

Abstract
The collagen VI-related muscular dystrophies in people include a broad spectrum of diseases ranging from the severe Ullrich congenital muscular dystrophy to the mild type Bethlem. Clinical features are attributable to both muscle and connective tissue and include progressive muscle weakness and respiratory failure, hyperlaxity of distal joints, and progressive contracture of large joints. Here we describe two different COL6A3 pathogenic variants in Labrador Retriever dogs that result in autosomal recessive or autosomal dominant congenital myopathies with hyperlaxity of distal joints and joint contracture, similar to the condition in people.
Keywords: canine, muscle, myopathy, collagen VI
1. Introduction
The collagen VI-related muscular dystrophies in people include a broad spectrum of severities ranging from the severe Ullrich congenital muscular dystrophy to the mild type Bethlem [1]. Ullrich congenital muscular dystrophy is rare but is among the most common type of congenital muscular dystrophies in several populations [1,2]. In northern England, the collagen VI-related myopathies have a point prevalence of 0.9 patients for 100,000 individuals [3]. These diseases are caused by both dominantly and recessively acting variants in the three major α-chains encoded by the collagen VI genes COL6A1, COL6A2 and COL6A3, which associate in equal stoichiometry to form a heterotrimeric monomer followed by further assembly to a tetrameric structure, which is then secreted into the extracellular matrix by fibroblastic cells in muscle and other connective tissues such as tendon. Clinical features are thus attributable to both muscle and connective tissue and include progressive muscle weakness, hyperlaxity of distal joints, progressive contracture of large joints, and respiratory failure in the more severe forms. In dogs, descriptions of collagen VI-related diseases have been limited to a recessively acting nonsense variant in COL6A1 described in young Landseer Newfoundland dogs [4] and a clinical, pathological and immunohistochemical description of subsarcolemmal collagen VI deficiency in a young Labrador Retriever without genetic confirmation [5].
Previously described inherited myopathies in young Labrador Retriever dogs include dystrophin deficient muscular dystrophy [6,7], X-linked myotubular myopathy [8,9], centronuclear myopathy [10,11], a congenital myasthenic syndrome attributable to a COLQ variant [12], and the above-mentioned sarcolemmal-specific collagen VI deficiency [5]. Here we describe causative recessively and dominantly acting variants in collagen VI-deficient congenital muscular dystrophy in a recently ascertained small family of Labrador Retrievers and in the case of previously recognized sarcolemmal-specific collagen VI deficiency [5].
2. Methods
2.1. Animals:
Dogs in this report were evaluated as clinical cases by practicing veterinary specialists. Owner approval for all diagnostic evaluations was obtained and IACUC approval was not required for evaluating diagnostic tissue specimens.
2.2. Histopathology and Immunofluorescence Staining
Unfixed muscle biopsies (biceps femoris, quadriceps and triceps brachii for Case 1, biceps femoris for Case 2) were either wrapped in a saline-dampened gauze sponge and chilled, or immersion-fixed in 10% neutral buffered formalin. Biopsies were shipped by an overnight service under refrigeration to the Comparative Neuromuscular Laboratory at the University of California, San Diego, immediately flash frozen in isopentane pre-cooled in liquid nitrogen and stored frozen at −80°C until further processed. Cryosections were stained or reacted with a standard panel of histochemical stains and reactions [13]. Additional cryosections (8 μm) were cut and stained by indirect immunofluorescence as previously described [14]. Sections were incubated with monoclonal or polyclonal antibodies against collagen VI (gift of Eva Engvall, 3G7, direct apply) [15], laminin α2 (gift of Eva Engvall, 1B4, direct apply) [16], and α-sarcoglycan (gift of Eva Engvall, 1:200) [17]. Fixed biopsies were paraffin embedded and processed by standard procedures.
2.3. Fibroblast Culture and Staining
Skin biopsies were collected using a punch biopsy procedure, placed in transport media (10% FBS/1% P/S/DMEM with ascorbic acid (final concentration 50 ng/μl) and shipped by an overnight service to the laboratory (NIH). Upon receipt, cells were cultured for 4 days in the same media until 80% confluence in an 8-well chamber slide. Cells were blocked at RT for 30 minutes in10% FBS/PBS with or without 0.1% triton X-100 (cell permeabilizing agent) to detect intracellular retention or extracellular matrix, respectively. Primary antibodies (COL6 rabbit antibody 1:500, ab6588, Abcam) were incubated at RT for 1.5 hr in 10% FBS/PBS followed by secondary antibodies (Alexa Fluor 488 goat anti-rabbit 1:500 (A11034, Invitrogen) at RT for 1 hr in the dark. After washes, the cells were incubated with DAPI 1:1000 at RT for 1–2 min in the dark. Following washes, the slides were cover slipped and stored at 4°C in the dark until analysis.
2.4. Whole Genome Sequencing and Variant Analysis: Case 1
Genomic DNA was extracted from muscle biopsies using the DNEasy kit standard protocol (Qiagen). Approximately 3 μg of DNA from each dog was submitted for library preparation and whole genome sequencing at Genewiz (Plainfield, NJ) using a 150 bp paired-end read configuration in separate lanes of an Illumina HiSeq 4000 high-throughput sequencing system. These reads have been made publicly available at NCBI’s Short Read Archive at https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP218260. WGS read alignment to CanFam3.1 and variant calling was performed using a standardized bioinformatics pipeline for all samples as described previously [18].
