

SUMMARY
Despite the remarkable microbial diversity found within humans, our ability to link genes to phenotypes is based upon a handful of model microorganisms. We report a comparative genomics platform for Eggerthella lenta and other Coriobacteriia, a neglected taxon broadly relevant to human health and disease. We uncover extensive genetic and metabolic diversity and validate a tool for mapping phenotypes to genes and sequence variants. We also present a tool for the quantification of strains from metagenomic sequencing data, enabling the identification of genes that predict bacterial fitness. Competitive growth is reproducible under laboratory conditions and attributable to intrinsic growth rates and resource utilization. Unique signatures of in vivo competition in gnotobiotic mice include an adhesin enriched in poor colonizers. Together, these computational and experimental resources represent a strong foundation for the continued mechanistic dissection of the Coriobacteriia and a template that can be applied to study other genetically intractable taxa.
Graphical Abstract
eToc blurb:
Bisanz et al. focus on an in-depth study of Eggerthella lenta and the Coriobacteriia class: highly prevalent members of the human gut microbiota which have been poorly described. Through construction of a paired isolate/genome library, they validate tools for comparative genomic approaches to uncover effectors of xenobiotic metabolism and fitness.
INTRODUCTION
A goal of the microbiome field is to leverage metagenomic sequencing datasets to generate testable hypotheses about the mechanisms through which the human microbiome shapes host health and disease. Follow-up experiments rely on the availability of representative isolates, prompting multiple groups to conduct large-scale culturing efforts to isolate and catalog human-associated bacterial strains (Lagier et al., 2016; Poyet et al., 2019). By necessity these efforts favor breadth of diversity over in-depth analysis of taxonomic groups of interest. Given extensive strain-level variation in the human microbiome (Greenblum et al., 2015; Zeevi et al., 2019), these culture collections likely underestimate the metabolic and phenotypic diversity of many species.
An alternative and complementary approach is the in-depth analysis of multiple strains within a clade as extensive gene loss and gain (Vos et al., 2015) contributes to qualitative phenotypic differences within bacterial species. The clinical relevance of these strain-level differences has been well-documented for enteric pathogens; for example, virulence factors produced by enterohemorrhagic Escherichia coli (Bai et al., 2018), and Bacteroides fragilis (Chung et al., 2018). However, pathogenicity islands are the tip of the iceberg: the core genome (genes shared by all strains within a bacterial species) generally comprises a fraction of the genome. Genome instability can also lead to phenotypically-relevant changes during the passaging of strains; for example, the loss of the mucin-binding pilin of Lactobacillus rhamnosus GG (Sybesma et al., 2013). Yet, functionally relevant strain-level variation within the human microbiome remains poorly understood, due in part to the lack of large and well-characterized strain collections outside of model species.
Here, we demonstrate the utility of conducting an in-depth analysis of a single class of neglected, but clinically relevant human gut bacteria for which genetic tools are lacking: the Coriobacteriia. Many of the Coriobacteriia, particularly Eggerthella and Paraeggerthella, are considered to be opportunistic pathogens because of their isolation from bacteremia patients (Gardiner et al., 2015). Coriobacteriia catalyze a wide range of biotransformations of drugs (Haiser et al., 2013; Koppel et al., 2018), dietary phytochemicals (Bess et al., 2020; Matthies et al., 2012), and endogenous compounds (Devlin et al., 2015; Harris et al., 2018; Rekdal et al., 2019). They are also associated with multiple chronic diseases including multiple sclerosis (Cekanaviciute et al., 2017) and rheumatoid arthritis (Chen et al., 2016). The numerous outstanding questions about the causal role and mechanisms through which Coriobacteriia impact the etiology and treatment of disease prompted us to develop a platform for their study.
In this manuscript, we describe the curation of a collection of paired isolates and genomes representing the Coriobacteriia with a focus on Eggerthella lenta. We then comprehensively analyze genomic and metabolic variation demonstrating how extensive strain-variability can be exploited as a tool for mechanistic research by mapping phenotypic variability to genetic determinants. We designed a graphical user interface for this purpose (ElenMatchR) and validate its usage on antibiotic resistance phenotypes. Next, we sought to examine determinants of competitive fitness which necessitated the development of a computational tool for quantification of highly related strains in metagenomic sequencing data (StrainR). We uncovered extensive strain competition in the mouse GI tract which was associated with an undescribed putative host adhesin molecule which may play a role in host tropism. Importantly, the computational and experimental framework established here could be readily extended to other lineages of interest or used to continue to address the numerous unanswered questions about the biology of gut Coriobacteriia.
RESULTS
Phylogenetic and taxonomic analysis.
Through public repositories and de novo isolation, we collected 95 genomes (n=48 E. lenta) including 42 newly sequenced genomes for analysis paired with 46 isolates for experimental analysis (Table S1). Newly sequenced genomes are high-quality drafts with a low number of contigs (59 [16–134]) and high N50 (135,856 [50,271–627,931]) (median [range]) and low rates of duplicated marker genes (Table S1). Our analysis also included 12 metagenome assembled genomes (MAGs) with 95.6% [92.0–99.2] estimated completion and 1.5% [0–4.4] estimated contamination (Nayfach et al., 2019).
