

Skeletal muscle shelters a group of T cells from systemic and chronic inflammation to protect T cell proliferative potential.
Abstract
CD8+ T cells become functionally impaired or “exhausted” in chronic infections, accompanied by unwanted body weight reduction and muscle mass loss. Whether muscle regulates T cell exhaustion remains incompletely understood. We report that mouse skeletal muscle increased interleukin (IL)–15 production during LCMV clone 13 chronic infection. Muscle-specific ablation of Il15 enhanced the CD8+ T cell exhaustion phenotype. Muscle-derived IL-15 was required to maintain a population of CD8+CD103+ muscle-infiltrating lymphocytes (MILs). MILs resided in a less inflamed microenvironment, expressed more T cell factor 1 (Tcf1), and had higher proliferative potential than splenic T cells. MILs differentiated into functional effector T cells after reentering lymphoid tissues. Increasing muscle mass via muscle-specific inhibition of TGFβ signaling enhanced IL-15 production and antiviral CD8+ T cell responses. We conclude that skeletal muscle antagonizes T cell exhaustion by protecting T cell proliferative potential from inflammation and replenishing the effector T cell progeny pool in lymphoid organs.
INTRODUCTION
Chronic viral infections and cancers frequently cause involuntary loss of body weight and muscle atrophy, also known as cachexia (1, 2). It has been well documented that T cells contribute to skeletal muscle mass loss (3–5). Whether and how skeletal muscle, in turn, influences CD8+ T cell functions are not clear. CD8+ T cells are functionally compromised and become “exhausted” in chronic infections and cancers (6, 7). Exhausted T cells are characterized by increased expression of inhibitory receptors [e.g., programmed cell death protein 1 (PD-1)] and decreased secretion of effector cytokines [e.g., interferon-γ (IFN-γ) and tumor necrosis factor–α (TNFα)] (8, 9). The underlying mechanisms through which effector T cells become exhausted remain incompletely understood. Previous studies have shown that antigen persistence, a lack of CD4+ T cell help, cytokines, endogenous metabolic stress, and transcription factors regulate T cell exhaustion (10). It remains largely unclear whether and how muscle regulates T cell exhaustion.
Through RNA-sequencing (RNA-seq) analysis of global skeletal muscle gene expression, we identified that muscle expressed high mRNA levels of Il15 during lymphocytic choriomeningitis virus (LCMV) clone 13 chronic infection. Interleukin-15 (IL-15) is known to be essential for memory CD8+ T cell homeostatic proliferation after acute infection (11, 12) but is not required for T cell homeostasis after viral clearance in the blood during chronic infection (13). Using a mouse strain with muscle-specific ablation of Il15, and a second model with a muscle-specific deficiency in transforming growth factor–β (TGFβ) receptor II [TGFβRII; involved in myopathies and muscle mass loss (14)], we observed that muscle protected CD8+ T cell proliferative potential by insulating systemic inflammation. This work has provided evidence that the spatial compartmentalization of a group of precursor cells in nonlymphoid tissues promotes the temporal continuity of antiviral immunity in lymphoid tissues.
RESULTS
Upregulation of IL-15/IL-15Rα complex production by skeletal muscle during chronic viral infection
To determine how chronic viral infection influences the global gene expression profile of skeletal muscle, we infected C57BL/6 mice with two LCMV strains: LCMV Armstrong and LCMV clone 13. LCMV Armstrong and LCMV clone 13 induce acute and chronic infections, respectively. We collected the mouse quadriceps muscle during the effector T cell phase [8 days postinfection (dpi)] for RNA extraction. The subsequent RNA-seq analysis revealed that 520 and 1506 protein-coding genes were significantly decreased or increased, respectively, by LCMV clone 13 infection compared with LCMV Armstrong infection (Fig. 1A). Muscle produces a group of soluble proteins known as myokines (15, 16). We hypothesized that muscle delivered myokines to lymphoid organs and remotely regulated cellular immune responses. We observed that the mRNA levels of four myokines, specifically Il15, Il15ra, Erfe, and Fndc5, were significantly changed between chronic and acute infections (Fig. 1B). Il15 and Il15ra encode the IL-15 and IL-15Rα proteins, respectively. IL-15–producing cells also express IL-15Rα to present IL-15 in trans to recipient cells (17, 18). Erythroferrone (encoded by Erfe) promotes liver iron release into the circulation after blood loss and supports erythropoiesis (19). Fndc5 encodes the membrane protein fibronectin type III domain–containing 5 (Fndc5). The Fndc5 protein is cleaved into a soluble form known as irisin, which is induced by exercise and promotes adipose tissue browning in mice (20). Among the four genes, Il15 and Il15ra immediately attracted our attention because the existing literature supports a role for IL-15 in regulating immune responses. We performed an enzyme-linked immunosorbent assay (ELISA) and found that the IL-15/IL-15Rα complex protein level in skeletal muscle, but not in cardiac muscle, serum, or white adipose tissue, was increased in mice infected with LCMV clone 13 compared with those infected with LCMV Armstrong (Fig. 1C). These results suggest that chronic infection increases IL-15/IL-15Rα complex production in skeletal muscles.
Fig. 1. Skeletal muscle increases IL-15/IL-15Rα complex production during chronic infection.
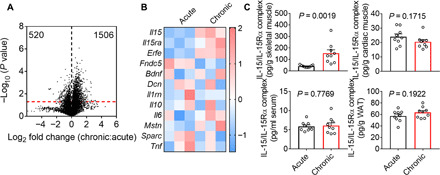
(A) A volcano plot shows the distribution of up- and down-regulated genes in the quadriceps muscle of mice at 8 days postinfection (dpi) with LCMV clone 13 (chronic infection) or LCMV Armstrong (acute infection). The red dashed line indicates the P value of 0.05, and the black dashed line separates the up- (1506) and down-regulated (520) genes. Data are representative of one experiment; n = 3 mice. (B) A heat map shows the mRNA expression z scores of the indicated myokine genes in the mouse quadriceps muscle at day 8 after acute or chronic LCMV infection. Data are representative of one experiment; n = 3 mice. (C) Bar graphs show the IL-15/IL-15Rα complex abundance in mouse hindlimb skeletal muscle, cardiac muscle, serum, and gonadal white adipose tissue (WAT) measured by ELISA after chronic or acute LCMV infection (8 dpi). Data are pooled from three independent experiments; n = 18 mice. The bars represent the mean, and the error bars represent the SD.
