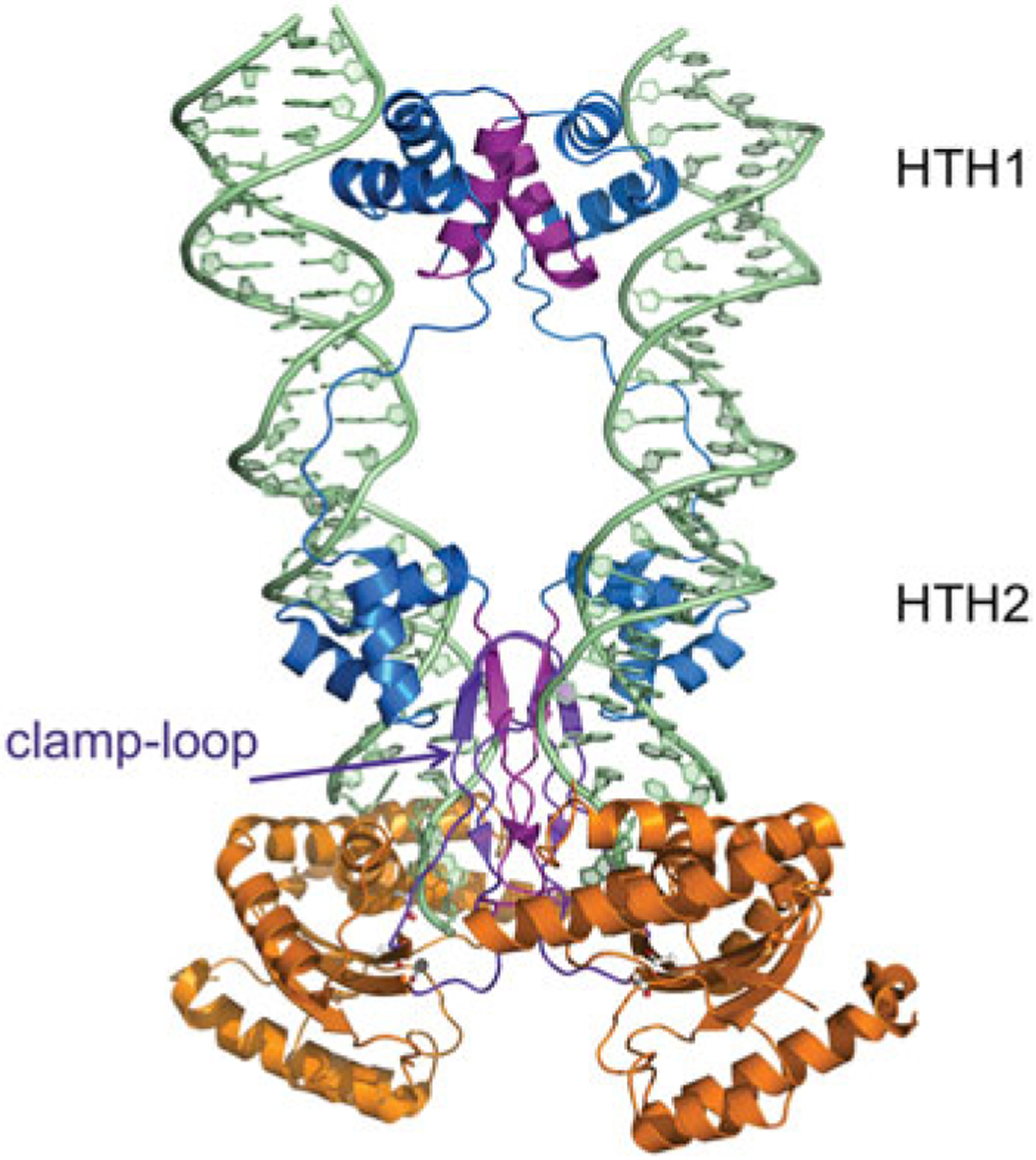
The dimeric Mos1 transpososome. The domain colors correspond to those shown in Fig. 2. The N-terminal DNA domain (two HTH domains connected by a long linker) is in blue and the RNaseH-like catalytic core is in orange. Residues involved in multimerization (amino acids 7–21, 112–125, and the clamp–loop residues 162–189) are shown in purple. The residues comprising the DDD motif (D156, D284, and D249) are shown in ball-and-stick representation. PDB code: 3HOT.
The overall architecture of the Mos1 transpososome bears very little similarity to that of Tn5. There are two small N-terminal site-specific DNA-binding domains (also collectively called the paired DNA-binding domain) each containing its own helix-turn-helix motif (HTH1 and HTH2; shown in blue in Fig. 7) that binds the major groove of the transposon end. In common with Tn5, this site-specific recognition is not immediately at the transposon end but rather subterminal, with HTH1 contacting nts 21–26 (again counting inwards from the transposon end) and HTH2 contacting nts 8–13. The HTH1- and HTH2-binding sites are thus separated by 7 bp and the protein loop connecting the two HTH domains binds the intervening AT-rich minor groove.
HTH1 is also a dimerization domain as two of its helices (which are shown in purple in Fig. 7) interact to form a two-fold symmetric interface, and it is likely that this interface is responsible for transposase dimerization before ITR binding (Augé-Gouillou et al. 2005a). The RNaseH-like catalytic domains (shown in orange in Fig. 7), similarly to those of the Tn5 transposase, interact with the ends of the transposon DNA in trans. The transposase protomer whose two HTH motifs bind one transposon end provides the catalytic domain that cleaves the other transposon end. The two transposon ends run almost parallel and approach ~10Å of each other. This is in sharp contrast to the Tn5 transpososome where the two transposon ends are oriented anti-parallel and far from each other (Figs 5 and 6).
The two catalytic domains within the transpososome also play important roles in holding the assembly together. One feature that appears crucial is the so-called ‘clamp-loop’, a short insertion in the catalytic domain (see Fig. 2). This 26 amino-acid long insertion (shown in purple in Fig. 7) was disordered in the uncomplexed (DNA-free) crystal structure of the Mos1 transposase (Richardson et al. 2006), and appears to be an important contributor to protein–protein interactions in the context of the active transpososome: the clamp–loops interact with each other and with the linker that connects HTH2 to the catalytic domain, and also form contacts with the TS. It has also recently been reported that the Mos1 nuclear localization signal (NLS) is found within this loop (Demattei et al. 2011), consistent with the notion that NLSs are typically located in unstructured, surface-accessible portions of proteins.
The key missing structural information is the configuration of the Mos1 transpososome at the initial NTS cleavage step, which might shed light onto the nature of the rearrangements that must take place to arrive at the configuration captured in the crystal structure. Furthermore, only an approximate model for the strand transfer step can be deduced from the post-TS cleavage transpososome configuration, as the two 3′-OH groups at the transposon ends are ~26Å away from each other. This is substantially further apart than the 18Å needed for the 2 bp separation between the staggered strand transfer sites on target DNA, assuming straight B form target DNA. Therefore, some conformational change involving the transpososome and/or bending of the target DNA will likely be needed.
2.4. The over-achieving cousin: PVF integrase and the retroviral integration pathway
Somewhat unexpectedly, the most complete view of a transposition-like reaction captured crystallographically at different transpososome stages comes from a system that is not strictly a DNA transposase but a retroviral integrase (Hare et al. 2010; Maertens et al. 2010; reviewed in Li et al. 2011). The strong mechanistic similarities between transposition and the integration of retroviral DNA are well-established (Engelman et al. 1991). As shown in Fig. 3a, from a biochemical perspective, retroviral integrases have a simpler task than cut-and-paste DNA transposases, as they do not have to remove the mobile element (in this case, the viral DNA) from its flanking DNA and therefore do not need to generate DSBs since the NTS arrives ‘pre-cut’.
