

Abstract
Quantitative systems pharmacology (QSP) has emerged as a transformative science in drug discovery and development. It is now time to fully rethink the biological functions of drug metabolizing enzymes (DMEs) and transporters within the framework of QSP models. The large set of DME and transporter genes are generally considered from the perspective of the absorption, distribution, metabolism, and excretion (ADME) of drugs. However, there is a growing amount of data on the endogenous physiology of DMEs and transporters. Recent studies—including systems biology analyses of “omics” data as well as metabolomics studies—indicate that these enzymes and transporters, which are often among the most highly expressed genes in tissues like liver, kidney, and intestine, have coordinated roles in fundamental biological processes. Multispecific DMEs and transporters work together with oligospecific and monospecific ADME proteins in a large multiorgan remote sensing and signaling network. We use the Remote Sensing and Signaling Theory (RSST) to examine the roles of DMEs and transporters in intratissue, interorgan, and interorganismal communication via metabolites and signaling molecules. This RSST‐based view is applicable to bile acids, uric acid, eicosanoids, fatty acids, uremic toxins, and gut microbiome products, among other small organic molecules of physiological interest. Rooting this broader perspective of DMEs and transporters within QSP may facilitate an improved understanding of fundamental biology, physiologically based pharmacokinetics, and the prediction of drug toxicities based upon the interplay of these ADME proteins with key pathways in metabolism and signaling. The RSST‐based view should also enable more tailored pharmacotherapy in the setting of kidney disease, liver disease, metabolic syndrome, and diabetes. We further discuss the pharmaceutical and regulatory implications of this revised view through the lens of systems physiology.
Quantitative systems pharmacology (QSP) is a rapidly developing transformative science applicable to drug discovery and development. In recent years, published examples describing the use of QSP to facilitate biomedical research have been increasing. Further, the pharmaceutical and biotechnology industries have rapidly adopted QSP to inform product development. Examples of QSP in regulatory decision making—such as defining postmarketing requirements and waiving certain clinical trials—are also appearing. 1 , 2
Because of the mechanistic nature of QSP models, they are well suited for the assessment of drug safety, and, in this context, may be applied as a tool to de‐risk drug development programs. 3 , 4 Late‐stage drug failures and postapproval withdrawals—which have huge costs for the pharmaceutical industry—are often secondary to safety concerns related to off‐target interactions or unforeseen metabolic changes. 5 Regulatory agencies, including the US Food and Drug Administration (FDA), have endorsed the development of model‐based predictive tools to increase drug safety. 6
Physiologically‐based pharmacokinetic (PBPK) models are increasingly viewed as a component of QSP that focus on how the body handles a drug or metabolite. 7 In contrast to population pharmacokinetic‐pharmacodynamic modeling, which generally focuses on the interaction of a single drug with a single target, PBPK‐QSP models aim to provide a comprehensive, integrated systems‐level description of drug exposure and effects at the molecular level.
An aim of QSP, as often presented, is to be rooted in a comprehensive understanding of all involved biological pathways, disease processes, and drug mechanisms of action. Yet, this assumes a reasonably accurate understanding and integration of the systems biology and systems physiology of drug metabolizing enzymes (DMEs) and transporters. We argue that this may not yet be the case. From the standpoint of “drug” metabolizing enzyme and “drug” transporter‐dependent biological pathways in QSP models, we believe it is time to reconceptualize the foundational science of drug metabolism and transport. Most available modeling tools, including PBPK and QSP, are based in fundamental biology of absorption, distribution, metabolism, and excretion (ADME) proteins to a limited extent. In other words, DMEs and drug transporters within the framework of QSP are usually only described in terms of their roles in the ADME of pharmaceuticals.
Nevertheless, there is a growing amount of data, often from outside “the field,” on the endogenous physiology of these “drug” metabolizing enzymes and “drug” transporters. Recent studies—including systems biology analyses of “omics” data from model systems, network analyses of ADME, and related genes, as well as human genetic data and metabolomic studies of complex diseases—indicate that these enzymes and transporters, often among the most highly expressed genes in tissues like liver, kidneys, and intestine, have vital, often coordinated roles in fundamental biological processes, 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 including remote interorgan and interorganismal communication via key metabolites and signaling molecules. 16 , 17 , 18 There is also a growing appreciation of how a "remote sensing and signaling network" of multispecific, oligospecific, and monospecific DMEs and transporters might aid in the capacity to (re)optimize levels of hundreds or thousands of small molecules in health and disease. 16 , 19 In addition, there is a need to develop biological models compatible with the proposed uses of endogenous biomarkers for hepatic and renal transporters as an alternative to traditional drug–drug interaction (DDI) studies that utilize probe drugs. 20
THE BIG PICTURE: REWRITING THE NARRATIVE FROM THE VIEWPOINT OF THE REMOTE SENSING AND SIGNALING THEORY
Here, we use the Remote Sensing and Signaling Theory (RSST) to detail the roles of DMEs and transporters in intratissue, interorgan, and interorganismal communication. We argue that this theory can lead to an improved understanding of the interactions between a drug and a physiological system at the level of cellular and biochemical networks. Below we explain the RSST, as well as its physiological and pharmaceutical implications, using a number of examples.
Rooting this broader RSST perspective of DMEs and transporters within QSP may allow for improved understanding and prediction of drug toxicities based upon the interplay of these proteins with metabolite/signaling networks. This may improve the prediction, mitigation, or circumvention of potential physiological aberrations by new drug entities, particularly small molecules that interact with ADME and related genes. 19
For example, if an investigational drug is identified as an inhibitor of the drug transporter organic anion transporter 1 (OAT1, SLC22A6), one might consider not only the impact on a co‐administered OAT1 drug substrate, or a single measurable metabolite known to be transported by OAT1, but also the indirect disruption of metabolic as well as sensing and signaling pathways that are facilitated through this transporter (e.g., purine metabolism, short chain fatty acids, gut microbiome products, or tricarboxylic acid (TCA) cycle intermediates). 21 , 22
From the perspective of metabolites and signaling molecules, this implies a thorough rewriting of the primary current narrative of how drug metabolizing enzymes and transporters function in the local and systemic physiology (and pathophysiology) of humans and other organisms. The data supporting this systems biology view of DMEs and transporters have come from a wide variety of sources, including in vivo metabolomics analyses of knockouts, in vitro studies demonstrating the formation and transport of physiologically important metabolites and signaling molecules, the effects of human mutations resulting in heritable metabolic disease, and the identification (via genomewide association study (GWAS) and other studies) of single nucleotide polymorphism in genes in common human metabolic abnormalities, and coexpression networks of ADME genes. 16 , 19
Thus, a reconceptualization of so‐called drug metabolizing enzymes and so‐called drug transporters is in order as the application of omics data and systems biology tools now supports their central roles in interorgan and interorganismal remote communication. This RSST‐based view may alter not only the drug discovery and development paradigm, but also improve understanding of fundamental disease biology.
OAT1 AND OTHER OATS AS EXEMPLARY SLC “DRUG” AND METABOLITE TRANSPORTERS
From a physiological perspective, OAT1 (SLC22A6), which was initially identified in 1996 as novel kidney transporter, is one of the best studied of the classic “drug” transporters 23 (Figure 1 ). At the time of its discovery, it was proposed to be either an organic anion transporter (OAT) or organic cation transporter (OCT). Subsequent studies have shown that OAT1 and the closely related OAT3 (SLC22A8) transport organic anions (e.g., nonsteroidal anti‐inflammatory drugs, and urate) as well as organic cations (e.g., cimetidine) and organic zwitterions (e.g., carnitine). Nevertheless, both OATs have, as implied by their names, a strong preference for organic anions; this is further borne out by recent machine‐learning analyses of molecular features of substrates interacting with OAT1, OAT3, OCT1, and OCT2. 24
Figure 1.