Effect of muscle-specific ablation of IL-15 on the CD8+ T cell exhaustion phenotype
The role of IL-15 in regulating T cells after acute infection has been well documented (11, 12, 21). How IL-15 influences CD8+ T cell survival, proliferation, and function during chronic infection remains largely unknown. To investigate the potential role of muscle-derived IL-15 in T cell exhaustion, we bred Il15Flox/Flox mice with mice expressing a skeletal and cardiac muscle–specific Cre enzyme to create the Il15Flox/FloxMck-Cre mouse strain (22). The deficiency in Il15 did not significantly influence the numbers of thymocytes, inguinal lymphocytes, total splenocytes, or splenic T cells (fig. S1A). The CD4+ and CD8+ T cell percentages in the thymocyte, splenocyte, and inguinal lymphocyte populations were comparable between Il15Flox/FloxMck-Cre+ mice and Il15Flox/FloxMck-Cre− control littermates (fig. S1B). Furthermore, splenic CD4+ and CD8+ T cells from the two mouse strains expressed similar levels of surface antigens, such as CD44, CD62L, CD69, and IL-7Rα (fig. S1, B and C). Together, these results suggest that Il15 deficiency in muscle does not affect thymic T cell development or peripheral T cell homeostasis under steady-state conditions.
To investigate whether muscle-derived IL-15 influences CD8+ T cell responses in chronic infection, we infected Il15Flox/FloxMck-Cre+ mice and control littermates with LCMV clone 13. The muscle-specific deficiency in Il15 significantly reduced the numbers of total CD8+ T cells and LCMV tetramer DbGP33–41–positive CD8+ T cells (Fig. 2A). The virus-specific CD8+ T cells in the Il15Flox/FloxMck-Cre+ mice expressed higher protein levels of inhibitory receptors, such as PD-1, CD244, LAG-3, and TIGIT, than those in the Il15Flox/FloxMck-Cre− control littermates (Fig. 2B). Furthermore, there were fewer CD8+ T cells producing the effector cytokines TNFα, IFN-γ, granzyme A, and granzyme B in the Il15Flox/FloxMck-Cre+ mice than in the control littermates (Fig. 2C). Overall, a muscle-specific deficiency in Il15 enhances the LCMV-specific CD8+ T cell exhaustion phenotype.
Fig. 2. Il15 deficiency in muscle affects antiviral CD8+ T cell responses.
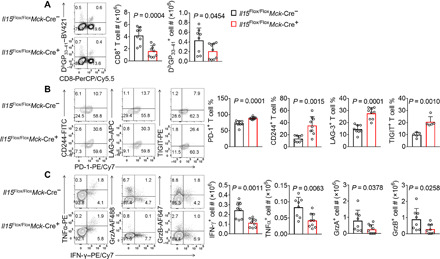
(A to C) Il15Flox/FloxMck-Cre+ mice and control littermates were infected with LCMV clone 13 and euthanized 21 days later. (A) Percentages and numbers of total CD8+ T cells and DbGP33–41 tetramer–positive CD8+ T cells. Data were pooled from two independent experiments; n = 8 for Il15Flox/FloxMck-Cre+ mice and 9 for control littermates. Percentages of DbGP33–41 tetramer–positive CD8+ T cells expressing PD-1, CD244, LAG-3, or TIGIT (B) and percentages or numbers of total splenic CD8+ T cells expressing effector cytokines (C). Data were pooled from three independent experiments; n = 8 for Il15Flox/FloxMck-Cre+ mice and 9 for control littermates. TIGIT staining results (B) were pooled from five pairs of mice from two independent experiments. The bars represent the mean, and the error bars represent the SD.
Phenotypic and functional characterization of CD8+ muscle-infiltrating lymphocytes
We reasoned that there were two possible mechanisms through which muscle-derived IL-15 antagonized antiviral CD8+ T cell exhaustion. First, muscle-produced IL-15 could diffuse through the circulation and remotely regulate the function of CD8+ T cells in lymphoid organs. However, the serum IL-15/IL-15Rα complex concentrations were very low and comparable between Il15Flox/FloxMck-Cre+ mice and control littermates (Fig. 1C), suggesting that muscle may not act through systemically increasing the IL-15 level in the circulation to antagonize T cell exhaustion. Second, instead of remotely regulating CD8+ T cells in lymphoid organs, muscle-derived IL-15 might act locally on CD8+ muscle-infiltrating lymphocytes (MILs). In support of this second possibility, CD8+ T cells were observed in the skeletal muscle tissue sections (Fig. 3A). These CD8+ T cells were in close proximity to the muscle cells (positive for dystrophin staining) and were not in blood vessels. Compared with splenic CD8+ T cells, these CD8+ MILs expressed higher levels of the tissue-tethering molecules CD103, CD69, and CD49a, presumably facilitating T cell residency in the muscle (Fig. 3B). CD8+ MILs expressed much higher protein levels of T cell factor 1 (Tcf1) and Eomes than splenic T cells, whereas T-bet protein expression was reduced (Fig. 3C). Compared with splenic CD8+ T cells, MILs expressed lower levels of inhibitory receptors, such as PD-1 and CD244. TIGIT and LAG-3 levels were not significantly different between MILs and splenic CD8+ T cells (Fig. 3D). Upon restimulation with the LCMV peptide GP33–41, MILs and splenic CD8+ T cells generated similar amounts of granzymes, with the CD8+ MILs producing more TNFα and IFN-γ (Fig. 3E). CD8+ MILs were not detectable in cardiac muscles, indicating that LCMV clone 13 infection did not cause general T cell infiltration into all types of muscles. Together, these results suggest that LCMV clone 13 infection–induced skeletal MILs are phenotypically distinct from their splenic counterparts.
Fig. 3. CD8+ MILs are phenotypically different from splenic CD8+ T cells.
