

Abstract
The multidrug resistance (MDR) system effectively expels antibiotics out of bacteria causing serious issues during bacterial infection. In addition to drug, indole, a common metabolic waste of bacteria, is expelled by MDR system of gram-negative bacteria for their survival. Experimental results suggest that AcrB, one of the key components of MDR system, undergoes large scale conformation changes during the pumping due to proton-motive process. However, due to extremely short time scale, it is difficult to observe (experimentally) those changes in the AcrB, which might facilitate the pumping process. Molecular simulations can shed light to understand the conformational changes for transport of indole in AcrB. Examination of conformational changes using all-atom simulation is, however, impractical. Here, we develop a hybrid coarse-grained force field to study the conformational changes of AcrB in presence of indole in the porter domain of monomer II. Using the coarse-grained force field, we investigated the conformational change of AcrB for a number of model systems considering the effect of protonation in aspartic acid (Asp) residues Asp407 and Asp408 in the transmembrane domain of monomer II. Our results show that in the presence of indole, protonation of Asp408 or Asp407 residue causes conformational changes from binding state to extrusion state in monomer II, while remaining two monomers (I and III) approach access state in AcrB protein. We also observed that all three AcrB monomers prefer to go back to access state in the absence of indole. Steered molecular dynamics simulations were performed to demonstrate the feasibility of indole transport mechanism for protonated systems. Identification of indole transport pathway through AcrB can be very helpful in understanding the drug efflux mechanism used by the MDR bacteria.
Keywords: multidrug resistance bacteria, AcrB, indole, coarse grained molecular simulation
1. INTRODUCTION
Multidrug efflux pumps in gram-negative bacteria often associated with drug resistance that helps to decrease drug accumulation inside the bacteria. The treatment of bacterial infections, as well as cancer chemotherapies, are hampered by the overexpression of efflux pumps, which is termed as multidrug resistance systems (MDRS).1,2 MDRS actively expel drug molecules from inside to outside of the bacterial cell. Additionally, these efflux proteins could also expel potentially harmful metabolic wastes produced by the bacteria themselves. One such example is indole, which is a metabolite produced from tryptophan amino acid. Since indole is toxic for all cells, Escherichia coli (E. coli) organisms defend themselves against the toxic action of indole by throwing indole out from the cell using efflux pumps.
MDRS is composed of AcrB, AcrA, and TolC 3–5 in Escherichia coli as shown in Figure 1a. Protein trimer AcrB spans across the inner and outer membranes of the cell, TolC is outer membrane channel,6 and AcrA is an adaptor protein that connects AcrB and TolC.7 Overexpression of the key AcrB component expels drug molecules out of the cell by antiport mechanism,3,8–12 during which the proton and substrate are transported in opposite directions. AcrB can be decomposed into three domains: the trans-membrane (TM) domain responsible for the proton transfer, the porter domain accounting for substrate recognition, binding, and extrusion, and the docking domain connected to TolC.13
Figure 1.
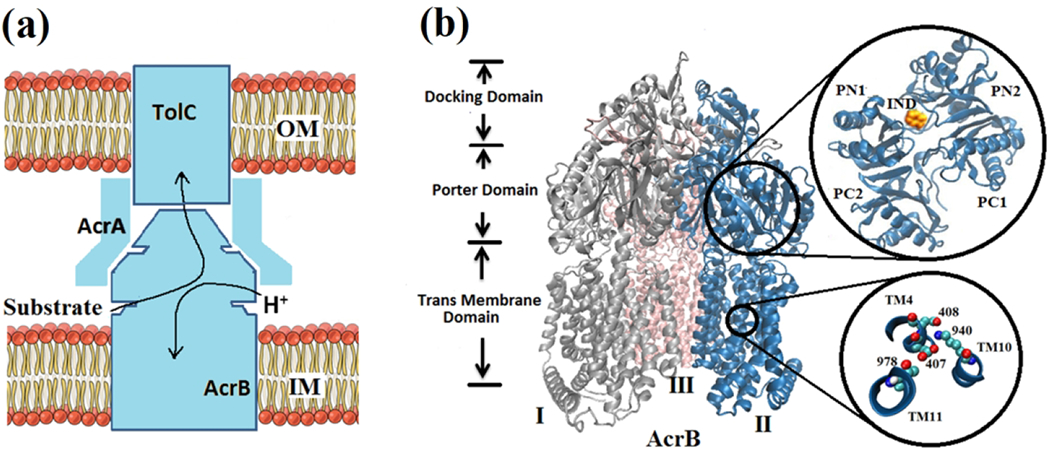
Multidrug resistance (MDR) system. (a) Schematic of the AcrB-AcrA-TolC tripartite efflux system. Arrows indicate the substrate/proton antiport directions. (b) New cartoon representation of the crystal structure of AcrB trimers: Monomer-I (silver color), Monomer-II (blue color) and Monomer-III (pink color). Each monomer can be decomposed into the transmembrane domain, porter domain, and docking domain. Porter domains are composed of four subdomains: PC1, PC2, PN1 and PN2 (top view of the monomer-II is magnified at the top-right by a black circle). Indole (IND) molecule has been shown in vdW representation. Ball and stick representation of the key residues in the TM domain (region magnified at the bottom-right by a black circle): Aspartic acid 407 (Asp407), aspartic acid 408 (Asp408), lysine 940 (Lys 940) and threonine 978 (Thr978).
AcrB trimer consists of three monomers, Monomer I, Monomer-II and Monomer III, as shown in Figure 1b. The crystal structure of symmetric monomers was first resolved in 2002.14 Later in 2006 and 2007,9,15,16 the asymmetric X-ray of AcrB has been captured. Based on the symmetric and asymmetric crystal structures of AcrB as well as their functionalities, three different states: access (A), binding (B) and extrusion (E) and a functional rotation mechanism have been proposed for substrate transport. Each of the AcrB monomers can be divided into four subdomains (PC1, PC2, PN1, and PN2). Top view of these four subdomains of monomer-II is magnified in the top-right of the Figure 1b.
Monomers undergo a series of structural changes from access to binding, then extrusion states during substrate transport. Although substrate translocation and the associated protein conformational change primarily occur in the porter domain, transduction energy of proton comes from the trans-membrane (TM) domain. From site-directed mutagenesis studies,17,18 four key residues in the TM domain have been identified as crucial components for the transport process. These key residues, Asp407, Asp408, Lys940, and Thr978, are magnified in the bottom-right of the Figure 1b with their corresponding helices.
To our knowledge, the exact transport mechanism of drug molecules, as well as indole, in AcrB is largely unclear. Recently all-atom molecular simulations have been performed to investigate the molecular mechanisms involved in drug binding and export of AcrB19–25, but the large-scale conformational changes of AcrB were mostly overlooked, or decoupled from the drug transport due to the expensive computational cost associated with the all-atom force fields. Lately, Vargiu et al. 26 have developed a computational protocol based on biased all-atom molecular simulations to investigate the important roles of the solvent for substrate transport during the binding to extrusion transition. In their method, the conformational change of AcrB during transition was simulated using targeted molecular dynamics, while the transport of the substrate (i.e. DOX) was studied using steered molecular dynamics. On the other hand, structure-based coarse-grained model has also been implemented to study the dynamics of AcrB27,28, but detailed and important molecular interactions were missing because of the coarse-graining. In one of our recent works29, we have successfully implemented a hybrid PACE force field30–36 to study the proton-dependent conformational changes of AcrB in microsecond simulations, but in absence of any substrate.
In this work, we investigate the conformational changes and the associated molecular mechanisms of AcrB in presence of indole using molecular simulations with PACE force field. We first develop the united-atom force field parameters for indole molecule using methodology similar to a sugar force field in Jewel et al. 37. After validation, we implement the force field to study the antiport transport process for indole. Here we are particularly interested in the effects of the protonation states of two key titratable residues: Asp407 and Asp408 in the TM domain of monomer II (aka binding monomer) to see large scale conformational changes in the porter domain of AcrB. Our results can explain a complete transport of indole from its distal binding pocket to the central funnel. We find sequential changes from binding state to extrusion state with important interactions of the protein with proton as well as indole. We believe the indole transport mechanism could be similar to drug transport through AcrB monomers for multidrug resistance.
