

The ongoing COVID-19 pandemic is the greatest health-care challenge of this generation. Early viral genome sequencing studies of small cohorts have indicated the possibility of distinct severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genotypes.1 If these subtypes result in an altered virus tropism or pathogenesis in infected hosts, this could have immediate implications for vaccine design, drug development, and efforts to control the pandemic. Therefore, the genomic surveillance and characterisation of circulating viral strains is a high priority for research and development. To facilitate the epidemiological tracking of SARS-CoV-2, researchers worldwide have created various web-portals and tools, such as the Johns Hopkins University COVID-19 dashboard.2 An unprecedented effort to make COVID-19-related data accessible in near real-time has resulted in more than 25 000 publicly available genome sequences of SARS-CoV-2 on Global Initiative on Sharing All Influenza Data (GISAID).3 Although platforms to survey epidemiological data are prevalent, tools that summarise publicly available viral genome data are scarce and those that are available do not offer users the ability to analyse in-house sequencing data. To address this gap, we have developed an accessible application, the COVID-19 Genotyping Tool (CGT). A video demonstration of CGT is available in appendix 1.
CGT uses publicly deposited SARS-CoV-2 consensus genome sequences from the GISAID EpiCoV™ database,3 and summarises relevant information through salient visualisations, including Uniform Manifold Approximation and Projection (UMAP), Minimum Spanning Trees (MST) of sequence networks, and allele frequencies of annotated high-prevalence non-synonymous Single-Nucleotide Polymorphisms (SNPs) within structural protein-coding genomic regions (envelope, membrane, nucleocapsid, and spike proteins; figure ). New sequencing data from GISAID are added to CGT once a week. Currently, three metadata types can be overlaid on the visualisations: the region of sample collection, the country of sample collection, and the sample collection date with respect to the start of the pandemic (heuristically defined as Dec 1, 2019; figure; appendix 2). Users can upload post-assembly consensus FASTA sequences of SARS-CoV-2 for interactive analysis. The 3' and 5' untranslated regions of genomes are trimmed because of low sequence identity, caused by difficulties in their amplification and sequencing. After sequence alignment, the DNA distance is calculated using the Kimura-80 model of nucleotide substitution.4 After the calculation of the distance and annotation of SNPs, visualisations are reactively reprocessed with user-uploaded data. We use UMAP and network analysis instead of phylogenetics because of a faster computation time and ease of interpretability for large datasets. UMAP reduces the high-dimensional representation of DNA sequences to a two-dimensional embedding, indicating a sequence similarity based on the distance between points;5 that is, SARS-CoV-2 genomes. The MST of the SARS-CoV-2 genome network is a metric used to define a network subset such that all the nodes are connected while minimising distance.6 MSTs have been used before in outbreak analysis to identify the most probable transmission events between hosts,7 and can therefore offer epidemiological insight into SARS-CoV-2 transmission. Lastly, heterogeneity within structural proteins is particularly notable in terms of the host immune response, and has direct implications for vaccine development. With this in mind, we present the most prevalent (based on minor allele frequency) non-synonymous SNPs in the genomic regions of structural proteins.
Figure.
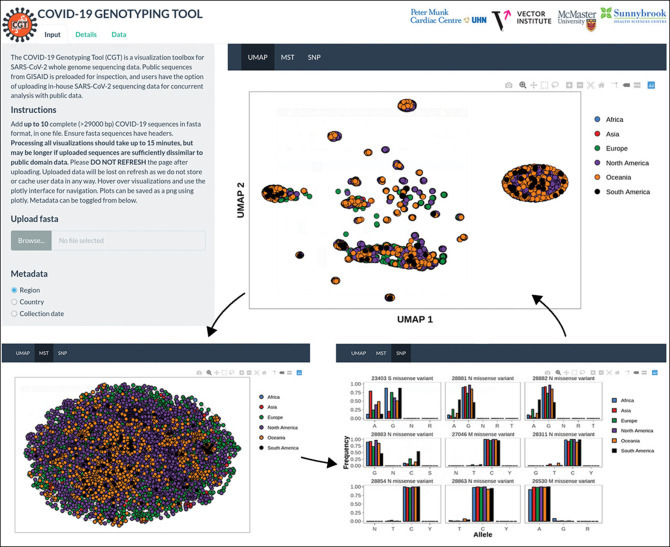
The COVID-19 Genotyping Tool user interface
There are three main tabs: Input, which allow users to upload SARS-CoV-2 genome sequences and adjust the visualisations; Details, which allows users to view details of the analysis algorithms; and Data, which allows users to view data sources and privacy information. The main panel consists of a tab interface with the three visualisations in the application (UMAP, MST, and SNP). Metadata corresponding to the region of sample origin, the country of sample origin, and the sample collection date can be toggled and overlaid on each visualisation. MST=minimum spanning trees. SARS-CoV-2=severe acute respiratory syndrome coronavirus 2. SNP=single-nucleotide polymorphisms. UMAP=uniform manifold projection and approximation.