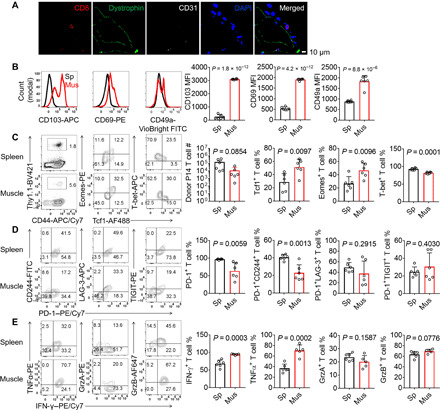
(A and B) LCMV clone 13–infected C57BL/6 mice were euthanized 8 days after infection. Distributions of CD8+ T cells in the gastrocnemius muscle tissue sections stained with an anti-CD8 antibody (red), an anti-dystrophin antibody (green, to stain muscle cells), an anti-CD31 antibody (white, to stain blood vessels), and 4′,6-diamidino-2-phenylindole (DAPI) (blue, to stain nuclei). Data are representative of 10 images of six mice from three independent experiments. Scale bar, 10 μm (A). Fluorescence-activated cell sorting (FACS) histograms and bar graphs showing the mean fluorescence intensity (MFI) of CD103, CD69, and CD49a in total CD8+ T cells in the spleen (Sp) or hindlimb muscles (Mus) (B). (C to E) Thy1.1+ T cell reporter (TCR) transgenic P14 CD8+ T cells (103 cells) were adoptively transferred into Thy1.2/1.2 recipient mice, which were subsequently infected with LCMV clone 13 and euthanized at 21 days after infection. Percentages and numbers of total donor P14 CD8+ T cells and P14 CD8+ T cells expressing Tcf1, Eomes, or T-bet (C). Percentages of P14 CD8+ T cells expressing PD-1, CD244, LAG-3, TIGIT (D), or effector cytokines (E). Data are representative of two independent experiments or cumulative; n = 6 mice (B to E). Data are means ± SD.
Because Tcf1 is required for T cells to maintain stemness and rapidly give rise to progeny cells (23–27), we examined whether MILs had higher proliferative potential than splenic CD8+ T cells. Briefly, we used Miltenyi Thy1.1 microbeads to purify Thy1.1+ P14 CD8+ T cells from the hindlimb skeletal muscle or spleen of LCMV clone 13–infected mice on 8 dpi and adoptively transferred 104 T cells into infection-matched recipient mice (Fig. 4A). One week later, the MILs generated significantly more progeny cells than the splenic donor T cells (Fig. 4B). Although the MIL-derived progeny cells showed a trend toward lower percentages of CD244+PD-1+, LAG-3+PD-1+, and TIGIT+PD-1+ cells among total donor CD8+ T cells, the progeny cells derived from the MILs and splenocytes generally expressed comparable protein levels of transcription factors, inhibitory receptors, and effector cytokines (Fig. 4, B to D). These results suggest that although the phenotypic differences between MILs and splenic CD8+ T cells are not imprinted on progeny cells, the MILs have a higher proliferative potential and generate more progeny cells in lymphoid organs than the splenic T cells.
Fig. 4. CD8+ MILs have a higher proliferative potential than splenic CD8+ T cells and give rise to phenotypically comparable progeny cells.
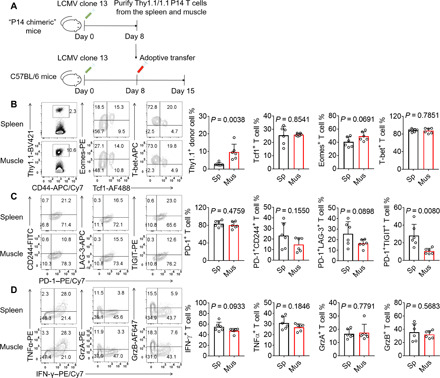
(A) Thy1.1+ TCR transgenic P14 CD8+ T cells (103) were adoptively transferred into Thy1.2/1.2 C57BL/6 recipients, which were subsequently infected with LCMV clone 13. Splenocytes and MILs were purified at 8 dpi and adoptively transferred into infection-matched recipients, which were euthanized 7 days later. (B to D) Percentages of total donor P14 CD8+ T cells in the spleen (Sp) and hindlimb muscles (Mus), and P14 CD8+ T cells expressing transcription factors (B), inhibitory receptors (C), or effector cytokines (D) are shown. Data are representative (FACS plots) of two independent experiments or cumulative (bar graphs); n = 6 mice. The bars represent the mean, and the error bars represent the SD.
Muscle preservation of CD8+ MIL stemness
The Tcf1 protein maintains T cell stemness and proliferative potential in chronic infections and cancers (23–25). Tcf1 protein expression is suppressed by the inflammatory cytokine IL-12 (28). We hypothesized that the maintenance of a higher protein Tcf1 level in MILs was because of a less inflamed microenvironment in the skeletal muscle than in the spleen. To test this hypothesis, we performed ELISA to measure the levels of several inflammatory cytokines. Compared with the spleen, the muscle had significantly lower protein levels of IL-12, IFN-β, and IFN-γ. IL-6 and TNFα concentrations were similar between the two tissues (Fig. 5A). In vitro IL-12 treatment potently suppressed Tcf1 expression in CD8+ MILs. IFN-β treatment moderately but significantly reduced Tcf1 protein levels in CD8+ MILs. We did not observe a change in Tcf1 protein expression in IFN-γ–treated CD8+ MILs (Fig. 5B). Because IL-12 is induced by IFN-γ in a manner dependent on signal transducer and activator of transcription 1 (STAT1) phosphorylation, we measured STAT1 phosphorylation at tyrosine-701 (pSTAT1Tyr701) in freshly harvested muscle and spleen tissue samples from LCMV clone 13–infected mice. We found that the spleen lysates had much higher levels of pSTAT1Tyr701 than the muscle lysates (Fig. 5C). One possible reason for the pSTAT1Tyr701 reduction was the decreased availability of IFN-γ (Fig. 5A). Another possibility was that muscle cells were less sensitive than splenocytes to the induction of IL-12 expression by IFN-γ, which was supported by the observation that in vitro–cultured muscle cells were resistant to IL-12 induction by IFN-γ (Fig. 5D). To test if the less inflamed environment in skeletal muscle preserved Tcf1 protein expression in CD8+ MILs, we treated CD8+ MILs with muscle or spleen lysates. The Tcf1 protein level was significantly higher in the muscle lysate–treated cells than in the spleen lysate–treated cells. Tcf1 expression in CD8+ MILs was reduced after the muscle lysates were supplemented with IL-12. In addition, anti–IL-12 antibody treatment increased the Tcf1 protein level in the spleen lysate–treated CD8+ MILs (Fig. 5E), echoing a previous report showing a suppressive role for IL-12 in Tcf1 protein expression (28). To further study whether the less inflamed environment in skeletal muscle preserved the CD8+ MIL proliferative potential, we treated CD8+ MILs with the aforementioned muscle and spleen lysates and adoptively transferred them into LCMV clone 13 infection–matched recipients. The muscle lysate–treated CD8+ MILs generated more progeny cells in different tissues of the recipient mice than the CD8+ MILs treated with the spleen lysates (Fig. 5, F and G). Collectively, these results suggest that the low inflammation in the muscle microenvironment maintains CD8+ MIL Tcf1 protein expression and proliferative potential.
Fig. 5. The low-inflammation microenvironment preserves the Tcf1 protein expression and high proliferative potential of CD8+ MILs.
