

Breeding exploits the phenomenon of heterosis for an improved productivity, but negative effects reduce heterosis in wide crosses.
Abstract
The genetics underlying heterosis, the difference in performance of crosses compared with midparents, is hypothesized to vary with relatedness between parents. We established a unique germplasm comprising three hybrid wheat sets differing in the degree of divergence between parents and devised a genetic distance measure giving weight to heterotic loci. Heterosis increased steadily with heterotic genetic distance for all 1903 hybrids. Midparent heterosis, however, was significantly lower in the hybrids including crosses between elite and exotic lines than in crosses among elite lines. The analysis of the genetic architecture of heterosis revealed this to be caused by a higher portion of negative dominance and dominance-by-dominance epistatic effects. Collectively, these results expand our understanding of heterosis in crops, an important pillar toward global food security.
INTRODUCTION
Plant and animal breeding successfully exploit the phenomenon of heterosis (1). Improved productivity due to heterosis contributes to feeding a growing human population. According to quantitative genetic theory, heterosis of a cross depends on genetic distance between its parents (2). Here, genetic distance refers to the squared difference in allele frequencies between parents, taking into account all quantitative trait loci (QTL) contributing to heterosis, i.e., heterotic loci (3). Heterosis can be explained by overdominant gene effects, where heterozygosity at individual loci leads to superior performance relative to that of either homozygous class (4). In the presence of partial to complete dominance, heterosis can arise because of the joint effects of multiple loci (4). If heterosis is caused by positive (over) dominance effects, breeders should strive to increase the genetic distance between parental populations to maximize heterosis. Even if dominance effects are absent, epistatic interactions between loci can contribute to heterosis (4). If heterosis results primarily from epistatic interactions, the optimum design of hybrid breeding programs is elusive. Even more so as Moll et al. (5) proposed that parents should not exceed an optimum genetic distance to avoid a reduction in grain-yield heterosis due to detrimental epistatic effects. The concept of a maximized fitness of individuals under an optimal genetic distance between parents has recently also been suggested by analyses in model organisms (6). To use heterosis most efficiently in breeding, it is critical to understand the contribution of (over)dominance and epistatic effects and the dependency on the genetic distance between parents.
A plethora of studies have investigated the relationship between heterosis and genetic distance in crops and livestock (7), estimated based on randomly sampled, genome-wide molecular markers instead of focusing on heterotic loci. Ignoring the genetic architecture of heterosis may have disguised potential trends between heterosis and genetic distance, particularly for diverse crosses (7). The latter are of relevance because it has been hypothesized that the genetic architecture of heterosis varies with the genetic distance between parents (5)—a hypothesis which, to date, could only be tested for the net effects across the genome.
To bridge this gap, we established three distinct hybrid wheat panels. Wheat was chosen because it is widely adapted, one of the most important crops worldwide, and an interesting target for hybrid breeding. Moreover, epistatic effects play a pivotal role for grain-yield heterosis (3). The Elite panel comprised 1655 elite wheat hybrids, the Historic×Elite set included 96 crosses between historic and elite lines, and the Exotic×Elite panel consisted of 152 hybrids established from crosses between elite wheat lines and exotic germplasm. We elaborated a heterotic genetic distance measure that gives special weight to dominance effects estimated in a Bayesian genome-wide prediction framework and showed that grain-yield heterosis increases steadily with heterotic genetic distance, albeit on a lower level for wider crosses. Genome-wide approaches applied to the Elite and Exotic×Elite panels revealed a change in the genetic architecture of grain-yield heterosis, with additive-by-additive epistatic effects driving heterosis in hybrids among elite lines, while in wide crosses, dominance and epistatic effects including dominance were more pronounced and more often negative, thus diminishing heterosis.
RESULTS
Broad parental sampling permits examining heterosis across a wide range of diversity
We produced 1903 hybrids, considering three different levels of global winter wheat diversity (figs. S1 and S2). Within the Elite set, we generated 1655 hybrids by crossing 177 female and 40 male parental lines from the current European elite wheat breeding pool. The Historic×Elite set consisted of crosses between two Central European elite wheat lines and 96 cultivars from the past five decades, with most having been developed in Europe. Although more challenging, we expanded our global wheat diversity collection by tapping into the winter wheat genetic resources maintained at the IPK genebank in Gatersleben and sampled 69 accessions, which were crossed with five Central European elite wheat lines to generate the Exotic×Elite set with 152 hybrids.
The parental lines were fingerprinted with a 15,000–single-nucleotide polymorphism (SNP) array that was established using sequence information of a global set of around 2000 diverse wheat lines. A subset of 40 genotypes from the Elite, 20 from the Historic×Elite, and 17 from the Exotic×Elite set were additionally fingerprinted using exome capture. We observed a tight correlation between the genetic distance matrices based on the exome capture and 15,000 SNP data, which shows the absence of ascertainment bias (r = 0.95, P < 0.001; fig. S3). The genetic diversity was highest within the group of Exotic lines (0.35), followed by the Historic (0.32) and Elite (0.32) lines. These differences in the within-group diversity also became apparent when inspecting the linkage disequilibrium (LD) that decayed more than twice as fast in the Exotic (r2 ≤ 0.1 at 3.11 cM) than in the Elite (r2 ≤ 0.1 at 6.80 cM) and Historic sets (r2 ≤ 0.1 at 6.27 cM; Fig. 1A). The fixation index of Weir and Cockerham (8) (FST) emphasized the pronounced differentiation between the Elite and Exotic lines [FST = 0.10 (0.09 to 0.13)95% CI; Fig. 1B], which was supported by the results of the principal coordinate and phylogenetic analyses (Fig. 1, C and D). This was further substantiated by the differences in persistence of the LD phase of the Exotic versus the Elite and Historic lines (Fig. 1A). Consistently, parents of the crosses of the Exotic×Elite set had an on average larger (P < 0.001) genetic distance (0.39) than parents of the Elite (0.32) and Historic×Elite sets (0.31) (fig. S4). Collectively, this underscores the broad parental sampling, and the hybrid sample is an appropriate panel for the study of the relationship between heterosis and genetic distance.
Fig. 1. Genetic diversity is maximized in the hybrid wheat parents.
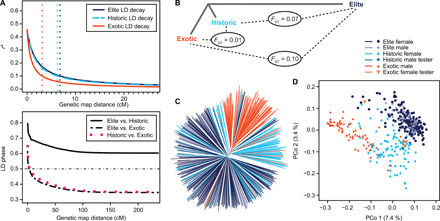
The 217 parents of the Elite set, 98 Historic parents, and 69 Exotic parents were analyzed for linkage disequilibrium (LD) and population structure parameters. (A) Decay of LD (r2) with genetic map distance in the three sets and persistence of the LD phase based on pairwise correlations of LD phase between the three sets. Vertical dotted lines show the decay of LD below 0.1. (B) Neighbor-joining tree based on the results of FST statistics for the three sets. (C) Neighbor-joining tree based on modified Rogers’ distances. (D) Principal coordinate analysis based on modified Rogers’ distances. Percentages in parentheses refer to the proportion of genotypic variance explained by the first and second principal coordinates.