Heterozygous or homozygous variants present in the probands were initially filtered against a population of 307 dogs of 48 different breeds whose entire genomes were sequenced as part of ongoing work in the laboratory. These breeds included: 34 Yorkshire Terriers, 22 Boxers, 20 Standard Poodles, 19 Miniature Schnauzers, 19 Great Danes, 13 Cavalier King Charles Spaniels, 12 Siberian Huskies, 11 Dachshunds, 11 Golden Retrievers, 10 Miniature Poodles, 10 Scottish Deerhounds, 10 Labrador Retrievers, and 116 dogs of 36 additional breeds. Whole genome sequences from these dogs were processed using a similar bioinformatics pipeline as described above. Variants that passed our filtering step and were unique to the proband were annotated using Variant Effect Predictor 91 [19], and evaluated based on the severity of the predicted effect. For the VEP analysis the Ensembl gene ID for COL6A3 is ENSCAFG00000012226, the ID for the transcript is ENSCAFT00000019411.5, and the ID for the protein is ENSCAFP00000018008.4. The prioritized variant was located at CanFam3.1 position 25:48014962_G/A. Variants we considered most impactful included frameshifts, in-frame insertions and deletions, premature-start, stop-gained, missense, and splice region changes. Candidate variants were then secondarily compared to those of control genomes from 640 dogs and 8 wolves found in the Dog Biomedical Variant Database Consortium (DBVDC) database and 668 dogs and 54 wild canids [20]. Accession numbers deposited in the European Nucleotide Archive (http://www.ebi.ac.uk/ena) are PRJEB10823, PRJEB13139, PRJEB13468, PRJEB13723, PRJEB14110, PRJEB14840, PRJEB16012, PRJEB4544, PRJEB5500, PRJEB5874, PRJEB5875, PRJEB6076, PRJEB6079, PRJEB7734, PRJEB7735, PRJEB7736, PRJEB7903, PRJEB9437, PRJEB9590, PRJEB9591, and PRJNA266585, in addition to those provided in Plassais et al [20].
2.5. RNA and DNA isolation – Case 2
RNA and DNA were isolated from the frozen archived muscle biopsy (biceps femoris) of Case 2. Sectioning of the muscle biopsy was carried out on a Microm HM550 cryostat (ThermoFisher Scientific, Waltham, MA). Thirty sections (10 um thickness each) of the muscle biopsy were collected in a pre-cooled microcentrifuge tube and were homogenized in one mL of Trizol reagent (ThermoFisher Scientific). Following the first phase separation, RNA was isolated from the aqueous phase, and DNA was isolated from the phenol/chloroform phase, according to manufacturer’s instructions. For the control dog, RNA and DNA were isolated from cultured dermal fibroblasts. After reaching confluence, cells were detached from the dish using 0.05% trypsin-EDTA (ThermoFisher Scientific) and were pelleted by centrifugation. After one wash in 1x phosphate buffered saline (ThermoFisher Scientific), the cell pellet was homogenized in one mL of Trizol reagent. Nucleic acid isolation was then carried out as described above.
2.6. Variant screening
Complementary DNA (cDNA) was prepared from one ug of RNA, using random primers and a Superscript III reverse transcriptase protocol, following manufacturer’s protocol (ThermoFisher Scientific). PCR amplification from the cDNA samples was carried out to screen the collagenous-encoding domains of COL6A1, COL6A2, and COL6A3 for the presence of potential pathogenic exon skips, using multiple overlapping PCR reactions to cover the entire regions, and from gDNA to screen the COL6A3 exon 16 region for genomic variants. PCRs were performed with the Advantage 2 PCR enzyme system (Takara Bio USA, Mountain View, CA), and the primers used are listed in Table S1. The cycling parameters used were as followed: initial denaturation at 95˚C for 1 min, 5 cycles of 95˚C for 30 sec, and 68˚C for 1 min, then 5 cycles of 95˚C for 30 sec, 64˚C for 30 sec, and 68˚C for 45 sec, followed by 25 or 30 cycles of 95˚C for 30 sec, 56˚ or 58˚C for 30 sec, and 68˚C for 45 sec, and a final extension at 68˚C for 1 min. PCR products were run on a 2.5% agarose gel containing SYBR Safe DNA Gel Stain (ThermoFisher) in Tris-Acetate EDTA (TAE) buffer using the RunOne electrophoresis system (Embi Tec, San Diego, CA). Fragments were detected using the ChemiDoc XRS+ molecular imager (Bio-Rad, Hercules, CA).
2.7. Genotyping
DNA samples of 105 Labrador Retrievers available in the University of Minnesota Canine Genetics Laboratory were genotyped for the COL6A3 exon 10 and exon 16 variants via Sanger sequencing. Amplicons of 399 bp and 231 bp respectively, were obtained from PCR primers as follows: For COL6A3 exon 10 forward primer 5’-AAGTCATACCTGGCCGTGAG-3’ and reverse primer 5’-GTCAGCTGAGGCACAGAGG-3’ that produce a 399 bp amplicon, and for COL6A3 exon 16 forward primer 5’-GGAAAATCACTGGAAATGCAA-3’ and reverse primer 5’-GGGGAGATGCTTGAAGGTAG-3’ that produce a 231 bp product. PCR products were sequenced at Genewiz (South Plainfield, NJ).