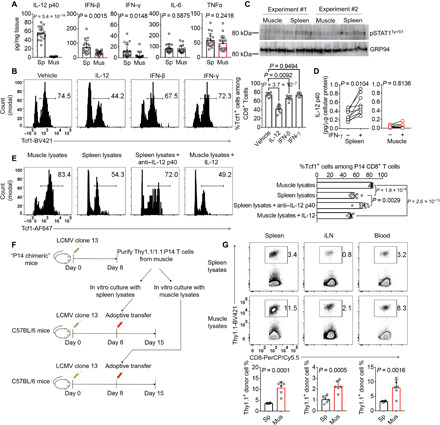
(A to D) LCMV clone 13–infected C57BL/6 mice were euthanized (8 dpi). The spleen and hindlimb muscles were homogenized for cytokine quantification by ELISA (A) or phospho-STAT1 detection by Western blotting (C). MILs were cultured with the indicated cytokines overnight before FACS analysis of Tcf1 expression in polyclonal CD8+ T cells (B). Splenocytes and hindlimb muscle cells were cultured with or without IFN-γ overnight. Supernatant IL-12 p40 levels were measured by ELISA (D). (E) C57BL/6 mice receiving 103 donor P14 CD8+ T cells were infected with LCMV clone 13. Splenic donor P14 CD8+ T cells were recovered (8 dpi) and cultured in complete medium supplemented with tissue lysates from infection-matched mice. The percentages of P14 CD8+ T cells expressing Tcf1 are shown in FACS histograms and a bar graph. (F and G) The P14 donor T cells treated with spleen or muscle lysates were adoptively transferred into infection-matched mice. Donor cells in different tissues were quantified 7 days later. Data are means ± SD and were pooled from two (B, C, and E to G) or three (A and D) independent experiments; n = 6 (B, C, and G), 8 (E), 9 (D), or 18 (A) mice.
In addition to measuring inflammatory cytokines in the skeletal muscle and spleen, we have also quantified viral titers in the two tissues by plaque assay. We found that Il15 deficiency did not influence the viral titers in both spleen and muscle on 8 dpi and 21 dpi but slowed down viral titer decrease on 45 dpi (fig. S2). The exacerbated T cell exhaustion phenotype on 21 dpi preceded the relative increase in viral titers in the Il15Flox/FloxMck-Cre+ mice compared with the wild-type (WT) littermates on 45 dpi, suggesting that muscle-specific deficiency of Il15 affects effector T cell responses on 21 dpi not because of higher viral loads (Fig. 2, B and C). We also observed that the viral titers in the muscle were three logs lower than that in the spleen (fig. S2), which is consistent with previous reports showing that skeletal muscle is resistant to LCMV infection (29, 30). Antigen persistence positively correlates with T cell effector function loss (8). The reduced antigen exposure of MILs helps to explain the lower levels of inhibitory receptors and higher levels of effector cytokine production in MILs (Fig. 3, D and E). It has been shown before that antigen stimulation does not decrease Tcf1 protein expression in CD8+ T cells (28). Together, skeletal muscle shelters a population of T cells in a microenvironment with less inflammation and antigen stimulation. This unique environment preserves Tcf1 protein expression and CD8+ T cell proliferative potential and protects against T cell functional impairment during chronic infection.
Essential role of muscle-derived IL-15 in CD8+ MIL accumulation
To investigate why muscle-derived IL-15 was required for the optimal differentiation of splenic effector CD8+ T cells (Fig. 2, A to C), we tested the hypothesis that muscle-derived IL-15 was required for the accumulation of CD8+ MILs, which had a high proliferative potential and could generate splenic effector CD8+ T cells (Fig. 4B). We generated “peripheral P14 chimeras” by adoptively transferring Thy1.1+ P14 CD8+ T cells into Thy1.2/1.2 Il15Flox/FloxMck-Cre+ mice and Il15Flox/FloxMck-Cre− control littermates. Those “P14 chimeras” were infected with LCMV clone 13, and the hindlimb muscles, including the quadriceps, tibialis anterior, gastrocnemius, and soleus muscles, were harvested for MIL isolation. Muscle-specific ablation of Il15 significantly reduced Thy1.1+ donor CD8+ MILs (Fig. 6A). Because CD8+ MILs expressed higher levels of the Tcf1 protein than splenic CD8+ T cells, we tested whether IL-15 increased Tcf1 protein expression. We found that IL-15 treatment in vitro did not significantly change Tcf1 expression in MILs, as determined by FACS (fluorescence-activated cell sorting) staining (Fig. 6B). We then tested whether IL-15 regulated CD8+ MIL survival or proliferation. We isolated CD8+ MILs from LCMV clone 13–infected mice and cultured them with or without IL-15. The CD8+ MILs rapidly underwent apoptosis without IL-15 supplementation, suggesting that IL-15 played an essential role in CD8+ MIL survival (Fig. 6B). Furthermore, a 5-bromo-2′-deoxyuridine (BrdU)–labeling assay revealed that CD8+ MIL proliferation was modestly but significantly enhanced by IL-15 addition (Fig. 6B). Overall, IL-15 antagonizes MIL apoptosis and promotes cell proliferation to support CD8+ MIL accumulation during LCMV clone 13 infection.
Fig. 6. Muscle-derived IL-15 promotes CD8+ MIL accumulation.
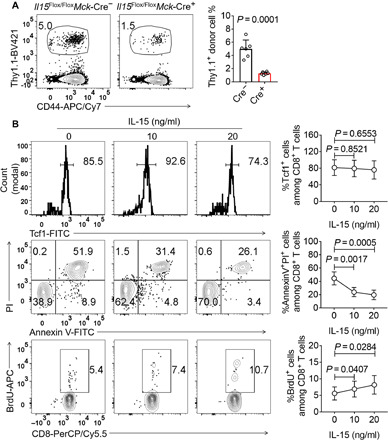
(A) FACS dot plots and a bar graph show the percentages of Thy1.1+ P14 donor T cells in the hindlimb muscles of Il15Flox/FloxMck-Cre+ mice and control littermates infected with LCMV clone 13 and euthanized on 8 dpi. Data are representative (FACS plots) of two independent experiments or cumulative (the bar graph); n = 6 mice. (B) C57BL/6 mice were infected with LCMV clone 13. Eight days later, MILs were isolated from the hindlimb muscles and treated with or without IL-15 for 24 hours. BrdU (10 μM) was added (bottom panel). FACS plots and line graphs show the Tcf1 expression (top panel), apoptosis (middle panel), and proliferation (lower panel) of the CD8+ T cells. Data are representative (FACS plots) of two independent experiments or cumulative (line graphs); n = 6 mice. Data are shown as the means ± SD.