Our results indicate that there are distinct viral isolate clusters for SARS-CoV-2 sequences uploaded to GISAID. Larger outbreak clusters and hubs from UMAP and MST probably reflect outbreak epicentres (figure). Smaller clusters might be indicative of isolated outbreaks, and singleton samples might be indicative of isolated cases (figure). The analysis of SNPs reveals variants with notable minor allele frequencies involving missense substitutions in structural protein-coding genome regions (figure). Our novel tool is accessible to those who might not be trained in bioinformatics and epidemiological analysis, and thus it serves as a platform for aiding in a pivotal aspect of the global research effort against the COVID-19 pandemic.
The current limitations of CGT include the sequence input limit and processing time, both of which are because of the size of the public data that must be concurrently processed with user-input sequences (>25 000 GISAID sequences). Our team is working to continuously integrate optimisations to the CGT data processing pipeline through code parallelisation and the refinement of deployment infrastructure. Future releases of the application will aim to decrease the processing time, increase the user-input sequence limit, incorporate the input and processing of raw SARS-CoV-2 sequencing data (eg, FASTQ files), and add additional information related to sequence epidemiology, such as travel history.
Complete documentation of the CGT analysis pipeline and application source-code are available in a GitHub repository. Our application does not store any user-uploaded sequence data on the server-side or client-side; CGT simply processes the data to create updated visualisations, and information does not persist after the user disconnects. SARS-CoV-2 genome sequence data and linked metadata from GISAID are not published on our website, as per the GISAID data usage policy. Up-to-date acknowledgments for the usage of GISAID uploaded sequences for analysis are available in the GitHub repository. Our team is grateful to all the researchers who have shared SARS-CoV-2 viral genome sequencing data on GISAID.
Acknowledgments
We declare no competing interests.
HMa, BW, and AGM conceptualised and designed the study. HMa and HMb collected and analysed the data. HMa, HMb, BW, AB, JAN, ARR, and AGM interpreted the initial results. HMa developed the application. HMa and ARR did the software testing. HMa and NK created the application documentation. HMa, HMb, ARR, AB, JAN, RAK, NK, SM, AGM, and BW tested the application. RAK, NK, SM, and AGM provided suggestions for application visualisations. HMa and HMb created the manuscript figures. HMa and BW wrote the manuscript. HMa, BW, HMb, AB, NK, SM, and AGM edited the manuscript.
Supplementary Materials
The COVID-19 Genotyping Tool tutorial
A short demonstration on using the COVID-19 Genotyping Tool application, understanding its features, and navigating the website
References
- 1.Tang X, Wu C, Li X. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev. 2020 doi: 10.1093/nsr/nwaa036. published online March 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020;20:533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elbe S, Buckland-Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall. 2017;1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 5.McInnes L, Healy J, Melville J. UMAP: Uniform Manifold Approximation and Projection for dimension reduction. arXiv. 2018 https://arxiv.org/abs/1802.03426 published online Feb 9. (preprint). [Google Scholar]
- 6.Mamun A, Rajasekaran S. An efficient Minimum Spanning Tree algorithm. 2016 IEEE Symposium on Computers and Communication; Messina, Italy; June 27–30, 2016 (abstr 1047–52).
- 7.Spada E, Sagliocca L, Sourdis J. Use of the Minimum Spanning Tree model for molecular epidemiological investigation of a nosocomial outbreak of hepatitis C virus infection. J Clin Microbiol. 2004;42:4230–4236. doi: 10.1128/JCM.42.9.4230-4236.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The COVID-19 Genotyping Tool tutorial
A short demonstration on using the COVID-19 Genotyping Tool application, understanding its features, and navigating the website