In contrast to the other two transpososomes described earlier, retroviral integrases assemble as tetramers in the retroviral intasome (the retroviral version of a transpososome). Tetrameric assembly appears mandatory for the key biological function of a retroviral integrase, the concerted integration of viral DNA into target chromosomal DNA of the infected cell (Li et al. 2006).
2.4.1. In complex with viral DNA ends
The configurational state of the intasome of the prototype foamy virus (PFV) just after 3k-processing reveals a tetramer bound to two 19-mer viral DNA ends (Fig. 8; Hare et al. 2010). PFV integrase consists of four domains: the ‘NTD extension domain’ (NED) (shown in dark blue in Fig. 8); the NTD (shown in light blue); the ‘catalytic core domain’ (CCD) that is the RNaseH-like domain (shown in shades of orange); and finally the CTD (shown in yellow). (The NED is not found in all retroviral integrases; for instance, it is not part of HIV-1 integrase.) All four domains and their linkers (shown in gray) interact with viral DNA. The terminal six base pairs of the viral ends appear to be the most important, and here the structure of the DNA significantly deviates from regular B form owing to the large number of protein–DNA contacts. Although the NED and the NTD bind the DNA in trans relative to their CCDs, two of the four CTDs interact with both DNA ends, essentially bridging the retroviral intasome.
Fig. 8.
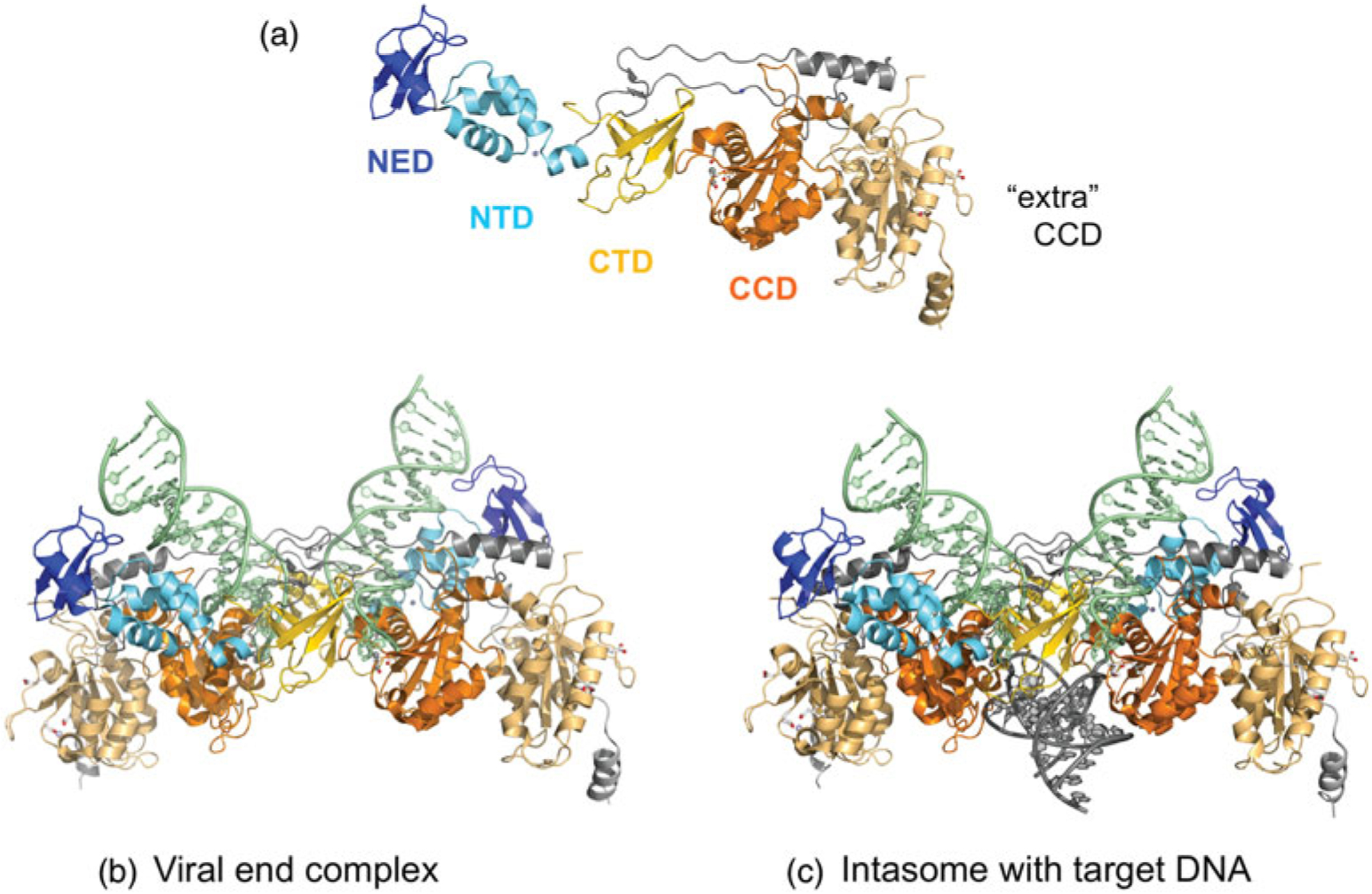
The PFV intasome. (a) The extended string of domains of one monomer of the intasome (domains in color) and its associated unused or ‘extra’ catalytic domain (in light orange). Below are the structures of (b) the intasome with two viral DNA ends and (c) the STC. The domain colors correspond to those shown in Fig. 2, where dark blue is the NED, light blue is the Zn-binding NTD (the Zn2+ ion is shown as a light blue sphere), orange is the CCD (there are two Mn2+ ions bound in the ‘used’ active site), and yellow is the CTD. The two additional CCDs with unused active sites on the periphery of the intasome contributed by outer subunits are shown in light orange in (b) and (c). PDB codes used to generate intasome figure with viral ends (3OY9) and with target DNA (3OS0).
One curious aspect of the structure is that there are four RNaseH-like catalytic subunits in the assembly, yet only two are arrayed to participate in the chemical steps of 3′ processing and strand transfer (Fig. 3a and b). Furthermore, the two protomers that participate in catalysis, known as inner subunits (darker orange in Fig. 8b and c), constitute most of the crystallographically visible part of the intasome: although there are two other integrase monomers present (‘outer’ subunits), only their CCDs with the apparently unused active sites (lighter orange) and connecting linker regions are sufficiently ordered to be visible. The other domains of these two extra integrases are evidently disordered and not involved in additional protein–protein or protein–DNA interactions. In fact, the requirement for tetrameric retroviral intasomes in catalyzing concerted integration is not currently understood, and the structures do not make the situation any clearer.
Even though two of the integrase protomers do not appear to be participating in the chemical process, they are bound to the functioning catalytic cores through extensive protein–protein interfaces (Fig. 8). Adding to the peculiarity is the fact that this particular CCD/CCD interface is present (with minor variations) in all crystal structures of retroviral integrases in which the catalytic domain was part of the crystallized assembly (reviewed in Jaskolski et al. 2009) and, therefore, was thought to be functionally relevant. For many years, this was a perplexing observation as it was clear from the very beginning that the active sites of these CCD/CCD dimers were not only too far apart but also oriented incorrectly (i.e., facing away from each other) to catalyze concerted integration 5 bp apart on target DNA. Nevertheless, the tetramer of the intasome is formed through this conserved interface.