2. MATERIALS AND METHODS
2.1. PACE Protein Model
PACE is a hybrid force field developed by Han et al.30–35 with a united-atom-based model (each heavy atom represents one site) for proteins coupled with the coarse-grained MARTINI38,39 water and lipid model (four heavy atoms represent one site). In protein models, each heavy atom with the attached hydrogen atoms is generally modeled with one site, but the hydrogens on backbone and side chain amide groups are also explicitly expressed for better accounting of H-bonding. Both bonded and nonbonded interactions are included in PACE and the total energy of the system can be expressed as:
| (1) |
The first four terms account for bonded interaction mediated by covalent bonds and the last four terms take care of the nonbonded interactions including water-water interactions (EW−W) water-protein interactions (EW−P), interactions between nonpolar protein sites (EvdW) and interactions between polar sites (Epolar). Eϕ,ψ,χ1 is for interactions between rotamers of the backbone (ϕ and ψ) and the side chains (χ1. The Lennard-Jones (LJ) potential is used for nonbonded interactions:
| (2) |
Here, εij represents the inter-particle binding energy and δij is the van der Waals radius. The distance between particles i and j is denoted by rij . For bonded interactions in Eq. (1), the equilibrium bond length and angle values were taken from the optimized geometries by quantum mechanics (QM) calculation. The dihedral parameters were obtained by fitting QM dihedral potential profiles of small molecules. Improper terms were used to maintain the planarity or chirality of groups. The interaction parameters for Eϕ,ψ,χ1 were obtained through iterative equilibrium simulations against side chain rotamer distributions and rotamer-dependent backbone conformations from a coil library. The parameters for water-protein interactions in Eq. (2) were optimized by fitting hydration free energies of 35 small organic molecules. Evdw parameters were obtained on the basis of densities of liquid states and free energies of evaporation of 8 organic compounds. The polar and charged sites interactions were optimized by fitting the potential mean forces (PMFs) from all-atom simulations with the OPLS-AA/L40 force field in explicit water. Details on the model development and parameter optimization can be found in Refs.32,34
2.2. The Indole Model
In this work, we have extended the original PACE force field to include indole molecule in our simulation. We have followed the same procedures as mentioned in the PACE to develop parameters for indole 32–35,37. A similar procedure has been implemented for glucose force field in our previous work 37. Briefly, indole molecule adopts the united-atom force field in which the hydrogen and the attached heavy atoms are combined to form one site. However, a hydrogen attached to nitrogen atom has been explicitly represented following the same procedures as the tryptophan molecule in the original PACE protein model. The bonded interactions parameters were obtained using the simplified Boltzmann inversion method.41 We have developed the indole-water interactions from the hydration free energy (HFE) calculations. In a water box of 30×30×30 Å3, an indole molecule was added both in coarse-grained and all-atom simulation to calculate HFE. The value for all-atom indole was calculated ~6.7 kcal/mol using free energy perturbation (FEP) methodology42. By changing the indole-water interaction parameters as well as by recalculating HFE for united-atom indole we have adjusted the HFE to ~6.4 kcal/mol, which is close to all-atom simulation value. By taking the identical simulation box, C-C parameters as well as N-N atom parameters of united-atom indole have been determined using PMF matching technique from all-atom simulations. Finally, for indole protein interactions, Lorentz-Berthelot rules have been applied.
Validation of united-atom indole force field has been performed in two consecutive steps. First, we have compared the binding process of indole molecule to the binding pocket of AcrB for both all-atom and united-atom model. Figure 2a shows the all-atom model of the indole molecule with the ball and stick representation. Indole binding site, as well as important amino acids, are also shown. After placing indole away from the binding site, we have performed all-atom as well as CG simulations only taking porter domain portion of monomer II in a periodic box 48× 48× 48 Å3 with water molecules. Figure 2b shows the distances between indole and the binding site (center of mass of 4 amino acids: phenylalanine 136 (Phe136), Phe178, Phe615, and Phe617) with respect to time. As shown, we have achieved almost the same binding dynamics between all-atom (red line) and united-atom (green line) indole. Next, we have compared the binding energy between indole with indole binding protein (PDB ID: 3SNM) using PMF calculations. Figure 2c shows our simulation system. The PMF profiles are calculated using adaptive biasing force (ABF) methodology. Indole has been constrained to move unidirectionally while calculating PMF in both all-atom and CG simulations. The comparison between all-atom PMF (red line) with hybrid PACE PMF (green line) has been presented in Figure 2d. The PMF profiles in general agree well between PACE and all-atom models. The binding energy of the PACE indole matched well (~lkcal/mol) with the all-atom model. Some deviations near the binding site are also observed, PACE model yields one energy minima while the all-atom model shows two. This is due to the coarse-graining in the PACE force field, such as the MARTINI water model and the united-atom protein/indole model.
Figure 2.
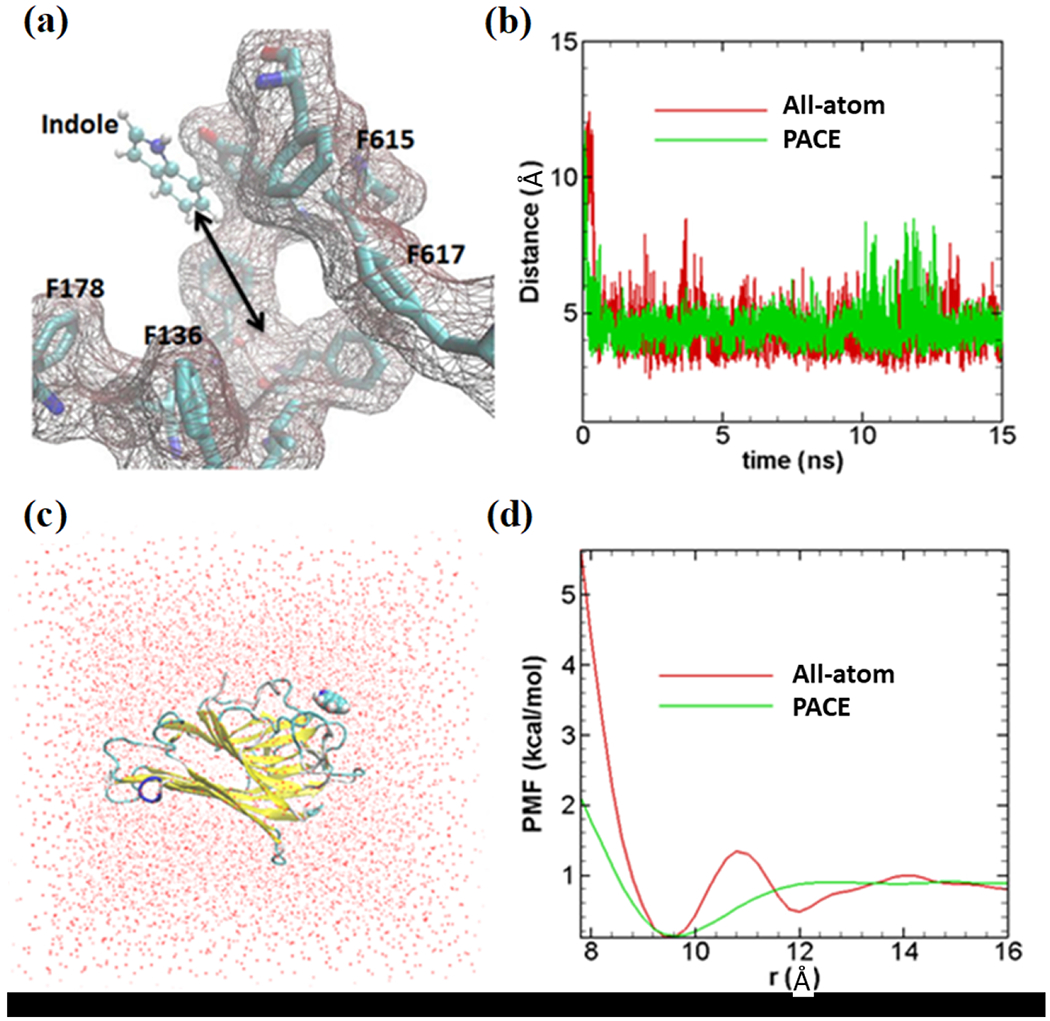
Validation of coarse-grained indole force field. (a) All-atom model of indole molecule has been shown in ball and stick representation; binding site with important amino acids are also shown. (b) Distances between indole center of mass and center of 4 amino acids shown in Figure 2a. All-atom indole binds to binding site very quickly (within ~1 ns) and stays close to the binding site as presented with a red line. United-atom indole (green line) shows similar dynamics as all-atom simulation. (c) All-atom simulation model (periodic box of 48×48×48 Å3) to calculate PMF; red dots are representing water molecules. Indole binding protein (PDB ID: 3SNM) is shown in new-cartoon representation. (d) PMF profiles for both methods. All-atom PMF, indicated by red lines, (~1kcal/mol) matches fairly well with PACE PMF (green line). Indole has been constrained to move unidirectionally while calculating PMF both in all-atom and PACE simulations.