As many isolates do not have an assigned species, taxonomy was refined first on the basis of 16S rRNA alignment and subsequently on whole genome-based phylogeny and average nucleotide identity (ANI; Figure 1AB, Figure S1A–C). This analysis confirmed the majority of the taxonomic assignments, while also highlighting taxonomic inconsistencies in the Coriobacteriia. Eggerthella sp. YY7918 was isolated from human feces; however, our analysis clearly places it outside both genera suggesting it may represent both an unnamed species and genus within the Eggerthellaceae. The inclusion of this species into common databases for taxonomic assignment of sequencing reads may skew accurate identification and quantification of the genus Eggerthella in metagenomic datasets (Truong et al., 2015). Other instances of inappropriate assignment of species are obvious based on examination of the distributions of ANI between assigned species (Figure 1C). The type strains of Gordonibacter faecihominis and G. urolithinfaciens exhibit 98.9% ANI and 98.4% 16S rRNA similarity. Similarly, the type strains of Asaccharobacter celatus and Adlercreutzia equolifaciens are consistent with a single species and genus (97.0% ANI, 99.9% 16S rRNA) (Nouioui et al., 2018). Newly isolated strains consistent with this clade were assigned to A. celatus based on their closest alignment to this type strain at the time of genome sequencing. Finally, Gordonibacter sp. 28C has not been assigned a species as it demonstrates only a 92.0% ANI to the nearest proposed type strain G. massiliensis Marseille-P2775 and thus it may be proposed to represent an undescribed species.
Figure 1. Phylogenetic and taxonomic consistency of Coriobacteriia genomes.
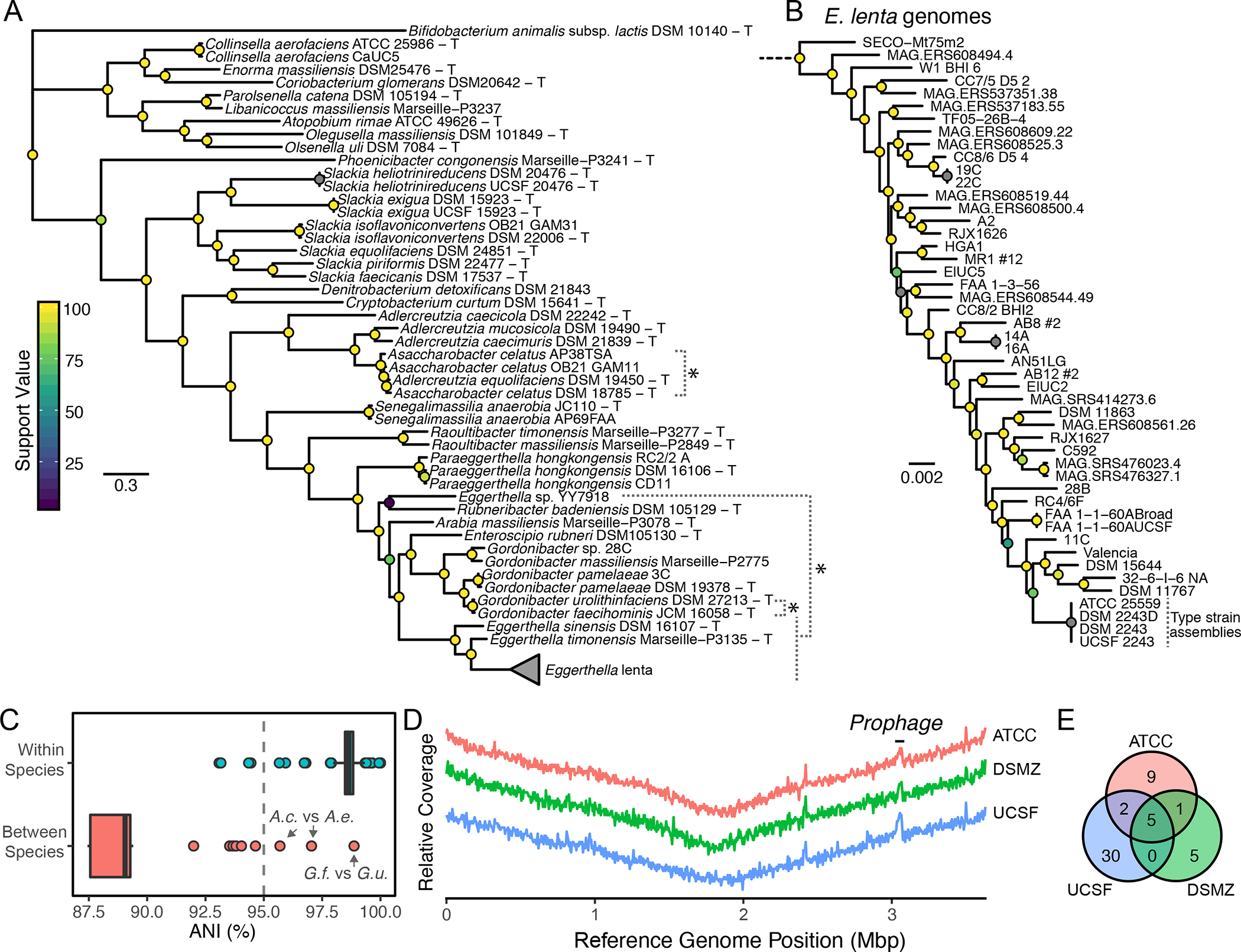
(A) Phylogenetic tree of Coriobacteriia genomes (T denotes a type or proposed-type strain). *Denotes taxonomic inconsistencies. (B) High-resolution phylogenetic tree of Eggerthella lenta strains based on SNPs in 195 core Eggerthella genes. (C) Whole genome average nucleotide identity (ANI) within and between species reveals consistent assignment of species based on a 95% speciation threshold. A.c. Assacharobacter celatus (2 strains) A.e. Adlercreutzia equolifaciens, G.f. Gordonibacter faecihominis,G.u. Gordonibacter urolithinfaciens. (D) Resequencing of the E. lenta type strain does not provide evidence of gene loss across contiguous reference assembly (CP001726.1). The y-axis denotes scaled relative sequencing depth for qualitative comparison. (E) Polymorphisms between E. lenta type strain isolates and the reference genome.