The oligomerization state of retroviral integrases before binding viral ends appears to vary. Although a double point mutant of HIV-1 integrase displays a dimer–tetramer equilibrium (Jenkins et al. 1996), Avian sarcoma virus integrase appears to be dimeric (Bojja et al. 2011). In contrast, PFV integrase is exclusively monomeric (Valkov et al. 2009). While the significance of these variations is not entirely clear, the CCD/CCD interface may be looser in the PFV intasome structure than in some of the other integrase CCD structures. This may explain why PFV integrase can be a soluble monomer when not bound to DNA.
As in the two previous transpososome examples, binding of the viral DNA ends is in trans, and the 3k-OH of the TS of the viral end that is bound by the NED and NTD of one monomer is located in the active site of the CCD of the other catalytically functional monomer. What we can therefore regard as the functional dimer is organized such that its two active sites (more precisely, the two 3′-OHs of the viral TS ends) are only 22.9Å apart and face toward each other, consistent with the 4 bp distance of the insertion points on each target DNA strand. The NTS of the viral ends are located away from the active sites, held safely in place in a groove between the trans catalytic core and the trans CTD.
A remarkable feature of the overall architecture of the intasome is that the NTDs form protein–protein interactions with the trans catalytic domain of the functional dimer, but none with the cis catalytic domain to which each is physically connected through the polypeptide. Augmenting these protein–protein interactions are the interactions that the viral ends forms with the NTD domains and the trans catalytic domain.
2.4.2. Target capture complexes
Three slightly different versions of the PFV intasome with bound viral ends which include target DNA (shown in dark green in Fig. 8c) have also been solved (Maertens et al. 2010): a ‘simple’ target complex where target DNA was added to the viral end complex; a STC generated in situ upon addition of Mg2+ in which the viral ends are covalently linked to target DNA; and a third in the presence of Mg2+ in which the viral ends are missing their 3′-OH groups thereby preventing strand transfer into target DNA. This immediately established that the crystallographically captured transpososome is enzymatically competent, at least for strand transfer.
When compared with the intasome containing only viral ends, there are no significant conformational changes upon target complex formation. Indeed, the crystal forms of the target complexes are isomorphous with that containing only the viral ends. It seems, therefore, that the retroviral intasome does not undergo major conformational changes during the reaction, possibly for the simple reason that it does not have to deal with the NTS. It is also possible that 3′-processing can also be accomplished without major rearrangements (unlike, e.g., Mos1) although the structure of the PFV intasome before 3′-processing has not yet been reported.
The main structural characteristic of the target capture configurations of the intasome is the conformation of the target DNA, which contains a significant bend widening the major groove. This results in a 55° negative roll between the 2 bp at the center of the integration site, a configuration that makes the appropriate scissile phosphates of the target DNA strands available to the active sites of the catalytic domains. These DNA distortions relative to standard B form DNA are achieved without disrupting target base pairing. Bending also widens the major groove, where a loop from the CTD enters and contacts bases, providing what seems to be an important stabilizing interaction of the observed configuration, and conferring one of the otherwise very sparse protein-target DNA base pair interactions in the intasome. When the viral TS terminal 3-OH groups are covalently attached to the respective target DNA strands, they are 21.8Å apart and this is only about 1Å different from the distance observed before target binding. However, due to the widened major groove, this distance is longer compared with the expected 18.9Å for standard B form DNA. On the other hand, the DNA backbone also makes a sharp turn at the insertion point, owing to the fact that the attacking 3′-OH has to attack the phosphate opposite from the direction of the bond broken during integration. This sharp turn renders the 3′-OH to 3′-OH distance shorter than the widened major groove would dictate.
2.5. Single-stranded transposition
A common feature of all the previously described transpososomes is that they each move double-stranded transposon DNA from one genomic location to another, and it was only recently that a mode of DNA transposition involving only ssDNA was discovered (Barabas et al. 2008; Guynet et al. 2008; Ton-Hoang et al. 2005). In ssDNA transposition, typified by members of the IS200/IS605 family of bacterial insertion sequences (Kersulyte et al. 2002), only one strand of the transposon is processed and moved into a single-stranded target site. Although at first glance, moving only one strand might imply that an ssDNA transpososome has an inherently easier task than those of cut-and-paste TEs, this is not the case.
There is mechanistic symmetry to the movement of many dsDNA transposons as the transposase generally recognizes related TIRs at the two ends of the mobile element, and identical chemical reactions can be carried out on both ends. In contrast, end asymmetry is an essential characteristic of ssDNA transposition, and is inherently intertwined with the notion that identical chemical cleavage events at two ends of a ssDNA transposon have different outcomes since DNA is directional. This concept is illustrated in Fig. 4b. Cleavage at the LE of a ssDNA transposon results in a 3′-OH group on the flanking DNA and a covalent 5′-phosphotyrosine linkage to the transposon end. However, cleavage at the RE results in a 3′-OH group on the transposon end and a covalent 5′-phosphotyrosine linkage to flanking DNA.
Structures of several different transpososomes from IS608 and ISDra2 have been determined which have captured many of the important steps along the ssDNA transposition pathway (Barabas et al. 2008; Hickman et al. 2010b; Ronning et al. 2005). In all of these structures, a transposase dimer is bound to two transposon ends. These are illustrated in Fig. 9, for the IS608 transposase bound to two LE (top) and two RE (bottom). As the authentic transpososome will contain one LE and one RE, this admittedly artificial structural situation has been dictated by the technical difficulties in assembling transpososomes containing one LE and one RE at the same time and subsequently obtaining X-ray diffraction quality crystals. The problem is that the transposase has nearly identical affinity for the different ends, and complexes formed in the presence of a mixture of LE and RE are a mixture of different complexes, only some of which contain one LE and one RE. Clearly, this situation is not conducive for crystallization. The situation is not dissimilar to that of dsDNA transpososomes where all the structures solved to date have involved artificially symmetrized ends. However, as asymmetry is an important characteristic of ssDNA transposomes, some loss of authenticity is inevitable. As many dsDNA transposomes are functional with symmetrized ends, the loss of information due to their use in structural experiments is arguably less serious.
Fig. 9.