Effects of increasing muscle mass by inhibiting TGFβ signaling on IL-15 production and CD8+ T cell functions
Because TGFβ signaling is linked to myopathies and muscle mass loss (14), we tested whether ablating TGFβ signaling increases muscle mass during LCMV clone 13 infection. We found that the mass of the muscle tissues in the hind limbs was significantly increased by TGFβ receptor II (encoded by Tgfbr2) deficiency (fig. S3A), suggesting that TGFβ promoted muscle mass loss during LCMV clone 13 infection. To investigate the potential role of muscle-specific TGFβ signaling in CD8+ T cell exhaustion, we adoptively transferred P14 CD8+ T cells into Tgfbr2Flox/FloxMck-Cre+ mice and Tgfbr2Flox/FloxMck-Cre− littermates before LCMV clone 13 infection. Muscle-specific ablation of Tgfbr2 increased IL-15 production, MIL numbers, and splenic CD8+ T cell numbers (fig. S3, B and C), echoing the role of muscle-derived IL-15 in maintaining MILs (Fig. 6A). Furthermore, splenic P14 CD8+ T cells expressed lower levels of inhibitory receptors and produced more effector cytokines in the Tgfbr2Flox/FloxMck-Cre+ mice than in the Tgfbr2Flox/FloxMck-Cre− littermates (fig. S3, D and E). Collectively, these results indicate that increasing muscle mass by inhibiting TGFβ signaling promotes IL-15 production, increases MIL numbers, and ameliorates the splenic T cell exhaustion phenotype.
DISCUSSION
T cell exhaustion and muscle mass loss are frequently observed in chronic infections and cancers. This study has provided a mechanistic link between these two seemingly isolated events. The interorgan communications between skeletal muscles and lymphoid organs suppress T cell exhaustion. Similar to muscle tissue, adipose tissue undergoes weight loss during chronic infections and cancers. One future direction extending from the current study is to test whether and how adipose tissue regulates T cell functions in chronic infectious diseases and cancers. These future studies, together with the current one, will enhance the understanding of the molecular mechanisms through which chronic infection– and cancer-associated cachexia influences antiviral and antitumor immunity.
Spatial compartmentalization is one mechanism to create diverse microenvironments in the immune system and to enable various immune cell differentiation pathways. Our study suggests that inflammatory cytokines are not evenly distributed among the skeletal muscles and lymphoid organs. The spatial compartmentalization of one population of CD8+ T cells in the relatively uninflamed microenvironment of the skeletal muscle shelters those T cells from systemic inflammation. The different degrees of exposure to inflammation diversify the Tcf1 expression levels and differentiation status of antiviral CD8+ T cells in various tissues. Hence, the T cells infiltrating skeletal muscle are less inflammation experienced, and the T cells exposed to more inflammation in the lymphoid organs express less Tcf1 and have a reduced T cell proliferative capacity. MILs, which express higher levels of Tcf1 and have a higher proliferative potential than T cells in the lymphoid organs, serve as precursors that give rise to progeny cells to replenish the antiviral T cell pool in the lymphoid organs. This mode of hierarchical T cell differentiation helps to prevent uncontrolled inflammation-mediated Tcf1 protein waning and a systemic loss of T cell stemness. By sheltering a population of less terminally differentiated T cells, skeletal muscle antagonizes T cell exhaustion and contributes to long-term antiviral immunity.
The origin of MILs remains to be identified. There are at least two possible ways through which effector CD8+ T cells accumulate within skeletal muscle during chronic infections. First, a small population of naïve CD8+ T cells migrates to the skeletal muscle and resides there before infection. Previous studies have shown that naïve mice have detectable T cells within their skeletal muscle under steady-state conditions (31), supporting the idea that naïve CD8+ T cells may survive in the skeletal muscle microenvironment. Upon LCMV clone 13 infection, antigen-specific T cells within this muscle-resident population expand in the low-inflammation environment and maintain high protein levels of Tcf1. Second, naïve CD8+ T cells become activated in lymphoid organs before migrating to skeletal muscle and establishing tissue residency. The chemotaxis pathways responsible for the muscle-tropic T cell migration during chronic infection remain to be determined. Identification of the MIL origin may help in the design of therapies to increase or decrease MIL accumulation to treat chronic infectious diseases or myositis.
The roles of IL-15 in T cell functions and differentiation after acute infection have been well characterized. Our study has suggested a role for muscle-derived IL-15 in sustaining antiviral T cell responses in viremic mice during chronic infection. In addition to IL-15, chronic infection also significantly influences the mRNA expression of genes encoding two myokines: erythroferrone and irisin. Erythroferrone increases the iron availability in the circulation, and irisin increases the browning of adipose tissue in mice (19, 20). Whether these two myokines regulate T cell exhaustion during chronic infection remains to be examined.
Together, our study results have revealed a role for skeletal muscle in compartmentalizing a population of less terminally differentiated T cells and protecting these cells from exposure to systemic and chronic inflammation. These T cell precursors replenish the effector T cell pool in lymphoid organs and sustain antiviral immunity.
MATERIALS AND METHODS
Mouse study approval
Mice were maintained in the German Cancer Research Center (DKFZ) specific pathogen–free facility. All the studies were performed in accordance with DKFZ regulations with approval by the German Regional Council at the Regierungspräsidium Karlsruhe.
Il15Flox/Flox mice [MGI; C57BL/6N-Il15tm1c(EUCOMM)Hmgu] with exon 5 flanked by two loxP sites were ordered from MRC Harwell (Oxfordshire, UK). Mck-Cre mice were from the Jackson laboratory (22). Tgfbr2Flox/Flox mice were provided by M. Bevan (Seattle) (32). WT C57BL/6N mice were from Janvier Labs. We used sex-matched 5- to 10-week-old mice (both female and male) for the experiments and subsequent comparisons. The sample collection and processing after LCMV infections were not blinded. No experimental results were excluded.
LCMV infection
In infection-related experiments, mice were infected with 2 × 105 plaque-forming units (PFU) LCMV Armstrong [intraperitoneally (ip)] or 2 × 106 PFU LCMV clone 13 (intravenously). Anti-CD4 (clone GK1.5) was injected (400 μg, ip) to promote T cell exhaustion after LCMV clone 13 infection. Where indicated, we generated the “P14 chimeric mice” by adoptively transferring 103 LCMV-specific P14 T cell reporter (TCR) transgenic CD8+ cells to C57BL/6 mice before LCMV clone 13 infection.