Finally, phylogenetic analysis and ANI revealed the presence of a set of genomes which were nearly identical to another genome in the dataset (n=8, ANI = 99.996 [99.990 – 99.999], median [range]). These isolates shared common history such as being isolated from the same host or represented resequencing of a publicly available genome. These clonal genomes were de-replicated before additional analysis and are identified in Table S1.
Polymorphisms in E. lenta type-strain stocks.
The type strain of E. lenta (DSM 2243 = ATCC 25559 = VPI 0255 = 1899 B) was sequenced and closed using a combination of Sanger and pyrosequencing (Saunders et al., 2009). Given our desire to map phenotypes to subtle genomic variation, we resequenced our lab isolate (termed UCSF 2243) and freshly obtained stocks from the DSMZ and ATCC collections using high-coverage Illumina sequencing (326 to 1,120-fold coverage; Figure 1D). Aligned sequence data covered the entire reference genome assembly (CP001726.1); however, we identified multiple shared and stock-specific variants (Figure 1E, Table S2). The highest number of unique variants were observed in our lab stock (n=30), followed by ATCC (n=9), and DSMZ (n=5). A missense variant (Gly52Asp) in a putative beta-lactamase was observed in the ATCC 25559 isolate which was consistent with its subtle but clinically relevant increase in penicillin G minimal inhibitory concentration (1 μg/mL) relative to the DSMZ and UCSF stocks (0.25–0.5 μg/mL; Table S3), although additional experimental validation is warranted. We also observed an 9.2±2.6% (mean±sd) increase in coverage at a putative prophage (3.03–3.06 Mbp) in all three stocks, suggesting that it may be replicative during in vitro growth. Together, these observations underlie the importance of routine resequencing of lab stocks to increase rigor and reproducibility.
Analysis of the E. lenta pan-genome.
To facilitate comparative genomic analysis, we analyzed gene content across dereplicated E. lenta genomes (n=42) by clustering all predicted coding sequences into orthologous groups (OGs) (Lechner et al., 2011). Rarefaction analysis demonstrates an open (unsaturated) pan-genome (Figure 2A). The core genome contains 771 OGs while the accessory genome contains 8,387 OGs. Every additional genome sequenced adds a median of 107 additional genes (range 21–325). Distributions of both OGs and functional annotations (KEGG orthologous groups [KOs]) show a high degree of strain variability with a number of genes and KOs being observed in a limited subset of strains (Figure 2B). Visualization of genome conservation relative to the closed reference E. lenta type strain genome (Figure 2C) revealed multiple large genomic islands indicative of horizontal gene transfer including a large ~150 Kbp region (HGT1) and a second region (HGT2) previously described due to its association with an 8 bp GAGTGGGA motif present recognized by P4 integrases (Song et al., 2012).
Figure 2. Gene and metabolite diversity in E. lenta.
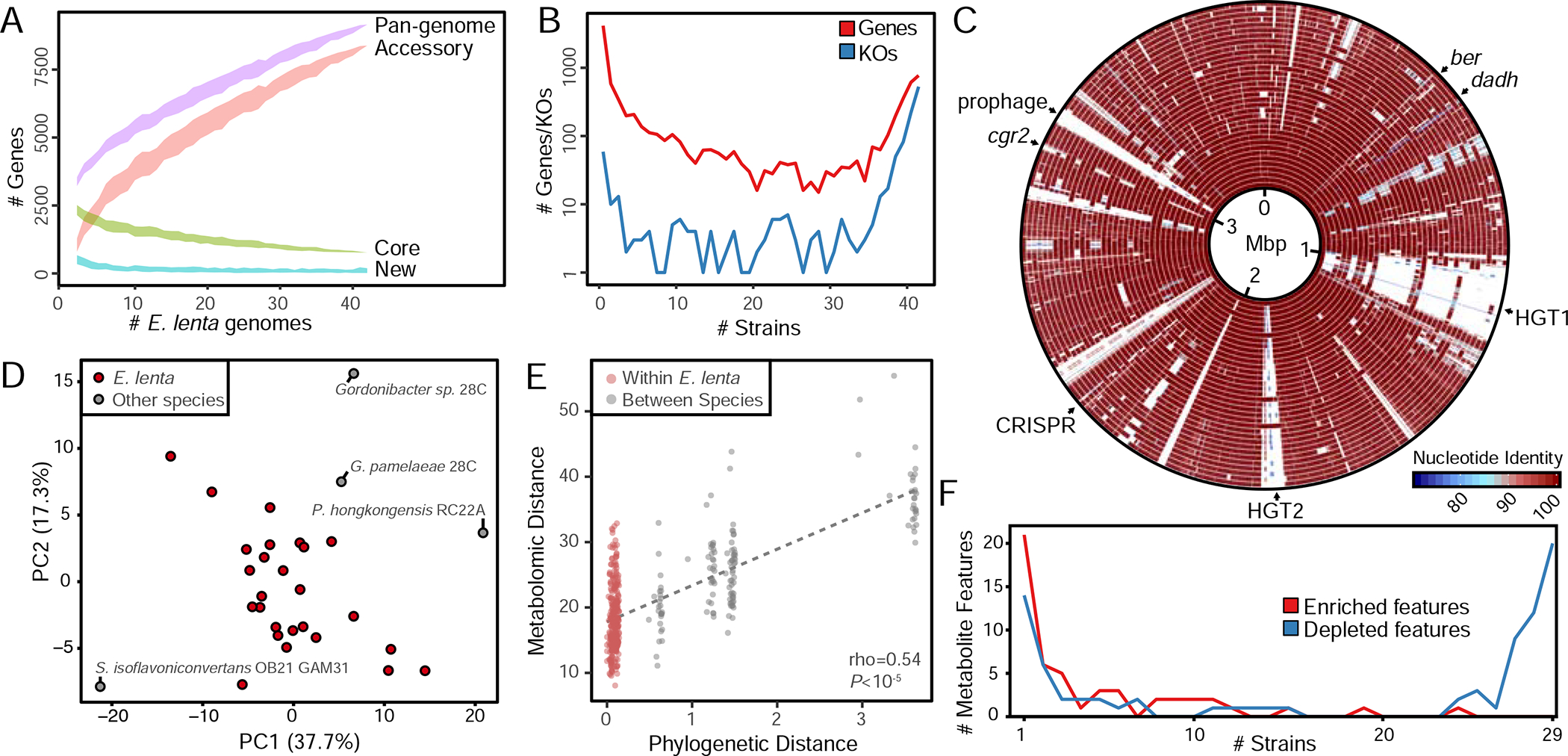
(A) Analysis of dereplicated isolate genomes (n=30) and MAGs (n=12) reveals an open pan-genome with a core genome of 771 genes. The ribbon denotes mean±SD. (B) Gene and KEGG orthologous group conservation across strains demonstrates functional diversity within E. lenta. (C) BLASTn comparison of all dereplicated E. lenta genomes (n=42) demonstrates localized regions of horizontal gene transfer including hot spots centered at ~1.1 Mb and ~1.9 Mb. (D) Principal components analysis of metabolite profiles within E. lenta species and close relatives. (E) Phylogeny (cophenetic distance) is correlated with metabolomic profiles. (F) Most of the 51 metabolite features enriched by any Coriobacteriia (counts shown in red) are strain specific, while strains have both a shared and unique set of features depleted (counts shown in blue, 47 features total).