The new indole force field was implemented with PACE to study indole/H+ antiport. All simulations were performed using the modified version of NAMD 2.10.43 PACE hybrid force field were adopted for protein, lipid, water and ion molecules. The simulation models were constructed using CHARMM-GUI44–47 and VMD.48 Figure 3 demonstrates our simulation system. As shown, an asymmetric AcrB trimer in absence of substrates (PDB ID: 2DHH)15 was initially embedded in a POPE lipid bilayer membrane. The missing parts of AcrB in the original crystal structure were modeled from another AcrB crystal structure (PDB ID: 2J8S).16 The whole system contains around 30,000 coarse-grained MARTINI waters and 415 lipids after removal of the lipids overlapping with the protein . A small number of chloride ions was added to neutralize the system.
Figure 3.
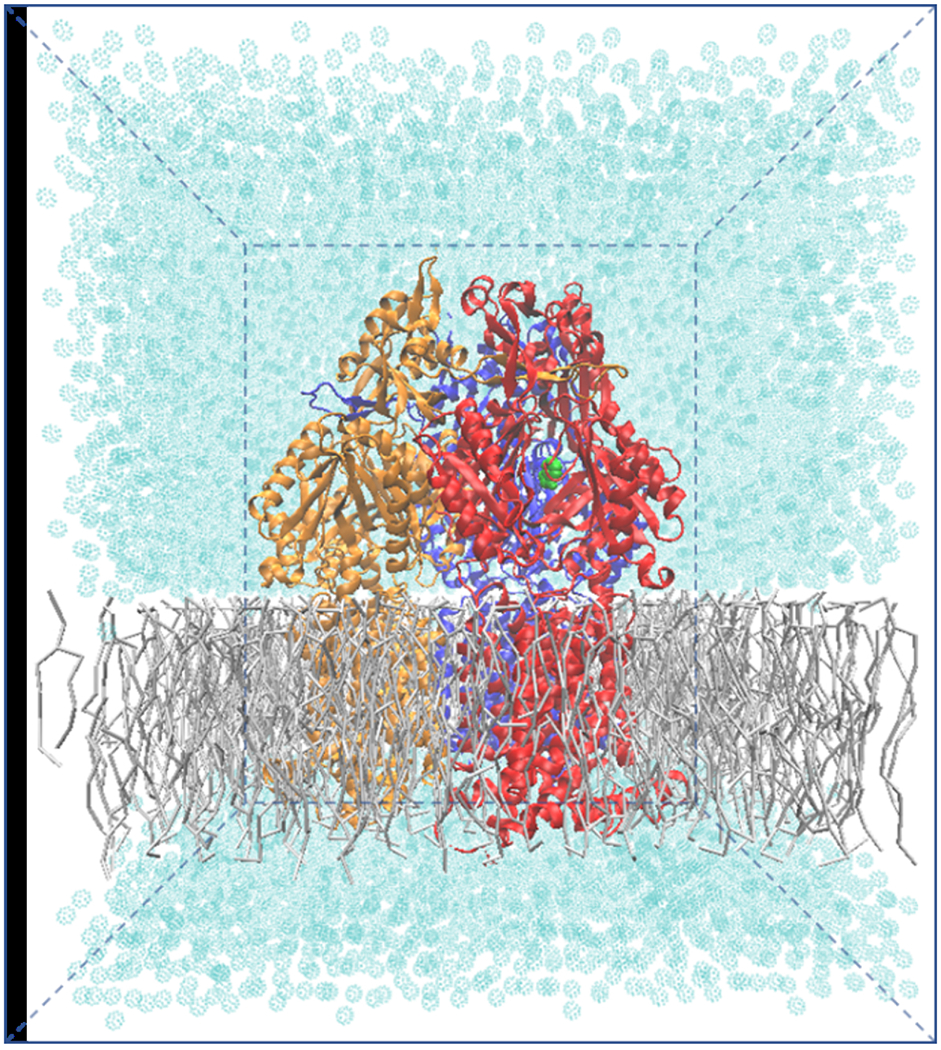
Our coarse-grained simulation model contains ~50,000 atoms with dimensions of ~140 × 140 × 160 Å3. Protein is placed near the center of the water-filled box. Gray colors are used for presenting lipid molecules with licorice representation. Cyan dots are presenting MARTINI waters (~30,000).
The lipid bilayer systems were equilibrated using the standard six-step equilibration process44–47 of gradually turning off the constraints at ~300 ps at a temperature of 300 K. In production simulations, periodic boundary conditions were applied in all three directions. The van der Waals interactions were calculated using LJ potential with a cutoff of 12 Å. Production simulations were carried out for ~2 μs using NPT (T=300 K, P=1 atm) ensembles. Since the protein force field is close to the all-atom model, a time-step of 5 fs was chosen. However, the total number of atoms is significantly reduced compared with all-atom simulations due to the use of MARTINI water and lipids. Graphical software VMD48 has been used for the analysis of atomic distances and taking snapshots.
To investigate the large-scale conformational changes in the porter domain of AcrB induced by indole and the different protonation states at TM domain, four model systems have been created. Since, we have added indole in the porter domain of monomer-II, effect of protonation in Asp407 and/or Asp408 on monomer II has been investigated in our simulations. Model details are presented in Table 1 for all four systems. Both Asp407 and Asp408 were deprotonated in the first model, Asp408 is protonated in the second model, Asp407 is protonated in the third model, and both Asp407 and Asp408 are deprotonated in the fourth model. Indole was added in model system 1, 2 and 3 at the distal binding pocket (i.e. similar position of the center of mass of DOX in PDB ID 2DHH) of monomer-II. Three independent simulations for each model were performed for statistical consistency. Since all three simulations for each model show similar results, only one of the simulation results are presented in the following sections for clarity.
Table 1.
List of the model systems used for molecular simulation. In all four models, the residues Asp407 and Asp408 in TM domain are kept deprotonated in monomer I and monomer III. For system 2, the Asp408 of monomer II is protonated, while for system 3 the Asp407 of monomer II is protonated. Indole molecule has been placed in monomer II for the first three systems.
| Model System | Indole | Monomer I | Monomer II | Monomer III |
|---|---|---|---|---|
| 1 | Yes | D407−/D408− | D407−/D408− | D407−/D408− |
| 2 | Yes | D407−/D408− | D407−/D408H | D407−/D408− |
| 3 | Yes | D407−/D408− | D407H/D408− | D407−/D408− |
| 4 | No | D407−/D408− | D407−/D408− | D407−/D408− |
3. RESULTS
3.1. Large-scale Conformation Changes in the Porter Domain
It has been proposed that the local conformational changes in the TM domain caused by the proton translocation can lead to the large-scale global movements in the porter domain. These global conformational changes are eventually responsible for the opening as well as the closing of the substrate transport channels to accommodate the substrate export. Based on the crystal structures of AcrB, in each monomer two important binding pockets: proximal and distal binding pockets have been identified.49,50 The proximal binding pocket is connected with several possible entrance channels including vestibule channel,15 cleft channel,9,16 and a channel from the central cavity.49 These entrance channels and proximal binding pockets are responsible for substrate uptake. An exit channel on the top of the porter domain is responsible for substrate export. In this section, we will focus on the large-scale structural changes in the porter domain of AcrB. We will monitor the conformational changes both at the cleft entrance channel (a preferred pathway for larger substrates) and at the exit channel.