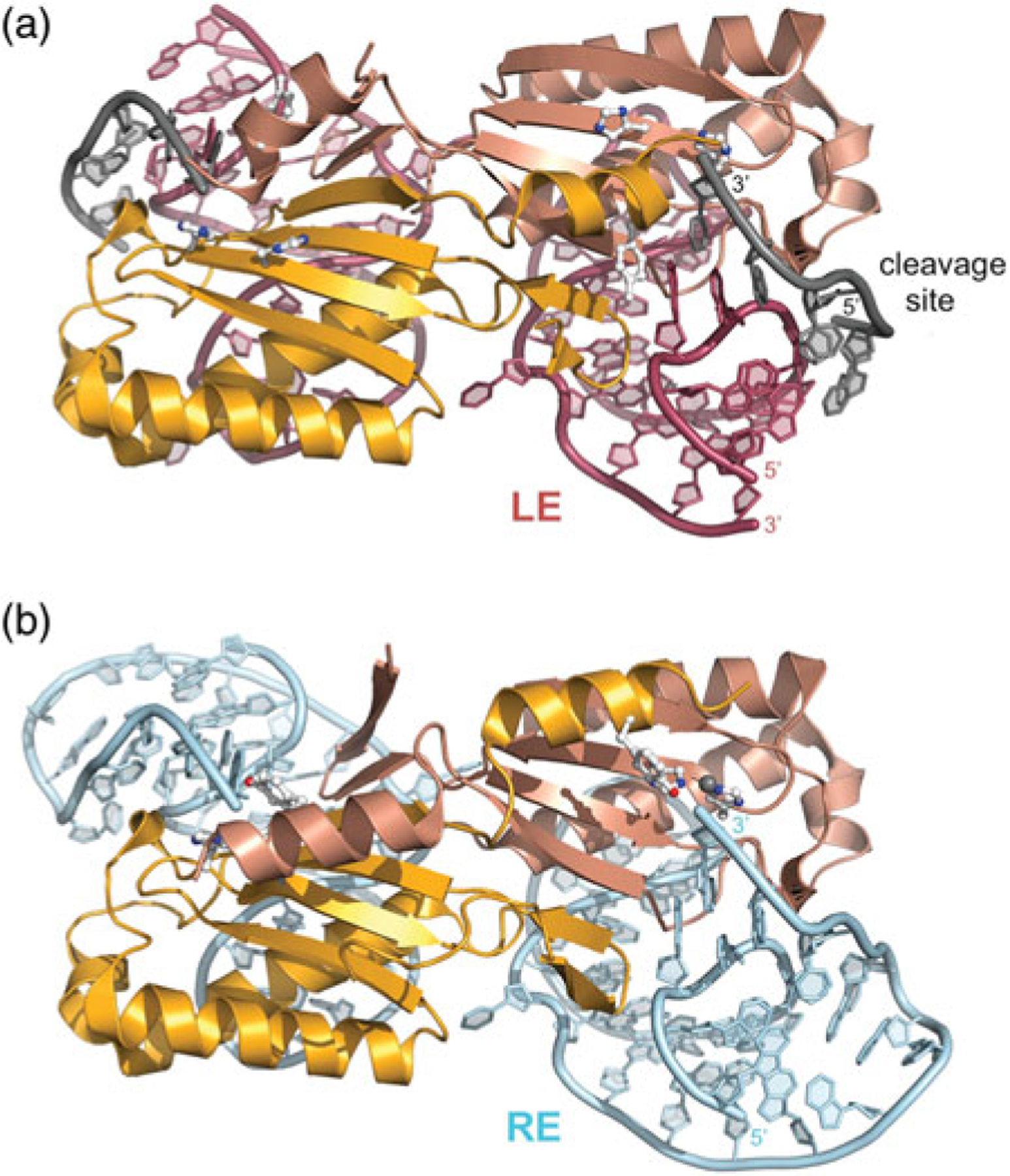
Dimeric IS608 TnpA transpososomes. (a) TnpA bound to an LE 26-mer (in red) and a 6-mer cleavage or target site (grey). The two monomers are shown in different shades of orange to illustrate the composite active site. Active site residues H64, H66, and Y127 are shown in ball-and-stick representation. PDB code: 2VJV. In this structure, Y127 has been replaced by Phe. (b) TnpA bound to a RE 35-mer (in blue). PDB code: 2VJU.
2.5.1. Recognizing the ends
ssDNA transposases recognize their DNA ends in a fundamentally different manner from dsDNA transposases. Although the latter use sequence-specific DNA-binding modules to recognize elements of their TIRs as we have seen in the Tn5, Mos1, and IN transpososome structures, ssDNA transposases recognize imperfect palindromic sequences that are essentially identical at both ends, but are located at different distances from the cleavage sites at the very ends of the mobile element (Fig. 4a). These imperfect palindromes form DNA hairpins that are recognized by the transposase (Fig. 9). Hairpin binding is not sequence-specific in a direct sense but structure-specific as most of the protein–DNA interactions are with the DNA sugar-phosphate backbone.
Both monomers within the transposase dimer contribute to the binding of each hairpin. The bulk of the non-specific protein–DNA interactions occur with one monomer and this same monomer contributes the only base-specific interaction that involves a single pyrimidine base in the hairpin loop. In contrast, the imperfections in the palindrome (a flipped out or extrahelical base in the case of IS608, a base mismatch in the case of ISDra2) are recognized by the other monomer within the dimer.
Since ssDNA transposases form dimers even in the absence of DNA (Ronning et al. 2005) and each monomer has one hairpin binding site, as long as they can locate each other, the problem of bringing the two ends of the transposon into each other’s proximity is solved. Similarly to the dsDNA transpososomes, there is no evidence that any cleavage occurs before both ends are bound (He et al. 2011). The ability to recognize DNA hairpins through an interaction involving the hairpin loop provides an ingenious way for the ssDNA transposases to distinguish between top and bottom strands of the transposon, only one of which is to be transposed. The tips of the hairpin loop contain pyrimidine bases (typically thymines) which are bound within protein pockets located on the transposase surface. The hairpin that forms on the bottom strand has a purine base in the corresponding location, and there is not enough room for it to fit into the transposase-binding pocket. This simple size test appears to be the basis for strand discrimination.
2.5.2. DNA cleavage at the transposon ends
Although the chemistry and polarity of strand cleavage is identical at the LE and RE of ssDNA transposons, always producing a 5′-phosphotyrosine linkage and a free 3′-OH end as products, the way in which this is coupled to hairpin binding is different at each end. This is because the LE cleavage site is located 5′ of the LE hairpin, whereas the RE cleavage site is located 3k of the RE hairpin (Fig. 4a). The transposase has adopted fundamentally the same approach to recognizing and cleaving the LE and RE, although the structural details differ because of the difference in directionality. The concept at the heart of ssDNA transposition is that cleavage sites, which differ at the two ends, are recognized by the transposase which co-opts a 4-nt long sequence just 5′ of the hairpins at either end as part of the active site. This tetranucleotide recognizes the cleavage site sequence through both canonical and non-canonical base-pairing interactions (see Fig. 4). Of course, this is possible only when the cleavage site is in the ssDNA form (the cleavage site at the LE is shown in gray in Fig. 9). Because of the differences in the relative orientations of the hairpin and the cleavage site on the two ends, the twists and turns that the ssDNA must adopt to accomplish this also differ, as illustrated in Fig. 9. One consequence of this strategy is that the recognition of two different cleavage sequences becomes possible without relying on sequence-specific DNA-binding domains (Barabas et al. 2008; Guynet et al. 2009).
As shown in Fig. 4b, LE and RE cleavage precede formation of the covalently closed single-stranded circular transposon, the transposition intermediate. This intermediate, the mechanistic equivalent of a dsDNA transposon that has been cut out of its original donor site, can only be generated when the 5′-phosphotyrosine-linked intermediates at each transposon end are resolved (i.e., the covalent bonds are broken). Although this stage of ssDNA transposition has not yet been captured crystallographically, it is believed that this is accomplished by the physical exchange of cleaved DNA strands from one active site to the other in conjunction with the movement of the two alpha-helices that bear the catalytic tyrosine – and hence the 5′-phosphotyrosine intermediates (for details, see Barabas et al. 2008). It is thought that these helices simply swap places with each other with respect to the active sites, bringing the phosphotyrosine-attached DNA with them and exchanging their positions. After this exchange, the 5′-phosphotyrosine linkage can be attacked by the 3′-OH group generated by cleavage on the opposite transposon end that has remained in the active site where it was created. This chemical step restores the DNA backbone, but because of the active site switch, the DNA connectivity is now different and a sealed donor site and an excised circular transposon have been generated.
2.5.3. Target binding and integration
Integration, which is sequence-specific, is organizationally very similar to the cleavage events that liberate the ssDNA transposon from flanking ssDNA. The cleavage site sequence on LE that is recognized through base pairing interactions with the short 5′ extension of the LE hairpin consists of the flanking DNA just 5′ of the cleavage site (this sequence is TTAC in the case of IS608). Integration occurs when the transpososome-bound ssDNA transposon circle finds a suitable TTAC target site, and binds it through precisely the same mechanism of base–base interactions as for the cleavage site. Two cleavages very similar to the ones that liberated the transposon will then accomplish integration, as shown in Fig. 4c.