To characterize the contribution of plasmids to the pan-genome of E. lenta, we searched for high coverage contigs within genome assemblies uncovering 13 such instances in 12 genomes, of which one represented a homologous prophage to that of DSM 2243 (90.0% global identity, Figure 1D). The remaining putative plasmids contained direct repeats on their termini indicative of being circular which was confirmed by PCR (Figure S2AB). The observed plasmids could be grouped into 3 families on the basis of BLASTN similarity without significant homology to previously isolated and sequenced plasmids (e-value<10; BLASTN against the RefSeq plasmid database; Figure S2C). Notably, these plasmids do not contain genes with obvious functions that may impart a selective advantage to these strains such as antibiotic resistance or virulence factors. As a step towards genetic engineering in E. lenta, we observed that kanamycin resistance is a rare trait in the species (n=3 resistant E. lenta, Table S3) and we validated a predicted aminoglycoside phosphotransferase which could serve as a selectable marker gene for cloning applications (Figure S2D).
Metabolomic diversity in E. lenta.
We further profiled the metabolic diversity of 30 strains grown in liquid culture using untargeted metabolomics (n=25 E. lenta). After quality filtering, we obtained a dataset describing 173 metabolite features, including 31 high-confidence identified compounds (18% of the high-quality features). We first investigated whether these metabolite profiles are a direct function of phylogenetic relatedness between strains, finding profiles partially separated strains according to phylogeny, with a distinct separation of taxa outside the Eggerthella and a significant correlation between phylogenetic and metabolic distance (Figure 2DE). Examining individual metabolite features, we identified 47 features (27.2% of total) that were significantly depleted in most (≥80%) strain cultures compared with media controls, including 20 assigned an identification. These 20 identified features were all various dipeptides of 8 amino acids, suggesting common utilization of these resources. In contrast, we found evidence of strain-specific metabolite production: of the 51 features significantly enriched in any culture, 36 were increased in 5 or fewer strains (Figure 2F, Figure S3A).
Next, we performed a preliminary analysis to assess whether cross-strain comparisons could inform the identification of the large majority of features (82%) not identified by library standards, as well as the inference of mechanistic links between genes and metabolites. Looking within 24 E. lenta strains, we found patterns of variable genes that coincided with large differential abundances in metabolite features (Figure S3BC). 27 highly differentially abundant features were associated with the presence or absence of gene family sets (Table S4). While only 4 of these metabolite features had an established identification, 22 were linked by this analysis to gene families with functional KEGG annotations, generating hypotheses on the metabolic roles of these features. For example, multiple identified dipeptide features were absent or substantially reduced in E. lenta 1-3-56 FAA cultures, which lacks two KOs involved in amino acid metabolism, indicating altered pathways for transport and/or metabolism of dipeptides. Unidentified variable metabolite features were associated with genes in several metabolic pathways, including iron and sulfur transport and metabolism, and gamma-polyglutamate biosynthesis. Interestingly, only one of the 27 feature-gene pairs were identified with low confidence by a metabolite-gene integration analysis based on homology search of enzyme databases (Erbilgin et al., 2019). Our analyses suggest that integrated cross-strain comparisons can be a source of hypotheses to guide follow-up studies on the identity and function of metabolites and gene families, particularly those that are poorly represented in reference databases, although further validation is needed to confirm the specific links reported here.
Natural product biosynthesis.