Primary cell cultures
Primary T cells derived from gender-matched mice (both male and female) were cultured in complete RPMI 1640 medium [plain RPMI 1640 medium with 10% fetal bovine serum (FBS), penicillin and streptomycin, 2-mercaptoethanol, l-glutamine, and nonessential amino acids]. In the BrdU-labeling assay, cells were pulsed with 10 μM BrdU for 24 hours before FACS staining. Antibodies, cytokines, peptides, and chemicals were supplemented as indicated. For the bulk muscle cell culture, we followed a previously published method (33). The C57BL/6 mouse extensor digitorum longus (EDL) muscle was collected from tendon to tendon. Then, EDL muscle was digested with 0.2% collagenase I in Dulbecco’s modified Eagle’s medium (DMEM) at 37°C for 45 min. The bulk muscle cells were collected and centrifuged at 1000 rpm for 5 min. Cell pellets were cultured in DMEM supplemented with 10% FBS, penicillin and streptomycin, 2-mercaptoethanol, l-glutamine, and nonessential amino acids. In parallel, spleens were collected, smashed into single cells, and cultured with the same DMEM medium. IFN-γ (10 ng/ml) was added to induce IL-12 production.
Antibodies used in flow cytometry and cell cultures
The following antibodies were ordered from BioLegend: CD16/32 (clone 93, catalog no. 101302, 1:500), CD8α-PerCP/Cy5.5 (clone 53-6.7, catalog no. 100734, 1:200 dilution), CD4-FITC (clone GK1.5, catalog no. 100406, 1:200 dilution), CD62L-PE/Cy7 (clone MEL-14, catalog no. 104418, 1:400 dilution), CD44-APC/Cy7 (clone IM7, catalog no. 103028, 1:400 dilution), IL-7Rα–PE (clone A7R34, catalog no. 135010, 1:200 dilution), PD-1–PE/Cy7 (clone 29F.1A12, catalog no. 135216, 1:200 dilution), CD244-FITC [clone m2B4(B6)458.1, catalog no. 133504, 1:200 dilution], LAG-3–APC (clone C9B7W, catalog no. 125210, 1:200 dilution), TIGIT-PE (clone 1G9, catalog no. 142104, 1:200 dilution), IFN-γ–PE/Cy7 (clone XMG1.2, catalog no. 505826, 1:400 dilution), TNFα-PE (clone MP6-XT22, catalog no. 506306, 1:400 dilution), granzyme A–AF488 (clone CB9, catalog no. 507212, 1:100 dilution), granzyme A–PE (clone 3G8.5, catalog no. 149704, 1:100 dilution), granzyme B–AF647 (clone GB11, catalog no. 515406, 1:100 dilution), Thy1.1-BV421 (clone OX-7, catalog no. 202529, 1:400 dilution), CD8β-PE (clone YTS156.7.7, catalog no. 126608, 4 μg per mouse, intravenous injection), CD103-APC (clone.2E7, catalog no. 121414, 1:200 dilution), anti–rabbit-AF488 (clone Poly4064, catalog no. 406416, 1:1000 dilution), and anti–rabbit-AF647 (clone Poly4064, catalog no. 406414, 1:1000 dilution). These following antibodies were from Miltenyi Biotec: annexin V–FITC (clone Poly4064, catalog no. 130-092-052, 1:100 dilution), CD49a-VioBright FITC (clone REA493, catalog no. 130-107-592, 1:100 dilution), Eomes-PE (clone REA116, catalog no. 130-102-378, 1:50 dilution), T-bet–APC (clone REA102, catalog no. 130-119-821, 1:50 dilution), CD69-VioBlue (clone H1.2F3, catalog no. 130-103-949, 1:200 dilution), and CD69-Vio770 (clone H1.2F3, catalog no. 130-103-944, 1:200 dilution). Tcf1 (clone C63D9, catalog no. 2203, 1:400 dilution) was from Cell Signaling Technology. DbGP33–41 monomers were from the National Institutes of Health (NIH). Tetramers were generated by mixing monomers with streptavidin-BV421 (catalog no. 405225, BioLegend) and used to stain cells at 1:200 dilution. Propidium iodide (catalog no. BMS500PI) was from Life Technologies.
Sample preparation and flow cytometry
Spleens were smashed using a syringe through a 70-μm cell strainer. Muscle tissues were cut into small pieces, digested using collagenase (0.2 mg/ml, 37°C, 45 min), and filtered using a 70-μm cell strainer. The single-cell suspension was spun down, resuspended in 40% Percoll diluted in RPMI 1640 medium, and loaded onto the 80% Percoll. After centrifugation (2000 rpm, 15 min), the middle layer containing leukocytes were collected for the following analysis. Cells were washed with FACS buffer before adding the fluorescently conjugated antibodies against the surface antigens. After incubating with antibodies for 30 min on ice, cells were washed with FACS buffer and fixed with the BioLegend fixation buffer containing 4% paraformaldehyde (PFA). For annexin V/propidium iodide (PI) staining, cells were washed with annexin V buffer before staining with anti–annexin V and PI. To stain cytokines, cells were fixed with 4% PFA and permeabilized using the eBioscience permeabilization buffer. To stain nuclear antigens, cells were fixed and permeabilized using the eBioscience Foxp3/transcription factor staining buffer set. We followed the manufacturer’s instructions of the Phase-Flow BrdU Kit to examine the BrdU incorporation into DNA. Samples were analyzed using an LSR II.
Western blot
Cells were harvested and lysed using radioimmunoprecipitation assay buffer [150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM tris (pH 8.0)] and boiled in 5× SDS sample buffer [10% SDS, 10 mM dithiothreitol, 20% glycerol, 200 mM tris-HCl (pH 6.8), and 0.05% bromophenol blue] for 10 min. Cell lysates were then loaded and resolved using SDS–polyacrylamide gel electrophoresis (120 V; until the blue indicator runs to the edge of the gel). Proteins were then transferred onto polyvinylidene difluoride (PVDF) membranes (25 V, 16 hours, 4°C). The membranes were then blocked with 1% bovine serum albumin (BSA) in phosphate-buffered saline supplemented with Tween 20 (PBST) for 1 hour at room temperature, followed by incubation overnight at 4°C with anti–phospho-STAT1Tyr701 (Cell Signaling Technology, clone 58D6, catalog no. 9167S, 1:500 dilution). The PVDF membrane was washed three times (5 to 10 min each time) with PBST and then incubated with horseradish peroxidase–conjugated secondary antibodies at room temperature for 1 hour. After extensive washing with PBST, the membrane was developed using the enhanced chemiluminesence method. The PVDF membranes were stripped, blocked, and reblotted with anti-GRP94 (Cell Signaling Technology, catalog no. 2104S, 1:1000 dilution) to assess protein loading.