3. Results
3.1. Clinical Evaluation of Two Cases of Congenital Myopathy in Labrador Retrievers
Case 1:
A 6-month-old male Labrador Retriever was evaluated for a 2-month history of angular limb deformity and carpal hyperflexion (Figure 1A). The serum creatine kinase (CK) activity was mildly increased at 866 IU/L (reference 10–200 IU/L). A neuromuscular disease was suspected and a clinical evaluation including muscle biopsies was performed. The dog was from a litter of 7 puppies resulting from a cross of 2 phenotypically normal dogs (Figure 1C). Three puppies (2 male and 1 female) were described as affected, 3 were phenotypically normal and 1 puppy died at birth. The dam was the aunt of the sire.
Figure 1. Clinical signs of congenital muscular dystrophy in two Labrador Retrievers.
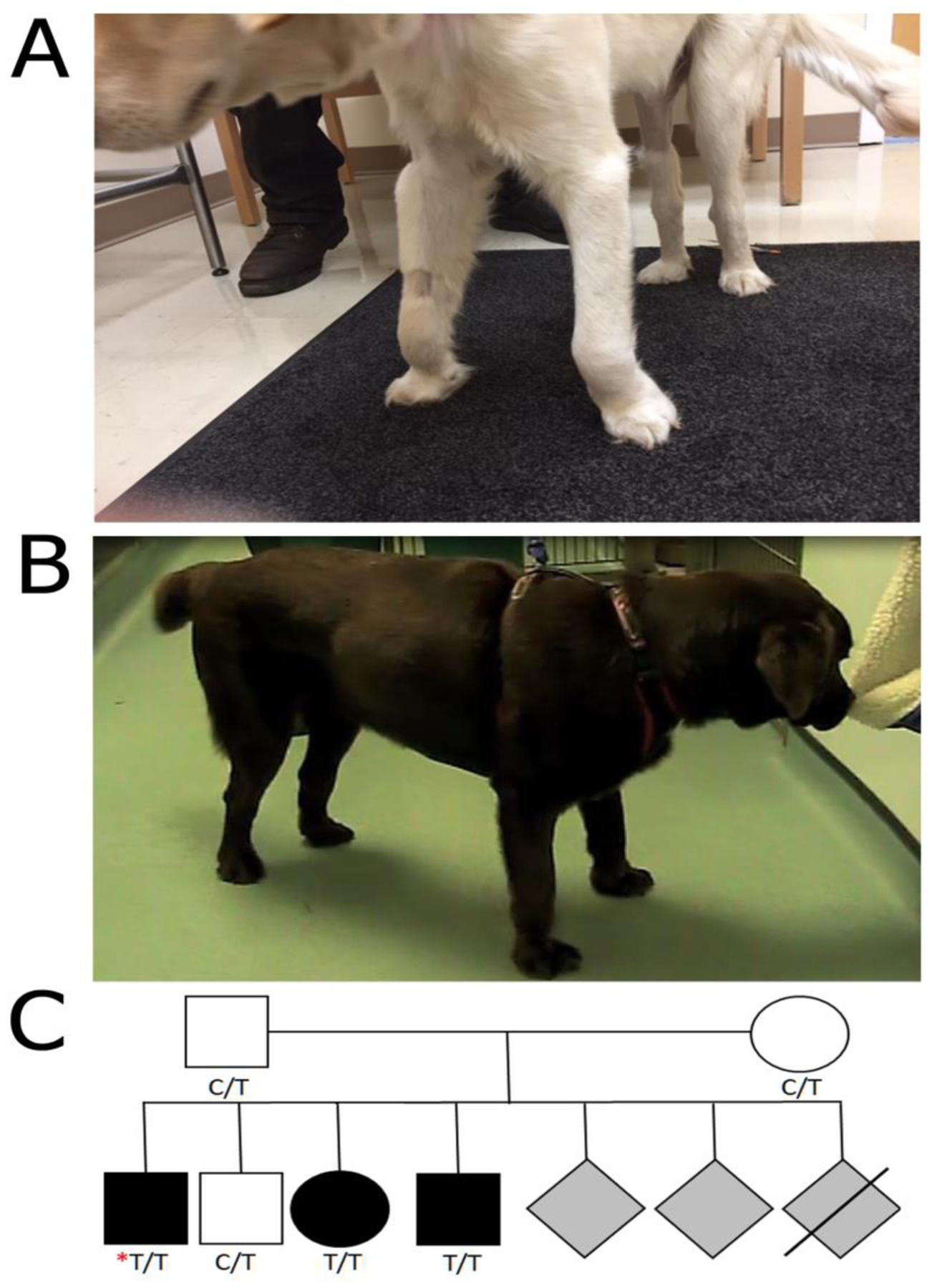
(A) Image of a 6 month-old male (Case 1) showing carpal hyperflexion and limb deformities. (B) Image of a 11 month-old female (Case 2) showing carpal and tarsal hyperflexion. (C) Limited pedigree of Case 1 (identified by the red asterisk). Genotypes at the COL6A3 c.4726C>T (p.R1576*) locus are indicated for individuals tested.
Case 2:
An 11-month-old female neutered Labrador Retriever was presented for evaluation of progressive gait abnormalities and carpal and tarsal hyperflexion of 6 months duration (Figure 1B). As the dog was adopted from a rescue shelter, previous history and information on the parents and littermates was not available. The clinical details, histopathology and immunostaining of muscle from this dog have been previously described [5].