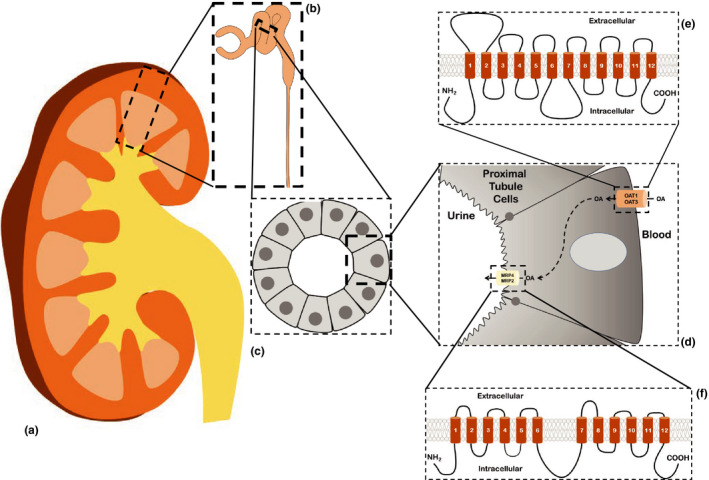
Transporter‐mediated clearance of organic anions in the kidney occurs in the proximal tubule and involves both SLC and ABC "drug" transporters. Illustration of the movement of organic anions out of the blood and into the tubular lumen occurring in cells of the proximal tubule. (a) Cross section through an adult kidney. (b) Enlarged area of renal cortex (indicated by dashed box in a) showing a partial nephron, including the glomerulus, proximal convoluted tubule, and a portion of the descending loop of Henle. (c) Enlarged cross section of the proximal tubule (indicated by dashed box in b), showing the arrangement of the epithelial cells around the tubular lumen. (d) Enlarged view of dashed box in c, showing a proximal tubule cell and the arrangement of SLC (OAT1, OAT3) and ABC (MRP2, MRP4) drug transporters. Organic anions (OAs) are moved from the blood and into the cell via SLC transporters (e.g., OAT1, OAT3) found on the basolateral membrane. These small anionic compounds are ultimately effluxed into the urine via ABC transporters (e.g., MRP2 and MRP4) found on the apical membrane. (e, f) Diagrams of the 12 transmembrane domain structure of the SLC e and ABC f drug transporters). [Colour figure can be viewed at wileyonlinelibrary.com]
OAT1 and OAT3 are two of the seven “drug” transporters initially highlighted by the FDA for focus on potential DDIs. 25 Although in vitro, murine knockout, and human studies clearly support their roles in the transport of drugs and toxins, 8 , 11 , 17 , 26 , 27 recent knockout mouse metabolomics and other studies have clarified the wide range of in vivo endogenous metabolites and signaling molecules capable of interacting with OATs. 15 , 21 , 22 , 28 , 29 , 30 The OATs and OAT‐like transporters constitute roughly half the of the SLC22 transporter family, which has over 30 members in humans and/or rodents, relatives of which can even be found in many invertebrates. 31 Members of the OAT family are expressed at high levels, if sometimes transiently, in many developing tissues, including the central nervous system (CNS), 32 , 33 before finally settling down, depending on the isoform, to particular adult expression patterns in the kidneys, liver, choroid plexus, blood‐brain barrier, olfactory epithelium, retina, pancreas, and/or other tissues. 34
All of this strongly supports key endogenous physiological functions of these “classical drug transporters” (OAT1 and OAT3), as well as other members of the OAT subfamily—and, indeed, the whole SLC22 transporter family. Physiologically important endogenous substrates of OAT1 and OAT3 include gut microbiome metabolites, prostaglandins (PGs), TCA cycle intermediates, odorant molecules, short chain fatty acids, bile acids, uric acid, cyclic nucleotides, and so‐called uremic toxins of chronic kidney disease (CKD). 15 , 30 , 35 Thus, apart from playing a key role in metabolism (e.g., TCA cycle and purine metabolism), OAT1 and OAT3 regulate levels of signaling molecules that act upon G‐protein coupled receptors and nuclear receptors, which are the basis of signaling and sensing mechanisms in physiology. 19 , 27
Furthermore, among the OAT subfamily members most closely related to the “multispecific” OAT1 and OAT3 are what seem to be more specific (“oligospecific”) transporters of cyclic GMP (OAT2), uric acid (URAT1, originally discovered as Rst in mouse), 36 odorants (OAT6), 37 and PGs (OAT‐PGs). 31 , 38 It now seems that one of the most studied drug transporter families (SLC22) is central to normal homeostasis and restoration of homeostasis after pathophysiological disturbances—a view strongly supported by the recent systems biology studies described below. 21
MRP4 AND BCRP AS EXEMPLARY ABC “DRUG” AND METABOLITE TRANSPORTERS
Several ABC efflux “drug” transporters, particularly the MRPs, have been shown to transport cyclic AMP and/or cyclic GMP in physiologically relevant contexts. Thus, these transporters, by affecting cellular levels of cyclic nucleotides, have the potential to regulate canonical second messengers involved in classical signaling pathways. 16 The functional case is strong for MRP4 as a cAMP efflux transporter that plays an important role in cellular behaviors mediated via cAMP, and the growing number of reports suggest that, in a sense similar to cAMP phosphodiesterase, egress through MRP4 might be considered a major mechanism for regulating cAMP levels. 39 , 40 , 41 If phosphodiesterase is considered part of classical signaling involving adenylate cyclase, it is not unreasonable to also include MRP4 and possibly MRP2.
Another ABC transporter, ABCG2 (BCRP)—perhaps the best‐known ABC drug transporter after MDR1 (P‐glycoprotein)—has, together with P‐glycoprotein, been extensively studied from the perspective of tumor resistance to chemotherapy; in certain resistant tumors, these two transporters are known to efflux chemotherapeutic agents. Although P‐glycoprotein continues to be generally thought of from the perspective of drug handling, it also participates in toxin and metabolite handling in the kidneys and other organs.
The view on BCRP is changing more rapidly, particularly because it is one of the major uric acid transporters in humans, as revealed by GWAS and other studies. 42 , 43 Thus, as with OAT1 and other “drug” transporters, BCRP (ABCG2) seems to have a dual character, functioning in drug and toxin transport as well as the regulation of physiologically (and pathophysiologically) important metabolites, such as uric acid—which is not only a natural antioxidant but, at high levels, is strongly associated with gout, kidney stones, progression of kidney disease, hypertension, and metabolic syndrome.
OVERVIEW OF THE ROLE OF PHASE I AND PHASE II DRUG METABOLIZING ENZYMES IN ENDOGENOUS METABOLISM
Since their discovery, cytochrome P450 (CYP450) enzymes—heme proteins that catalyze numerous so‐called phase I chemical reactions, such as hydroxylation, oxidation, and reduction—have been very often described as drug and toxicant metabolizing enzymes (phase I DMEs). The growth of information regarding CYP450‐mediated drug metabolism, coupled with knowledge of phase II conjugation pathways, including the UDP‐glucuronosyltransferases (UGTs) that catalyze the covalent linkage of glucuronic acid to substrates, has allowed for the prediction of drug interactions at the level of metabolism, leading to greater safety in the prescription of certain drugs. 44 Further individualization of drug therapy is achieved through understanding genetic polymorphisms in DMEs that affect drug metabolism and are thereby linked to response or toxicity. 45 Although it is beyond the scope of this paper to synthesize the huge amount of data regarding phase I and phase II DMEs in physiology, particularly as it pertains to links with transporters, we will highlight some examples of how the endogenous roles of these enzymes connect to SLC and ABC transporters in certain physiological contexts.
A number of endogenous functions of phase I and phase II enzymes have been elucidated and point to a central role for DMEs in local and remote sensing and signaling pathways. The endogenous functions of phase I enzymes include bile acid synthesis, steroid hormone synthesis, bioactivation of vitamin D via hydroxylation, metabolism of retinoic acid, 46 , 47 , 48 and bioactivation of arachidonic acid via hydroxylation and epoxidation. 49 Endogenous roles for phase II DMEs include metabolism and homeostasis of bilirubin, thyroid hormones, fatty acids, and amino acids.
Additional insight into the relevance and endogenous roles of metabolic enzymes can be gained from genetic polymorphisms associated with loss or gain of function. To illustrate, CYP2D6 is a polymorphically expressed P450 enzyme with differential functional activity in various people. 50 , 51 Multiple allelic variants of the CYP2D6 gene have been identified, which manifest clinically in patients as poor, intermediate, extensive, or ultra‐rapid metabolizers. 52 Interestingly, early research suggested that poor CYP2D6 metabolizers—determined on the basis of the clearance of the prototypical CYP2D6 probe drug debrisoquine—had lower incidence of psychasthenia as compared with extensive metabolizers. 53 Subsequent studies have shown that CYP2D6 expression and activity is related to psychological and neurocognitive functioning, including associations with disorders involving impulsivity, 54 schizophrenia, 55 eating disorders, 56 and suicide. 57 These findings suggest that CYP2D6 is involved in the metabolism of endogenous neuroactive substrates. 58 Indeed, CYP2D6—which is highly expressed in the cortex, hippocampus, and cerebellum—is known to be involved in the metabolism of several neuroactive signaling molecules, such as monoamines, endocannabinoids, and endomorphines. 58 Thus, as with “drug” transporters, evidence connecting DMEs to complex endogenous physiological functions is now quite widespread.
CONNECTIONS BETWEEN DRUG METABOLIZING ENZYMES AND SLC AND ABC TRANSPORTERS IN PHYSIOLOGY
It is well established that many compounds are substrates for both DMEs and transporters. Transporter‐mediated influx and efflux modulates the extent of drug accumulation in the cell, thereby directly affecting exposure to DMEs localized to the mitochondria or endoplasmic reticulum. From a pharmacokinetic perspective, the “interplay” between DMEs and transporters was first recognized between CYP3A and P‐glycoprotein, based in part upon coregulation via the pregnane X receptor, considerable drug substrate overlap, colocalization in intestinal epithelia, and hepatocytes, as well as adjacent gene locations. 59 , 60 Defining this link had key consequences for conceptualization of drug disposition, particularly in the prediction of bioavailability and DDIs. So‐called drug transporters began to be termed “phase III” of drug elimination. 61
As a comparable overlap of substrate specificity, expression patterns and regulation by similar transcription factors also exist between DMEs and transporters in the case for endogenous substrates, the same kinds of linkages can be generalized to posit a role for all categories of these proteins in the context of normal physiology. That is, in order for DME‐derived endogenous signaling molecules (e.g., eicosanoids) to bind to target receptors, they are usually effluxed from the cells where they are synthesized and transported across other biological membranes (in a nearby or remote location) via SLC and/or ABC “drug” and other transporters. This mechanism is likely to be very important not only in the liver but in nonhepatic tissues, such as the kidney proximal tubule, brain capillary endothelium, and choroid plexus that take up DME‐derived signaling molecules.
All categories of DMEs are highly expressed not only in the liver but also in the kidneys, gut, choroid plexus, as well as other tissues, 60 although they are only beginning to be functionally studied in nonhepatic physiological contexts. Furthermore, hepatic phase I or phase II modifications could generate a signaling molecule that is then transported via “drug” transporters into the choroid plexus or kidney proximal tubule whereupon it is potentially acted upon by phase I or phase II DMEs in the remote tissue. In this scenario, DMEs in one tissue would communicate remotely with DMEs in another tissue via influx and efflux transporters. This general notion has been recently supported by the development, analysis, and partial validation of a large gut‐liver‐kidney network of DMEs, and SLC and ABC transporters, as well as interacting regulatory genes. 19
What is now required are thorough clinical and basic data‐supported, systems biology approaches to understanding unifying underlying principles of what otherwise might seem to be unrelated domains and subdomains of pharmacology, metabolism, and endocrine (and other) physiology—despite being well‐developed fields in and of themselves.
THE RSST: INTEGRATING THE SYSTEMS PHYSIOLOGY OF SLC AND ABC TRANSPORTERS AND DRUG METABOLIZING ENZYMES
The RSST has been described elsewhere in considerable detail 16 , 31 and will be reviewed here (Figure 2 ). It was formulated in multiple papers in 2006–2009 based on what then seemed disparate observations on the roles of OATs and other SLC and ABC drug transporters in metabolism, signaling, organ development, and disease. 16 , 17 , 18 , 27 , 62 As more RNA profiling, quantitative polymerase chain reaction, and in situ hybridization data became available, it began to be appreciated that SLC and ABC transporters, as well as closely related isoforms, were expressed in tissues throughout the body, including cells not conventionally considered to be crucial to drug disposition.