Higher mean heterosis for grain yield in Elite than in Exotic×Elite hybrids
We evaluated the Elite set at six and the Historic×Elite and Exotic×Elite sets at five agro-ecologically diverse locations in Germany for grain yield and obtained high estimates of heritability, above 0.8 (table S1). The broad-sense heritabilities of heterosis were comparable to previous estimates for wheat (3) and ranged between 0.66 for the Elite and 0.78 for the Exotic×Elite set (fig. S5), reflecting the high-quality phenotypic data.
Unexpectedly, the Elite, Historic×Elite, and Exotic×Elite hybrids all showed a comparable amount of mean heterosis relative to the midparent performance with 9.1, 9.5, and 9.2%, respectively (fig. S6 and table S1). The average absolute values of heterosis, by contrast, decreased gradually from the Elite to the Historic×Elite and to the Exotic×Elite set and were significantly (P < 0.01) lower for the Exotic×Elite (0.72 Mg ha−1) than for the Elite (0.83 Mg ha−1) crosses (Fig. 2A). The female parents of the Exotic×Elite hybrids were low yielding due to a lack of intensive breeding and adaptation issues (figs. S7 and S8). This is illustrated by the substantially stronger negative correlations between grain yield and plant height (r = −0.60) as well as the adaptive trait heading date (r = −0.44), compared to the Elite lines (fig. S8). As midparent performance was negatively associated with heterosis (Fig. 2B), it might have been expected that this low performance would result in an increased heterosis in the Exotic×Elite compared to the Elite hybrids. However, heterosis was, on average, 5.1% lower for Exotic×Elite than for Elite crosses when comparing a similar level of midparent performance. Thus, the extent of heterosis was specific for each hybrid set, which can be explained either by differences in genetic distance among parents or by differing genetic architectures of heterosis in the Elite and Exotic×Elite hybrids.
Fig. 2. Associations between grain-yield heterosis and midparent value for grain yield or genetic distance.
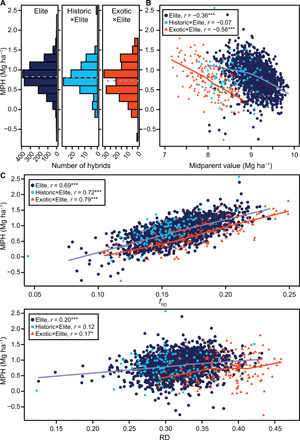
(A) Distribution of midparent heterosis (MPH) for grain yield for the Elite (1655), Historic×Elite (96), and Exotic×Elite (152) hybrids. The vertical dashed line indicates the mean. (B) Association between MPH and midparent value for grain yield. (C) Association between MPH and heterotic genetic distance (fRD) or Rogers’ distance (RD). The colored trendlines are locally weighted regression lines for the Elite (blue) and Exotic×Elite sets (red).
Heterosis for grain yield increases linearly with heterotic genetic distance
When assuming the absence of epistatic interactions, heterosis of a cross is genetically determined as the product between dominance effects and the genetic distance between parents, summed across all heterotic loci (2). On the basis of this quantitative genetic framework, we implemented a heterotic genetic distance measure (fRD) that permits incorporation of information on the genetic architecture of heterosis (Supplementary Note). Genome-wide prediction (9, 10) was performed to obtain dominance effects, which were then applied to weight the marker loci. In accordance with the expectation under a dominance model, heterosis increased monotonically with increasing heterotic genetic distance of the parents of the hybrids and a linear fit explained 45% of the variation of this association (Fig. 2C and fig. S9). The unexplained proportion of variation may be due to noise in the phenotypic data and/or epistatic effects. The latter was supported by partitioning of variance of heterosis into its components, which must be interpreted cautiously (11), but revealed that epistasis explained 92% of the total genetic variation. The comparatively low contribution of dominance effects to the total genetic variance of heterosis and the tight association between heterotic genetic distance and heterosis are not mutually contradictory. Rather, the estimated dominance effects also capture different types of epistatic interactions, as indicated by the correlations between the marker-derived kinship matrices for dominance and the three types of digenic epistatic effects (tables S2 to S5). The linear trend between heterosis and heterotic genetic distance was verified using fivefold cross-validation (r = 0.60 P < 0.001; fig. S10). In accordance with previous findings (12, 13), as well as with quantitative genetic theory (2), the trend was only weakly apparent when ignoring the genetic architecture of heterosis in estimating the genetic distances (Fig. 2C and Supplementary Note).
We defined the heterotic genetic distance such that loci with negative dominance effects reduce the genetic distance (Supplementary Note). Alternatively, loci can be weighted with their absolute dominance effects, which, in our study, also resulted in a continuous increase of heterosis with genetic distance (fig. S11). Nevertheless, as expected on the basis of quantitative genetic theory (2), this association was less pronounced. Thus, none of these analyses in the total population of wheat hybrids provided an indication for a breakdown of heterosis under maximized genetic distance.
Reduced heterosis in Exotic×Elite hybrids
The continuous increase of heterosis with heterotic genetic distance is in contrast to our finding that the average absolute heterosis among elite lines significantly outperformed that of the Exotic×Elite set, although they were on average less distant. We therefore studied the association between heterosis and heterotic genetic distance separately for the Elite and Exotic×Elite sets. The trendlines run nearly in parallel, thus resulting in an overall linear trend (Fig. 2C). However, for a given heterotic genetic distance, heterosis was lower for the Exotic×Elite than for the Elite hybrids. This may be explained by the violation of the assumption of homogeneous genetic effects across the hybrid sets made when estimating the heterotic genetic distances. We therefore applied a quantitative genetic framework (3) to elucidate the genetic basis of grain-yield heterosis separately in the Elite and in the Exotic×Elite hybrids.
Detrimental dominance-by-dominance effects contribute to the reduction of heterosis
In accordance with a previous finding in European wheat (3), we found that heterosis in the Elite set was, to a large extent, caused by additive-by-additive epistasis: It accounted for 61% of the genetic variance of grain-yield heterosis, and 111 of the 213 significant marker effects were additive-by-additive effects (Fig. 3, table S6, and fig. S12). Eight of the 12 identified heterotic QTL were reported previously in another European hybrid wheat panel (3) that had six hybrids in common, pointing to a low probability of ‘‘apparent epistasis” (14, 15) and underlining the general significance for elite hybrid wheat. By contrast, only one heterotic QTL overlapped between the Elite and Exotic×Elite sets. Moreover, in the Exotic×Elite set, dominance and dominance-associated epistatic effects mainly determined heterosis, explaining 69% of the genetic variance and accounting for 136 of the 143 significant marker effects. According to quantitative genetic theory (3), dominance-by-dominance epistasis can cause directed effects on heterosis, which is in contrast to other digenic epistatic effects. All of the significant dominance-by-dominance effects were negative. We observed more than three times as many significant dominance-by-dominance effects in the Exotic×Elite (106) than in the Elite (30) set (table S6), which explains, in part, the lower heterosis of the Exotic×Elite hybrids.