Cytokine measurements by ELISA
Mouse spleen, hindlimb muscle (including quadriceps, tibialis anterior, gastrocnemius, and soleus muscles), cardiac muscle, serum, and white adipose tissue were collected. Tissues were homogenized using a Dounce homogenizer (100 strokes per tissue sample) on ice in prechilled buffer containing 100 mM tris (pH 7.4), 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, and proteinase inhibitor. The tissue lysate was centrifuged for 10 min at 13,000 rpm at 4°C. The tissue lysate supernatants, together with the mouse serum collected using sodium citrate tubes, were analyzed using the mouse IL-15/IL-15Rα uncoated ELISA Kit (88-7215-22, Life Technologies), IL-12 p40 DuoSet ELISA Kit (DY2398-05, R&D Systems), IFN-β DuoSet ELISA Kit (DY8234-05, R&D Systems), IFN-γ DuoSet ELISA Kit (DY485-05, R&D Systems), IL-6 DuoSet ELISA Kit (DY406-05, R&D Systems), and TNFα DuoSet ELISA Kit (DY410-05, R&D Systems) following the manufacturer’s instructions. The protein levels in the tissues and serum were normalized to the tissue weight and serum volume, respectively. Where indicated, IL-12 p40 in cell culture supernatants was analyzed and normalized to the protein levels of the cell pellets.
Muscle tissue section immunofluorescence
Mouse gastrocnemius muscles were harvested, embedded in O.C.T. (optimal cutting temperature) compound, and immediately frozen on dry ice. Tissues were cut into 5-μm sections in a cryomicrotome (Leica CM1950). Sections were blocked with 5% BSA and then stained with anti–CD8β-PE (BioLegend, clone YTS156.7.7, catalog no. 126608, 1:100 dilution). Sections were then fixed and permeabilized with eBioscience Foxp3/transcription factor staining buffer set (catalog no. 00-5523) and stained with anti-dystrophin [Abcam, polyclonal rabbit immunoglobulin G (IgG), catalog no. ab15277, 1:50 dilution] and anti-mouse CD31-FITC (BioLegend, clone MEC13.3, catalog no. 102506, 1:50 dilution). After washing with PBST, the sections were stained with a secondary antibody (BioLegend, anti-rabbit IgG AF647, catalog no. 406414, 1:200 dilution) and DAPI (4′,6-diamidino-2-phenylindole). We imaged the sections using a Zeiss LSM710 confocal microscope. The fluorescent images were processed with the Zeiss ZEN microscope software.
RNA-sequencing
Quadriceps muscle from LCMV Armstrong– and LCMV clone 13–infected C57BL/6 mice was collected and homogenized. We extracted RNA from the skeletal muscle using the QIAGEN RNeasy Mini Kit and removed genomic DNA using the QIAGEN RNase-Free DNase Set. Library was prepared by the German Cancer Research Center High Throughput Sequencing Unit. Then, libraries were pooled with six samples in each lane and sequenced [Illumina HiSeq 2000 v4 Single-Read 50 base pairs (bp)]. For all samples, low-quality bases were removed with fastq_quality_filter from the FASTX-Toolkit 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit/index.html) with 90% of the read needing a quality phred score >20. Homertools 4.7 (34) was used for Poly-A tail trimming, and reads with length <17 were removed. Picard Tools 1.78 (https://broadinstitute.github.io/picard/) was used to compute the quality metrics with CollectRnaSeqMetrics. With STAR 2.3 (35), the filtered reads were mapped against mouse genome 38 (mm10) using default parameters. Count data were generated using featureCounts (36) (parameters --minReadOverlap 3 -T 3 -M -O) for the genes annotated in the gencode.vM8.gtf file. For the comparison with DESeq2 (37), the input tables containing the replicates for groups to compare were created by a custom perl script. For DESeq2, DESeqDataSetFromMatrix was applied, followed by estimateSizeFactors, estimateDispersions, and nbinomWald testing. The result tables were annotated with gene information (gene symbol) derived from the gencode.vM8.gtf file. We then further analyzed the RNA-seq results with cutoffs of <0.5 or >2-fold change using the Ingenuity Pathway Analysis (IPA; QIAGEN). Accession numbers of the RNA-seq data are GSE134928 and are available at the Genome Expression Omnibus.
Statistical analysis
All the data were presented as means ± SD (error bar) unless otherwise specified. Where indicated, P values were determined by a two-tailed Student’s t test. We used GraphPad Prism (version 7.0.3) to perform the statistical analysis. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank the DKFZ core facility colleagues [animal work units (W410 and W450), flow cytometry unit (W220), biosafety unit (V155), and Genomics and Proteomics Core Facility] for assistance to our work. We thank I. Hofmann and colleagues [GPCF Unit Antibodies (W170)] for technical assistance. We thank the NIH core facility for providing the LCMV tetramer. Funding: G.C. is supported by a Helmholtz Young Investigator Award (#VH-NG-1113), German Research Foundation (DFG; #CU375/5-1, #CU375/7-1, #CU375/9-1, and #259332240/RTG 2099), German Cancer Aid Foundation (DKH; #70113343), Helmholtz Zukunftsthema Aging and Metabolic Programming (AMPro; #ZT0026), Rare Disease Foundation, and BC Children’s Hospital Foundation (#2286, #2604, and #3272). Author contributions: J.W. and G.C. designed the experiments. J.W., N.W., A.Ma., S.M., A.Mi., M.H., K.M., T.S., and H.B. performed the experiments. A.H.-W. performed the bioinformatic analysis. G.C. wrote the manuscript. Competing interests: G.C. receives research funding from Baye AG and Boehringer Ingelheim, but that funding was not relevant to this study. The rest of the authors declare that they have no competing interests. Data and materials availability: The GEO accession number for the RNA-seq data is GSE134928. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaba3458/DC1
REFERENCES AND NOTES
- 1.Morley J. E., Thomas D. R., Wilson M.-M. G., Cachexia: Pathophysiology and clinical relevance. Am. J. Clin. Nutr. 83, 735–743 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Deans C., Wigmore S. J., Systemic inflammation, cachexia and prognosis in patients with cancer. Curr. Opin. Clin. Nutr. Metab. Care 8, 265–269 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Baazim H., Schweiger M., Moschinger M., Xu H., Scherer T., Popa A., Gallage S., Ali A., Khamina K., Kosack L., Vilagos B., Smyth M., Lercher A., Friske J., Merkler D., Aderem A., Helbich T. H., Heikenwälder M., Lang P. A., Zechner R., Bergthaler A., CD8+ T cells induce cachexia during chronic viral infection. Nat. Immunol. 20, 701–710 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doherty P. C., Hou S., Southern P. J., Lymphocytic choriomeningitis virus induces a chronic wasting disease in mice lacking class I major histocompatibility complex glycoproteins. J. Neuroimmunol. 46, 11–17 (1993). [DOI] [PubMed] [Google Scholar]