3.2. Cleft Entrance Channel
As shown in Figure 4a, we monitor the changes in the cleft entrance by measuring the center of mass distance between Thr676 and Phe563. Phe563 is located at the TM domain with negligible global movement throughout our simulations. The Thr676 adopts different orientations to control the intake of substrates underneath of Phe617-loop. The distance between residues Phe563 and Thr676 for all simulated systems are monitored and presented with respect to time in Figure 4b. For model system 1, it has been observed that drug entry gate does not show any conformational changes (very small changes). However, for model system 2, entry gate closes as Thr676 moves towards Phe617-loop. The model system 3 shows similar behavior like the model system 2 as the Thr676 goes towards the Phe617-loop. Similar to model system 1, no conformational changes (or minimal conformational changes) has been observed in case of model system 4 in which no indole or proton has been added to the system.
Figure 4.
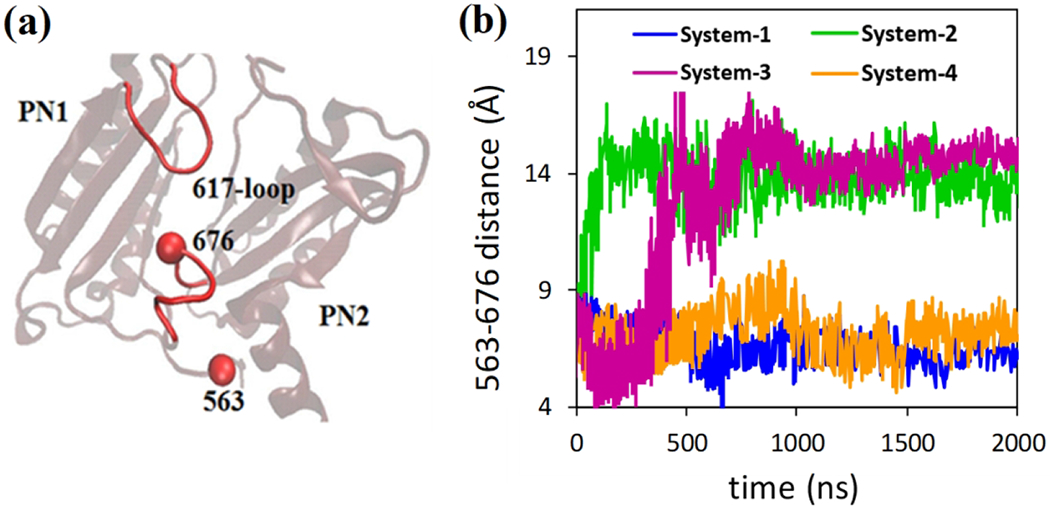
Conformational changes at the entry gate for all systems. a) Phe617-loop resides between proximal and distal binding pocket. Conformations of residue Thr676 are monomer specific, and it has been measured here by taking phenylalanine 563 (Phe563) residue as a reference. b) Distances between residues Phe563 and Thr676 are plotted with respect to time. In the model systems 1 and 4, the entry gate (distances between Thr676 and Phe617 loop) remains unchanged while for the model system 2 and 3, the entry gate closes.
3.3. Exit Channel
The exit gate is formed by residues glutamine 124 (Gln124) and tyrosine 758 (Tyr758) to control the passage or blockage of the substrate molecules.16 Therefore to monitor the changes of the exit channel, we measured the center of mass distances between Gln124 and Tyr758 (see Figure 5a) in our simulations. Figure 5b shows the time evolution of the Gln124-Tyr758 distances. For model system 1 it has been observed that exit gate opens as Tyr758 loop moves away from residue Gln124. Similar conformational changes have been observed for model system 2 and 3 in which exit gate also opens. However, no conformational changes (or minimal conformational changes) has been observed in case of model system 4 in which no indole/proton has been added to the system. The opening/closure of the exit gate is also confirmed by the following pore radius measurements.
Figure 5.
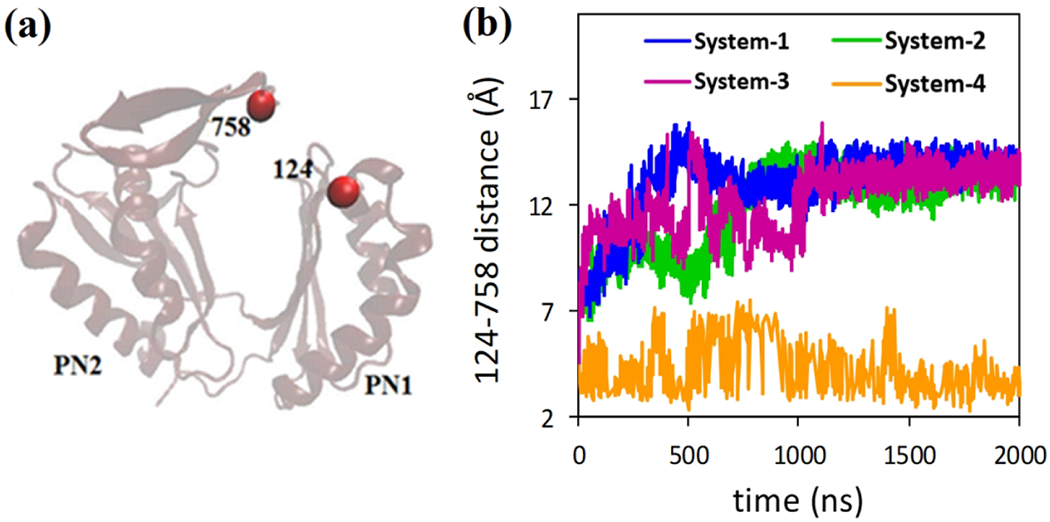
Conformational changes at the exit gate for all systems. a) Residue Gln124 and Tyr758 control the substrate extrusion by a close/open motion of the gate. b) Distances between residues Gln124 and Tyr758 are plotted with respect to time. In model systems 1, 2 and 3 (with indole) the exist gate opens for substrate extrusion, while no changes are observed for model system 4 (no indole and protonation).
3.4. Pore Radius Change
To further investigate the conformational transition of the systems, we measured cavity radius of channel 1 and 2 for each monomer as shown in Figure 6. The pore radius are determined using CAVER 2.0,51 at the end of the simulations. Figure 7 shows the pore radius profile for all four systems presented in Table 1. The pore radius profile for each model system is calculated by averaging over 100 frames of the last 100 ns simulation data and over three independent simulations. (We do not show the statistical error bars in Figure 7 for clarity purpose, however, the information regarding the error bars can be found in Supporting Information, Figure S1 and S2). For comparison, the pore radius profiles are also presented for crystal structure (dashed lines) of each monomer.
Figure 6.