3.2. Histopathology, Muscle and Fibroblast Immunofluorescence Staining
Biopsies were collected from the biceps femoris, quadriceps and triceps brachii muscles of Case 1 by an open biopsy procedure under general inhalational anesthesia. Similar pathological changes were observed in all three muscles including excessive variability in myofiber size, mild endomysial fibrosis and occasional internalized nuclei (Figure 2A, biceps femoris muscle shown). Immunofluorescence staining of the biceps femoris and triceps muscles for localization of collagen VI showed an absence of staining in both the interstitium and sarcolemma compared to archived control muscle (Figure 2B). An antibody against α-sarcoglycan highlighted the outline of the myofibers in both the case and control muscles (Figure 2B). For comparison, similar myopathic changes were present histologically in the biopsies from Case 2 [5]. However, immunofluorescence staining in Case 2 for localization of collagen VI showed that staining was present in the interstitium but not on the sarcolemma, indicative of a sarcolemmal-specific collagen VI deficiency [5].
Figure 2. Abnormal muscle histology and absence of collagen VI staining in Case 1.
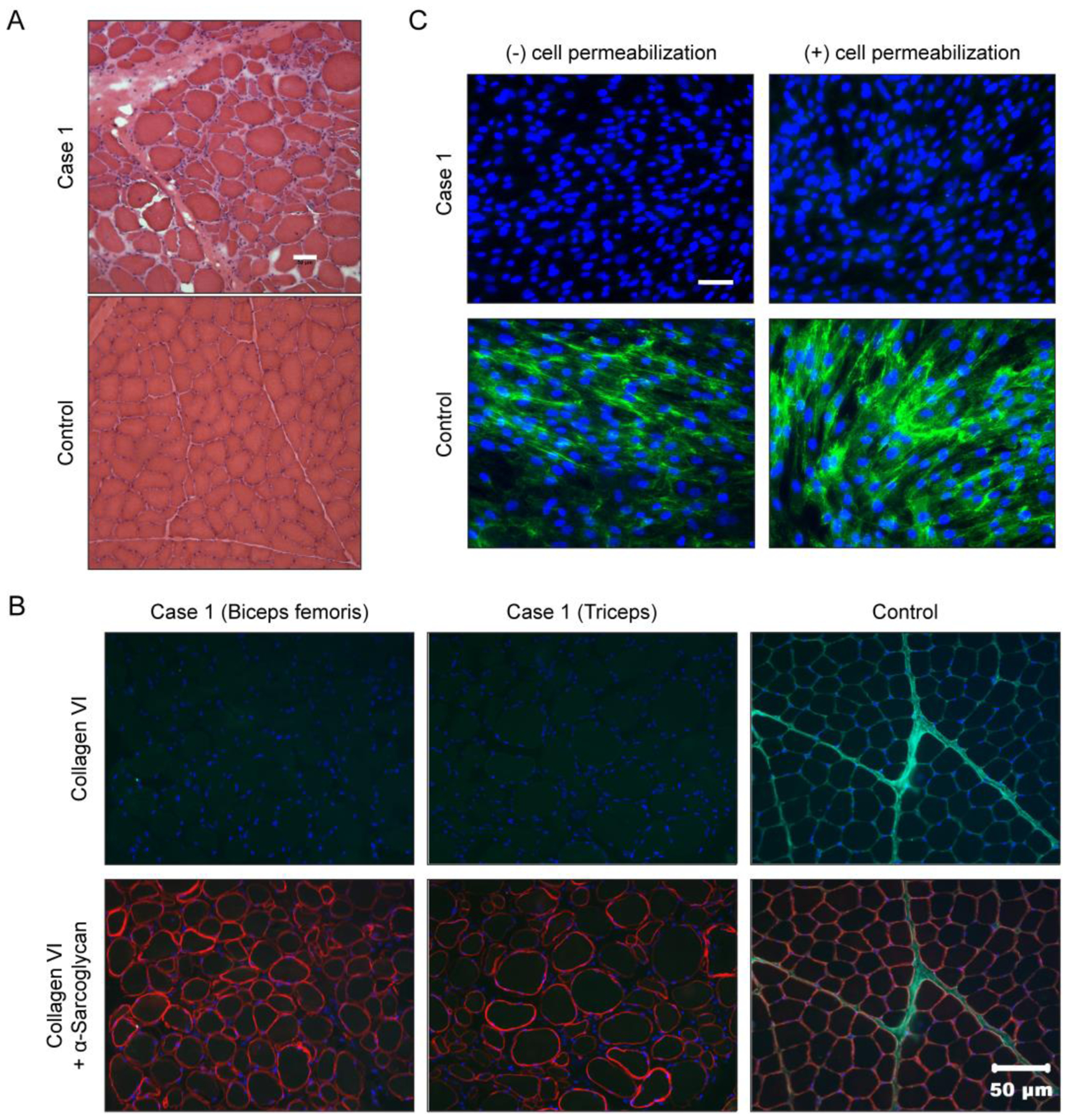
(A) Hematoxylin and eosin (H&E) staining of cryosections from the biceps femoris muscle from Case 1 showing a dystrophic phenotype including excessive variability in myofiber size, endomysial fibrosis and excessive internal nuclei. Scale bar = 50 μm. (B) Muscle biopsy sections co-stained for collagen VI (green) and α-sarcoglycan (red). Scale bar = 50 μm. (C) Primary dermal fibroblasts cultured and stained for collagen VI (green), and nuclei (DAPI, blue), in absence (−) or presence (+) of a cell permeabilizing agent. Scale bar = 50 μm.