3. Transposomes: the architectures of controlled DNA remodeling
As the number of high-resolution transpososome structures is very limited, it is clear that there is little that seems to conform to overall architectural paradigms (Fig. 5). Of course, one common feature is that there are always two DNA transposon ends bound, whether in dsDNA or ssDNA form, but this is clearly dictated by the fact that transposable genetic elements must have two ends. Given that there are a number of common functional requirements, it is quite remarkable how these are satisfied by complex assemblies that are so architecturally different.
One common functional requirement appears to be the control of nuclease activity. There is no convincing evidence indicating that a transposase catalyzes cleavage when only one end is bound i.e., without synapsis. As far as we are aware, the only known exception among systems that are closely related to transposases is the initial nicking step in V(D)J recombination catalyzed by the RAG1/2 complex. Using a truncated version of RAG1, it has been shown that nicking can take place before the two recombination signal sequences are synapsed (Yu et al. 2000). Among transposases, however, the requirement of binding of both ends before any catalysis seems to be true not only for the so-called cut-and-paste transposons but also for at least some prokaryotic replicative transposons as well (reviewed in Kobryn et al. 2002). One way to achieve this regulatory control is well illustrated in the transpososome structures solved to date, which show how catalysis in trans is mandated, and how multimerization sometimes depends on binding two DNA ends.
It is also true that the case for Mos1 is not yet settled, as currently available data can be – and have been – interpreted in different ways. This ambiguity can arise only because of the apparent ability of the Mos1 transposase to dimerize before ITR binding, which means that the trans active site is available in the SEC2, which is significant only if we assume that first strand cleavage also occurs in trans. Unfortunately, there is no convincing evidence for this. However, it does not appear that multimerization before TE end binding is an exception and true only in the mariner family of transposons. For instance, there are prokaryotic transposases that contain what appears to be mandatory (i.e., not regulatory, such as in the case of Tn5) multimerization domains, such as the OrfAB transposase of IS911 (Haren et al. 1998). It is also clear that the eukaryotic DNA transposase of the Hermes transposon, forms obligatory multimers before end binding (Hickman et al. 2005). Although some retroviral integrases exist as multimers before viral end binding, the dominant multimerization interface driving this is the CCD/CCD interface. The structural evidence of the retroviral intasome is clear about the trans activity both for 3′-processing and for strand transfer, and it also appears that productive multimerization occurs only after the viral ends have been bound. For the ssDNA transpososomes, while dimerization is obligatory, the structures suggest that communication between active sites is likely, assuring that nuclease activity can occur only once both are occupied.
Taken collectively, one of the major outstanding mechanistic questions is how strand cleavage is controlled, if at all, for those transpososomes where productive multimerization seem to occur before transposon end binding. As unproductive single-ended cleavages would result in genomic damage, it would be comforting to assume that mechanisms exist to prevent it. Only future efforts can tell whether this is indeed the case and, if so, what its mechanism is.
Another recurring theme in all systems is the accurate geometric positioning of the two 3′-OH groups at the end of the TE before strand transfer. Again, this seems mandatory since the number of nucleotides involved in TSD is a characteristic of each system. Inherent in this reproducibility is that target positioning must also be accurate, as the correct target strand has to be attacked by the correct 3′-OH group. At this point, only the PFV intasome structure speaks clearly about this. Both for Tn5 and Mos1, some reconfiguration is clearly necessary to form the STC; its exact nature is not yet clear.
It is worth pointing out the inherent difficulties of structural experiments involving target DNA. As target binding for the cut-and-paste transposases has little or no specificity, experiments involving oligonucleotides representing target DNA molecules run into the problem that the many different target binding modes which can occur cause difficulties in crystallization. Of course, the case for ssDNA transposition is very different where integration is site-specific and mechanistically equivalent to LE cleavage. Therefore, there are no configurational differences between an LE cleavage complex and the integration complex, as it involves the same set of interactions. This is probably why the first complete structural characterization of a transposition pathway via transpososome crystal structures and biochemical experiments was captured and understood for ssDNA transposition.
Another key question, left unanswered by the currently available data, is the fate of the transpososome after strand transfer is complete. In some cases, such as bacteriophage Mu, an ATP-dependent protein-unfolding machine, ClpX, is required (Abdelhakim et al. 2010; Burton & Baker, 2003; Mhammedi-Alaoui et al. 1994; North & Nakai, 2005). This is entirely consistent with the notion that the transposition reaction is an energetically downhill pathway, forming a sequence of complexes with increased stability. Taking apart the MuA/DNA to allow replication to proceed needs energy input from sources such as ATP. It would be very interesting to see whether a MuA transpososome crystal structure could reveal something about the initiation of this process beyond what has been biochemically established. Also, it would be exciting to understand whether ClpX is involved in the disassembly of other transpososome. Although ClpX involvement in transposition is certainly a huge step forward in understanding transpositional DNA remodeling, there is currently a total absence of information regarding any such factors for eukaryotic systems. It is certain that more mechanistic work would need to be done to understand more completely these fascinating molecular processes.
4. Acknowledgments
This work was supported at the NIH by the Intramural Program of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), Bethesda MD and in France by continuous intramural funding from the CNRS (France) and, in its later stages, by ANR grant Mobigen (M.C.), and by European contract LSHM-CT-2005-019023 (M.C.).
5. References
- Abdelhakim AH, Sauer RT & Baker TA (2010). The AAA+ ClpX machine unfolds a keystone subunit to remodel the Mu transpososome. Proceedings of the National Academy of Sciences of the United States of America 107, 2437–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arciszewska LK, Drake D & Craig NL (1989). Transposon Tn7. cis-acting sequences in transposition and transposition immunity. Journal of Molecular Biology 207, 35–52. [DOI] [PubMed] [Google Scholar]
- Arciszewska LK, Mckown RL & Craig NL (1991). Purification of TnsB, a transposition protein that binds to the ends of Tn7. Journal of Biological Chemistry 266, 21736–21744. [PubMed] [Google Scholar]
- Augé-Gouillou C, Hamelin MH, Demattei MV, Periquet M & Bigot Y (2001). The wild-type conformation of the Mos-1 inverted terminal repeats is suboptimal for transposition in bacteria. Molecular Genetics and Genomics 265, 51–57. [DOI] [PubMed] [Google Scholar]
- Augé-Gouillou C, Brillet B, Germon S, Hamelin MH & Bigot Y (2005a). Mariner Mos1 transposase dimerizes prior to ITR binding. Journal of Molecular Biology 351, 117–130. [DOI] [PubMed] [Google Scholar]
- Augé-Gouillou C, Brillet B, Hamelin MH & Bigot Y (2005b). Assembly of the mariner Mos1 synaptic complex. Molecular and Cellular Biology 25, 2861–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azaro MA & Landy A (2002). λ Integrase and the λ Int family In Mobile DNA II (eds. Craig NL, Craigie R, Gellert M & Lambowitz A), pp. 118–148. Washington: American Society of Microbiology. [Google Scholar]