Fig. 3. Genetic architecture of midparent heterosis for grain yield in wheat differs between Elite and Exotic×Elite hybrids.
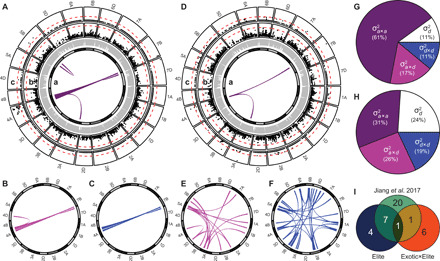
Results are shown for the Elite (A to C and G) and Exotic×Elite hybrids (D to F and H). (A to F) The 21 chromosomes are indicated as bars in the inner circle; gray shadings differentiate the A, B, and D genomes. The genetic map positions of SNPs are given by gray connector lines. Colored links in the centers of the circles represent significant digenic epistatic interactions: additive-by-additive interactions (Aa and Da), additive-by-dominance interactions (B) and E), and dominance-by-dominance interactions (C and F). Manhattan plots for the dominance effects (Ab and Db) and the heterotic effects (Ac and Dc) from genome wide association mapping (GWAS). Significance thresholds are indicated as red dashed lines. (G and H) Partitioning of variance of heterosis for grain yield into its components (, dominance variance; , additive-by-additive variance; , additive-by-dominance variance; , dominance-by-dominance variance). (I) Venn diagram showing the number of overlapping heterotic QTL between the Elite and Exotic×Elite sets and the study by Jiang et al. (3).
Negative dominance effects also diminish heterosis in the Exotic×Elite hybrids
Genome-wide association mapping detected one significant dominance effect in the Elite and three in the Exotic×Elite set, all being positive except one in the Exotic×Elite set. When relaxing the significance threshold to P < 0.001, which corresponds to a false discovery rate of 0.17 in the Exotic×Elite set, we found that only 1 of the 15 (7%) dominance effects in the Elite hybrids was negative (Fig. 4). By contrast, 18 of the 57 (32%) dominance effects were negative in the Exotic×Elite set. Analysis of these 72 SNPs revealed that their dominance effects differed between both sets of hybrids. The higher portion of negative dominance effects is thus a further explanation for the lower extent of heterosis in the Exotic×Elite compared to the Elite hybrids. In addition, we evaluated widely used phenology loci and found no significant (P < 0.001) negative dominance effects for the photoperiod sensitivity gene Ppd-B1, the vernalization gene Vrn-A1, or the Reduced height (Rht)-B1 and Rht-D1 semidwarfing genes.
Fig. 4. Negative dominance effects are more abundant in Exotic×Elite hybrids.
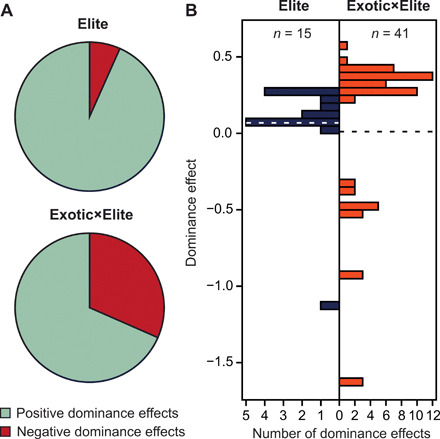
(A) Portion of positive and negative dominance effects in the Elite and Exotic×Elite hybrids, identified at a threshold of P < 0.001. (B) Histograms of these dominance effects.
DISCUSSION
A lower heterosis of crosses between genetically distant individuals than between more closely related ones concerns hybrid breeding but also adaptive differentiation in natural populations (2, 16). While we observed no breakdown of heterosis with maximized genetic distance in either the Elite or the Exotic×Elite hybrids, we did find a consistently lower grain-yield heterosis of the Exotic×Elite hybrids throughout the entire genetic distance space. This suggests that intensive selection through breeding and potentially also genetic drift have created divergence between current elite wheat germplasm and lines subject to less intensive breeding, resulting in a reduced heterosis in crosses among them. We therefore applied a quantitative genetic framework (3) to dissect the underlying genetic cause. This revealed a larger number of negative dominance effects contributing to the lower heterosis in Exotic×Elite versus Elite hybrids, which mirrors results from a recent study focusing on the net genetic effects across the genome underlying fitness and its components in an Arabidopsis thaliana cross of lines separated by a wide geographic distance (17). Besides negative dominance effects, a larger number of negative dominance-by-dominance effects also reduced heterosis of Exotic×Elite compared to Elite hybrids. This can be interpreted as analogous to the Bateson-Dobzhansky-Muller model of interspecific incompatibility (18), according to which hybrid sterility and inviability are caused by epistatic incompatibilities between alleles at two or more loci. Purifying selection against negative dominance and dominance-by-dominance effects in elite wheat germplasm seems unexpected at first, as the parental lines of this study were developed in inbred line and not in hybrid breeding programs. However, wheat breeders routinely apply pedigree breeding for which early generations are characterized by high levels of heterozygosity, thereby making selection against such loci possible. Thus, intensive pedigree breeding appears to have purged negative dominance effects from elite germplasm.
Knowledge on how to maximize hybrid performance is of paramount importance for efficient design of hybrid breeding programs. Our findings clearly revealed that to maximize the exploitation of heterosis, it is crucial to increase the genetic distance between parental populations to a certain extent. Heterosis, however, is only one component of hybrid performance, and the determinant role of midparent value makes high-yielding elite lines favorable for hybrid breeding (fig. S13). We observed a negative correlation between midparent values and heterosis (Fig. 2B). This raises the question as to whether heterosis will be lost by a further improvement of the per se performance of the lines and, thus, of the midparent value (19). The additive and dominance effects estimated in the genome-wide prediction model were not correlated (r = 0.006, P > 0.1), which suggests that the negative association between the line per se performance and heterosis is not due to pleiotropy. Further studies are needed to assess whether a concomitant increase of midparent value and midparent heterosis (MPH) will be possible in future hybrid wheat breeding. In conclusion, the observed hybrid performances are encouraging and underscore the potential of hybrid wheat breeding to increase grain yield levels globally.