Fibroblasts isolated from a skin biopsy of Case 1 were cultured and stained to assess for collagen VI secretion and matrix assembly. Under normal staining conditions (i.e. without cell permeabilization), we observed that fibroblasts from Case 1 did not produce any formed collagen VI matrix, as opposed to a control sample in which the matrix was visible (Figure 2C, left panels). Absence of collagen VI reactivity in matrix cultures is typical for recessive mutations that cause Ullrich congenital muscular dystrophy [1]. Collagen VI immunostaining with cell permeabilization can detect if collagen VI tetramers are retained intracellularly as a result of dominant-negatively acting mutations. Here, fibroblasts from Case 1 showed no retention (Figure 2C, right panel), consistent with the presence of mutations leading to loss of collagen VI expression.
3.3. Genome Sequencing and Variant Analysis on Case 1
COL6A3 has 43 exons, encoding 3169 amino acids (Figure 3A). Initial screening of the exonic sequences encoding the triple helical domains of COL6A1, COL6A2, and COL6A3 of Case 1 by cDNA sequencing did not identify any pathogenic mutations (data not shown). However, whole genome sequencing identified a premature stop codon variant in exon 10 of the COL6A3 gene (CanFam3.1 chr25:48,014,962G>A; NM_001103215.1 c.4726C>T, p.R1576*) (Figure 3A, B) which would truncate about half the open reading frame. Pathogenicity is also highly supported by the fact that this variant is not found in any of the > 1300 dogs across WGS databases (DBVDC, [19], UMN internal WGS database). Follow-up analysis of the dam and sire showed that both were heterozygous for this variant (Figure 3B) and clinically normal. An affected female littermate was also homozygous mutant, while an unaffected male littermate was heterozygous (Figure 1C), demonstrating that the genotypes segregated with the phenotype in this pedigree and were consistent with a recessive collagen VI-associated muscular dystrophy. No carriers of the R1576* variant were found in a population of 105 unrelated Labrador Retrievers.
Figure 3. Muscular dystrophy in Labrador retriever dogs caused by variants in COL6A3.
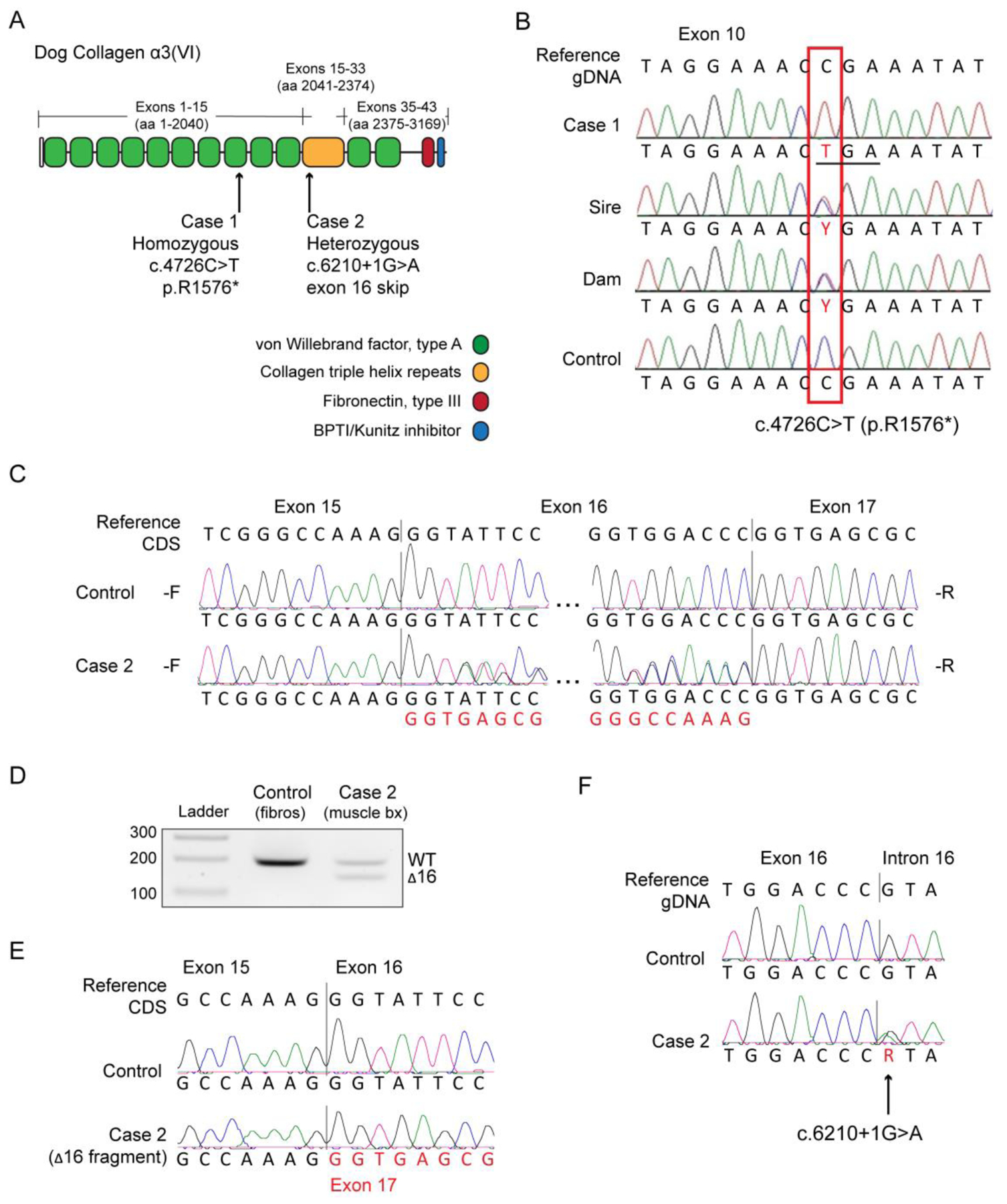
(A) Schematic of the main collagen a3(VI) chain protein domains in the dog, and the relative positions of the recessive and dominant variants found in the affected Labrador retriever dogs Case 1 and Case 2, respectively. aa = amino acid. (B) Alignments of exon 10 sequences of Case 1, the sire, the dam, and a control obtained by Sanger sequencing of a PCR amplicon. The box indicates the location where a homozygous C nucleotide in the control genome is replaced with a homozygous T nucleotide in the case. Both the sire and dam are heterozygous. The variant changes the codon for an arginine residue (CGA) to TCA to the stop codon (TGA). (C) Complementary DNA (cDNA) forward (-F) and reverse (-R) sequencing chromatograms of a control dog and of Case 2, spanning exon 16 of the COL6A3 gene. CDS = coding sequence. (D) cDNA amplification and electrophoresis of COL6A3 transcripts. The fragments in Case 2 lane show an approximately equal expression of normal transcripts (wild-type, WT) and transcripts lacking exon 16 (Δ16), which is 54 nucleotides in size. (E) Sequencing chromatograms of the PCR fragments isolated from the electrophoresis gel in (B) showing the junction of exon 15 to exon 17. (F) Sequencing chromatograms from genomic DNA show that Case 2 is a heterozygous carrier of the COL6A3 c.6210+1G>A variant.