Figure 2.
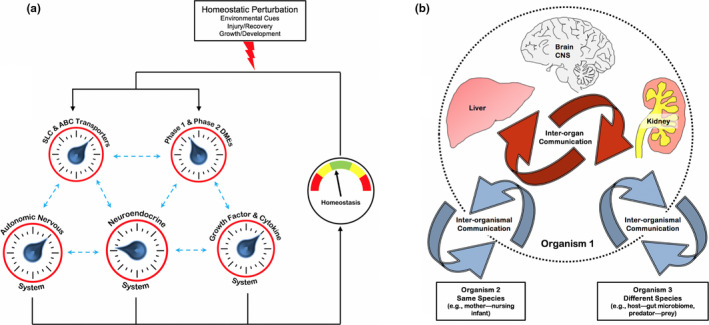
Remote sensing and signaling via ABC and SLC drug transporters and phase I and phase II drug metabolizing enzymes (DMEs). (a) Schematic representation of how SLC and ABC “multispecific drug” transporters and DMEs (expressed in many tissues) (b) are linked to form a small molecule communication system. A "remote sensing and signaling network" (RSSN)—consisting of multispecific transporters and DMEs functioning together with oligospecific and monospecific counterparts as well as regulatory proteins—helps modulate levels of many thousands of metabolites in tissues and fluids throughout the body. 19 This remote sensing and signaling system works in parallel with other regulatory systems to maintain homeostasis and has a complex organization and emergent properties, particularly after perturbation. (b) “Drug” transporters consist of SLC and ABC transporters expressed in all epithelial, as well as many nonepithelial, tissues throughout the body. The remote sensing and signaling theory not only emphasizes interorgan communication (red arrows) but also interorganismal communication (blue arrows), such as the movement of small molecules across the intestine (host‐gut microbiome), and/or into breast milk (mother—nursing infant), or across the placental barrier (mother–baby). CNS, central nervous system (adapted from reference 16). [Colour figure can be viewed at wileyonlinelibrary.com]
As discussed above, these transporters are capable of transporting, in vitro and in vivo, a broad range of key metabolites (e.g., citric acid cycle intermediates and short chain fatty acids) and signaling molecules (e.g., cyclic nucleotides, PGs, thyroxine, odorants, and bile acids) known to activate classical signaling pathways mediated by G‐protein coupled receptors and nuclear receptors. In addition, human mutations in SLC and ABC “drug” transporters were increasingly being shown to result in metabolic disease affecting lipid metabolism, levels of uric acid, carnitine, and other endogenous metabolites. 13 , 63 Furthermore, the SLC and ABC transporters were shown to be expressed early in mammalian development, sometimes transiently at high levels in particular cells during organogenesis (e.g., aortic arch and developing neural tissue), before “settling down” to be largely or solely expressed in a mature organ, such as the liver, kidneys, or intestine. Intriguingly, despite the overwhelming focus on human drug transport, it was clear that these multispecific SLC and ABC transporters were quite ancient from an evolutionary standpoint—with orthologs found in flies, worms, and other model organisms.
The RSST argues that SLC and ABC “drug” transporters in epithelial, endothelial, and other cells (barrier and nonbarrier) are central to the regulation of a systemwide small molecule remote communication network—shuttling key metabolites and signaling molecules into and between particular tissues and body fluid compartments (e.g., blood, cerebrospinal fluid, amniotic fluid, breast milk, urine, and bile) lined by the polarized epithelial or endothelial cells of those tissues. A tentative remote sensing and signaling network involving SLC and ABC transporters, as well as DME and regulatory genes in the gut‐liver‐kidney axis, has been recently proposed. 19
The SLC and ABC transporters in question are hypothesized to be not only essential to remote interorgan communication but also to interorganismal (e.g., gut microbiome and nursing infant) communication. 16 , 17 , 18 , 19 , 64 If SLC and ABC drug transporters and DMEs regulate metabolic and signaling networks within cells and organs—as knockout and other studies discussed here have shown—then transporters regulating the influx and efflux of key metabolites, signaling molecules, nutrients, and antioxidants in specific tissues can essentially connect multiple metabolic and signaling networks among organs, thereby enabling remote communication. This can be through remote effects on metabolism, signaling, redox state, and other key cellular processes. Because of overlapping substrate specificities for organic anions—which include a variety of key metabolites and signaling molecules—OATPs and MRPs in the liver can communicate with OATs in the kidneys, enabling remote communication functions to be performed by different organ‐selective transporters (e.g., kidney OATs vs. liver OATPs). Thus, these sets of transporters of “organic anions with high informational content” can essentially link up metabolic and signaling networks in different organs, in this case, the liver and kidneys. Once the intracellular compartments of cells in different organs are linked, so, potentially, are organelle compartments (e.g., mitochondria), which themselves contain SLC and ABC transporters.
Thus, a hierarchical interconnected architecture is established: Interorganismal to whole organism to organ system to organ to tissue to cell to organelle. 18 , 19 , 64 This architecture need not be top‐down or bottom‐up; depending on the circumstance (e.g., organ injury and disease), its center can be anywhere within this architecture. The potential for independent transporter and DME modulation at nearly every layer of this architecture makes for a robust, highly flexible, and adaptable system that is resilient to perturbation and has emergent properties (e.g., organ differentiation and regeneration). Various combinations of multispecific, oligospecific, and monospecific transporters and DMEs can work together in the RSSN to acheive equivalent effects, potentially enhancing the system's adaptability. It is worth emphasizing that a number of multispecific ADME genes appear to be "hubs" in the RSSN. Importantly, after perturbation, the system resets—although as a result of altered expression and localization of transporters, DMEs, and other gene products (as well as epigenetic modifications) due to injury, the specific relationships between different layers of architecture may not be the same. 17 , 64 In the long run, for instance, after multiple bouts of organ injury, a kind of “hysteresis” may prevail, leading to chronic changes in organ physiology.
This small molecule remote communication system functions beside, and together with, the neuro‐hormonal and growth factor‐cytokine systems to maintain and restore homeostasis (Figure 2 ); judging from effects of human and nonhuman mutations on local and system physiology—as well as on morphogenesis in model organisms—this SLC and ABC transporter RSST system (functioning in conjunction with phase I and phase II DMEs) might be just as important as the neuro‐hormonal and growth factor‐cytokine systems in maintaining health and, particularly, in reacting to disease. 16 , 17 , 18 , 64
In the setting of acute or chronic perturbation of organ function, such as liver or kidney injury, this small molecule communication mediated through SLC and ABC transporters as well as DMEs in numerous tissues seems to take on special significance in the organism’s attempt to regulate local and systemic concentrations of multiple metabolites and signaling molecules. 16 , 17 , 18 , 27 In interorgan communication mediated by SLC and ABC drug transporters as well as DMEs in the RSSN, one tissue “reacts” to the state (distress) of another tissue, presumably by sensing—with the help of nuclear receptors and G‐protein‐coupled receptors (GPCRs) as well as mechanisms possibly directly involving the transporter itself 65 —the local small molecule milieu, by altering transporter and DME expression and function. This would presumably require regulation of transcription, protein levels, and trafficking, including recycling. This alteration of the RSSN topology may almost completely resolve once homeostasis is restored. On the other hand, a drug that alters the expression and activity of an enzyme and transporter may cause similar downstream effects, yet these are poorly understood.
EXAMPLES OF REMOTE SENSING AND SIGNALING IN ORGAN AND SYSTEMIC PHYSIOLOGY AND PATHOPHYSIOLOGY
What follows are three examples that demonstrate aspects of how SLC and ABC “drug” and other transporters work with phase I and phase II DMEs to regulate/modulate metabolism and signaling involving small molecules in normal physiology where remote interorgan communication and/or interorganismal communication is important.
Example 1: Bile acid transporters and DMEs through the lens of remote sensing and signaling
Applying the lens of the remote sensing and signaling theory to a well‐described set of pathways regulating bile acid physiology provides insight into systems biology principles applicable to less‐studied pathways. In particular, the bile acid pathways provide an example of how the coordination between “multispecific” SLC and ABC transporters with “oligospecific” and “monospecific” transporters, as well as DMEs, leads to interorgan and interorganismal communication—involving activation of classical signaling pathways involving GPCRs and nuclear receptors as well as other mechanisms.
Bile acids, well‐known cholesterol‐derived compounds that act as physiologic detergents to facilitate the intestinal absorption of dietary fats and fat‐soluble vitamins, 66 , 67 are now recognized as versatile signaling molecules involved in the regulation of lipid, glucose, and energy homeostasis as well as inflammatory responses. 68 , 69 The signaling functions of bile acids are dependent upon the size and composition of the bile acid pool as well as intracellular modification of bile acids—both of which are tightly regulated by DMEs and transporters (Figure 3 ).
Figure 3.