Examination of the E. lenta pan-genome identified several widespread natural product biosynthetic gene clusters (Figure 3AB). A subset of the strains encode the non-ribosomal peptide synthetase (NRPS) machinery required to produce a compound belonging to the 2-hydroxyphenylthiazoline family (Figure 3C), members of which are also present in other Actinobacteria (Seipke et al., 2011). Given the environment in which these species were found, it is likely that as with the similar metallophore yersiniabactin, this compound may not be a simple siderophore but may play a more complex role involving multiple metals and responses to oxidative stress as well as metal homeostasis in the host environment (Paauw et al., 2009; Robinson et al., 2018). A nanocompartment-forming encapsulin (annotated as a bacteriocin) and its rubrerythrin cargo are also likely to be involved in the cellular response to iron deficiency or oxidative stress (Figure 3D; Giessen and Silver, 2017). Most strains contain gene clusters that encode a set of hypervariable ribosomally produced post-translationally modified peptides (RiPPs, Figure 3E) that are related to (but distinct from) sactipeptides and ranthipeptides (Hudson et al., 2019). The core biosynthetic enzyme in these RiPP pathways is a radical SAM enzyme which constitutes a previously unidentified family (Figure 3F) and is predicted to mediate sulfur-carbon bond formation in at least 7 significantly different precursor peptides (Figure 3G), while a combined protease/exporter cleaves the leader peptide to release the mature RiPP (Håvarstein et al., 1995). Finally, the components necessary to produce the carotenoid lycopene and its precursors via the deoxyxylulose 5-phosphate/methylerythritol 4-phosphate (MEP/DOXP) pathway (Paniagua-Michel et al., 2012) are encoded in all of the genomes which we investigated (Figure 3H).
Figure 3. Identification of natural products produced by E. lenta strains.
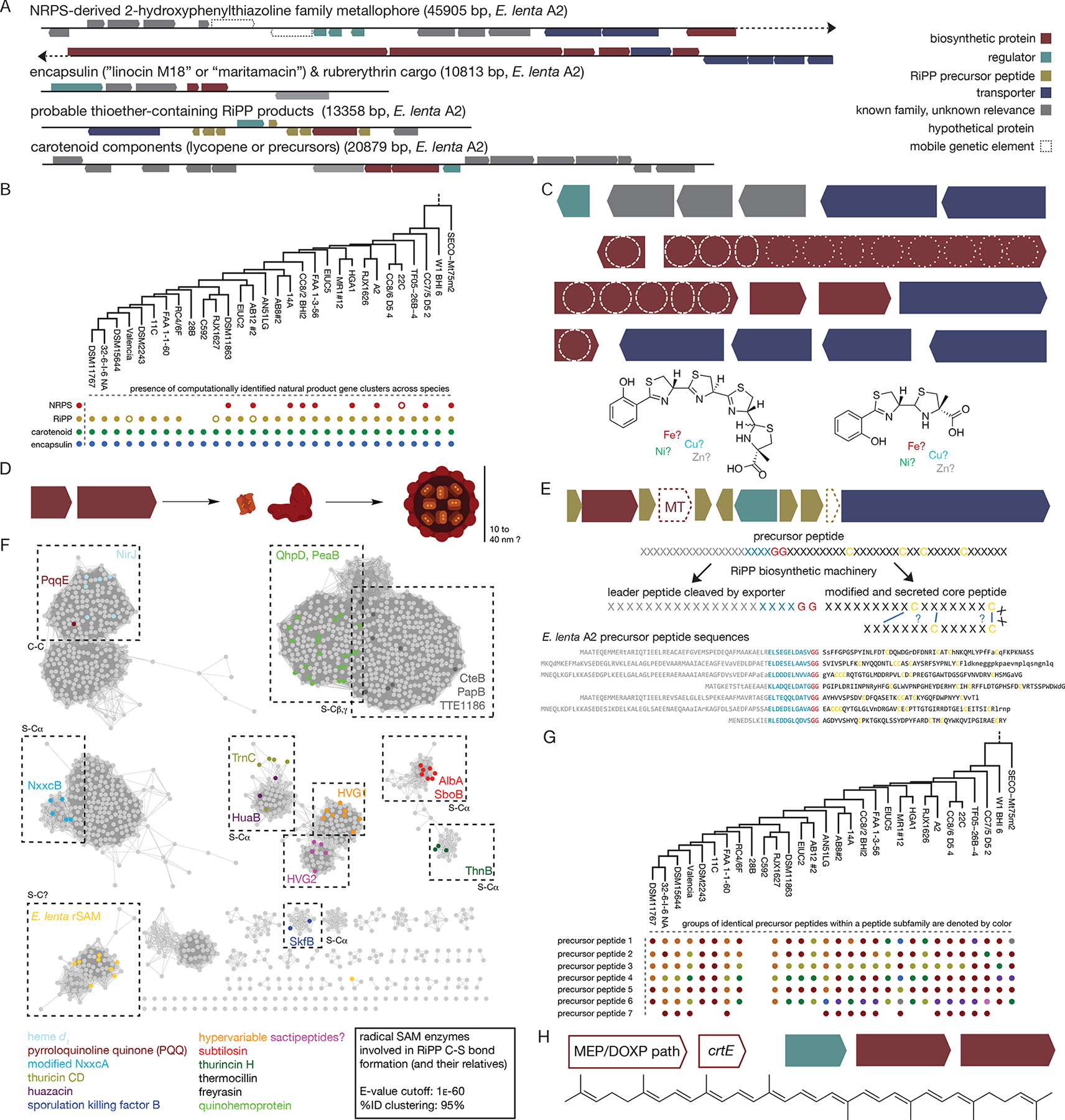
(A) 4 biosynthetic gene clusters are consistently identified in E. lenta predicted to generate: a non-ribosomal peptide synthetase (NRPS)-derived compound, a carotenoid, a series of ribosomally synthesized and post-translationally modified peptides (RiPP), and an encapsulin. (B) The carotenoid and encapsulin gene clusters are found in all E. lenta strains while RiPP and NRPS gene clusters are variable. Empty circles represent variant clusters (truncated NRPS clusters or RiPP clusters with methyltransferases.) (C) Detailed analysis of the NRPS gene cluster. The NRPS cluster encodes a probable metallophore from the 2-hydroxyphenylthiazoline family, which includes the known metallophores pyochelin and yersiniabactin. Most strains encode a larger compound with up to four thiazoline-derived heterocycles; two appear to encode a smaller compound more similar to pyochelin or watasemycin (predicted structures shown underneath). NRPS domains include A (adenylation), Cy (cyclization/condensation), E* (non-canonical epimerase, also found in pyochelin), P (peptidyl carrier protein), MT (methyltransferase), Sal (salicylate activation domain), and TE (thioesterase). (D) The encapsulin (encaps.) is predicted to form a nanocompartment with the rubrerythrin (Rb.) its predicted cargo. (E) The RiPP gene cluster contains only two biosynthetic enzymes: a radical SAM enzyme (rSAM) and in some cases a methyltransferase (MT). A regulator and a LagD-family exporter (which is expected to cleave the leader peptide) are also found, as are 7 or more precursor peptides. The core peptides are hypervariable, but consistently