3.4. Genome Sequencing and Variant Analysis on Case 2
The clinical diagnosis in Case 2 of sarcolemmal-specific collagen VI deficiency [5] prompted the search for the causative variant in this dog. The exons encoding the triple helical domains of COL6A1, COL6A2, and COL6A3 were screened by cDNA sequencing. Sequencing results for COL6A1 and COL6A2 were negative (data not shown). In COL6A3, we detected the presence of double peaks in both the forward (-F) and reverse (-R) sequences flanking exon 16, denoting the presence of two mRNA species: one normal and one lacking exon 16 (Δ16) (Figure 3C). To confirm the expression of these two species in muscle tissue from Case 2, we separated reverse-transcription PCR (RT-PCR) products by size on an agarose gel (Figure 3D), and sequenced the products isolated from the gel. Sequencing confirmed the skipping of exon 16 in the shorter products, as shown by the junction of exon 15 to exon 17 (Figure 3E). In addition, the seemingly equal expression levels of normal (WT) and Δ16 mRNA (Figure 3D) were suggestive of the presence of a heterozygous variant affecting exon 16 splicing, such as splice site variants or genomic deletions.
To identify the genomic variant underlying the exon 16 skipping, we sequenced genomic DNA in the vicinity of exon 16. The sequencing chromatograms showed a heterozygous guanine to adenine variant at the first position of intron 16 (CanFam3.1 chr25:48,007,994C>T; NM_001103215.1 c.6210+1G>A), changing the immediate splice donor site motif from CCgt to CCat (Figure 3F). COL6A3 exon 16 is 54 nucleotides in length, and its removal from the mature messenger RNA (mRNA) does not affect the reading frame, thus suggesting that this splice variant is translated and acting in a dominant manner because it would be predicted to be assembled into the tetrameric state. The amino acids encoded by this exon are located at the N-terminus of the triple helical domain, a hot-spot for dominant-negative assembly-competent variants [1] (Figure 3A). Indeed, in-frame skipping of COL6A3 exon 16 is one of the more common consequences of dominantly acting variants identified in individuals with Ullrich congenital muscular dystrophy [21,22], and has been shown to act as dominant-negative [21–23]. The genetic findings in Case 2 were thus consistent with a pathogenic COL6 variant acting as a dominant-negative and causing the sarcolemmal-specific collagen V deficiency previously identified in this dog [5]. No carriers of this COL6A3 c.6210+1G>A variant were found in the population of 105 unrelated Labrador Retrievers, nor in any of the > 1300 dogs across WGS databases (DBVDC, [19], UMN internal WGS database).
4. Discussion
Here we report two COL6A3 variants causing a muscular dystrophy in Labrador Retriever dogs that has resemblance to human COLVI-related muscular dystrophies, including Ullrich type congenital muscular dystrophy. The clinical presentations in affected Labrador Retrievers with variants in COL6A3 were similar to that of human patients in that there was an early onset of joint deformities, laxity at 5 to 6 months of age and slow progression lasting into adulthood. Case 1 is still alive at this writing (at 31 months of age) and the dog has maintained ambulation. By 50 months post-diagnosis, Case 2 showed significant bilateral symmetrical atrophy of the appendicular and masticatory muscles, and corneal opacities. The dog became unable to support weight and was euthanized 80 months post-diagnosis.