- Aziz RK, Breitbart M & Edwards RA (2010). Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Research 38, 4207–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Brennan P, Rizzu P, Smith S, Fell M, Talbot RT, Gustincich S, Freeman TC, Mattick JS, Hume DA, Heutink P, Carninci P, Jeddeloh JA & Faulkner GJ (2011). Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479, 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainton RJ, Kubo KM, Feng J & Craig NL (1993). Tn7 transposition: target DNA recognition is mediated by multiple Tn7-encoded proteins in a purified in vitro system. Cell 72, 931–943. [DOI] [PubMed] [Google Scholar]
- Barabas O, Ronning DR, Guynet C, Hickman AB, Ton-Hoang B, Chandler M & Dyda F (2008). Mechanism of IS200/IS605 family DNA transposases: activation and transposon-directed target site selection. Cell 132, 208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsoum E, Martinez P & Åström SU (2010). α3, a transposable element that promotes host sexual reproduction. Genes and Development 24, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry C, Malinsky S, Restituito M, Kapusta A, Rosa S, Meyer E & Bétermier M (2009). PiggyMac, a domesticated piggyBac transposase involved in programmed genome rearrangements in the ciliate Paramecium tetraurelia. Genes and Development 23, 2478–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhasin A, Goryshin IY, Steiniger-White M, York D & Reznikoff WS (2000). Characterization of a Tn5 pre-cleavage synaptic complex. Journal of Molecular Biology 302, 49–63. [DOI] [PubMed] [Google Scholar]
- Biémont C (2010). A brief history of the status of transposable elements: from junk DNA to major players in evolution. Genetics 186, 1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beese LS & Steitz TA (1991). Structural basis for the 3′−5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO Journal 10, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojja RS, Andrake MD, Weigand S, Merkel G, Yarychkivska O, Henderson A, Kummerling M & Skalka AM (2011). Architecture of a full-length retroviral integrase monomer and dimer, revealed by small angle X-ray scattering and chemical cross-linking. Journal of Biological Chemistry 286, 17047–17059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braam LAM, Goryshin IY & Reznikoff WS (1999). A mechanism for Tn5 inhibition – carboxyl-terminal dimerization. Journal of Biological Chemistry 274, 86–92. [DOI] [PubMed] [Google Scholar]
- Britten RJ (2004). Coding sequences of functioning human genes derived entirely from mobile element sequences. Proceedings of the National Academy of Sciences of the United States of America 101, 16825–16830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouuaert CC & Chalmers R (2010). Transposition of the human Hsmar1 transposon: rate-limiting steps and the importance of the flanking TA dinucleotide in second strand cleavage. Nucleic Acids Research 38, 190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouuaert CC, Liu DX & Chalmers R (2011). A simple topological filter in a eukaryotic transposon as a mechanism to suppress genome instability. Molecular and Cellular Biology 31, 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundock P & Hooykaas P (2005). An Arabidopsis hAT-like transposase is essential for plant development. Nature 436, 282–284. [DOI] [PubMed] [Google Scholar]
- Burton BM & Baker TA (2003). Mu transpososome architecture ensures that unfolding by ClpX or proteolysis by ClpXP remodels but does not destroy the complex. Chemistry and Biology, 10, 463–472. [DOI] [PubMed] [Google Scholar]
- Campos-Olivas R, Louis JM, Clérot D, Gronenborn B & Gronenborn AM (2002). The structure of a replication initiator unites diverse aspects of nucleic acid metabolism. Proceedings of the National Academy of Sciences of the United States of America 99, 10310–10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C, Smidansky ED, Arnold JJ, Maksimchuk KR, Moustafa I, Uchida A, Götte M, Konigsberg W & Cameron CE (2009). Nucleic acid polymerases use a general acid for nucleotidyl transfer. Nature Structural and Molecular Biology 16, 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchward G (2002). Conjugatve transposons and related mobile elements In Mobile DNA II (eds. Craig NL, Craigie R, Gellert M & Lambowitz A), pp. 177–191. Washington: American Society of Microbiology. [Google Scholar]
- Copeland NG & Jenkins NA (2010). Harnessing transposons for cancer gene discovery. Nature Reviews. Cancer 10, 696–706. [DOI] [PubMed] [Google Scholar]
- Cordaux R, Udit S, Batzer MA & Feschotte C (2006). Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proceedings of the National Academy of Sciences of the United States of America 103, 8101–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O’shea KS, Moran JV & Gage FH (2009). L1 retrotransposition in human neural progenitor cells. Nature 460, 1127–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craigie R (2001). HIV integrase, a brief overview from chemistry to therapeutics. Journal of Biological Chemistry 276, 23213–23216. [DOI] [PubMed] [Google Scholar]
- Curcio MJ & Derbyshire KM (2003). The outs and ins of transposition: from Mu to kangaroo. Nature Reviews. Molecular Cell Biology 4, 865–877. [DOI] [PubMed] [Google Scholar]
- Datta S, Larkin C & Schildbach JF (2003). Structural insights into single-stranded DNA binding and cleavage by F factor TraI. Structure 11, 1369–1379. [DOI] [PubMed] [Google Scholar]
- Davies DR, Goryshin IY, Reznikoff WS & Rayment I (2000). Three-dimensional structure of the Tn5 synaptic complex transposition intermediate. Science 289, 77–85. [DOI] [PubMed] [Google Scholar]
- Dawson A & Finnegan DJ (2003). Excision of the Drosophila mariner transposon Mos1: comparison with bacterial transposition and V(D)J recombination. Molecular Cell 11, 225–235. [DOI] [PubMed] [Google Scholar]
- Demattei M, Hedhili S, Sinzelle L, Bressac C, Casteret S, Moiré N, Cambefort J, Thomas X, Pollet N, Gantet P & Bigot Y (2011). Nuclear importation of/Mariner/transposases among eukaryotes: motif requirements and homo-protein interactions. PLoS One 6, e23693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire KM, Kramer M & Grindley NDF (1990). Role of instability in the cis action of the insertion sequence IS903 transposase. Proceedings of the National Academy of Sciences of the United States of America, 87, 4048–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Cohen J, Jubin C, Dessen P, Goût J-F, Mousset S, Aury J-M, Jaillon O, Noël B, Arnaiz O, Bétermier M, Wincker P, Meyer E & Sperling L (2008). Analysis of sequence variability in the macronuclear DNA of Paramecium tetraurelia: a somatic view of the germline. Genome Research 18, 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval-Valentin G & Chandler M (2011). Co-translational control of DNA transposition: a window of opportunity. Molecular Cell 44, 989–996. [DOI] [PubMed] [Google Scholar]
- Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R & Davies DR (1994). Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 266, 1981–1986. [DOI] [PubMed] [Google Scholar]
- Engelman A, Mizuuchi K & Craigie R (1991). HIV-1 DNA integration – mechanism of viral-DNA cleavage and DNA strand transfer. Cell 67, 1211–1221. [DOI] [PubMed] [Google Scholar]
- Finnegan DJ (1989). Eukaryotic transposable elements and genome evolution. Trends in Genetics 5, 103–107. [DOI] [PubMed] [Google Scholar]
- Gratias A, Lepére G, Garnier O, Rosa S, Duharcourt S, Malinsky S, Meyer E & Bétermier M (2008). Developmentally programmed DNA splicing in Paramecium reveals short-distance crosstalk between DNA cleavage sites. Nucleic Acids Research 36, 3244–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasch A, Lucas M, Moncalián G, Cabezas M, Pérez-Luque R, Gomis-Rüth FX, De La Cruz F & Coll M (2003). Recognition and processing of the origin of transfer DNA by conjugative relaxase TrwC. Nature Structural Biology 10, 1002–1010. [DOI] [PubMed] [Google Scholar]