contain 4+ cysteines spread throughout the sequence. The leader peptides, while also variable, contain a conserved recognition region and cleavage site for the LagD exporter and the precursor peptide genes are sometimes annotated as members of the Nif11-like precursor peptide family. Representative sequences from E. lenta A2 are depicted with universally conserved residues in capital letters. (F) Sequence similarity network for the E. lenta RiPP rSAM. This radical SAM biosynthetic enzyme is distinct from previously observed families of cysteine-modifying rSAM enzymes. (G) Variability of RiPP precursor peptide types between strains. Some precursor peptide subfamilies exhibit more sequence diversity, and some possibly defunct precursor peptides (containing poorly conserved LagD cleavage sites or cysteines) are also found in most species. Groups of identical precursor peptides are highlighted in different colors. (H) E. lenta strains encode the requisite machinery for the production of lycopene (or its precursors) via the MEP/DOXP pathway, although the genes are scattered across multiple genomic locations.
Gene-phenotype matching via comparative genomics.
A major motivation for assembling this collection of strains and genomes was to facilitate the discovery of genetic determinants of the metabolism of pharmaceutical, dietary, and endogenous compounds. Drawing on the logic of tools using random forest classifiers for correlating genes with phenotypes (Bayjanov et al., 2012), we developed ElenMatchR, a purpose built tool with built-in databases for the discovery of effector genes in our strain collection (Figure S4A). Briefly, user-provided phenotypes are used as the input to a classifier with binary gene presence or k-mer content as predictors. Features are then extracted from the model and ranked on variable importance with a number of helpful graphical outputs to aid the user in interpreting the data.
As an initial validation case, we sought to further examine antibiotic resistance in E. lenta. Tetracycline resistance displayed a bimodal distribution of minimum inhibitory concentrations (MIC) in both broth microdilution (Figure 4A) and E-test assays (Figure 4B) suggesting the presence of a resistance mechanism (n=14 resistant and sensitive). Using ElenMatchR with a clustering threshold of 30% amino acid identity and 50% coverage, we uncovered a single gene cluster (OG2477) with elevated importance (Figure 4C) which is only observed in resistant strains (Figure 4D). This gene, annotated as tetW in E. lenta DSM 11767, is predicted to be a ribosomal protection protein. We validated that this gene imparts tetracycline resistance through heterologous expression in E. coli (Figure 4EF).
Figure 4. Validation of comparative genomics methodology using tetracycline resistance.
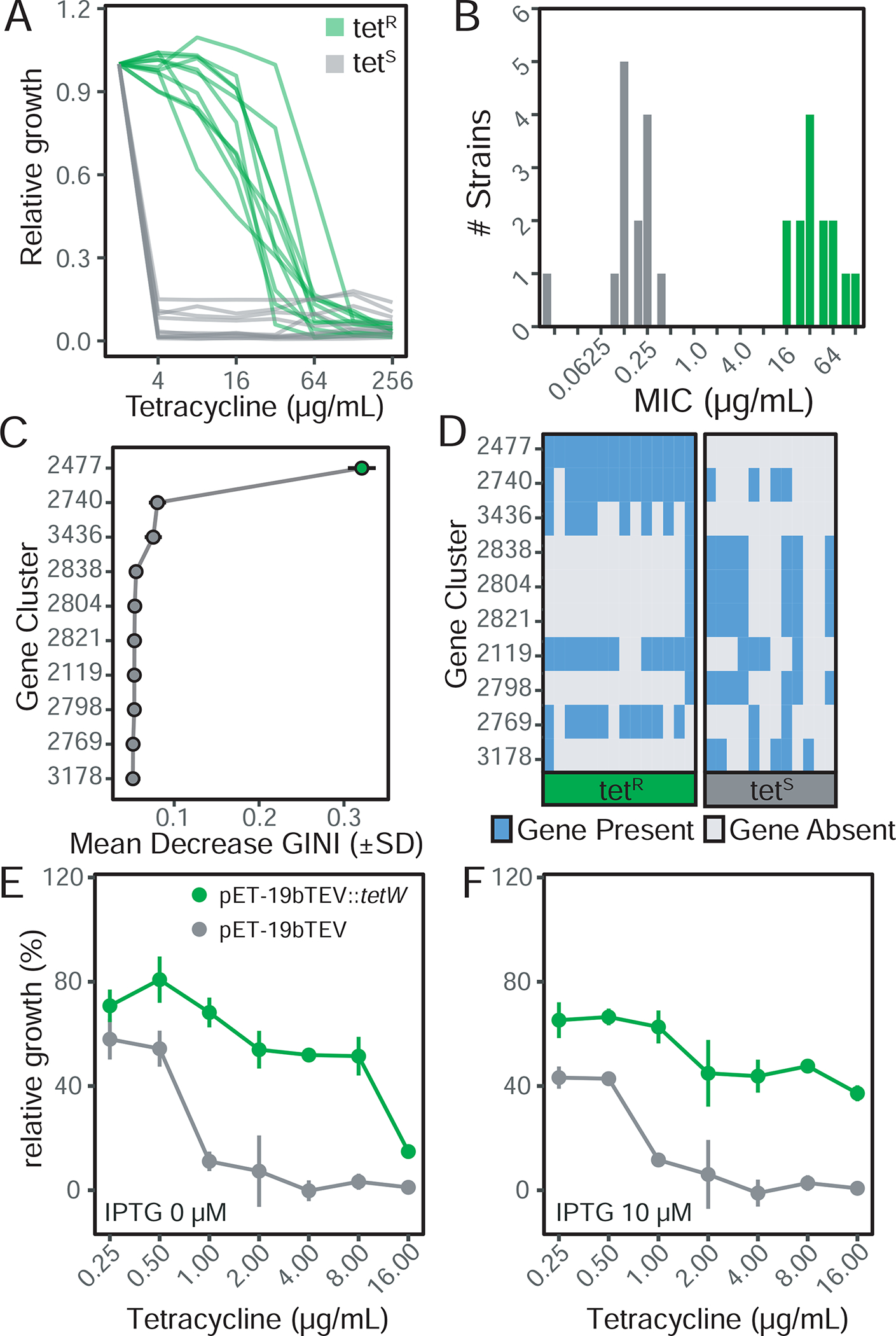
(A) Broth minimum inhibitory concentration (MIC) testing reveals a bimodal distribution in resistance (n=22 strains). (B) Epsilometer test on solid media confirms bimodal resistance profiles (n=28 strains). All 22 of the retested strains showed the same phenotype based on solid media. Panels A,B: green denotes tetracycline resistant strains. (C) Gene clusters ranked on random forest classifier importance (mean decrease in GINI coefficient). (D) Visualization of gene presence/absence in TetR and TetS strains reveals a single gene (2477) present in all TetR strains and absent in all TetS. Heterologous expression of the putative tetW resistance protein from E. lenta DSM 11767 in E. coli increases resistance to tetracycline under induced (E) and basal expression (F). Data in panel E and F represents mean±sem.