We identified both autosomal recessively and dominantly acting variants in COL6A3, closely resembling the underlying genetic mechanism in human patients. Specifically, the heterozygous exon 16 (COL6A3) variant identified in Case 2 in its consequence is identical to human mutations with the same exon skipping outcome that have been shown to act via a dominant-negative mechanism leading to a severe Ullrich type phenotype and typically arise de novo. In fact, COL6A3 skipping of exon 16 is a recurrent human dominant Ullrich variant mechanism [21,22]. In particular, the c.6210+1G>A variant that was found as the cause for the exon 16 skip in Case 2 is the same variant that is recognized as the most frequent cause of exon 16 skips in patients in the literature [21, 22], and in public databases (ClinVar, LOVD Leiden Muscular Dystrophy Pages). As Case 2 was obtained by the owner as a rescue, the dam and sire were not available for genotyping. As this is a relatively common variant class in human patients, we have recently demonstrated proof-of principle for the therapeutic validity of mutant allele-specific knockdown of the exon 16 (COL6A3) deleted transcript using RNA interference [23].
In Case 1, the homozygous premature stop codon (p.R1576*) variant in exon 10 of the 43 exon (3169 amino acid) COL6A3 gene would result in truncation of approximately half of the protein, and ablation of the triple helical domain, which would be predicted to be nonfunctional and not capable of assembly. Premature termination variants are also frequently associated with non-sense mediated mRNA decay, resulting in no translation into protein. The fibroblast staining revealed a complete absence of a formed collagen VI matrix which is consistent with both of these mechanisms and supportive of a complete Null situation. Of interest, this variant was reported in a single patient [24], and in a public database (ClinVar). In both instances the variant was identified in a heterozygous state. The consequence of the variant is significantly different however: in dogs, Arg1576 is encoded by the codon “CGA”, whereas in human, it is encoded by “CGG”. In both cases, nucleotide 4726 (NM_004369.3 in human) corresponds to the first nucleotide of the codon, but while it produces a termination codon in the dog (CGA>TGA), in the human it results in a tryptophan codon (CGG>TGG). As a consequence, p.R1576W is still classified as a variant of unknown significance, and it is still to be determined whether it is pathogenic. Nevertheless, it is noteworthy that those two particular genomic locations (c.4726C and c.6210+1G) are variant targets in the different species.
The clinical presentation was similar in both affected Labrador Retrievers with variants in COL6A3 but differed from that previously reported in Landseer dogs with a variant in COL6A1 [4]. In the Landseer dogs, severe weakness and prolonged sleeping were apparent at a few weeks of age with euthanasia by 5 to 15 months of age. Laxity of distal joints and angular contractures of proximal joints as found in the Labradors were not described in the Landseers, although the stifle and tarsal joints were held in a slight flexed position. A decreased range of motion of the joints was described [4] as opposed to hyperlaxity. The phenotypic differences between the two breeds of dogs with COL6 variants is also in contrast to what is found in human patients, as the clinical presentation and severity is independent of whether the variants lie in COL6A1, COL6A2, or COL6A3. These phenotypic differences may be a result of the different genetic backgrounds, the effect of modifier genes, or the consequence of compensatory mechanisms such as the expression of additional collagen VI chains that could more effectively compensate for the lack of α3(VI) than of α1(VI) chains [25,26].
Conclusions
This study expands the spectrum of congenital neuromuscular diseases affecting dogs with direct correlations to the corresponding human conditions. Indeed, both autosomal recessive and dominant variants in genes encoding collagen VI that occur in humans have also been confirmed in dogs. Phenotypic differences between dog breeds may be a result of differences in genetic background or the effect of modifying genes. It is likely that the pace of discovery of novel mutations underlying canine neuromuscular disorders will continue to increase as whole genome sequencing is more widely applied.
Supplementary Material
Highlights.
Whole genome and Sanger sequencing resulted in the discovery of new collagen VI variants
Autosomal recessive and dominant pathogenic variants identified affecting COL6A3
Hyperlaxity and contracture of distal joints were similar to the human phenotype
Canine condition has direct correlations to corresponding human condition
Acknowledgements
SGF is supported in part by an NIH Special Emphasis Research Career Award (1 K01 OD027058) in Pathology and Comparative Medicine sponsored by the Division of Comparative Medicine, Office of Research Infrastructure Programs. This research was supported (in part) by the Intramural Research Program of the NIH, NINDS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bonneman CG. The collagen VI-related myopathies: muscle meets matrix. Nat Rev Neurol 2011;7(7):379–90. doi: 10.1038/nrneurol.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Allamand V, Briñas L, Richard P, Stojkovic T, Quijano-Roy S, Bonne G. Col VI myopathies: where do stand, where do we go. Skelet Muscle 2011. September 23;1:30. doi: 10.1186/2044-5040-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 2009; 32(11): 3175–86. doi: 10.1093/brain/awp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Steffen F, Bilzer T, Brands J, Golini L, Jagannathan V, Weidmer M, et al. A nonsense variant in COL6A1 in Landseer dogs with muscular dystrophy. G3 (Bethesda). 2015; 5(12):2611–7. doi: 10.1534/g3.115.021923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Marioni-Henry K, Haworth P, Scott H, Witte P, Guo LT, Shelton GD. Sarcolemmal specific collagen VI deficient myopathy in a Labrador Retriever. J Vet Intern Med 2014; 28:243–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Smith BF, Kornegay JN, Duan D. Independent canine models of Duchenne muscular dystrophy due tointronic insertions of repetitive DNA. Mol Ther. 2007; 15(Suppl 1):S51. [Google Scholar]