MATERIALS AND METHODS
Plant materials and field trials
This study was based on three different sets of wheat hybrids, termed Elite, Historic×Elite, and Exotic×Elite. In the terminology used in maize heterosis research, the Elite set mimics a convergent population and the Exotic×Elite hybrids a divergent population (20, 21). The Elite set initially comprised 434 potential female lines provided by the following 13 wheat breeding companies: Bayer CropScience AG, Deutsche Saatveredelung AG, KWS LOCHOW GmbH, Limagrain GmbH, Pflanzenzucht Oberlimpurg, RAGT2n, Saatzucht Bauer GmbH, Saatzucht Josef Breun GmbH & Co. KG, Saatzucht Streng-Engelen GmbH & Co. KG, Secobra Saatzucht GmbH, Strube Research GmbH & Co. KG, Syngenta Seeds GmbH, and W. von Borries-Eckendorf GmbH & Co. KG (fig. S1A). We genotyped those lines with 22 simple sequence repeats (SSR) markers and, based on genetic distances among them (Rogers’ distance > 0.2) and by maximizing the allelic diversity (>97% of alleles maintained after selection), selected 189 of the 434 as female parents for the hybrids. Forty-one male lines were selected on the basis of suitable floral characteristics (22) and were provided by the two wheat breeding companies Limagrain GmbH and Nordsaat Saatzucht GmbH (fig. S1B). Within the Elite set, we were able to produce enough seeds for 1750 elite wheat hybrids by crossing the 189 elite female lines and the 41 elite male lines in an incomplete factorial mating design using chemical hybridization agents (fig. S2A). For all three sets of hybrids, the sterility of the female parents after chemical hybridization agent application was checked by bagging one to three plants. The 1750 hybrids, their 189 female and 41 male parental lines, as well as 11 commercial varieties as checks, i.e., eight line varieties (quality class A: JB Asano, Julius, RGT Reform; quality class B: Colonia, KWS Loft, Rumor, Tobak; quality class C: Elixer), and three hybrids (quality class B: Hybred, Hystar; quality class C: LG Alpha), were evaluated for grain yield (Mg ha−1), heading date (days from January 1st), and plant height (cm) at six agro-ecologically diverse locations in Germany in the growing season 2015–2016. The locations were Asendorf, Biendorf, Gatersleben, Hadmersleben, Rosenthal, and Seligenstadt (table S7). Owing to the large number of entries in the experiment, groups of 495 genotypes and all 11 checks were made, as these can be harvested on a single day. The experimental design for each group of 506 entries was an α-design with a size of the incomplete blocks of 11. Sowing density was 200 seeds per m−2 for all entries and plot size at the different locations ranged from 7.2 to 11 m2. Plots were treated with fertilizers, fungicides, and herbicides according to best agronomic practices.
The Historic×Elite set consisted of 96 wheat lines sampled on the basis of a temporal and spatial selection strategy (fig. S1, C and D). About half of these wheat varieties were released between the 1960s and 1980s, but some more recently released varieties were included (fig. S1C). The spatial component mainly focused on Western European origins. More than 80% of the varieties were released in Germany, France, or Great Britain, but also cultivars from regions with a more continental climate were included. The Historic×Elite material is part of the winter wheat panel described recently by Boeven et al. (23). All of these lines were used as female components with an elite male tester mix in a topcross mating design using chemical hybridization agents (fig. S2C). The elite male tester mix was provided by Nordsaat Saatzucht GmbH and comprised two unreleased breeding sibling lines with known good male floral characteristics. We were able to produce enough seeds for 96 hybrids derived from crosses of 96 ‘‘Historic” female lines with the male tester mix.
For the Exotic×Elite set, a random sample of 1500 gene bank accessions obtained from the gene bank of the IPK Gatersleben were screened in observation plots on site for their male floral characteristics. Last, 101 accessions were selected and crossed as male components in an incomplete factorial mating design with nine German elite varieties (Famulus, Franz, Glaucus, JB Asano, Patras, RGT Reform, Rumor, Tabasco, and Türkis) used as female testers that were emasculated by a chemical hybridization agent (fig. S2B). According to available passport data and information obtained from the genetic resources information and analytical system for wheat and triticale (http://wheatpedigree.net), the acquisition date of more than 50% of these lines predates the year 1970, and more than 20 worldwide origins were represented by this random gene bank sample (fig. S1, E and F). Although the generation of these Exotic×Elite hybrids is challenging, we were able to produce enough seeds for 200 hybrids.
The Historic×Elite and Exotic×Elite hybrids, as well as their female and male parental lines, were evaluated for grain yield (Mg ha−1), heading date (days from January 1st), and plant height (cm) at five agro-ecologically diverse locations in Germany in the growing season 2015–2016. The locations were Stuttgart-Hohenheim, Renningen, Gatersleben, Schackstedt, and Böhnshausen (table S1). Heading date was only recorded at two locations. The experimental designs were unreplicated α-designs. To avoid neighboring effects of the much taller gene bank accessions, the material was split into two adjacent trials according to plant height. The ‘‘tall trial” (243 entries) included all Exotic lines of >100 cm and their hybrids, while the ‘‘short trial” (378 entries) included all Exotic lines of <100 cm, their hybrids and all their female testers, all Historic×Elite hybrids, the male tester mix and also its single components, and the same common elite checks as used in the Elite trial, and 95 further genotypes not considered in this study. The two adjacent trials were linked by 10 commercial medium-long check varieties [Bernstein, Capo, Discus, Hybery (hybrid), Hymack (hybrid), KWS Milaneco, Midas, Naturastar, and Xantippe] at each location. Sowing density was 220 seeds m−2 for all entries, and plot size at the different locations ranged from 7.56 to 12 m2. Plots were treated according to best agronomic practices, but N-fertilizers were reduced by 25% and an additional application of growth regulators was applied to prevent lodging.
In all trials, data were recorded for heading date as the number of days from January 1st to the day when half of the heads of a plot had emerged from the flag leaves, for plant height in centimeters from the ground to the tip of the erected ears, excluding awns, at the growth stage of dough development of the kernels, and for grain yield in Mg ha−1 with a moisture content of 140 g H2O kg−1. Homogeneity of hybrids was visually assessed on a scale from 1 (uniform) to 9 (50/50 mix of two genotypes) about 2 to 3 weeks after flowering. In addition, hybridity of the Exotic×Elite hybrids was visually assessed on a scale from 1 (more than 90% selfing) to 9 (only hybrids) in observation plots of two rows and 1.25 m in length at the IPK Gatersleben, where each hybrid and its parents were sown side by side. We identified 53 hybrids of the Elite set and 48 hybrids of the Exotic×Elite set, which did not meet these criteria and were therefore not considered for the subsequent heterosis analyses. In turn, this affected 12 Elite females and 36 Exotic parents that were only used in the discarded hybrid combinations.
Phenotypic data analyses
Data for both experiments were analyzed by a two-stage approach, where first data of each experiment was analyzed separately, and then, means across experiments were calculated. All data were screened for outliers using the method 4 ‘‘Bonferroni-Holm with re-scaled median absolute deviation standardized residuals” as suggested by Bernal-Vasquez et al. (24). Our mixed model description follows the syntax suggested by Patterson (25), where crossed effects are denoted with a dot operator, and fixed and random effects are separated by a colon, with fixed effects appearing first. The phenotypic data of the Elite set were analyzed on the basis of the following linear mixed model
| (1) |
where G, Loc, Exp, and Block denote the effects of the genotypes, locations, trials, and incomplete blocks, respectively. Error, block, and experiment variances were assumed to be heterogeneous among locations. Genotype had 1991 levels, 1750 for hybrids, 189 and 41 for female and male parents, respectively, and 11 for checks.