This test case also demonstrates the utility of Random Forests given their ability to detect the combined predictive power of interacting features (Breiman, 2001). The tetracycline resistance protein is present as two homologs (tetO and tetW) with 67.3% amino acid identity to each other. These two homologs occur in a mutually exclusive pattern within resistant strains (Figure S4B). When running at more stringent clustering thresholds, these proteins are separated into two orthologous gene clusters; however, both homologs are present in the default outputs as predictive features (Figure S4B).
As additional demonstration cases, we have packaged ElenMatchR with phenotypic data for three clinically relevant biotransformations: digoxin, pinoresinol reduction, and dopamine dehydroxylation. In the case of digoxin metabolism, we were able to map metabolism to a single locus of genes termed the cgr-associated gene cluster (Figure S5A–C). Combining these observations with transcriptomic data led to a single gene termed cardiac glycoside reductase 2 (cgr2) which was expressed in Rhodococcus erythropolis, confirming activity (Koppel et al., 2018). We mapped pinoresinol metabolism to a two-gene locus containing benzyl-ether reductase (ber) and its putative transcriptional regulator (Figure S5D–F). The ber gene was cloned into E. coli and confirmed to be active (Bess et al., 2020). The metabolism of dopamine proved to be more complicated as no single gene was predictive of metabolic activity. Through transcriptomic analysis, we uncovered dopamine dehydroxylase (dadh) and determined activity is conferred by a missense SNP variant (Rekdal et al., 2019). This led to the refinement of ElenMatchR to also detect SNPs by applying 31-mers as the predictors instead of gene content. This method reveals a set of overlapping k-mers associated with perfect predictive accuracy covering the explanatory SNP (Figure S5G–I). Taken together, these results provide strong support for the utility of ElenMatchR for rapidly identifying causal genes responsible for antibiotic resistance and small molecule biotransformations of interest.
Culture-independent quantification of E. lenta.
Given the difficulty in selectively isolating and enumerating E. lenta in culture, we leveraged our pan-genome analysis to design an assay for quantitative analysis of E. lenta in mixed communities. We identified a single copy gene, termed elenmrk1 (Genbank: WP_009608299.1), a putative luxR-family transcriptional regulator which is highly conserved (>98.5% nucleotide identity) in all E. lenta strains, but undetectable in the other analyzed Eggerthella and Coriobacteriia genomes. We then designed a double-dye qPCR assay to facilitate multiplexed quantification with a second assay targeted to specific E. lenta effector genes including cardiac glycoside reductase (cgr). Benchmarked against metagenomic data, dilution series, and mono- and mock community-colonized gnotobiotic mice, we were able to detect E. lenta at as little as 1,400 genome copies/g with detection robust to the presence of other highly abundant organisms (Figure 5AB, Table S5). We also confirmed E. lenta’s ability to colonize across the murine small and large intestines (Figure 5C). Finally, we used this assay to examine E. lenta prevalence and abundance in human populations, increasing the prevalence estimate from 41.5% based on metagenomic sequencing to 81.6% and have previously reported this finding but report its development here (Koppel et al., 2018).