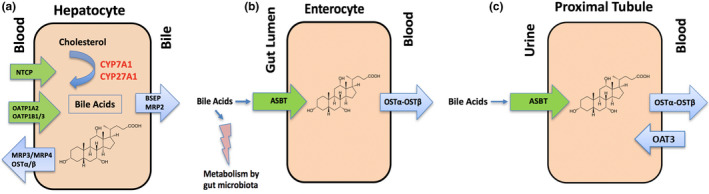
Interplay of bile acids with transporters, metabolizing enzymes, and the microbiome in the liver, intestine, and kidney. (a) In hepatocytes, bile acids control their own biosynthesis through negative feedback on CYP7A1 and CYP27A1. Cholesterol‐derived bile acids are negatively charged at physiologic pH and require carrier‐mediated transport to cross membranes. Blood to bile transport is governed by Na + taurocholate cotransporting peptide (NTCP), OATP1A2, OATP1B1, and OATP1B3. Secretion of bile acids from the liver into the bile canaliculi occurs primarily by BSEP and MRP2 to a lesser extent. MRP3, MPR4, and OSTα‐OSTβ play a compensatory role in bile acid efflux under cholestatic conditions. (b) Following the delivery of bile acids to the intestinal lumen through the bile ducts, bile acids are actively transported across the apical intestinal brush border membrane by ASBT, followed by efflux across the basolateral membrane and into the portal circulation by OSTα‐OSTβ. Additionally, bile acids in the gut lumen interact with intestinal microbes. Bile acids have direct antimicrobial effects on gut microbes. Conversely, deconjugation of bile acids by gut microbes prevents ASBT‐directed intestinal reuptake. (c) Bile acids are reclaimed in the kidney by active reabsorption in the proximal tubules. The transport mechanisms involved in renal tubular reabsorption involve ASBT and OSTα‐OSTβ expressed on the apical and basolateral membranes, respectively, while OAT3 contributes to secretion. Throughout these pathways, bile acids act as signaling molecules as ligands for G protein–coupled receptors and nuclear receptors. [Colour figure can be viewed at wileyonlinelibrary.com]
Bile acids control their own biosynthesis through negative feedback on hepatic cytochrome p450 enzymes including cholesterol‐7α‐hydroxylase (CYP7A1) and sterol 27‐hydroxylase (CYP27A1). 70 Synthesis pathways are complex and extensively reviewed elsewhere. 71 , 72 Negatively charged at physiologic pH, bile acids require carrier‐mediated transport to cross biological membranes. 73 SLC and ABC transporters in the liver, intestine, and kidneys are critical in driving their recirculation and regulating the levels of the bile acid pool. Thus, the physiological signaling roles of bile acids are inextricably linked to the expression and function of DMEs and transporters.
The apical and basolateral carrier mechanisms involved in bile acid transport have generated significant physiological and pharmaceutical interest. 73 , 74 , 75 , 76 In hepatocytes, the blood to bile transport of bile acids is principally governed by the basolateral located Na + taurocholate cotransporting peptide (NTCP) and OATPs, namely OATP1A2, OATP1B1, and OATP1B3 (the latter two being classical "drug" transporters). 75 The secretion of bile acids from the liver into the bile canaliculi occurs predominantly by the bile salt export pump (BSEP; ABCB11), 77 whereas the ATP Binding Cassette Protein C2 (MRP2) plays a minor role transporting bile acid conjugates. 78 In addition, MRP3, MPR4, and the heteromeric organic solute transporter OSTα‐OSTβ (SLC51) seem to play a compensatory role in bile acid efflux under cholestatic conditions 79 (Figure 3 ).
Over 90% of bile acids delivered to the intestinal lumen are re‐absorbed across the intestinal epithelium and returned to the liver by the portal venous circulation. Bile acids are actively transported across the apical intestinal brush border membrane by ASBT (SLC10A2), followed by efflux across the basolateral membrane and into the portal circulation by OSTα‐OSTβ 74 (Figure 3 ). Bile acids are also reclaimed in the kidneys by re‐absorption in the proximal tubules involving renal tubular reabsorption similar to that in enterocytes (i.e., ASBT and OSTα‐OSTβ are expressed on the apical and basolateral membranes, respectively), whereas OAT3 contributes to renal secretion (Figure 3 ). 29 , 80
The signaling properties of bile acids are achieved predominantly through their ability to act as ligands for GPCRs, most notably the membrane receptor TGR5, and the nuclear receptor farnesoid X receptor (FXR), 81 both of which are highly expressed in tissues that are exposed to bile acids. 82 Bile acid activation of FXRs alters the expression of aforementioned uptake and efflux transporters involved in their disposition. For example, the binding of bile acids to FXR decreases NTCP expression and increases BSEP expression. 83 , 84 , 85 Further, FXR activation has a variety of physiologic consequences, and, in the liver, FXR activation by bile acids leads to a reduction in plasma triglyceride concentrations via the inhibition of de novo lipogenesis and very low‐density lipoprotein overproduction. 82 In Fxr knockout mice, total cholesterol and triglycerides are elevated; consistent with this, activation of FXR by the synthetic agonist GW4064 lowers cholesterol and triglyceride levels in wild‐type and diabetic mice, but not in FXR‐/‐ mice. 86 , 87
Thus, DMEs and transporters, including classic “drug” transporters like SLCO1B1 and SLCO1B3, are key determinants of bile acid exposure within cells and target organs. The diverse signaling functions of bile acids can be viewed as the net result of (i) autoregulation of bile acid biosynthesis via hepatic DMEs and (ii) coordinated expression of transporter genes to modify the intracellular bile acid exposure in key organs (e.g., liver, intestine, and kidneys), whereby bile acids subsequently interact with specific GPCRs and/or nuclear receptors in order to trigger a specific biochemical event (e.g., gluconeogenesis).
It is to be emphasized how the remote interorgan communication that regulates the myriad of signaling and metabolic events highlighted above relies upon “multispecific” SLC (e.g., OATPs) and ABC (e.g., MRPs) “drug” transporters functioning in series and in parallel with transporters of much more limited specificity (monospecificity and/or oligospecificity; e.g., BSEP, OSTs, NTCP, and ASBT). This theme recurs in small molecule homeostasis, for instance, in the case of uric acid (discussed below).
In addition, bile acids also serve as signaling molecules promoting remote interorganismal communication between the gut microbiota and the host (Figure 3 ). Emerging evidence points to significant crosstalk between the intestinal microbiome and the host, which is partly mediated through bioconversion of bile acids. 88 , 89 For example, bile acids have direct antimicrobial effects on gut microbes. 90 Moreover, deconjugation of bile acids by gut microbes prevents ASBT‐directed intestinal reuptake, thereby altering signaling through FXR. 90 Dysregulation of this balance has been shown in diabetic and obese phenotypes, which have been linked to enterohepatic diseases, including cancer. 91 Secondary bile acids derived from the gut flora are absorbed by the intestine, and apart from potentially affecting metabolism and signaling in ways described above, also seem to act as endotoxins affecting a variety of cellular processes and are associated with the development of colon cancer. 92
Viewed from the perspective of the RSST, bile acids are a particularly well‐studied example of small molecule remote interorgan and interorganismal communication that, when gone awry, leads to disease. Bile acids are also particularly interesting because, while traversing the remote interorgan and interorganismal communication pathways via SLC and ABC drug and other transporters, these endogenous small molecules are currently being used as drugs to treat gall stones. 93 Thus, here is one rather obvious example of the potential of co‐opting a local remote sensing and signaling system in the service of drug efficacy. Although this drug happens to be an endogenous molecule, the general principle applies to metabolite‐like drugs as well.
Example 2: Uric acid homeostasis as a model of remote communication between organs and within tissues
One of the more common metabolic abnormalities worldwide is a high plasma uric acid. Uric acid, considered a major antioxidant in the body, is the byproduct of purine metabolism and, under basal conditions, it is largely, but not exclusively, eliminated by net transport from blood to urine by proximal tubule cells of the kidneys. The role of intestinal extrusion into the gut lumen is, however, increasingly appreciated, particularly in the setting of compromised kidney function. 42 Hyperuricemia is associated with gout, kidney stones, CKD progression, and hypertension, 94 and is part of the “metabolic syndrome,” sometimes called Syndrome X—one of the most common diseases in the world. 95
One of the important successes in the GWAS heyday was the identification of several loci associated with hyperuricemia in different ethnic populations, ultimately leading to the identification of unexpected genes in humans. 96 Knockout animal and in vitro studies implicated other genes regulating uric acid handling by the kidneys. 9 Nearly all of these genes are transporters, and a remarkable fraction of these are “drug” transporters or their close relatives: ABCG2 (BCRP), SLC22A6 (OAT1), SLC22A8 (OAT3), SLC22A12 (URAT1), SLC22A11 (OAT4), and possibly ABCC2 (MRP2) and ABCC4 (MRP4). Other key uric acid transporters include SLC2A9, which is a member of an SLC family of sugar transporters, and members of the phosphate transporter family, NPT1‐3 (SLC17 family). 12 Of the SLC and ABC transporters mentioned, most have very high expression in the kidneys, but some, such as ABCG2, are highly expressed in intestinal epithelium. 97 , 98
Thus, if viewed from the limited lens of the important clinical syndrome of human hyperuricemia, a reasonable and supportable case might be made for the argument that several major ABC and SLC “drug” transporters regulate uric acid homeostasis; from this (limited) perspective, the ability of these transporters to absorb, distribute, and eliminate NSAIDs, chemotherapeutic agents, antivirals, and other drugs might seem incidental. Indeed, multispecific “drug” transporters, like ABCG2, OAT1, and OAT3, seem to function in concert with uric acid transporters of much more limited specificity, such as SLC2A9 and URAT1, which transport only a few other molecules apart from urate.
When the rodent kidney is partly resected in a well‐established model of progressive kidney disease (5/6 nephrectomy), intestinal efflux increases markedly, and intestinal ABCG2 expression increases. 99 This suggests some sort of remote interorgan communication between the kidneys and the intestine in the setting of compromised renal function aimed at lowering uric acid levels toward normal levels when the function of the organ primarily involved in uric acid excretion, the kidneys, declines. The phenomenon seems operative in humans as well. In patients with CKD, single nucleotide polymorphisms in ABCG2, presumably in the intestine, became several orders of magnitude more important for regulation of plasma uric acid. 10
These human and rodent studies suggest that the injury to the kidney—leading to hyperuricemia as well as many other metabolic abnormalities—generates a signal that results in remote communication with the intestine, leading to increased activity of (apparently) the major intestinal uric acid efflux (also drug) transporter, ABCG2. This results in net movement of uric acid from the plasma to the gut lumen, thereby mitigating the effect of low renal function on plasma uric acid levels.