- [7].Vieira NM, Guo LT, Estrela E, Kunkel LM, Zatz M, Shelton GD. Muscular dystrophy in a family of Labrador Retrievers with no muscle dystrophin and a mild phenotype. Neurouscul Disord 2015:25;363–70. [DOI] [PubMed] [Google Scholar]
- [8].Beggs AH, Böhm J, Snead E,Kozlowski M, Maurer M, Minor K, et al. MTM1 mutation associated with X-linked myotubular myopathy in Labrador retrievers. Proc Natl Acad Sci U S A. 2010. August 17;107(33):14697–702. doi: 10.1073/pnas.1003677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Snead EC, Taylor SM, van der Kooij M, Cosford K, Beggs AH, Shelton GD. Clinical phenotype of X-linked myotubular myopathy in Labrador rretriever puppies. J Vet Intern Med. 2015. January;29(1):254–60. doi: 10.1111/jvim.12513. Epub 2015 Jan 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pelé M, Tiret L, Kessler JL, Blot S, Panthier JJ. SINE exonic insertion in the PTPLA gene leads to multiple splicing defects and segregates with the autosomal recessive centronuclear myopathy in dogs. Hum Mol Genet. 2005. June 1;14(11):1417–27. Epub 2005 Apr 13. Erratum in: Hum Mol Genet. 2005 Jul 1;14(13):1905–6. [DOI] [PubMed] [Google Scholar]
- [11].Maurer M, Mary J, Guillaud L, Fender M, Pele M, Bilzer T, et al. Centronuclear myopathy in Labrador retrievers: a recent founder mutation in the PTPLA gene has rapidly disseminated worldwide. PLoS One. 2012;7(10):e46408. doi: 10.1371/journal.pone.0046408. Epub 2012 Oct 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rinz CJ, Levine J, Minor KM, Humphries HD, Lara R, Starr-Moss AN, et al. A COLQ missense mutation in Labrador Retrievers having congenital myasthenic syndrome. PLoS One. 2014. August 28;9(8):e106425. doi: 0.1371/journal.pone.0106425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dubowitz V, Sewry CA, Oldfors A. Histological and histochemical stains and reactions In: Dubowitz V, Sewry CA, Oldfors A eds. Muscle Biopsy: a practical approach. 4th ed. 2013; Oxford: Saunders Elsevier, 16–27. [Google Scholar]
- [14].Guo LT, Moore SA, Forcales S, Engvall E, Shelton GD. Evaluation of commercial dysferlin antibodies on canine, mouse and human skeletal muscle. Neuromuscul Disord 2010; 20:820–5. [DOI] [PubMed] [Google Scholar]
- [15].Hessle H, Engvall E. Type VI collagen. Studies on its localization, structure, and biosynthetic form with monoclonal antibodies. J Biol Chem 1984;259:3955–61 [PubMed] [Google Scholar]
- [16].Leivo I, Engvall E. Merosin, a protein specific for basement membranes of Schwann cells, straited muscle and trophoblast, is expressed late in nerve and muscle development. Proc Natl Acad Sci USA 1988;85:1544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liu LA, Engvall E. Sarcoglycan isoforms in skeletal muscle. J Biol Chem 1999; 274:38171–6. [DOI] [PubMed] [Google Scholar]
- [18].Friedenberg SG, Meurs KM. Genotype imputation in the domestic dog. Mamm Genome. April 2016. doi: 10.1007/s00335-016-9636-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–70. doi: 10.1093/bioinformatics/btq330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Plassais J, Kim J, Davis BW, Karyadi DM, Hogan AN, Harris AC, et al. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat Commun 2019,April 2;10(1) 1489/ doi: 10.1038/s41467-019-09373-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Baker NL, Morgelin M, Peat R, Goemans N, North KN, Bateman JF, et al. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet 2005;14:279–93. 10.1093/hmg/ddi025. [DOI] [PubMed] [Google Scholar]
- [22].Lampe AK, Zou Y, Sudano D, O’Brien KK, Hicks D, Laval SH, et al. Exon skipping mutations in collagen VI are common and are predictive for severity and inheritance. Hum Mutat 2008. June;29(6):809–22. doi: 10.1002/humu.20704. [DOI] [PubMed] [Google Scholar]
- [23].Bolduc V, Zou Y, Ko D, Bönnemann CG. siRNA-mediated allele-specific silencing of a COL6A3 mutation in a cellular model of dominant Ullrich musculardystrophy. Mol Ther Nucleic Acids. 2014. February 11;3:e147. doi: 10.1038/mtna.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nallamilli BRR, Chakravorty S, Kesari A, Tanner A, Ankala A, Schneider T, et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol 2018. December 1;5(12):1574–87. doi: 10.1002/acn3.649. eCollection 2018 Dec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sabatelli P, Gara SK, Grumati P, Urciuolo A, Gualandi F, Curci R, et al. Expression of the collagen VI α5 and α6 chains in normal human skin and in skin of patients with collagen VI-related myopathies. J Invest Derm 2011. January;131 (1):99–107. 131(1)99–107. doi: 10.1038/jid.2010.284. Epub 2010 Sep 30. [DOI] [PubMed] [Google Scholar]
- [26].Fitzgerald J, Rich C, Zhou FH, Hansen U. Three novel collagen VI chains, alpha4(VI), alpha 5 (VI), and alpha6 (VI). J Biol Chem 2008; 283(29):20170–80. doi: 10.1074/jbc.M710139200. Epub 2008 Apr 9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.