- Grabundzija I, Irgang M, Mátés L, Belay E, Matrai J, Gogol-Döring A, Kawakami K, Chen W, Ruiz P, Chuah MKL, Vandendriessche T, Izsvák Z & Ivics Z (2010). Comparative analysis of transposable element vector systems in human cells. Molecular Therapy 18, 1200–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindley NDF & Joyce CM (1980). Analysis of the structure and function of the kanamycin-resistance transposon Tn903. Cold Spring Harbor Symposia on Quantitative Biology 45, 125–133. [DOI] [PubMed] [Google Scholar]
- Grundy GJ, Ramón-Maiques S, Dimitriadis EK, Kotova S, Biertümpfel C, Heymann JB, Steven AC, Gellert M & Yang W (2009). Initial stages of V(D)J recombination: the organization of RAG1/2 and RSS DNA in the postcleavage complex. Molecular Cell 35, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guynet C, Hickman AB, Barabas O, Dyda F, Chandler M & Ton-Hoang B (2008). In vitro reconstitution of a single-stranded transposition mechanism of IS608. Molecular Cell 29, 302–312. [DOI] [PubMed] [Google Scholar]
- Guynet C, Achard A, Ton-Hoang B, Barabas O, Hickman AB, Dyda F & Chandler M (2009). Resetting the site: redirecting integration of an insertion sequence in a predictable way. Molecular Cell 34, 612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haniford DB (2006). Transpososome dynamics and regulation in Tn10 transposition. Critical Reviews in Biochemistry and Molecular Biology 41, 407–424. [DOI] [PubMed] [Google Scholar]
- Hare S, Gupta SS, Valkov E, Engelman A & Cherepanov P (2010). Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464, 232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haren L, Polard P, Ton-Hoang B & Chandler M (1998). Multiple oligomerization domains in the IS911 transposase: a leucine zipper motif is essential for activity. Journal of Molecular Biology 283, 29–41. [DOI] [PubMed] [Google Scholar]
- He S, Hickman AB, Dyda F, Johnson NP, Chandler M & Ton-Hoang B (2011). Reconstitution of a functional IS608 single-strand transpososome: role of non-canonical base pairing. Nucleic Acids Research 39, 8503–8512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman AB, Li Y, Mathew SV, May EW, Craig NL & Dyda F (2000). Unexpected structural diversity in DNA recombination: the restriction endonuclease connection. Molecular Cell 5, 1025–1034. [DOI] [PubMed] [Google Scholar]
- Hickman AB, Ronning DR, Kotin RM & Dyda F (2002). Structural unity among viral origin binding proteins: crystal structure of the nuclease domain of adeno-associated virus Rep. Molecular Cell 10, 327–337. [DOI] [PubMed] [Google Scholar]
- Hickman AB, Perez ZN, Zhou LQ, Musingarimi P, Ghirlando R, Hinshaw JE, Craig NL & Dyda F (2005). Molecular architecture of a eukaryotic DNA transposase. Nature Structural and Molecular Biology 12, 715–721. [DOI] [PubMed] [Google Scholar]
- Hickman AB, Chandler M & Dyda F (2010a). Integrating prokaryotes and eukaryotes: DNA transposases in light of structure. Critical Reviews in Biochemistry and Molecular Biology 45, 50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman AB, James JA, Barabas O, Pasternak C, Ton-Hoang B, Chandler M, Sommer S & Dyda F (2010b). DNA recognition and the precleavage state during single-stranded DNA transposition in D. radiodurans. EMBO Journal 29, 3840–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder JW & Craig NL (2010). Architecture of the Tn7 posttransposition complex: an elaborate nucleoprotein structure. Journal of Molecular Biology 401, 167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics Z, Li MA, Mátés L, Boeke JD, Nagy A, Bradley A & Izsvák Z (2009). Transposon-mediated genome manipulation in vertebrates. Nature Methods 6, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain C & Kleckner N (1993a). IS10 mRNA stability and steady state levels in Escherichia coli: indirect effects of translation and role of rne function. Molecular Microbiology 9, 233–247. [DOI] [PubMed] [Google Scholar]
- Jain C & Kleckner N (1993b). Preferential cis action of IS10 transposase depends upon its mode of synthesis. Molecular Microbiology 9, 249–260. [DOI] [PubMed] [Google Scholar]
- Jaskolski M, Alexandratos JN, Bujacz G & Wlodawer A (2009). Piecing together the structure of retroviral integrase, an important target in AIDS therapy. FEBS Journal 276, 2926–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins TM, Engelman A, Ghirlando R & Craigie R (1996). A soluble active mutant of HIV-1 integrase – involvement of both the core and carboxyl-terminal domains in multimerization. Journal of Biological Chemistry 271, 7712–7718. [DOI] [PubMed] [Google Scholar]
- Jones JM & Gellert M (2004). The taming of a transposon: V(D)J recombination and the immune system. Immunological Reviews 200, 233–248. [DOI] [PubMed] [Google Scholar]
- Kapitonov VV & Jurka J (2005). RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biology 3, e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersulyte D, Velapatiño B, Dailide G, Mukhopadhyay AK, Ito Y, Cahuayme L, Parkinson AJ, Gilman RH & Berg DE (2002). Transposable element ISHp608 of Helicobacter pylori: nonrandom geographic distribution, functional organization, and insertion specificity. Journal of Bacteriology 184, 992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenchin VA, Czyz A, Goryshin IY, Gradman R, Lovell S, Rayment I & Reznikoff WS (2008). Phosphate coordination and movement of DNA in the Tn5 synaptic complex: role of the (R)YREK motif. Nucleic Acids Research 36, 5855–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobryn K, Watson MA, Allison RG & Chaconas G (2002). The Mu three-site synapse: a strained assembly platform in which delivery of the L1 transposase binding site triggers catalytic commitment. Molecular Cell 10, 659–669. [DOI] [PubMed] [Google Scholar]
- Koonin EV & Ilyina TV (1993). Computer-assisted dissection of rolling circle DNA replication. Biosystems 30, 241–268. [DOI] [PubMed] [Google Scholar]
- Kunze R & Starlinger P (1989). The putative transposase of transposable element Ac from Zea mays L. interacts with subterminal sequences of Ac. EMBO Journal 8, 3177–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko I, Luo L & Baker TA (1995). Disassembly of the Mu transposase tetramer by the ClpX chaperone. Genes and Development 9, 2399–2408. [DOI] [PubMed] [Google Scholar]
- Li M, Mizuuchi M, Burke TR & Craigie R (2006). Retroviral DNA integration: reaction pathway and critical intermediates. EMBO Journal 25, 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Krishnan L, Cherepanov P & Engelman A (2011). Structural biology of retroviral DNA integration. Virology 411, 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Bischerour J, Siddique A, Buisine N, Bigot Y & Chalmers R (2007). The human SETMAR protein preserves most of the activities of the ancestral Hsmar1 transposase. Molecular and Cellular Biology 27, 1125–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell S, Goryshin IY, Reznikoff WR & Rayment I (2002). Two-metal active site binding of a Tn5 transposase synaptic complex. Nature Structural Biology 9, 278–281. [DOI] [PubMed] [Google Scholar]
- Maertens GN, Hare S & Cherepanov P (2010). The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature 468, 326–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mátés L, Chuah MKL, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A, Grzela DP, Schmitt A, Becker K, Matrai J, Ma L, Samara-Kuko E, Gysemans C, Pryputniewicz D, Miskey C, Fletcher B, Vandendriessche T, Ivics Z & Izsvák Z (2009). Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nature Genetics 41, 753–761. [DOI] [PubMed] [Google Scholar]
- Mcclintock B (1950). The origin and behavior of mutable loci in maize. Proceedings of the National Academy of Sciences of the United States of America 36, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahillon J & Chandler M (1998). Insertion sequences. Microbiology and Molecular Biology Reviews 62, 725–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhammedi-Alaoui A, Pato M, Gama M-J & Toussaint A (1994). A new component of bacteriophage Mu replicative transposition machinery: the Escherichia coli ClpX. Molecular Microbiology 11, 1109–1116. [DOI] [PubMed] [Google Scholar]