How might this occur? Certain “drug” transporters undergo substrate induction, 11 and one signal that could alter transcriptional mechanisms in intestinal epithelial cells is uric acid itself. 12 However, uric acid is also an antioxidant, and rising levels could modify the redox state of the intestinal epithelial cell, thereby affecting redox‐dependent signaling that might increase the expression and/or activity of intestinal ABCG2. On the other hand, the expression of ABCG2 is increased in enterocytes cultured in the presence of indoxyl sulfate. Indeed, ABCG2 is regulated by transcription factors and nuclear receptors like HNF1, HNF4, and AHR, 8 some of which are sensitive to uremic toxins (e.g., indoxyl sulfate) that accumulate in CKD or substrates, such as fatty acids that might have otherwise been eliminated by OATs in the normal kidney but now accumulate in the setting of renal dysfunction. 13 Defining the mechanism of remote sensing and signaling to maintain uric acid homeostasis is of tremendous basic and clinical importance. Drugs that can increase intestinal extrusion of uric acid in the setting of progressive renal disease may not only lower plasma uric acid but also slow the progression of CKD, in which uric acid is thought to play a causative role. 94
Example 3: Prostaglandin homeostasis involving DMEs and drug transporters as remote sensing and signaling
Multiple cytochrome P450 families, including CYP1, CYP2, CYP3, and CYP4, participate in several steps in the biosynthesis and degradation of eicosanoids—diverse signaling molecules that include PGs, prostacyclins, thromboxanes, leukotrienes, hydroxyeicosatetraenoic (HETE) acids, and epoxyeicosatetraenoic (EET) acids. 14 Collectively, eicosanoids are central to a variety of critical biological processes, such as inflammation, fever, blood pressure regulation, clotting, immune modulation, renal blood flow, tissue growth, and vascular autoregulation. 15
Along with cyclooxygenase and lipoxygenase, CYP450 enzymes—principally CYP2C, CYP2J, CYP4A, and CYP4F isoforms—control the generation of arachidonic acid metabolites to bioactive eicosanoids. 106 , 107 , 108 , 109 The diverse signaling compounds generated by these pathways include vasoactive EET acids and HETE acids. EET acids are produced by CYP2C and CYP2J isoforms, which produce anti‐inflammatory, vasodilatory, and neuroprotective effects. 110 , 111 Conversely, the terminal hydroxylation of arachidonic acid by CYP4A and CYP4F isoforms produce 20‐HETE, which is a highly potent vasoconstrictive, pro‐angiogenic, hypertensive, and pro‐inflammatory metabolite that is a known mediator of injury after various brain insults. 112 , 113 , 114 , 115 The paradoxical opposing effects of EET vs. 20‐HETE metabolites, suggests that isoform specific regulation of CYP activity may underlie both basal vascular autoregulation as well as resultant damage after brain injury.
Vasodilation by glutamate requires nitric oxide inhibition of 20‐HETE to allow for PGE2 dilation at the microvascular level. 113 This finding was particularly important given that the inhibition of nitric oxide synthase prevented glutamate vasodilation, and the use of a 20‐HETE inhibitor restored vasodilation even in the presence of the nitric oxide synthase inhibitor. This work is consistent with prior studies implicating 20‐HETE inhibition as a mechanism of nitric oxide dilation and autoregulatory control. 116 , 117 , 118 Furthermore, these studies illustrate the counterbalancing actions between CYP and cyclooxygenase enzymes in regulation of cerebrovascular tone and autoregulation.
At the systemic and organ level PGs play a key role in regulating blood flow and sodium excretion. “Drug” transporters seem crucial in the renal excretion of PGE2, involving multiple basolateral transporters in the proximal tubule, including OAT1, OAT2, OAT3, OATPs, and possibly OCTs, 119 as well as apical transport due to OAT4. 120 Phase II metabolism via glucuronidation (including UGT2B7 and UGT1A9) also play a role in bioinactivation of HETE, including 12‐HETE, 15‐HETE, and 20‐HETE, thereby limiting the availability of these compounds for cellular processes and promoting renal excretion of HETE conjugated metabolites (Figure 4 ). 121 , 122 EET metabolites undergo subsequent conversion to lesser activity dihydroxyeicosatrienoic acid metabolites by soluble epoxide hydroxylase. Thus, there is considerable evidence that drug transporters and DMEs coordinate systemic and local levels of these paradigmatic signaling molecules, thereby playing a major role in regulating local and systemic physiology.
Figure 4.
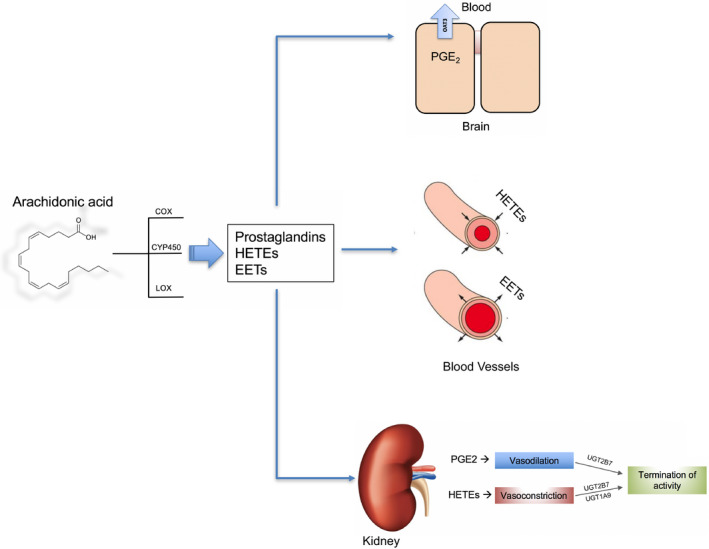
Prostaglandin biosynthesis and transport. Illustration of the synthesis of bioactive eicosanoids including prostaglandin E2 (PGE2), epoxyeicosatrienoic acids (EETs), and hydroxyeicosatetraenoic acids (HETEs). These arachidonic acid metabolites are generated principally by cyclooxygenase (COX), lipooxygenase (LOX), and CYP450 enzymes (CYP2C, CYP4A, and CYP4F isoforms). HETEs and EETs have vasoconstrictive and vasodilatory effects, respectively, in blood vessels and the kidney, and phase II metabolism via glucuronidation (including UGT2B7 and UGT1A9) plays a role in bioinactivation, limiting the availability of these compounds for cellular processes. Transporters, including "drug" transporters like OAT3, regulate intratissue and intracellular uptake and efflux of various eicosanoids in the brain and elsewhere. [Colour figure can be viewed at wileyonlinelibrary.com]
Given that 20‐HETE has been implicated as a mediator of brain injury, whereas EET metabolites demonstrate neuroprotection and anti‐inflammatory effects, therapeutic strategies to decrease 20‐HETE formation by CYP4 inhibition and/or increase EET metabolites by soluble epoxide hydroxylase inhibition have been evaluated in preclinical models of neuronal injury, pain, and hypertension. 112 , 114 , 123 , 124
Apart from the signaling properties of eicosanoids, some studies have given special attention to local concentrations, for example in the CNS. PGE2 in the CNS plays a role in regulating the sleep/wake cycle, and cerebrospinal fluid concentrations of PGE2 are a determinant of the progression of neuroinflammation in stroke. 125 , 126 As drug transporters regulate intratissue and intracellular uptake and efflux of various eicosanoids, these proteins participate jointly with DMEs in regulating local concentrations. 127 , 128 Thus, the examples of neuroactive signaling molecules and PGE2 illustrate how remote (intratissue, interorgan, and interorganismal) communication via small molecules involves a close connection between DMEs with “drug” transporters.
THE SYSTEMS BIOLOGY VIEW OF DMES AND “DRUG” TRANSPORTERS WITHIN QSP: PHARMACEUTICAL AND REGULATORY IMPLICATIONS
Due to safety concerns stemming from transporter and metabolism‐mediated drug interactions, the FDA, European Medicines Agency (EMA), and the Ministry of Health, Labour, and Welfare of Japan (MHLW) have published guidance documents containing recommendations for the evaluation of DDI potential of investigational drugs. 25 , 129 , 130 These regulatory recommendations almost exclusively emphasize pharmacokinetic issues. That is, how does the interaction of one drug with a DME or transporter change the exposure (concentration) of another drug?
This narrow view ignores the diverse endogenous physiological functions of transporters and DMEs presented in this paper. Furthermore, largely as a result of recent systems biology studies that apply omics methodologies to knockouts of the aforementioned “drug” transporters (and DMEs), development and analysis of an ADME gene network, and re‐evaluation of in vitro transport studies—as well as metabolic reconstructions from “omics” data and analyses of human mutations and polymorphisms associated with rare and common metabolic diseases—a very different picture emerges than implied by the focus of regulatory agencies. In the case of SLC22 transporters, this new picture has little, if anything, to do with transport of drugs or even the transport of exogenous toxins (e.g., organic mercurials). 131
Within the conceptual framework of the RSST, an entirely different and biologically well‐supported narrative about these DMEs and transporters can be written based on the accumulating genetic, “omics,” and systems biology evidence. Again, this need not have anything to with drugs: When incorporated into QSP and PBPK models, this revised narrative of DMEs and transporters may facilitate an improved understanding of fundamental biology and pathophysiology and may lead to a clinically relevant re‐interpretation of much that remains unexplained in the field of multispecific SLC and ABC transporters, as well as those with more limited (mono or oligo) specificity.
Notably, much work remains to be done on fleshing out this conceptual framework and validating it in nonhuman models, much less clinical contexts. But that is not to say that an RSST‐based view cannot be incorporated into current thinking. To the extent that this system’s view of enzymes and transporters ultimately becomes integrated within QSP models, drug therapies for pathological states can be designed to either restore homeostasis, particularly of small organic molecules, or minimize drug‐metabolite interactions, broadly interpreted. It may be possible to quantitatively model the selective inhibition or activation of transporter and/or DME expression and/or function by pharmaceutical or other means in one or more tissues to predictably ameliorate syndromes associated with these pathological states (i.e., the uremic syndrome of CKD) by favorably altering the distribution and/or elimination of key endogenous molecules. Again, much work remains to be done on building out this conceptual framework within PBPK/QSP models, including model qualification and verification practices. This is a new perspective in a developing field, and such steps will be critical to improving model confidence in order to form the basis for “go/no‐go” decision making in industry.