The phenotypic data of the Historic×Elite and Exotic×Elite sets were analyzed by a model analogous to model (1). Here, genotype had 621 levels, 200 for Exotic×Elite hybrids, 101 for Exotic male parental lines, 9 for Exotic female testers, 96 for Historic×Elite hybrids and female parental lines, respectively, 3 for Historic male tester and its components, 11 for elite checks, 96 for additional genotypes, and 20 for the 10 medium-long check varieties coded separately for the tall and ‘‘short” trials. The reason for the latter was a plausibility check before analyses. We tested for identical yield of the commercial medium-long check varieties in both experiments using the following linear mixed model
| (2) |
where MLchecks, Trial, and Loc denote the effects of the medium-long checks, trials, and locations, respectively. Extending the data and the model by including all data did not change the result that there was a significant and consistent overestimation of medium-tall genotypes in the ‘‘short trial” compared to the ‘‘tall trial” (on average, 0.53 Mg ha−1). As neighboring effects are one plausible explanation, we tried to account for this by treating differences in plant height of the neighboring plots as covariable. However, this covariable was not significant, leaving a still significant systematic yield effect of the ‘‘tall trial”. We, therefore, decided not to rely on the checks for connecting the two trials and rather coded the medium-long checks of each trial as different genotypes. Thus, trial adjustment was performed across locations but within the same group of genotypes.
In a second step, the Elite, Historic×Elite, and Exotic×Elite sets were analyzed in a joint analysis based on the common elite checks. Here, adjusted entry means and corresponding SEs of genotypes from the first step were used in the following linear mixed model
| (3) |
where G and Set denote the genotypes and sets, respectively. Owing to the use of one divided by the squared SEs of means as weighting factor, we set the residual variance to one applying method 3 suggested by Möhring and Piepho (26).
We assumed fixed genotypic effects to obtain best linear unbiased estimations (BLUEs) of the genotypic values of hybrids, females, and males (data S1). BLUEs were used to calculate MPH for each hybrid as MPH = F1 − MP, where F1 refers to the performance of a hybrid and MP refers to the midparent value of the two parental lines P1 and P2. Relative MPH (MPH %) was calculated for each hybrid as MPH (%) = (MPH/MP) × 100. Better-parent heterosis (BPH) was calculated as BPH = F1 − PBetter, where PBetter refers to the performance of the better performing parental line. Relative BPH was calculated as BPH (%) = (BPH/MP) × 100. Correlations based on BLUEs were tested with Pearson’s product moment correlation coefficients. BLUEs of different genotypic groups were compared by Student’s t tests.
Variance components were estimated by the restricted maximum likelihood method treating all effects as random except for the group effect. Binary dummy variables were used to estimate variance components for each group. The Elite set was analyzed with the following mixed model
| (4) |
where Group refers to the genetic group effects (hybrids, females, males, and each check have a separate level), Checks refer to the effect of the checks, Females refer to the effect of the female parents, Males refer to the effect of the male parents, GCA denotes general combining ability effects, and SCA denotes the specific combining ability effect. For sake of simplicity, dummy variables were suppressed in the model stated above. We assumed group-specific error variances. Similarly, the Historic and Exotic sets were analyzed with the following linear mixed model
| (5) |
where Group refers to the genetic group effects (hybrids Historic×Elite and Exotic×Elite, female parents Historic, male parents Exotic, elite varieties including all checks and Exotic female parents, and Rest), EliteCultivars refers to the effects of the elite checks and Exotic female parents, and Rest refers to the effect of genotypes not further considered in this study. The male tester within the Historic×Elite set was considered as fixed effect. We assumed group-specific error variances. Broad-sense heritability (h2) on an entry-mean basis was estimated as the ratio of genotypic to phenotypic variance, , where and refer to the total genotypic variance of the different genetic groups and their interaction with location, respectively, L refers to the number of locations, R refers to the number of replications, and refers to the residual variance. Except for checks within the Elite set and GCA of hybrids produced in an incomplete factorial mating design, was confounded with . Genotypic variance of hybrids was assumed to be the sum of GCA and SCA variances. Heritability of MPH was defined as a measure of how much heterosis can be inherited and was estimated as described by Jiang et al. (3). Briefly, MPH was calculated on the basis of block-corrected values of hybrids and their parents at each location. We then used these values and fitted a linear mixed model including random genotype and location effects. Heritability was estimated as , where is the mean variance of a difference of two best linear unbiased predictions (BLUPs) (27). All statistical analyses were performed within the R environment (28) and with the software package ASReml-R 3.0 (29).
Genotypic data and analyses
All parental lines were fingerprinted using a 15 k Infinium SNP array that contains a subset of markers from the 90 k Illumina Infinium assay (30). The development of the 15 k SNP chip and genotyping was performed by TraitGenetics GmbH (www.traitgenetics.com) and resulted in a total of 13,006 polymorphic SNP markers. In the rare event of missing marker data, imputation was performed by Random Forest regression (31). After quality tests, 9763 high-quality SNPs with available map position remained that were used for all subsequent analyses. Genotyping of one Elite male line failed, which was consequently excluded from the subsequent analyses. This line was the parent of 42 Elite hybrids, which could not be considered for further analyses, too.
The FST statistic for each pair of subpopulations among Elite, Historic×Elite, and Exotic×Elite was estimated using the method of Weir and Cockerham (8) as implemented in the R package ‘‘diveRsity” (32) and visualized by a neighbor-joining tree using the R package ‘‘ape” (33). The 95% confidence intervals for the FST statistic were obtained from 1000 bootstrap replicates. Population structure among parental lines was analyzed by principal coordinate analysis, and cluster analysis was based on modified Rogers’ distances. The neighbor-joining tree was generated using the R package ‘‘ape” (33). Genetic distance was measured by Rogers’ distance (34). In addition, a heterotic genetic distance, implemented as a function of the Rogers’ distance, was calculated by incorporating estimated dominance effects of the SNPs as
| (6) |
where X and Y represent two genotypes under consideration, Xuj and Yuj are allele frequencies of the j-th allele at the u-th locus, nu is the number of alleles at the u-th locus, L refers to the number of loci, and wu is the dominance weight for the u-th locus. The dominance weight wu was calculated as the dominance effect of the u-th locus du divided by the mean of all absolute dominance effects. Dominance effects of SNPs were estimated based on the BayesCπ approach previously outlined in detail by Zhao et al. (13). We performed a fivefold cross-validation with 100 runs to estimate dominance effects and to assess the association between heterosis and fRD. The trends between the different measures of genetic distance and heterosis were visualized by fitting natural smoothing splines.