- Miskey C, Papp B, Mátés L, Sinzelle L, Keller H, Izsvák Z & Ivics Z (2007). The ancient mariner sails again: transposition of the human Hsmar1 element by a reconstructed transposase and activities of the SETMAR protein on transposon ends. Molecular and Cellular Biology 27, 4589–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuuchi K (1992). Transpositional recombination: mechanistic insights from studies of Mu and other elements. Annual Review of Biochemistry 61, 1011–1051. [DOI] [PubMed] [Google Scholar]
- Montaño SP & Rice PA (2011). Moving DNA around: DNA transposition and retroviral integration. Current Opinion in Structural Biology 21, 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namgoong SY & Harshey RM (1998). The same two monomers within a MuA tetramer provide the DDE domains for the strand cleavage and strand transfer steps of transposition. EMBO Journal 17, 3775–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann TA & Reznikoff WS (2000). Trans catalysis in Tn5 transposition. Proceedings of the National Academy of Sciences of the United States of America 97, 8944–8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normand C, Duval-Valentin G, Haren L & Chandler M (2001). The terminal inverted repeats of IS911: requirements for synaptic complex assembly and activity. Journal of Molecular Biology 308, 853–871. [DOI] [PubMed] [Google Scholar]
- North SH & Nakai H (2005). Host factors that promote transpososome disassembly and the PriA-PriC pathway for restart primosome assembly. Molecular Microbiology 56, 1601–1616. [DOI] [PubMed] [Google Scholar]
- Nowacki M, Higgins BP, Maquilan GM, Swart EC, Doak TG & Landweber LF (2009). A functional role for transposases in a large eukaryotic genome. Science 324, 935–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE & Craig NL (2001). Tn7: smarter than we thought. Nature Reviews. Molecular Cell Biology 2, 806–814. [DOI] [PubMed] [Google Scholar]
- Reznikoff WS (2008). Transposon Tn5. Annual Review of Genetics 42, 269–286. [DOI] [PubMed] [Google Scholar]
- Richardson JM, Dawson A, O’hagan N, Taylor P, Finnegan DJ & Walkinshaw MD (2006). Mechanism of Mos1 transposition: insights from structural analysis. EMBO Journal 25, 1324–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JM, Colloms SD, Finnegan DJ, & Walkinshaw MD (2009). Molecular architecture of the Mos1 paired-end complex: the structural basis of DNA transposition in a eukaryote. Cell 138, 1096–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers KK, Villey IJ, Ptaszek L, Corbett E, Schatz DG & Coleman JE (1999). A dimer of the lymphoid protein RAG1 recognizes the recombination signal sequence and the complex stably incorporates the high mobility group protein HMG2. Nucleic Acids Research 27, 2938–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronning DR, Guynet C, Ton-Hoang B, Perez ZN, Ghirlando R, Chandler M & Dyda F (2005). Active site sharing and subterminal hairpin recognition in a new class of DNA transposases. Molecular Cell 20, 143–154. [DOI] [PubMed] [Google Scholar]
- Rosta E, Nowotny M, Yang W & Hummer G (2011). Catalytic mechanism of RNA backbone cleavage by ribonuclease H from quantum mechanics/molecular mechanics simulations. Journal of the American Chemical Society 133, 8934–8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth DB, Menetski JP, Nakajima PB, Bosma MJ & gellert M (1992). V(D)J recombination: broken DNA molecules with covalently sealed (hairpin) coding ends in scid mouse thymocytes. Cell 70, 983–991. [DOI] [PubMed] [Google Scholar]
- Rousseau P, Normand C, Loot C, Turlan C, Alazard R, Duval-Valentin G & Chandler M (2002). Transposition of IS911 In Mobile DNA II (eds. Craig NL, Craigie R, Gellert M & Lambowitz A), pp. 366–383. Washington: American Society of Microbiology. [Google Scholar]
- Sakai J, Chalmers RM & Kleckner N (1995). Identification and characterization of a pre-cleavage synaptic complex that is an early intermediate in Tn10 transposition. EMBO Journal 14, 4374–4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnovsky RJ, May EW & Craig NL (1996). The Tn7 transposase is a heteromeric complex in which DNA breakage and joining activities are distributed between different gene products. EMBO Journal 15, 6348–6361. [PMC free article] [PubMed] [Google Scholar]
- Schoeberl UE & Mochizuki K (2011). Keeping the soma free of transposons: programmed DNA elimination in ciliates. Journal of Biological Chemistry 286, 37045–37052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinzelle L, Izsvák Z & Ivics Z (2009). Molecular domestication of transposable elements: from detrimental parasites to useful host genes. Cellular and Molecular Life Sciences 66, 1073–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelding Z, Sarnovsky R & Craig NL (2002). Formation of a nucleoprotein complex containing Tn7 and its target DNA regulates transposition initiation. EMBO Journal 21, 3494–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiniger-White M, Bhasin A, Lovell S, Rayment I & Reznikoff WS (2002). Evidence for “unseen” transposase-DNA contacts. Journal of Molecular Biology 322, 971–982. [DOI] [PubMed] [Google Scholar]
- Tang M, Cecconi C, Kim H, Bustamante C & Rio DC (2005). Guanosine triphosphate acts as a cofactor to promote assembly of initial P-element transposase-DNA synaptic complexes. Genes and Development 19, 1422–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ton-Hoang B, Turlan C & Chandler M (2004). Functional domain of the IS1 transposase: analysis in vivo and in vitro. Molecular Microbiology 53, 1529–1543. [DOI] [PubMed] [Google Scholar]
- Ton-Hoang B, Guynet C, Ronning DR, Cointin-Marty B, Dyda F & Chandler M (2005). Transposition of ISHp608, member of an unusual family of bacterial insertion sequences. EMBO Journal 24, 3325–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkov E, Gupta SS, Hare S, Helander A, Roversi P, Mcclure M & Cherepanov P (2009). Functional and structural characterization of the integrase from the prototype foamy virus. Nucleic Acids Research 37, 243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, Sanmiguel P & Schulman AH (2007). A unified classification system for eukaryotic transposable elements. Nature Reviews. Genetics 8, 973–982. [DOI] [PubMed] [Google Scholar]
- Williams TL, Jackson EL, Carritte A & Baker TA (1999). Organization and dynamics of the Mu transpososome: recombination by communication between two active sites. Genes and Development 13, 2725–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagihara K & Mizuuchi K (2003). Progressive structural transitions within Mu transpositional complexes. Molecular Cell 11, 215–224. [DOI] [PubMed] [Google Scholar]
- Yin ZQ, Suzuki A, Lou Z, Jayaram M & Harshey RM (2007). Interactions of phage mu enhancer and termini that specify the assembly of a topologically unique interwrapped transpososome. Journal of Molecular Biology 372, 382–396. [DOI] [PubMed] [Google Scholar]
- York D & Reznikoff WS (1996). Purification and biochemical analyses of a monomeric form of Tn5 transposase. Nucleic Acids Research 24, 3790–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu KF & Lieber MR (2000). The nicking step in V(D)J recombination is independent of synapsis: implications for the immune repertoire. Molecular and Cellular Biology 20, 7914–7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan JF, Beniac DR, Chaconas G, & Ottensmeyer FP (2005). 3D reconstruction of the Mu transposase and the Type 1 transpososome: a structural framework for Mu DNA transposition. Genes and Development 19, 840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y & Wessler SR (2011). The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proceedings of the National Academy of Sciences of the United States of America 108, 7884–7889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A & Craig NL (2011). A hyperactive piggyBac transposase for mammalian applications. Proceedings of the National Academy of Sciences of the United States of America 108, 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbib D, Jakowec M, Prentki P, Galas DJ & Chandler M (1987). Expression of proteins essential for IS1 transposition: specific binding of InsA to the ends of IS1. EMBO Journal 6, 3163–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Mitra R, Atkinson PW, Hickman AB, Dyda F & Craig NL (2004). Transposition of hAT elements links transposable elements and V(D)J recombination. Nature 432, 995–1001. [DOI] [PubMed] [Google Scholar]