Nevertheless, these pathways are now beginning to be revealed by “omics”‐based metabolic reconstructions, such as the OAT1‐centered metabolic network and the building of gene‐protein "remote sensing and signaling networks" aimed at recapitulating the ADME genes among themselves and with other genes, including regulatory genes. 19 , 21 These “maps” will surely be revised as more data becomes available and systems biology methods improve. Although the complexities of extrapolating in vitro or in vivo animal data to human must be carefully considered, including differences in DME and transporter gene expression, protein content, activity, and substrate specificity, this systems‐based view may help to predict drug toxicities and off‐target effects, particularly in the many dynamic settings encountered clinically—which are often the most unpredictable aspect of drug discovery and development. For instance, given that OATs seem to be major transporters of uremic toxins in kidney disease, it seems prudent to not only consider the effect of an investigational drug on the levels of these individual OAT‐transported uremic toxins but also other “distal” metabolites in the OAT‐1 centered network (e.g., purine metabolism, short chain fatty acids, or TCA cycle intermediates). The time dimension and spectrum of organ dysfunction (i.e., mild acute kidney injury to late CKD) indicates how complex these issues may be. However, they are beginning to seem tractable with current and evolving “omics” data and systems biology tools.
From the perspective of drug discovery and development, the recognition that pharmaceuticals affect complex biological pathways independent of the target of interest—many of which involve interactions with DMEs and transporters—calls for the integration of the systemic biology of DMEs and transporters, such as described here in the context of the RSST, into QSP. If this integration can be achieved, it may better facilitate target selection, dose optimization, the prediction of clinical efficacy and toxicity (including off‐target effects), and patient stratification (precision medicine).
Funding
This work was funded in part by grants to S.K.N. from the National Institutes of Health (NIH, Eunice K. Shriver National Institute of Child Health and Human Development (NICHD) U54HD090259, National Institute of Diabetes, Digestive and Kidney Disease (NIDDK) R01DK109392, and National Institute of General Medical Sciences (NIGMS) R01GM132938).
Conflicts of Interest
The authors declared no competing interests for this work.
References
- 1. Zineh, I. Quantitative systems pharmacology: a regulatory perspective on translation. CPT Pharmacometrics Syst. Pharmacol. 8, 336–339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bai, J.P.F. , Earp, J.C. & Pillai, V.C. Translational quantitative systems pharmacology in drug development: from current landscape to good practices. AAPS J. 21, 72 (2019). [DOI] [PubMed] [Google Scholar]
- 3. Nijsen, M. et al Preclinical QSP modeling in the pharmaceutical industry: an IQ Consortium Survey examining the current landscape. CPT Pharmacometrics Syst. Pharmacol. 7, 135–146 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Workgroup, E.M. et al Good practices in model‐informed drug discovery and development: practice, application, and documentation. CPT Pharmacometrics Syst. Pharmacol. 5, 93–122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Onakpoya, I.J. , Heneghan, C.J. & Aronson, J.K. Post‐marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC Med. 14, 10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abernethy, D.R. , Woodcock, J. & Lesko, L.J. Pharmacological mechanism‐based drug safety assessment and prediction. Clin. Pharmacol. Ther. 89, 793–797 (2011). [DOI] [PubMed] [Google Scholar]
- 7. Rostami‐Hodjegan, A. Reverse translation in PBPK and QSP: going backwards in order to go forward with confidence. Clin. Pharmacol. Ther. 103, 224–232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burckhardt, G. , & Burckhardt, B.C . In vitro and in vivo evidence of the importance of organic anion transporters (OATs) in drug therapy In Drug Transporters. Handbook of Experimental Pharmacology, Vol. 201 (eds. Fromm M. & Kim R.) 29–104 (Springer, Berlin, Heidelberg, 2011). [DOI] [PubMed] [Google Scholar]
- 9. Eraly, S.A. et al Multiple organic anion transporters contribute to net renal excretion of uric acid. Physiol. Genomics 33, 180–192 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gottesman, M.M. & Ambudkar, S.V. Overview: ABC transporters and human disease. J. Bioenerg. Biomembr. 33, 453–458 (2001). [DOI] [PubMed] [Google Scholar]
- 11. Lepist, E.I. & Ray, A.S. Beyond drug‐drug interactions: effects of transporter inhibition on endobiotics, nutrients and toxins. Expert Opin. Drug Metab. Toxicol. 13, 1075–1087 (2017). [DOI] [PubMed] [Google Scholar]
- 12. Lin, L. , Yee, S.W. , Kim, R.B. & Giacomini, K.M. SLC transporters as therapeutic targets: emerging opportunities. Nat. Rev. Drug Discov. 14, 543–560 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tarling, E.J. , de Aguiar Vallim, T.Q. & Edwards, P.A. Role of ABC transporters in lipid transport and human disease. Trends Endocrinol. Metab. 24, 342–350 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. VanWert, A.L. , Gionfriddo, M.R. & Sweet, D.H. Organic anion transporters: discovery, pharmacology, regulation and roles in pathophysiology. Biopharm. Drug Dispos. 31, 1–71 (2010). [DOI] [PubMed] [Google Scholar]
- 15. Wikoff, W.R. , Nagle, M.A. , Kouznetsova, V.L. , Tsigelny, I.F. & Nigam, S.K. Untargeted metabolomics identifies enterobiome metabolites and putative uremic toxins as substrates of organic anion transporter 1 (Oat1). J. Proteome Res. 10, 2842–2851 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug. Discov. 14, 29–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ahn, S.Y. & Nigam, S.K. Toward a systems level understanding of organic anion and other multispecific drug transporters: a remote sensing and signaling hypothesis. Mol. Pharmacol. 76, 481–490 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu, W. , Dnyanmote, A.V. & Nigam, S.K. Remote communication through solute carriers and ATP binding cassette drug transporter pathways: an update on the remote sensing and signaling hypothesis. Mol. Pharmacol. 79, 795–805 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rosenthal, S.B. , Bush, K.T. & Nigam, S.K. A network of SLC and ABC transporter and DME genes involved in remote sensing and signaling in the gut‐liver‐kidney axis. Sci. Rep. 9, 11879 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chu, X. , Chan, G.H. & Evers, R. Identification of endogenous biomarkers to predict the propensity of drug candidates to cause hepatic or renal transporter‐mediated drug‐drug interactions. J. Pharm. Sci. 106, 2357–2367 (2017). [DOI] [PubMed] [Google Scholar]
- 21. Liu, H.C. et al An organic anion transporter 1 (OAT1)‐centered metabolic network. J. Biol. Chem. 291, 19474–19486 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jansen, J. et al Remote sensing and signaling in kidney proximal tubules stimulates gut microbiome‐derived organic anion secretion. Proc. Natl. Acad. Sci. USA 116, 16105–16110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lopez‐Nieto, C.E. , You, G. , Bush, K.T. , Barros, E.J. , Beier, D.R. & Nigam, S.K. Molecular cloning and characterization of NKT, a gene product related to the organic cation transporter family that is almost exclusively expressed in the kidney. J. Biol. Chem. 272, 6471–6478 (1997). [DOI] [PubMed] [Google Scholar]
- 24. Liu, H.C. et al Molecular properties of drugs interacting with SLC22 transporters OAT1, OAT3, OCT1, and OCT2: a machine‐learning approach. J. Pharmacol. Exp. Ther. 359, 215–229 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. US Food and Drug Administration . Guidance for Industry: Drug Interaction Studies—Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations, (US Department of Health and Human Services, US Food and Drug Administration, Center for Drug Evaluation and Research, Silver Spring, MD, 2012). [Google Scholar]
- 26. Sweet, D.H. , Bush, K.T. & Nigam, S.K. The organic anion transporter family: from physiology to ontogeny and the clinic. Am. J. Physiol. Renal Physiol. 281, F197–F205 (2001). [DOI] [PubMed] [Google Scholar]
- 27. Nigam, S.K. et al The organic anion transporter (OAT) family: a systems biology perspective. Physiol. Rev. 95, 83–123 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu, W. et al Multispecific drug transporter Slc22a8 (Oat3) regulates multiple metabolic and signaling pathways. Drug Metab. Dispos. 41, 1825–1834 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bush, K.T. , Wu, W. , Lun, C. & Nigam, S.K. The drug transporter OAT3 (SLC22A8) and endogenous metabolite communication via the gut‐liver‐kidney axis. J. Biol. Chem. 292, 15789–15803 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu, W. , Bush, K.T. & Nigam, S.K. Key role for the organic anion transporters, OAT1 and OAT3, in the in vivo handling of uremic toxins and solutes. Sci. Rep. 7, 4939 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nigam, S.K. The SLC22 transporter family: a paradigm for the impact of drug transporters on metabolic pathways, signaling, and disease. Annu. Rev. Pharmacol. Toxicol. 58, 663–687 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pavlova, A. , Sakurai, H. , Leclercq, B. , Beier, D.R. , Yu, A.S. & Nigam, S.K. Developmentally regulated expression of organic ion transporters NKT (OAT1), OCT1, NLT (OAT2), and Roct. Am. J. Physiol. Renal. Physiol. 278, F635–F643 (2000). [DOI] [PubMed] [Google Scholar]
- 33. Pavlova, A. , Stuart, R.O. , Pohl, M. & Nigam, S.K. Evolution of gene expression patterns in a model of branching morphogenesis. Am. J. Physiol. 277, F650–F663 (1999). [DOI] [PubMed] [Google Scholar]