- 5.Stamm A., Valentine L., Potts R., Premenko-Lanier M., An intermediate dose of LCMV clone 13 causes prolonged morbidity that is maintained by CD4+ T cells. Virology 425, 122–132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baitsch L., Baumgaertner P., Devêvre E., Raghav S. K., Legat A., Barba L., Wieckowski S., Bouzourene H., Deplancke B., Romero P., Rufer N., Speiser D. E., Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Invest. 121, 2350–2360 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moskophidis D., Lechner F., Pircher H., Zinkernagel R. M., Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 362, 758–761 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Wherry E. J., Blattman J. N., Murali-Krishna K., van der Most R., Ahmed R., Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77, 4911–4927 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barber D. L., Wherry E. J., Masopust D., Zhu B., Allison J. P., Sharpe A. H., Freeman G. J., Ahmed R., Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006). [DOI] [PubMed] [Google Scholar]
- 10.McLane L. M., Abdel-Hakeem M. S., Wherry E. J., CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 37, 457–495 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Becker T. C., Wherry E. J., Boone D., Murali-Krishna K., Antia R., Ma A., Ahmed R., Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J. Exp. Med. 195, 1541–1548 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldrath A. W., Sivakumar P. V., Glaccum M., Kennedy M. K., Bevan M. J., Benoist C., Mathis D., Butz E. A., Cytokine requirements for acute and Basal homeostatic proliferation of naive and memory CD8+ T cells. J. Exp. Med. 195, 1515–1522 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin H., Blackburn S. D., Blattman J. N., Wherry E. J., Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J. Exp. Med. 204, 941–949 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burks T. N., Cohn R. D., Role of TGF-β signaling in inherited and acquired myopathies. Skelet. Muscle 1, 19 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henningsen J., Rigbolt K. T. G., Blagoev B., Pedersen B. K., Kratchmarova I., Dynamics of the skeletal muscle secretome during myoblast differentiation. Mol. Cell. Proteomics 9, 2482–2496 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pedersen B. K., Muscles and their myokines. J. Exp. Biol. 214, 337–346 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Lodolce J. P., Burkett P. R., Boone D. L., Chien M., Ma A., T cell–independent interleukin 15rα signals are required for bystander proliferation. J. Exp. Med. 194, 1187–1194 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubois S., Mariner J., Waldmann T. A., Tagaya Y., IL-15Rα recycles and presents IL-15 in trans to neighboring cells. Immunity 17, 537–547 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Kautz L., Jung G., Valore E. V., Rivella S., Nemeth E., Ganz T., Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 46, 678–684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boström P., Wu J., Jedrychowski M. P., Korde A., Ye L., Lo J. C., Rasbach K. A., Boström E. A., Choi J. H., Long J. Z., Kajimura S., Zingaretti M. C., Vind B. F., Tu H., Cinti S., Højlund K., Gygi S. P., Spiegelman B. M., A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481, 463–468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kennedy M. K., Glaccum M., Brown S. N., Butz E. A., Viney J. L., Embers M., Matsuki N., Charrier K., Sedger L., Willis C. R., Brasel K., Morrissey P. J., Stocking K., Schuh J. A. C. L., Joyce S., Peschon J. J., Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15–deficient mice. J. Exp. Med. 191, 771–780 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brüning J. C., Michael M. D., Winnay J. N., Hayashi T., Hörsch D., Accili D., Goodyear L. J., Kahn C. R., A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 2, 559–569 (1998). [DOI] [PubMed] [Google Scholar]
- 23.Siddiqui I., Schaeuble K., Chennupati V., Fuertes Marraco S. A., Calderon-Copete S., Pais Ferreira D., Carmona S. J., Scarpellino L., Gfeller D., Pradervand S., Luther S. A., Speiser D. E., Held W., Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Utzschneider D. T., Charmoy M., Chennupati V., Pousse L., Ferreira D. P., Calderon-Copete S., Danilo M., Alfei F., Hofmann M., Wieland D., Pradervand S., Thimme R., Zehn D., Held W., T cell factor 1-expressing memory-like CD8+ T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Im S. J., Hashimoto M., Gerner M. Y., Lee J., Kissick H. T., Burger M. C., Shan Q., Hale J. S., Lee J., Nasti T. H., Sharpe A. H., Freeman G. J., Germain R. N., Nakaya H. I., Xue H.-H., Ahmed R., Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeannet G., Boudousquié C., Gardiol N., Kang J., Huelsken J., Held W., Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl. Acad. Sci. U.S.A. 107, 9777–9782 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou X., Yu S., Zhao D.-M., Harty J. T., Badovinac V. P., Xue H.-H., Differentiation and persistence of memory CD8+ T cells depend on T cell factor 1. Immunity 33, 229–240 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Danilo M., Chennupati V., Silva J. G., Siegert S., Held W., Suppression of Tcf1 by inflammatory cytokines facilitates effector CD8 T cell differentiation. Cell Rep. 22, 2107–2117 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Fazakerley J. K., Southern P., Bloom F., Buchmeier M. J., High resolution in situ hybridization to determine the cellular distribution of lymphocytic choriomeningitis virus RNA in the tissues of persistently infected mice: Relevance to arenavirus disease and mechanisms of viral persistence. J. Gen. Virol. 72, 1611–1625 (1991). [DOI] [PubMed] [Google Scholar]
- 30.Iwasaki M., Urata S., Cho Y., Ngo N., de la Torre J. C., Cell entry of lymphocytic choriomeningitis virus is restricted in myotubes. Virology 458-459, 22–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burzyn D., Kuswanto W., Kolodin D., Shadrach J. L., Cerletti M., Jang Y., Sefik E., Tan T. G., Wagers A. J., Benoist C., Mathis D., A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levéen P., Carlsén M., Makowska A., Oddsson S., Larsson J., Goumans M. J., Cilio C. M., Karlsson S., TGF-β type II receptor–deficient thymocytes develop normally but demonstrate increased CD8+ proliferation in vivo. Blood 106, 4234–4240 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Pasut A., Jones A. E., Rudnicki M. A., Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J. Vis. Exp. 2013, e50074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaba3458/DC1