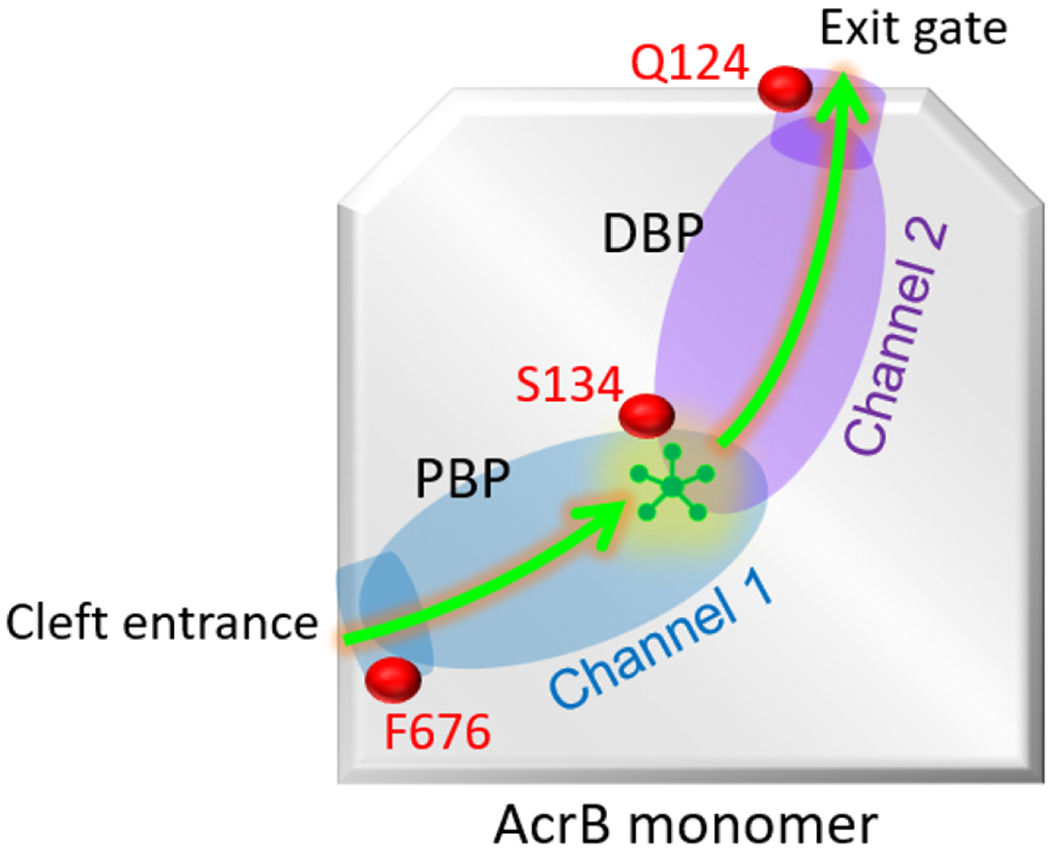
Schematic of the substrate (indole) transport pathway inside AcrB monomer. The pathway starts with the cleft entrance and ends at the exit gate. Channel 1 consists of cleft entrance and proximal binding pocket (PBP), while the channel 2 consists of exit gate and distal binding pocket (DBP). The relative location of important residues such as Phe676 (F676), Serine 134 (S134), and Glutamine 124 (Q124) are shown along the channel.
Figure 7.
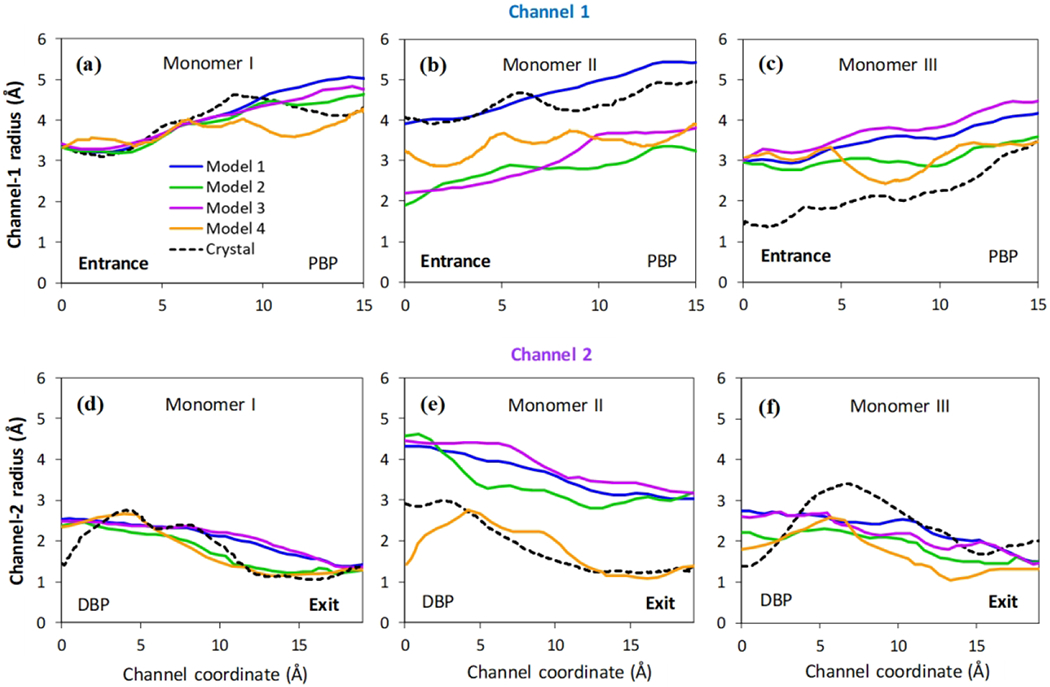
Pore radius profiles. The pore radius profiles calculated by CAVER 2.06 for channel 1 (a, b and c) and channel 2 (d, e and f) of all three monomers for four models considered here. Solid lines present the averages from 100 frames of the last 100 ns simulations and three independent realizations. The pore radius profiles from the original crystal structure (PDB ID: 2DHH) are also included as black dash lines for comparison.
For model system 2, the pore radius profile is very similar to that of crystal structure in monomer I for both channel 1 and 2, suggesting that the monomer I remains in access state. The pore radius profile for monomer III is similar to monomer I, indicating a transition from extrusion state to access state after 2 μs. On the other hand, for monomer II, the pore radius measurements clearly demonstrate the closing at the entrance region and opening at the exit region. This strong evidence suggests that the monomer II is transitioning from native binding state to extrusion state. In summary, upon protonation of Asp408 in transmembrane domain of monomer II, AcrB can facilitates substrate (indole) transport through monomer II, while monomer I and III are ready for substrate intake.
Like model system 2, the entrance gate opens and exit gate closes for monomer I in model system 3. Moreover, the pore radius profile in monomer I does not changes significantly in channel 1, but minor deviation is observed in channel 2 close to the distal binding pocket (DBP). However, the overall trend follows the native crystal structure of monomer I, implying that monomer I will remain in the access state. For monomer II, the pore radius profile is similar to system 2 suggesting that monomer 2 changes from the binding state to extrusion state. On the other hand, for monomer III, the entrance gate opens and exit gate closes for system 3. Moreover, the radius profile is very similar to native crystal structure of monomer I, suggesting transition to access state for monomer III.
Similar to model system 2 and 3, substrate is present in the binding pocket of monomer II for model system 1, but none of the aspartic acid is protonated in any monomers. For this non-protonated case, both entrance and exit gate stay open for monomer II, and does not provide any known conformation. However, radius data of monomer I and III closely mimic the native crystal state of monomer I, indicating transition to access state. In the absence of any substrate and/or protonation (model system 4), all three monomers approach the native crystal structure of monomer I in both channel 1 and 2. In other words, in the absence of the substrate, all three monomers transition to access state. Table 2 summarizes the conformational change results from our molecular simulation.
Table 2.
Conformational change of the considered systems for all three monomers.
| Model System | Indole | Monomer I | Monomer II | Monomer III |
|---|---|---|---|---|
| 1 | Yes | A to A | Unclear transition | E to A |
| 2 | Yes | A to A | B to E | E to A |
| 3 | Yes | A to A | B to E | E to A |
| 4 | No | A to A | B to A | E to A |
3.5. Indole Transport
To monitor the indole movement, we have measured indole positions with respect to residue Phe563 as shown in Figure 8a. We have started our indole simulations (i.e. model system 1, 2 and 3) from the position indicated as 1. In model system 2 and 3, indole moves close to residue Phe563 at ~1.5 μs, and the position is indicated by 2. Distances between residue Phe563 and indole center have been plotted in Figure 8b with respect to time. For model system 1, indole moves slightly towards residue 563 and becomes stabilized. The porter domain of monomer-II displayed different conformations for model system 2 and 3, where indole moved much closer to residue 563 compared to model system 1. However, regardless of the protonation states of 407 and 408, indole obtained stable positions near the binding site for all three systems after ~2 μs.
Figure 8.