- 34. You, G. & Morris, M.E. (eds.), Overview of drug transporter families In Drug Transporters: Molecular Characterization and Role in Drug Disposition, 2nd edn., 1–6 (Wiley‐Blackwell, Hoboken, NJ, 2014). [Google Scholar]
- 35. Vanholder, R.C. , Eloot, S. & Glorieux, G.L. Future avenues to decrease uremic toxin concentration. Am. J. Kidney Dis. 67, 664–676 (2016). [DOI] [PubMed] [Google Scholar]
- 36. Mori, K. et al Kidney‐specific expression of a novel mouse organic cation transporter‐like protein. FEBS Lett. 417, 371–374 (1997). [DOI] [PubMed] [Google Scholar]
- 37. Monte, J.C. , Nagle, M.A. , Eraly, S.A. & Nigam, S.K. Identification of a novel murine organic anion transporter family member, OAT6, expressed in olfactory mucosa. Biochem. Biophys. Res. Commun. 323, 429–436 (2004). [DOI] [PubMed] [Google Scholar]
- 38. Shiraya, K. et al A novel transporter of SLC22 family specifically transports prostaglandins and co‐localizes with 15‐hydroxyprostaglandin dehydrogenase in renal proximal tubules. J. Biol. Chem. 285, 22141–22151 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carozzo, A. et al Dual role of cAMP in the transcriptional regulation of multidrug resistance‐associated protein 4 (MRP4) in pancreatic adenocarcinoma cell lines. PLoS One 10, e0120651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Copsel, S. et al Multidrug resistance protein 4 (MRP4/ABCC4) regulates cAMP cellular levels and controls human leukemia cell proliferation and differentiation. J. Biol. Chem. 286, 6979–6988 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Decouture, B. et al Impaired platelet activation and cAMP homeostasis in MRP4‐deficient mice. Blood 126, 1823–1830 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hosomi, A. , Nakanishi, T. , Fujita, T. & Tamai, I. Extra‐renal elimination of uric acid via intestinal efflux transporter BCRP/ABCG2. PLoS One 7, e30456 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wen, C.C. et al Genome‐wide association study identifies ABCG2 (BCRP) as an allopurinol transporter and a determinant of drug response. Clin. Pharmacol. Ther. 97, 518–525 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Foti, R.S. & Dalvie, D.K. Cytochrome P450 and non‐cytochrome P450 oxidative metabolism: contributions to the pharmacokinetics, safety, and efficacy of xenobiotics. Drug Metab. Dispos. 44, 1229–1245 (2016). [DOI] [PubMed] [Google Scholar]
- 45. Maliepaard, M. et al Pharmacogenetics in the evaluation of new drugs: a multiregional regulatory perspective. Nat. Rev. Drug Discov. 12, 103–115 (2013). [DOI] [PubMed] [Google Scholar]
- 46. Nebert, D.W. & Russell, D.W. Clinical importance of the cytochromes P450. Lancet 360, 1155–1162 (2002). [DOI] [PubMed] [Google Scholar]
- 47. Nebert, D.W. , Wikvall, K. & Miller, W.L. Human cytochromes P450 in health and disease. Philos. Trans. R Soc. Lond. B Biol. Sci. 368, 20120431 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nebert, D.W. & Dalton, T.P. The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat. Rev. Cancer 6, 947–960 (2006). [DOI] [PubMed] [Google Scholar]
- 49. Imig, J.D. , Simpkins, A.N. , Renic, M. & Harder, D.R. Cytochrome P450 eicosanoids and cerebral vascular function. Expert Rev. Mol. Med. 13, e7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou, S.F. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part II. Clin. Pharmacokinet. 48, 761–804 (2009). [DOI] [PubMed] [Google Scholar]
- 51. Zhou, S.F. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part I. Clin. Pharmacokinet. 48, 689–723 (2009). [DOI] [PubMed] [Google Scholar]
- 52. Teh, L.K. & Bertilsson, L. Pharmacogenomics of CYP2D6: molecular genetics, interethnic differences and clinical importance. Drug Metab. Pharmacokinet. 27, 55–67 (2012). [DOI] [PubMed] [Google Scholar]
- 53. Bertilsson, L. , Alm, C. , De Las Carreras, C. , Widen, J. , Edman, G. & Schalling, D. Debrisoquine hydroxylation polymorphism and personality. Lancet 1, 555 (1989). [DOI] [PubMed] [Google Scholar]
- 54. Penas, L.E.M. , Dorado, P. , Pacheco, R. , Gonzalez, I. & LLerena, A. Relation between CYP2D6 genotype, personality, neurocognition and overall psychopathology in healthy volunteers. Pharmacogenomics 10, 1111–1120 (2009). [DOI] [PubMed] [Google Scholar]
- 55. Llerena, A. , Dorado, P. , Penas‐Lledo, E.M. , Caceres, M.C. & De la Rubia, A. Low frequency of CYP2D6 poor metabolizers among schizophrenia patients. Pharmacogenomics J. 7, 408–410 (2007). [DOI] [PubMed] [Google Scholar]
- 56. Penas‐Lledo, E.M. et al High risk of lifetime history of suicide attempts among CYP2D6 ultrarapid metabolizers with eating disorders. Mol. Psychiatry 16, 691–692 (2011). [DOI] [PubMed] [Google Scholar]
- 57. Zackrisson, A.L. , Lindblom, B. & Ahlner, J. High frequency of occurrence of CYP2D6 gene duplication/multiduplication indicating ultrarapid metabolism among suicide cases. Clin. Pharmacol. Ther. 88, 354–359 (2010). [DOI] [PubMed] [Google Scholar]
- 58. Penas‐Lledo, E.M. & Llerena, A. CYP2D6 variation, behaviour and psychopathology: implications for pharmacogenomics‐guided clinical trials. Br. J. Clin. Pharmacol. 77, 673–683 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Christians, U. , Schmitz, V. & Haschke, M. Functional interactions between P‐glycoprotein and CYP3A in drug metabolism. Expert Opin. Drug Metab. Toxicol. 1, 641–654 (2005). [DOI] [PubMed] [Google Scholar]
- 60. Wacher, V.J. , Wu, C.Y. & Benet, L.Z. Overlapping substrate specificities and tissue distribution of cytochrome P450 3A and P‐glycoprotein: implications for drug delivery and activity in cancer chemotherapy. Mol. Carcinog. 13, 129–134 (1995). [DOI] [PubMed] [Google Scholar]
- 61. Xu, C. , Li, C.Y. & Kong, A.N. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 28, 249–268 (2005). [DOI] [PubMed] [Google Scholar]
- 62. Kaler, G. et al Structural variation governs substrate specificity for organic anion transporter (OAT) homologs. Potential remote sensing by OAT family members. J. Biol. Chem. 282, 23841–23853 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schmitz, G. , Kaminski, W.E. & Orso, E. ABC transporters in cellular lipid trafficking. Curr. Opin. Lipidol. 11, 493–501 (2000). [DOI] [PubMed] [Google Scholar]
- 64. Nigam, A.K. et al Unique metabolite preferences of the drug transporters OAT1 and OAT3 analyzed by machine learning. J. Biol. Chem. 295, 1829–1842 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wu, W. , Bush, K.T. , Liu, H.C. , Zhu, C. , Abagyan, R. & Nigam, S.K. Shared ligands between organic anion transporters (OAT1 and OAT6) and odorant receptors. Drug Metab. Dispos. 43, 1855–1863 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hofmann, A.F. & Hagey, L.R. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell. Mol. Life Sci. 65, 2461–2483 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Reshetnyak, V.I. Physiological and molecular biochemical mechanisms of bile formation. World J. Gastroenterol. 19, 7341–7360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 3, 1191–1212 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mertens, K.L. , Kalsbeek, A. , Soeters, M.R. & Eggink, H.M. Bile acid signaling pathways from the enterohepatic circulation to the central nervous system. Front. Neurosci. 11, 617 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen, W. & Chiang, J.Y. Regulation of human sterol 27‐hydroxylase gene (CYP27A1) by bile acids and hepatocyte nuclear factor 4alpha (HNF4alpha). Gene 313, 71–82 (2003). [DOI] [PubMed] [Google Scholar]
- 71. Chiang, J.Y. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. J. Hepatol. 40, 539–551 (2004). [DOI] [PubMed] [Google Scholar]
- 72. Chiang, J.Y. Bile acids: regulation of synthesis. J. Lipid. Res. 50, 1955–1966 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kosters, A. & Karpen, S.J. Bile acid transporters in health and disease. Xenobiotica 38, 1043–1071 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dawson, P.A. , Lan, T. & Rao, A. Bile acid transporters. J. Lipid Res. 50, 2340–2357 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Alrefai, W.A. & Gill, R.K. Bile acid transporters: structure, function, regulation and pathophysiological implications. Pharm. Res. 24, 1803–1823 (2007). [DOI] [PubMed] [Google Scholar]
- 76. Shneider, B.L. Intestinal bile acid transport: biology, physiology, and pathophysiology. J. Pediatr. Gastroenterol. Nutr. 32, 407–417 (2001). [DOI] [PubMed] [Google Scholar]
- 77. Halilbasic, E. , Claudel, T. & Trauner, M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J. Hepatol. 58, 155–168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Akita, H. et al Characterization of bile acid transport mediated by multidrug resistance associated protein 2 and bile salt export pump. Biochim. Biophys. Acta 1511, 7–16 (2001). [DOI] [PubMed] [Google Scholar]
- 79. Yang, K. , Kock, K. , Sedykh, A. , Tropsha, A. & Brouwer, K.L. An updated review on drug‐induced cholestasis: mechanisms and investigation of physicochemical properties and pharmacokinetic parameters. J. Pharm. Sci. 102, 3037–3057 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ballatori, N. et al OSTalpha‐OSTbeta: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 42, 1270–1279 (2005). [DOI] [PubMed] [Google Scholar]
- 81. Thomas, C. , Pellicciari, R. , Pruzanski, M. , Auwerx, J. & Schoonjans, K. Targeting bile‐acid signalling for metabolic diseases. Nat. Rev. Drug. Discov. 7, 678–693 (2008). [DOI] [PubMed] [Google Scholar]
- 82. Li, T. & Chiang, J.Y. Bile acid signaling in metabolic disease and drug therapy. Pharmacol. Rev. 66, 948–983 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Denson, L.A. et al The orphan nuclear receptor, shp, mediates bile acid‐induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology 121, 140–147 (2001). [DOI] [PubMed] [Google Scholar]