The extent of pairwise LD was assessed using the squared allele-frequency correlations (r2). Decay of LD with genetic map distance for the Elite and Exotic×Elite sets was assessed by fitting natural smoothing splines to the r2 values. The persistence of linkage phase between the Elite, Historic×Elite, and Exotic×Elite sets was inferred by analyzing how similar or dissimilar the correlations between pairs of markers are. As the r2 values do not allow to differentiate between a positive and a negative correlation, we calculated LD as the correlation coefficient r, where r can take values between −1 and 1. The correlation of r between two different sets was defined as LD phase and plotted against the genetic map distance and was again assessed by fitting natural smoothing splines. The LD parameters r and r2 were calculated using the software package PLINK (35).
To test the relevance of a potential ascertainment bias using a 15 k Infinium SNP array we generated exome capture sequencing data for a subset of 96 genotypes. Seeds were germinated and grown in the green house for 2 weeks. The Sequence Capture Sequencing Libraries were prepared as described in SeqCap EZ Library SR User’s Guide version 5.3 (http://technical-support.roche.com/_layouts/net.pid/Download.aspx?documentID=ee33953e-0bf0-e711-4ebf-00215a9b3428&fileName=RSS_SeqCap_EZ_UGuide_v5.4&extension=pdf&mimeType=application%2Fpdf&inline=False). Samples were multiplexed in pools of four samples (preindexing) to save probes. The Sequence Capture Sequencing Libraries were quantified using KAPA Library Quantification Kit (Universal) from Kapa Biosystems. The sequencing of the libraries was performed in two steps: As part of the quality control, the samples were sequenced on an Illumina MiSeqTM (Illumina Inc., San Diego, CA, USA) to check the insert sizes and index distribution. The final sequencing was performed on an Illumina HiSeqTM 2500 (Illumina Inc., San Diego, CA, USA) using one TruSeq SBS Kit v3-HS (200 cycles; FC-401-3001) and four HiSeq Rapid SBS Kit v2 (500 cycles; FC-402-4023). Through the sequencing runs, the sequence yield per sample was monitored, and sample sheets and sample concentrations were corrected to get equal yields per sample. Sequence raw data are available under The European Molecular Biology Laborator (EMBL), European Nucleotide Archive (ENA) accession PRJEB28165.
Obtained sequencing reads were adapter trimmed using Cutadapt (36) (version 1.9.1) removing reads after trimming with lengths less than 30 nt. Afterward, reads were mapped against the reference genome sequence of wheat cultivar Chinese Spring with BWA-MEM (37) (version 0.7.13, default parameters) and transformed into binary alignment map (BAM) format with SAMtools (38) (version 1.9). NovoSort (version V3.02.12, http://novocraft.com/products/novosort/) was used for sorting and detection of duplicates. A realignment step was performed using the Genome Analysis Toolkits (GATK) (39) (version 3.8) programs RealignerTargetCreator and IndelRealigner to reduce the number of miscalls of INDELs.
BAM files from different Illumina flow cells were merged with NovoSort by sample name. Read groups were corrected using Picard Tools (version 1.84, http://broadinstitute.github.io/picard/). Three samples did not generate substantial amount of read data and were, therefore, excluded from all subsequent analysis. SAMtools mpileup and BCFtools (40) were applied for SNP calling. Resulting variant call format (VCF) files were filtered using the following criteria: (i) sites with a read depth less than one to three times for homozygous/heterozygous genotype calls were removed, (ii) sites with a query quality (QUAL) score below 40 were removed, (iii) sites with total read depth over all samples below 50 times were removed, (iv) sites with less than one homozygous genotype call or when the proportion of homozygous genotype calls was below 90% were removed, (v) sites with a minor allele frequency below 5% were removed, (vi) sites with less than 5% of samples with a genotype call were removed, and (vii) sites with less than one single occurrence of the minor allele in all samples were removed.
Missing data was imputed using FILLIN (41) from the TASSEL software suite (42) (version 5.0) with default parameters. Results were loaded into the R statistical environment (28) (version 3.4.2), converted into corearray genomic data structure (GDS) format with package ‘‘SeqArray” (43) and further filtering was performed. SNP sites located on ‘‘chrUn” were removed. Furthermore, only sites were retained with at least half of all samples with present genotype calls. Rogers’ distance (34) was calculated among the 77 genotypes based on the exome capture sequencing data. Distance matrix was compared to the one estimated based on the 15 k SNP array data applying a Mantel test.
Partitioning of genetic variance components for MPH
Genetic variance components for MPH were estimated by fitting an extended genomic best linear unbiased prediction model (44, 45) including dominance and digenic epistatic effects. Briefly, the model can be described as follows
| (7) |
where y is the vector of MPH values for all hybrids, gd, gaa, gad, and gdd are vectors of genetic values contributed by dominance, additive-by-additive, additive-by-dominance and dominance-by-dominance effects, respectively, and e is a residual term. In the model, we assume , , , and , where Kd, Kaa, Kad and Kdd are marker-derived kinship matrices for the different genetic effects. T is a r × (r + s) matrix of linear transformation from the vectors of the original trait (grain yield) to the vectors of MPH, where r is the number of hybrids and s is the number of parental lines. Following (3), we used the F∞-metric to calculate the marker-derived kinship matrices, which leads to a nonorthogonal parametrization of the model. Thus, it is not unexpected that the estimated kinship matrices for different genetic effects were correlated (table S3 to S5), indicating that a certain extent of confounding exists in the partition of variance components. However, it is difficult to obtain an orthogonal parametrization in the mixed-model setting and with the presence of LD among markers.
Note that in the model (7), the residual term is not assumed to be independently distributed. The reason is that we usually assume independent residual terms for the original trait grain yield, but the MPH values are derived from the original trait values in the form of the linear transformation T. The marker-derived kinship matrices are also specific to MPH instead of the original trait. We refer to Jiang et al. (3) for more details on the implementation of the model. The variance components , , , and were estimated by the multikernel method in the R package BGLR (46). We estimated the posterior distribution of the covariances between dominance and the three types of digenic epistatic effects, i.e., Cov(gs, gt), for s, t ∈ {d, aa, ad, dd}, where gd, gaa, gad, and gdd are the different genetic effects as in (Eq. 7). This was performed by fitting the genomic prediction model (Eq. 7) once with the full dataset and extracting the estimated genetic effects in each sampling round of the Bayesian iteration procedure (21).
Definition of heterotic effects
The heterotic effect of a locus is the genetic contribution of the locus to MPH, which is a complex combination of the dominance effect of the locus itself and the epistatic interaction effects with the entire genetic background (3). The precise definition is described as follows:
Let Q be the set of all QTL for the original trait. QTL were coded as 0, 1, or 2, depending on the number of a chosen allele at each locus. Considering one hybrid, we denote by Rkl (k, l = 0 or 2) the subset of loci where the female parent has genotype k and the male parent has genotype l. For i, j ∈ Q and i ≠ j, let di be the dominance effect of the i-th QTL, aaij is the additive-by-additive epistatic effect between the i-th and the j-th QTL, adij is the additive-by-dominance epistatic effect between the i-th and the j-th QTL, and ddij is the dominance-by-dominance epistatic effect between the i-th and the j-th QTL. The heterotic effect of the i-th locus was defined as
| (8) |
With this definition, the MPH value of each hybrid is the sum of heterotic effects across all loci, i.e., .