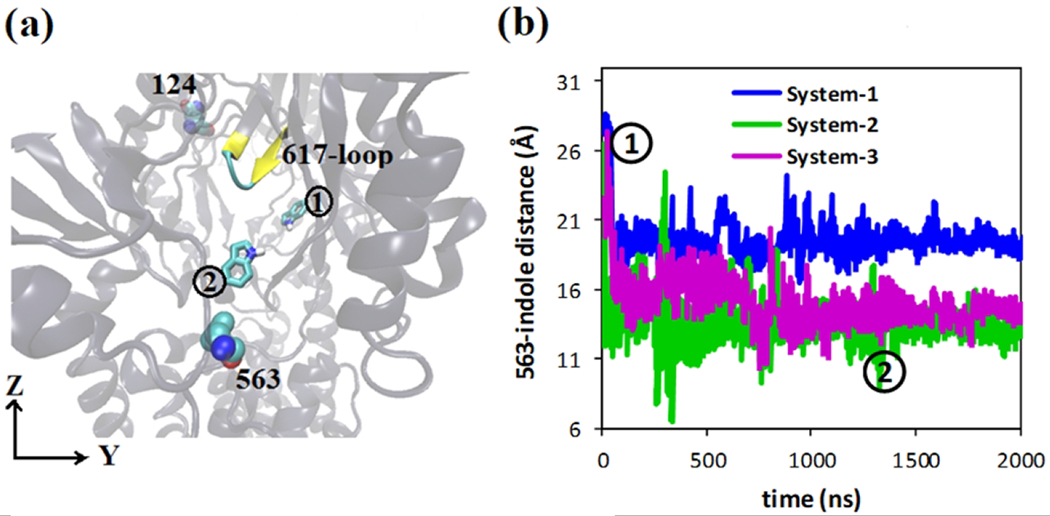
Indole equilibration. a) Initial position of indole (presented in licorice representation) at the binding pocket as indicated by 1; stable residues Phe563 and Gln124 are presented in vdW representation; Phe617-loop is presented in New Cartoon representation. At the end of the simulation (~2 μs) indole moved to new position near to residue Phe563 as indicated by 2 observed in model system 2 and 3. b) Distances between residues 563 and indole are plotted with respect to time; positions 1 and 2 are also indicated.
To explore the indole transport, after the equilibrium simulations for model system 3 we have applied a pulling force force to indole molecule toward the exit gate (residues Gln124 and Tyr758) to facilitate the movement of indole. Using steered molecular dynamics (SMD) simulations, we have forced indole to move with a constant velocity 0.0005 A/ts (spring constant k=5 kcal/mol/A2) as shown in Figure 9(a). Figure 9(b) shows the magnitude of the pulling force (Fpull) as a function of the indole displacement (drel) relative to its initial position in DP. For comparison purpose, we also performed SMD simulations by pulling the indole out of AcrB with initial crystal structure (PDB ID: 2DHH) mimicking a binding state when the exit gate is closed. As shown, when the exit gate is open (Extrusion state), the pulling force profile is relatively smooth and no large forces are observed. However, when the exit gate is closed (Binding state), a clear peak force of ~8 kcal/(mol · Å) is required for the indole to overcome bottleneck around the exit gate. The results demonstrate the importance of the conformational transition, i.e. from Binding to Extrusion for model system 3, in facilitating the indole transport. Moreover, the indole pathway, as well as important residues (i.e. residue Val672, Gly179, Ser135, Lys292, Ser132, Ser46, Gln176, Ser128, Glu130, Ser180, Val129, Ser48 and Gln120), have been identified and illustrated in Figure 9(c) with ball and stick representations.
Figure 9.
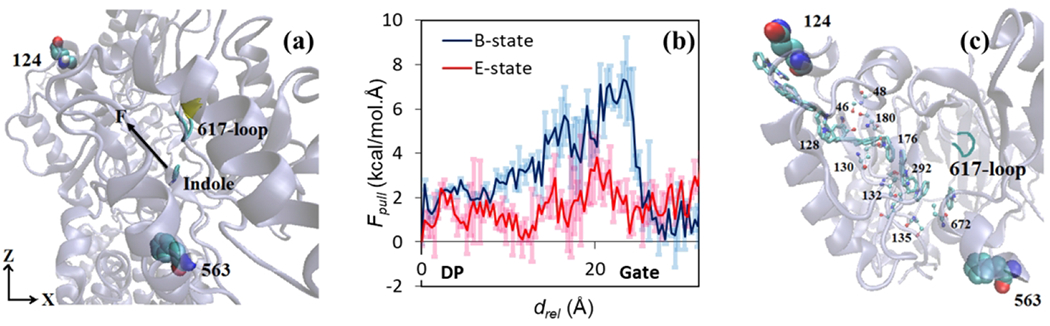
Steered MD simulations of indole transport. (a) Additional pulling force has been applied starting from indole equilibrium position towards the exit gate (Gln124 and Tyr758 (not shown)). (b) Profiles of the pulling force along the indole pathway. (c) Important lining residues along the indole pathway presented in a ball and stick representation. Indoles (presented in licorice representation) are imposed throughout the transport pathway of the porter domain of monomer-II.
The hydration of the transport channel play key roles in mediating the indole-protein interactions and indole transport.26 As shown in Figure 10(a) for model system 3, we observe a smooth and continuous water distribution when the exit gate is open (Extrusion state). However, at the initial binding state when the gate is closed, the water distribution is discontinuous at the exit gate. This is also clearly reflected in Figure 10(b) when we calculate the number of water along the channel coordinate. The continuous water distribution, i.e. hydration of indole and AcrB internal surface, effectively screen the indole-AcrB interactions and promote the indole transport.
Figure 10.
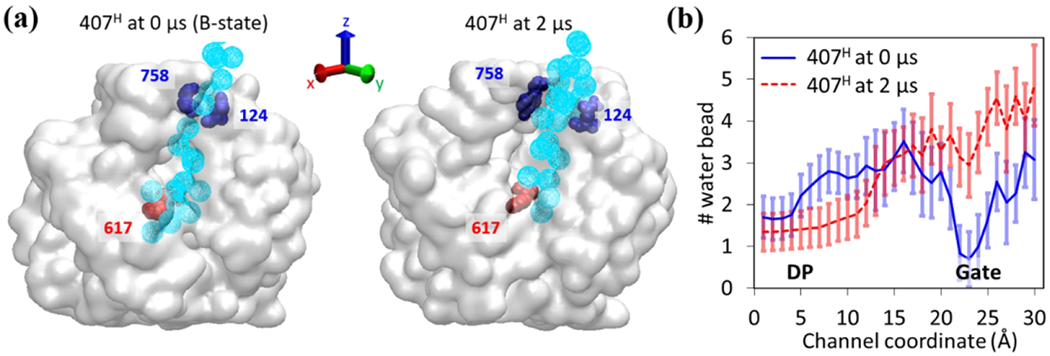
Hydration of the transport channel for model system 3 (407H). (a) Water distribution maps along the transport channel from DP (Phe617) to exit gate (Tyr758 and Gln124) at the initial (Binding) state (left) and the final (Extrusion) state (right). The water molecules are shown in light blue spheres. (b) The number of MARTINI water beads along the transport channel. The data are averaged over 100 frames during the initial and last 100 ns simulations and over three independent realizations.
4. DISCUSSION
We have monitored the conformational changes at entry and exit gates of the porter domain for monomer-II to quantify indole dependent changes. For model system 1 and model system 4, Phe676 loop does not move towards Phe617-loop, indicating open conformation of the drug entry gate. We have monitored the closure of entry gate similar to extrusion state structure for model system 2 and 3 as shown in Figure 4. Additionally, for model system 1, 2 and 3, exit gate (distance between residue Gln124 and residue Tyr758) was found to open like the extrusion state structure. No changes have been observed in the exit gate for the model system 4 as shown in Figure 5.
Our results also demonstrate that Indole is only able to change its host monomer (monomer II in our case) with protonation, as shown in model system 2 and 3, from binding state to extrusion state. Thus, the fully functional rotating mechanism ceases to exist if only one of three monomers host the substrate. It has been found that in the absence of indole in the monomer I or III, the corresponding monomer remains in the access state and disrupts the functional rotation.