- 84. Ananthanarayanan, M. , Balasubramanian, N. , Makishima, M. , Mangelsdorf, D.J. & Suchy, F.J. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J. Biol. Chem. 276, 28857–28865 (2001). [DOI] [PubMed] [Google Scholar]
- 85. Plass, J.R. et al Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology 35, 589–596 (2002). [DOI] [PubMed] [Google Scholar]
- 86. Kim, I. et al Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 48, 2664–2672 (2007). [DOI] [PubMed] [Google Scholar]
- 87. Sinal, C.J. , Tohkin, M. , Miyata, M. , Ward, J.M. , Lambert, G. & Gonzalez, F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102, 731–744 (2000). [DOI] [PubMed] [Google Scholar]
- 88. Wahlstrom, A. , Kovatcheva‐Datchary, P. , Stahlman, M. , Backhed, F. & Marschall, H.U. Crosstalk between bile acids and gut microbiota and its impact on farnesoid X receptor signalling. Dig. Dis. 35, 246–250 (2017). [DOI] [PubMed] [Google Scholar]
- 89. Wahlstrom, A. , Sayin, S.I. , Marschall, H.U. & Backhed, F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 24, 41–50 (2016). [DOI] [PubMed] [Google Scholar]
- 90. Ridlon, J.M. , Kang, D.J. , Hylemon, P.B. & Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 30, 332–338 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tsuei, J. , Chau, T. , Mills, D. & Wan, Y.J. Bile acid dysregulation, gut dysbiosis, and gastrointestinal cancer. Exp. Biol. Med. (Maywood) 239, 1489–1504 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ajouz, H. , Mukherji, D. & Shamseddine, A. Secondary bile acids: an underrecognized cause of colon cancer. World J. Surg. Oncol. 12, 164 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lanzini, A. & Northfield, T.C. Pharmacological treatment of gallstones. Practical guidelines. Drugs 47, 458–470 (1994). [DOI] [PubMed] [Google Scholar]
- 94. van den Brand, J.A. et al Uremic solutes in chronic kidney disease and their role in progression. PLoS One 11, e0168117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kaur, J. A comprehensive review on metabolic syndrome. Cardiol. Res. Pract. 2014, 943162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 96. Kottgen, A. et al Genome‐wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 45, 145–154 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ni, Z. , Bikadi, Z. , Rosenberg, M.F. & Mao, Q. Structure and function of the human breast cancer resistance protein (BCRP/ABCG2). Curr. Drug Metab. 11, 603–617 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mao, Q. & Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport–an update. AAPS J. 17, 65–82 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yano, H. , Tamura, Y. , Kobayashi, K. , Tanemoto, M. & Uchida, S. Uric acid transporter ABCG2 is increased in the intestine of the 5/6 nephrectomy rat model of chronic kidney disease. Clin. Exp. Nephrol. 18, 50–55 (2014). [DOI] [PubMed] [Google Scholar]
- 100. Bhatnagar, V. et al Analysis of ABCG2 and other urate transporters in uric acid homeostasis in chronic kidney disease: potential role of remote sensing and signaling. Clin. Kidney J. 9, 444–453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Williamson, B. , Dooley, K.E. , Zhang, Y. , Back, D.J. & Owen, A. Induction of influx and efflux transporters and cytochrome P450 3A4 in primary human hepatocytes by rifampin, rifabutin, and rifapentine. Antimicrob. Agents Chemother. 57, 6366–6369 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chen, M. et al Soluble uric acid increases PDZK1 and ABCG2 expression in human intestinal cell lines via the TLR4‐NLRP3 inflammasome and PI3K/Akt signaling pathway. Arthritis Res. Ther. 20, 20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nigam, S.K. & Bush, K.T. Uraemic syndrome of chronic kidney disease: altered remote sensing and signalling. Nat. Rev. Nephrol. 15, 301–316 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Capdevila, J.H. , Harris, R.C. & Falck, J.R. Microsomal cytochrome P450 and eicosanoid metabolism. Cell Mol. Life Sci. 59, 780–789 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Goetzl, E.J. , An, S. & Smith, W.L. Specificity of expression and effects of eicosanoid mediators in normal physiology and human diseases. FASEB J. 9, 1051–1058 (1995). [DOI] [PubMed] [Google Scholar]
- 106. Fan, F. , Muroya, Y. & Roman, R.J. Cytochrome P450 eicosanoids in hypertension and renal disease. Curr. Opin. Nephrol. Hypertens. 24, 37–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fan, F. & Roman, R.J. Effect of cytochrome P450 metabolites of arachidonic acid in nephrology. J. Am. Soc. Nephrol. 28, 2845–2855 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Maier, K.G. & Roman, R.J. Cytochrome P450 metabolites of arachidonic acid in the control of renal function. Curr. Opin. Nephrol. Hypertens. 10, 81–87 (2001). [DOI] [PubMed] [Google Scholar]
- 109. Kroetz, D.L. & Zeldin, D.C. Cytochrome P450 pathways of arachidonic acid metabolism. Curr. Opin. Lipidol. 13, 273–283 (2002). [DOI] [PubMed] [Google Scholar]
- 110. Wang, L. , Luo, G. , Zhang, L.F. & Geng, H.X. Neuroprotective effects of epoxyeicosatrienoic acids. Prostaglandins Other Lipid Mediat. 138, 9–14 (2018). [DOI] [PubMed] [Google Scholar]
- 111. Spector, A.A. Arachidonic acid cytochrome P450 epoxygenase pathway. J. Lipid Res. 50 (suppl), S52–S56 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Poloyac, S.M. , Zhang, Y. , Bies, R.R. , Kochanek, P.M. & Graham, S.H. Protective effect of the 20‐HETE inhibitor HET0016 on brain damage after temporary focal ischemia. J. Cereb. Blood Flow. Metab. 26, 1551–1561 (2006). [DOI] [PubMed] [Google Scholar]
- 113. Donnelly, M.K. et al 20‐HETE is associated with unfavorable outcomes in subarachnoid hemorrhage patients. J. Cereb. Blood Flow Metab. 35, 1515–1522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Renic, M. et al Effect of 20‐HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 29, 629–639 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Poloyac, S.M. Chapter 8: Drug and Fatty Acid Cytochrome P450 Metabolism In Critical Care In: Drug Metabolism in Diseases (Xie W., ed.), pp. 115–138. (Elsevier, Philadelphia, PA, 2016). [Google Scholar]
- 116. Wink, D.A. , Osawa, Y. , Darbyshire, J.F. , Jones, C.R. , Eshenaur, S.C. & Nims, R.W. Inhibition of cytochromes P450 by nitric oxide and a nitric oxide‐releasing agent. Arch. Biochem. Biophys. 300, 115–123 (1993). [DOI] [PubMed] [Google Scholar]
- 117. Alonso‐Galicia, M. , Sun, C.W. , Falck, J.R. , Harder, D.R. & Roman, R.J. Contribution of 20‐HETE to the vasodilator actions of nitric oxide in renal arteries. Am. J. Physiol. 275, F370–F378 (1998). [DOI] [PubMed] [Google Scholar]
- 118. Sun, C.W. , Falck, J.R. , Okamoto, H. , Harder, D.R. & Roman, R.J. Role of cGMP versus 20‐HETE in the vasodilator response to nitric oxide in rat cerebral arteries. Am. J. Physiol. Heart Circ. Physiol. 279, H339–H350 (2000). [DOI] [PubMed] [Google Scholar]
- 119. He, X. , Garza, D. , Nigam, S.K. & Chang, G. Multispecific organic cation transporter 1 (OCT1) from Bos taurus has high affinity and slow binding kinetics towards prostaglandin E2. PLoS One 11, e0152969 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kimura, H. , Takeda, M. , Narikawa, S. , Enomoto, A. , Ichida, K. & Endou, H. Human organic anion transporters and human organic cation transporters mediate renal transport of prostaglandins. J. Pharmacol. Exp. Ther. 301, 293–298 (2002). [DOI] [PubMed] [Google Scholar]
- 121. Little, J.M. et al Glucuronidation of oxidized fatty acids and prostaglandins B1 and E2 by human hepatic and recombinant UDP‐glucuronosyltransferases. J. Lipid Res. 45, 1694–1703 (2004). [DOI] [PubMed] [Google Scholar]
- 122. Turgeon, D. et al Glucuronidation of arachidonic and linoleic acid metabolites by human UDP‐glucuronosyltransferases. J. Lipid Res. 44, 1182–1191 (2003). [DOI] [PubMed] [Google Scholar]
- 123. Shaik, J.S. et al Soluble epoxide hydrolase inhibitor trans‐4‐[4‐(3‐adamantan‐1‐yl‐ureido)‐cyclohexyloxy]‐benzoic acid is neuroprotective in rat model of ischemic stroke. Am. J. Physiol. Heart Circ. Physiol. 305, H1605–H1613 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Morisseau, C. & Hammock, B.D. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 53, 37–58 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Matsumura, H. , Honda, K. , Choi, W.S. , Inoue, S. , Sakai, T. & Hayaishi, O. Evidence that brain prostaglandin E2 is involved in physiological sleep‐wake regulation in rats. Proc. Natl. Acad. Sci. USA 86, 5666–5669 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Takemiya, T. Prostaglandin E2 produced by microsomal prostaglandin E synthase‐1 regulates the onset and the maintenance of wakefulness. Neurochem. Int. 59, 922–924 (2011). [DOI] [PubMed] [Google Scholar]
- 127. Kawashima, H. & Strobel, H.W. cDNA cloning of three new forms of rat brain cytochrome P450 belonging to the CYP4F subfamily. Biochem. Biophys. Res. Commun. 217, 1137–1144 (1995). [DOI] [PubMed] [Google Scholar]
- 128. Tachikawa, M. , Ozeki, G. , Higuchi, T. , Akanuma, S. , Tsuji, K. & Hosoya, K. Role of the blood‐cerebrospinal fluid barrier transporter as a cerebral clearance system for prostaglandin E(2) produced in the brain. J. Neurochem. 123, 750–760 (2012). [DOI] [PubMed] [Google Scholar]
- 129. European Medicines Agency . Guideline on the investigation of drug interactions <https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐investigation‐drug‐interactions‐revision‐1_en.pdf> (2012). Accessed March 16, 2020. [Google Scholar]
- 130. Ishiguro, A. , Sato, R. & Nagai, N. Development of a new Japanese guideline on drug interaction for drug development and appropriate provision of information. Drug Metab Pharmacokinet. 35, 12–17 (2020). [DOI] [PubMed] [Google Scholar]
- 131. Engelhart, D.C. et al Systems biology analysis reveals eight SLC22 transporter subgroups, including OATs, OCTs, and OCTNs. Int. J. Mol. Sci. 21, 1791 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]