Genome-wide scan for significant heterotic effects
We applied the following three-step procedure developed by Jiang et al. (3) to detect significant heterotic effects: First, genome-wide association mapping was performed to identify significant component effects (i.e., dominance and digenic epistatic effects). We used a standard linear mixed model with a marker-derived kinship matrix controlling for the structure of multiple levels of relatedness and polygenic background effects (47). Since the presence of epistasis was assumed, it is necessary to apply a model controlling the polygenic background effects consisting of both main and epistatic effects (48). The model can be described as follows
| (9) |
where y, gd, gaa, gad, gdd, and e are the same as in (7). In particular, α is the genetic effect being tested, m is the corresponding coefficient. More precisely, α is the dominance effect of any marker or the epistatic interaction effect for any pair of markers. We assumed that α is an unknown fixed parameter. The other assumptions are the same as for (7). For computational efficiency, the model was transformed to a standard linear regression model in which only the residual terms are random. The transformed model is equivalent to the original one, provided that the influence of different α on the estimation of variance components is negligible (48, 49). After the transformation, the significance of the effect α can be assessed by an F test. We refer to Jiang et al. (3) for more details on the implementation of the model.
In the second step, the significant component effects were integrated into the heterotic effects according to (8). All nonsignificant effects were set to zero.
Last, the heterotic effect hi of each locus was tested by a permutation test. More precisely, for each locus, the MPH values of all hybrids can be predicted using the heterotic effect of this particular locus. Then, the Pearson correlation coefficient between the predicted and observed MPH values was calculated, and a permutation test for the correlation coefficient was performed.
In the first and third steps, the genome-wide threshold for P values was determined in the following way: For the Elite panel, the threshold was P < 0.05 after Bonferroni-Holm correction for multiple testing (50). For the Exotic×Elite panel, the power of detecting significant epistatic effects was hindered by the small population size. We therefore used a modified Bonferroni correction method based on the effective number of independent markers pe, which was obtained by performing principal component analysis for the marker LD matrix (51). The threshold for dominance and heterotic effects was P < 0.05/pe, for additive-by-additive, dominance-by-dominance epistatic effects P < 0.05/(pe(pe − 1)/2), and for additive-by-dominance effects P < 0.05/(pe(pe − 1)).
Supplementary Material
Acknowledgments
We thank GFPi and proWeizen for project coordination. Funding: This work was funded by BMEL through BLE within the ZUCHTWERT project (Grant ID: 2814604113). Author contributions: J.C.R., C.F.H.L., P.H.G.B., Y.Z., and T.W. conceived and designed the study. P.H.G.B., C.F.H.L., J.C.R., M.G., A.P., R.S., J.S., E.E., E.K., V.M., J.D., S.K., R.H., H.C., J.Ho., A.J., L.R., C.R., N.S., P.V., A.S., and F.S. acquired and contributed data. P.H.G.B., Y.J., F.L., and Y.Z. processed the data, performed the analyses, and analyzed the results. S.B. and U.S. did data management. H.P.M. did SSR analyses. J.Ha. and P.T. supported in data analyses. P.H.G.B., Y.J., Y.Z., J.C.R., and T.W. interpreted the results and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The phenotypic data that supported the findings of this study are available as data S1. Sequence raw data are available under EMBL ENA accession PRJEB28165. The genomic data are available at the electronic data archive library e!dal (52). 15 k SNP data: http://dx.doi.org/10.5447/ipk/2020/12. Exome capture SNP matrix: http://dx.doi.org/10.5447/ipk/2020/13. Sample R code for implementing the heterotic genetic distance is provided as a supplementary text file (Supplementary R code). R codes for implementing genomic prediction for MPH, genome-wide association mapping for dominance and epistatic effects, and genome-wide scan for heterotic effects were published previously (3).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaay4897/DC1
REFERENCES AND NOTES
- 1.Schnable P. S., Springer N. M., Progress toward understanding heterosis in crop plants. Annu. Rev. Plant. Bio. 64, 71–88 (2013). [DOI] [PubMed] [Google Scholar]
- 2.D. S. Falconer, T. F. C. Mackay, Introduction to quantitative genetics (Longman, ed. 4, 1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang Y., Schmidt R. H., Zhao Y., Reif J. C., A quantitative genetic framework highlights the role of epistatic effects for grain-yield heterosis in bread wheat. Nat Genet 49, 1741–1746 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Lippman Z. B., Zamir D., Heterosis: Revisiting the magic. Trends Genet. 23, 60–66 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Moll R. H., Longquist J. H., Fortuna J. V., Johnson E. C., The relation of heterosis and genetic divergence in maize. Genetics 52, 139–144 (1965). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei X., Zhang J., The optimal mating distance resulting from heterosis and genetic incompatibility. Sci. Adv. 4, eaau5518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.A. E. Melchinger, in The Genetics and Exploitation of Heterosis in Crops, J. G. Coors, S. Pandey, Eds. (CSSA, 1999), pp. 99–118. [Google Scholar]
- 8.Weir B. S., Cockerham C. C., Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984). [DOI] [PubMed] [Google Scholar]
- 9.Habier D., Fernando R. L., Kizilkaya K., Garrick D. J., Extension of the bayesian alphabet for genomic selection. BMC Bioinformatics 12, 186 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Y., Zeng J., Fernando R., Reif J. C., Genomic prediction of hybrid wheat performance. Crop. Sci. 53, 802–810 (2013). [Google Scholar]
- 11.Huang W., Mackay T. F. C., The genetic architecture of quantitative traits cannot be inferred from variance component analysis. PLOS Genet. 12, e1006421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dreisigacker S., Melchinger A. E., Zhang P., Ammar K., Flachenecker C., Hoisington D., Warburton M. L., Hybrid performance and heterosis in spring bread wheat, and their relations to SSR-based genetic distances and coefficients of parentage. Euphytica 144, 51–59 (2005). [Google Scholar]
- 13.Zhao Y., Li Z., Liu G., Jiang Y., Maurer H. P., Würschum T., Mock H. P., Matros A., Ebmeyer E., Schachschneider R., Kazman E., Schacht J., Gowda M., Longin C. F. H., Reif J. C., Genome-based establishment of a high-yielding heterotic pattern for hybrid wheat breeding. Proc. Natl. Acad. Sci. U.S.A. 112, 15624–15629 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wood A. R., Tuke M. A., Nalls M. A., Hernandez D. G., Bandinelli S., Singleton A. B., Melzer D., Ferrucci L., Frayling T. M., Weedon M. N., Another explanation for apparent epistasis. Nature 514, E3–E5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de los Campos G., Sorensen D. A., Toro M. A., Imperfect linkage disequilibrium generates phantom epistasis (& perils of big data). G3 (Bethesda) 9, 1429–1436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lynch M., The genetic interpretation of inbreeding depression and outbreeding depression. Evolution 45, 622–629 (1991). [DOI] [PubMed] [Google Scholar]