Since AcrB operate with a substrate/proton antiport mechanism, the substrate translocation and the protein conformational changes in the porter domain must be tightly coupled with the proton transport and the helices movement at the trans-membrane (TM) domain. To gain the fundamental insights about the transport mechanism of TM domain of AcrB, Eicher et al. 52 have systematically analyzed the TM domain helices movement from an improved atomic structure of wildtype AcrB 50. Two 5-helix parallel repeats have been identified at the TM domain and each repeat is coupled with the porter domain by a single TM helix, TM2 and TM8, respectively. TM2 is connected to the PN2/PC2 units and TM8 is connected to PN1/PC2 units in the porter domain. Structural overlays and interpolations of the conformations from binding to extrusion states indicated that the TM2 moved upward, and TM8 moved closer to TM10 leading to a more compact state in the associated repeat. Here, we also examine the helices movements in TM domain from our simulation results. As shown in Figure 11, we focus on the model system 3, in which the Asp407 is protonated and as a result, the monomer II has been transitioned from binding to extrusion state after 2 μs simulation. Figure 11(a) shows the comparison between the initial binding state to the final extrusion state for several key helices. We do observe global movements during the transition. To quantify the movements, we calculate the center of mass distance between TM8 and TM10 (Figure 11(b)), and the vertical coordinate of the center of mass for TM2 (Figure 11(c)) as a function of time. As clearly shown, the TM8 moves closer to TM10, and TM2 moves upward within 500 ns. Our simulation results for helices movement in TM domain are consistent with the analysis from crystal structures. 52
Figure 11.
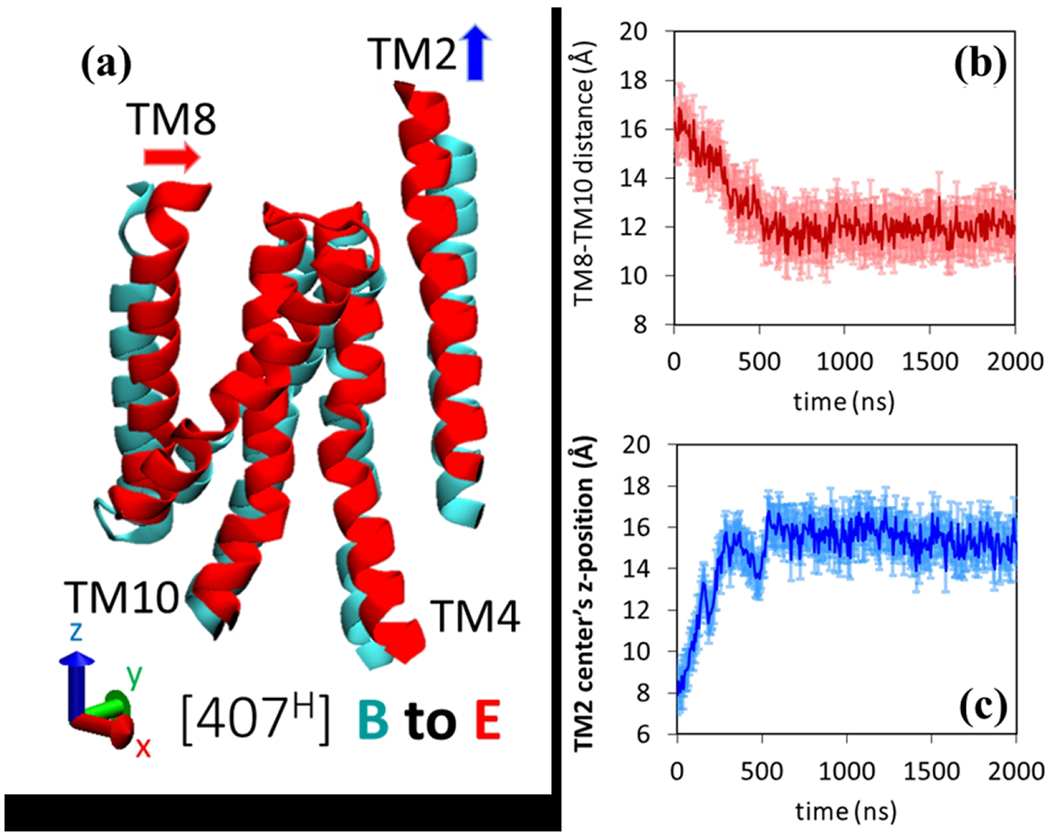
Helical movements in the TM domain for model system 3 (407H). (a) Comparison of the key helices in TM domain between initial (Binding: Cyan) state and final (Extrusion: Red) state. Time evolution of the (b) center of mass distance between TM8 and TM10 and (c) center of mass vertical z-coordinate TM2. The statistical error bars are calculated based on three independent realizations.
5. CONCLUSIONS
In this work, we have investigated the substrate transport mechanism of AcrB protein complex. Indole has been chosen as it is a metabolic waste of bacteria and its extrusion through AcrB protein of gram-negative bacteria is needed for survival. Using hybrid coarse-grained simulations, we have observed indole and proton-dependent conformational changes of AcrB, which can explain a complete transport of indole from its distal binding pocket to the central funnel. We have identified that only the presence of indole and protonation at 408 or 407 residues facilitate conformational changes from binding state to extrusion state for the host monomer. We also observed that, without indole or protonation, AcrB monomer will tend to stay at the access-state. It has been found that the functional rotation mechanism can be disrupted, if the access to substrate is blocked in any monomer. Steered molecular dynamics simulations and hydration analysis of the transport channel clarified the possible indole transport mechanism as well as the pathway. We have shown important residues involved in indole transport. We believe drugs, as well as other macromolecules, may follow a similar pathway for being transported through AcrB.
Supplementary Material
ACKNOWLEDGMENTS
The research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM122081 and the National Science Foundation under CBET-1604211. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Computational resources were provided in part by the Extreme Science and Engineering Discovery Environment (XSEDE) under grant No. MCB170012.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
References
- 1.Li XZ, Nikaido H. Efflux-mediated drug resistance in bacteria. Drugs 2004;64(2):159–204. [DOI] [PubMed] [Google Scholar]
- 2.Nikaido H Multidrug Resistance in Bacteria. Annu Rev Biochem 2009;78:119–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zgurskaya HI, Nikaido H. Bypassing the periplasm: Reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc Natl Acad Sci U S A 1999;96(13):7190–7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sulavik MC, Houseweart C, Cramer C, Jiwani N, Murgolo N, Greene J, DiDomenico B, Shaw KJ, Miller GH, Hare R, Shimer G. Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes. Antimicrob Agents Chemother 2001;45(4):1126–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tikhonova EB, Zgurskaya HI. AcrA, AcrB, and TolC of Escherichia coli form a stable intermembrane multidrug efflux complex. J Biol Chem 2004;279(31):32116–32124. [DOI] [PubMed] [Google Scholar]
- 6.Paulsen IT, Park JH, Choi PS, Saier MH. A family of Gram-negative bacterial outer membrane factors that function in the export of proteins, carbohydrates, drugs and heavy metals from Gram-negative bacteria. FEMS Microbiol Lett 1997;156(1):1–8. [DOI] [PubMed] [Google Scholar]
- 7.Dinh T, Paulsen IT, Saier MH. A family of extracytoplasmic proteins that allow transport of large molecules across the outer membranes of gram-negative bacteria. J Bacteriol 1994;176(13):3825–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zgurskaya HI, Nikaido H. Multidrug resistance mechanisms: drug efflux across two membranes. Mol Microbiol 2000;37(2):219–225. [DOI] [PubMed] [Google Scholar]
- 9.Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 2006;313(5791):1295–1298. [DOI] [PubMed] [Google Scholar]
- 10.Seeger MA, Diederichs K, Eicher T, Brandstaetter L, Schiefner A, Verrey F, Pos KM. The AcrB Efflux Pump: Conformational Cycling and Peristalsis Lead to Multidrug Resistance. Curr Drug Targets 2008;9(9):729–749. [DOI] [PubMed] [Google Scholar]
- 11.Seeger MA, von Ballmoos C, Eicher T, Brandstatter L, Verrey F, Diederichs K, Pos KM. Engineered disulfide bonds support the functional rotation mechanism of multidrug efflux pump AcrB. Nat Struct Mol Biol 2008;15(2):199–205. [DOI] [PubMed] [Google Scholar]
- 12.Takatsuka Y, Nikaido H. Covalently Linked Trimer of the AcrB Multidrug Efflux Pump Provides Support for the Functional Rotating Mechanism. J Bacteriol 2009;191(6):1729–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lomovskaya O, Zgurskaya HI, Nikaido H. It takes three to tango. Nat Biotechnol 2002;20(12):1210–1212. [DOI] [PubMed] [Google Scholar]
- 14.Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 2002;419(6907):587–593. [DOI] [PubMed] [Google Scholar]
- 15.Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 2006;443(7108):173–179. [DOI] [PubMed] [Google Scholar]
- 16.Sennhauser G, Amstutz P, Briand C, Storchenegger O, Gruetter MG. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol 2007;5(1):106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guan L, Nakae T. Identification of essential charged residues in transmembrane segments of the multidrug transporter MexB of Pseudomonas aeruginosa. J Bacteriol 2001;183(5):1734–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takatsuka Y, Nikaido H. Threonine-978 in the transmembrane segment of the multidrug efflux pump AcrB of Escherichia coli is crucial for drug transport as a probable component of the proton relay network. J Bacteriol 2006;188(20):7284–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vargiu AV, Nikaido H. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc Natl Acad Sci U S A 2012;109(50):20637–20642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer N, Kandt C. Porter domain opening and closing motions in the multi-drug efflux transporter AcrB. BBA-Biomembranes 2013;1828(2):632–641. [DOI] [PubMed] [Google Scholar]
- 21.Schulz R, Vargiu AV, Collu F, Kleinekathoefer U, Ruggerone P. Functional Rotation of the Transporter AcrB: Insights into Drug Extrusion from Simulations. PLoS Comput Biol 2010;6(6):e1000806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuo Z, Wang B, Weng J, Wang W. Stepwise substrate translocation mechanism revealed by free energy calculations of doxorubicin in the multidrug transporter AcrB. Sci Rep 2015;5:13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuo Z, Weng J, Wang W. Insights into the Inhibitory Mechanism of D13–9001 to the Multidrug Transporter AcrB through Molecular Dynamics Simulations. J Phys Chem B 2016;120(9):2145–2154. [DOI] [PubMed] [Google Scholar]
- 24.Schulz R, Vargiu AV, Ruggerone P, Kleinekathoefer U. Role of Water during the Extrusion of Substrates by the Efflux Transporter AcrB. J Phys Chem B 2011;115(25):8278–8287. [DOI] [PubMed] [Google Scholar]
- 25.Vargiu AV, Collu F, Schulz R, Pos KM, Zacharias M, Kleinekathoefer U, Ruggerone P. Effect of the F610A Mutation on Substrate Extrusion in the AcrB Transporter: Explanation and Rationale by Molecular Dynamics Simulations. J Am Chem Soc 2011;133(28):10704–10707. [DOI] [PubMed] [Google Scholar]
- 26.Vargiu AV, Ramaswamy VK, Malvacio I, Malloci G, Kleinekathofer U, Ruggerone P. Water-mediated interactions enable smooth substrate transport in a bacterial efflux pump. Biochimica Et Biophysica Acta-General Subjects 2018;1862(4):836–845. [DOI] [PubMed] [Google Scholar]
- 27.Yao X-Q, Kenzaki H, Murakami S, Takada S. Drug export and allosteric coupling in a multidrug transporter revealed by molecular simulations. Nat Commun 2010;1:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao XQ, Kimura N, Murakami S, Takada S. Drug Uptake Pathways of Multidrug Transporter AcrB Studied by Molecular Simulations and Site-Directed Mutagenesis Experiments. J Am Chem Soc 2013;135(20):7474–7485. [DOI] [PubMed] [Google Scholar]
- 29.Jewel Y, Liu J, Dutta P. Coarse-grained simulations of conformational changes in multidrug efflux transporter AcrB. Mol Biosyst 2017;13:2006–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han W, Wu Y-D. Coarse-grained protein model coupled with a coarse-grained water model: Molecular dynamics study of polyalanine-based peptides. J Chem Theory Comput 2007;3(6):2146–2161. [DOI] [PubMed] [Google Scholar]
- 31.Han W, Wan C-K, Wu Y-D. Toward a Coarse-Grained Protein Model Coupled with a Coarse-Grained Solvent Model: Solvation Free Energies of Amino Acid Side Chains. J Chem Theory Comput 2008;4(11):1891–1901. [DOI] [PubMed] [Google Scholar]
- 32.Han W, Wan C-K, Jiang F, Wu Y-D. PACE Force Field for Protein Simulations. 1. Full Parameterization of Version 1 and Verification. J Chem Theory Comput 2010;6(11):3373–3389. [DOI] [PubMed] [Google Scholar]
- 33.Han W, Wan C-K, Wu Y-D. PACE Force Field for Protein Simulations. 2. Folding Simulations of Peptides. J Chem Theory Comput 2010;6(11):3390–3402. [DOI] [PubMed] [Google Scholar]
- 34.Han W, Schulten K. Further Optimization of a Hybrid United-Atom and Coarse-Grained Force Field for Folding Simulations: Improved Backbone Hydration and Interactions between Charged Side Chains. J Chem Theory Comput 2012;8(11):4413–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wan C-K, Han W, Wu Y-D. Parameterization of PACE Force Field for Membrane Environment and Simulation of Helical Peptides and Helix-Helix Association. J Chem Theory Comput 2012;8(1):300–313. [DOI] [PubMed] [Google Scholar]
- 36.Jewel Y, Dutta P, Liu J. Coarse-grained simulations of proton-dependent conformational changes in lactose permease. Proteins 2016;84:1067–1074. [DOI] [PubMed] [Google Scholar]
- 37.Jewel Y, Dutta P, Liu J. Exploration of conformational changes in lactose permease upon sugar binding and proton transfer through coarse-grained simulations. Proteins: Struct, Funct, Bioinf 2017;85(10):1856–1865. [DOI] [PubMed] [Google Scholar]
- 38.Marrink SJ, de Vries AH, Mark AE. Coarse grained model for semiquantitative lipid simulations. J Phys Chem B 2004;108(2):750–760. [Google Scholar]
- 39.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. The MARTINI force field: Coarse grained model for biomolecular simulations. J Phys Chem B 2007;111(27):7812–7824. [DOI] [PubMed] [Google Scholar]
- 40.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B 2001;105(28):6474–6487. [Google Scholar]
- 41.Reith D, Putz M, Muller-Plathe F. Deriving effective mesoscale potentials from atomistic simulations. J Comput Chem 2003;24(13):1624–1636. [DOI] [PubMed] [Google Scholar]
- 42.Liu P, Dehez F, Cai WS, Chipot C. A Toolkit for the Analysis of Free-Energy Perturbation Calculations. J Chem Theory Comput 2012;8(8):2606–2616. [DOI] [PubMed] [Google Scholar]
- 43.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem 2005;26(16):1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jo S, Kim T, Iyer VG, Im W. Software news and updates - CHARNIM-GUI: A web-based graphical user interface for CHARMM. J Comput Chem 2008;29(11):1859–1865. [DOI] [PubMed] [Google Scholar]
- 45.Jo S, Lim JB, Klauda JB, Im W. CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys J 2009;97(1):50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila-Contreras EM, Qi YF, Lee JM, Monje-Galvan V, Venable RM, Klauda JB, Im W. CHARMM-GUI Membrane Builder Toward Realistic Biological Membrane Simulations. J Comput Chem 2014;35(27):1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qi Y, Cheng X, Han W, Jo S, Schulten K, Im W. CHARMM-GUI PACE CG Builder for Solution, Micelle, and Bilayer Coarse-Grained Simulations. J Chem Inf Model 2014;54(3):1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph Model 1996;14(1):33–38. [DOI] [PubMed] [Google Scholar]
- 49.Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 2011;480(7378):565–569. [DOI] [PubMed] [Google Scholar]
- 50.Eicher T, Cha HJ, Seeger MA, Brandstatter L, El-Delik J, Bohnert JA, Kern WV, Verrey F, Grutter MG, Diederichs K, Pos KM. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc Natl Acad Sci U S A 2012;109(15):5687–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petrek M, Otyepka M, Banas P, Kosinova P, Koca J, Damborsky J. CAVER: a new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinformatics 2006;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eicher T, Seeger MA, Anselmi C, Zhou W, Brandstaetter L, Verrey F, Diederichs K, Faraldo-Gomez JD, Pos KM. Coupling of remote alternating-access transport mechanisms for protons and substrates in the multidrug efflux pump AcrB. Elife 2014;3:e03145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.