- 17.Oakley C. G., Ågren J., Schemske D. W., Heterosis and outbreeding depression in crosses between natural populations of Arabidopsis thaliana. Heredity 115, 73–82 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Orr H. A., Dobzhansky, Bateson, and the genetics of speciation. Genetics 144, 1331–1335 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Troyer A. F., Wellin E. J., Heterosis decreasing in hybrids: Yield test inbreds. Crop Sci. 49, 1969–1976 (2009). [Google Scholar]
- 20.Technow F., Schrag T. A., Schipprack W., Bauer E., Simianer H., Melchinger A. E., Genome properties and prospects of genomic prediction of hybrid performance in a breeding program of maize. Genetics 197, 1343–1355 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alves F. C., Granato Í. S. C., Galli G., Lyra D. H., Fritsche-Neto R., de los Campos G., Bayesian analysis and prediction of hybrid performance. Plant Methods 15, 14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boeven P. H. G., Longin C. F. H., Leiser W. L., Kollers S., Ebmeyer E., Würschum T., Genetic architecture of male floral traits required for hybrid wheat breeding. Theor. Appl. Genet. 129, 2343–2357 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Boeven P. H. G., Longin C. F. H., Würschum T., A unified framework for hybrid breeding and the establishment of heterotic groups in wheat. Theor. Appl. Genet. 129, 1231–1245 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Bernal-Vasquez A.-M., Utz H.-F., Piepho H.-P., Outlier detection methods for generalized lattices: A case study on the transition from ANOVA to REML. Theor. Appl. Genet. 129, 787–804 (2016). [DOI] [PubMed] [Google Scholar]
- 25.H. D. Patterson, in Statistical Methods for plant variety evaluation, R. A. Kempton, P. N. Fox, M. Cerezo, Eds. (Chapman & Hall, ed. 1, 1997), pp. 139–161. [Google Scholar]
- 26.Möhring J., Piepho H.-P., Comparison of weighting in two-stage analysis of plant breeding trials. Crop Sci. 49, 1977–1988 (2009). [Google Scholar]
- 27.Piepho H.-P., Möhring J., Computing heritability and selection response from unbalanced plant breeding trials. Genetics 177, 1881–1888 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.R Core Team, R: A Language and Environment for Statistical Computing (Vienna, Austria, 2015); http://R-project.org/.
- 29.D. G. Butler, B. R. Cullis, A. R. Gilmour, B. J. Gogel, Mixed models for S language environments. ASReml-R reference manual, Release 3.0. Technical report, ASReml estimates variance components under a general linear mixed model by residual maximum likelihood (REML) (Technical report, Queensland Department of Primary Industries, 2009); https://vsni.co.uk/software/asreml.
- 30.Wang S., Wong D., Forrest K., Allen A., Chao S., Huang B. E., MacCaferri M., Salvi S., Milner S. G., Cattivelli L., Mastrangelo A. M., Whan A., Stephen S., Barker G., Wieseke R., Plieske J.; International Wheat Genome Sequencing Consortium, Lillemo M., Mather D., Appels R., Dolferus R., Brown-Guedira G., Korol A., Akhunova A. R., Feuillet C., Salse J., Morgante M., Pozniak C., Luo M.-C., Dvorak J., Morell M., Dubcovsky J., Ganal M., Tuberosa R., Lawley C., Mikoulitch I., Cavanagh C., Edwards K. J., Hayden M., Akhunov E., Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 12, 787–796 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He S., Zhao Y., Mette M. F., Bothe R., Ebmeyer E., Sharbel T. F., Reif J. C., Jiang Y., Prospects and limits of marker imputation in quantitative genetic studies in European elite wheat (Triticum aestivum L.). BMC Genomics 16, 168 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keenan K., Mcginnity P., Cross T. F., Crozier W. W., Prodöhl P. A., diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 4, 782–788 (2013). [Google Scholar]
- 33.Paradis E., Claude J., Strimmer K., APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (2004). [DOI] [PubMed] [Google Scholar]
- 34.J. S. Rogers, in Studies in genetics VII. Publ. 7213 (University of Texas, Austin, 1972), pp. 145–153. [Google Scholar]
- 35.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A. R., Bender D., Maller J., Sklar P., de Bakker P. I. W., Daly M. J., Sham P. C., PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 17, 10–12 (2011). [Google Scholar]
- 37.H. Li, Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:13033997 (2013).
- 38.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The Sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H., A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swarts K., Li H., Navarro J. A. R., An D., Romay M. C., Hearne S., Acharya C., Glaubitz J. C., Mitchell S., Elshire R. J., Buckler E. S., Bradbury P. J., Novel methods to optimize genotypic imputation for low-coverage, next-generation sequence data in crop plants. Plant Genome 7, 1–12 (2014). [Google Scholar]
- 42.Bradbury P. J., Zhang Z., Kroon D. E., Casstevens T. M., Ramdoss Y., Buckler E. S., TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Zheng X., Gogarten S. M., Lawrence M., Stilp A., Conomos M. P., Weir B. S., Laurie C., Levine D., SeqArray-a storage-efficient high-performance data format for WGS variant calls. Bioinformatics 33, 2251–2257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu S., Zhu D., Zhang Q., Predicting hybrid performance in rice using genomic best linear unbiased prediction. Proc. Natl. Acad. Sci. U.S.A. 111, 12456–12461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang Y., Reif J. C., Modeling epistasis in genomic selection. Genetics 201, 759–768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pérez P., de Los Campos G., Genome-wide regression and prediction with the BGLR statistical package. Genetics 198, 483–495 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu J., Pressoir G., Briggs W. H., Bi I. V., Yamasaki M., Doebley J. F., McMullen M. D., Gaut B. S., Nielsen D. M., Holland J. B., Kresovich S., Buckler E. S., A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Xu S., Mapping quantitative trait loci by controlling polygenic background effects. Genetics 195, 1209–1222 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lippert C., Listgarten J., Liu Y., Kadie C. M., Davidson R. I., Heckerman D., FaST linear mixed models for genome-wide association studies. Nat. Methods 8, 833–835 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Holm S., A simple sequentially rejective multiple test procedure. Scand. J. Stat. 6, 65–70 (1979). [Google Scholar]
- 51.Gao X., Starmer J., Martin E. R., A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 32, 361–369 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Arend D., Lange M., Chen J., Colmsee C., Flemming S., Hecht D., Scholz U., e! DAL-a framework to store, share and publish research data. BMC Bioinformatics 15, 214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaay4897/DC1