Figure 5. Validation of a duplexed qPCR assay for quantifying Eggerthella lenta and specific genes of interest.
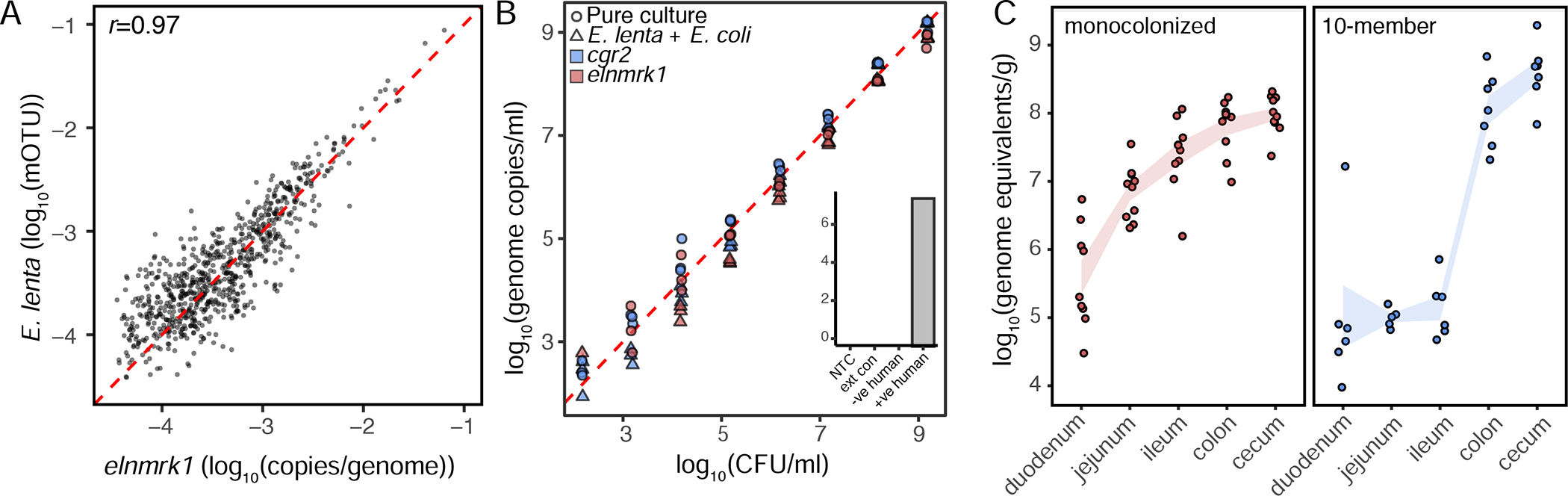
(A) Correlation of elnmrk1 and E. lenta abundances in metagenomes reveals a high degree of correlation (p<0.001, r=0.97) and little evidence of an elnmrk1-negative E. lenta population in 760 human GI tracts (red dashed line x=y). (B) Validation of assay sensitivity demonstrates a practical quantification limit of 1400 genome copies and that the presence of a second more abundant, and easily lysed, organism (4E9 CFU/mL E. coli K12 MG1655) does not prevent the recovery and quantification of E. lenta (inset controls: no-template control (NTC), blank DNA extraction control (ext con), and negative and positive human controls. (C) Quantification of E. lenta DSM 2243 in gnotobiotic mice monocolonized or colonized with a 7-member synthetic community. E. lenta is detectable along the entire length of the gastrointestinal tract with highest levels observed in the colon and cecum. Points represent individual animals (n=6–10/group) and lines represent mean±sem.
Mapping genetic determinants of bacterial fitness.
While inter-species competition in the gut microbiota has been extensively studied (Patnode et al., 2019; Theriot and Young, 2015; Verster and Borenstein, 2018), the mechanisms that govern strain-level (intra-species) competition remain poorly understood. Part of the reason for this knowledge gap is the technical challenge in quantifying competitive growth at the strain-level. Culture-based enumeration would require individual sequencing of colonies to determine strain identity and thus provide a shallow sampling of diversity. Quantitative PCR (qPCR) is a viable candidate; however, the assay development and reaction number make this experiment cost and time-prohibitive. Finally, shotgun sequencing is high-throughput and relatively affordable, but computational methods for accurately quantifying closely related strains are not widely available requiring the development of an applicable tool.
To test our ability to quantitatively distinguish E. lenta strains using shotgun sequencing data, we simulated a 22-strain mixed population with equal abundances (Figure S6). We were able to map 8.1% ± 3.7e−5% of reads conclusively to their strain of origin (n=3 replicate simulations; mean ± sd); however, the composition of these pools was highly skewed as a function of the divergence from other strains in the pool (Figure S6A–C). We reasoned that we could correct for the number of potential mapping sites by quantifying uniqueness in terms of read-sized k-mers which we found was correlated with mapping rate and thus the apparent relative abundance (Figure S6A–C). Given the low rate of quantitatively informative reads, we sought to maximize their use: as opposed to quantifying only unique gene islands or core gene SNPs, we devised a method of normalization (FKM; fragments per thousand unique k-mers per million reads mapped) to adjust for this bias. In preliminary testing, we observed a bias leading to inflated abundances in plasmid-carrying strains (Figure S2). To prevent the undue influence of multicopy elements, varied quality in assemblies, and inherent noise in coverage over the genome, we segmented each genome into smaller 50kb bins in silico and treated each of these as an independent unit of measurement. This allowed for multiple point estimates of any given strain’s abundance wherein the median value is highly stable and taken forward as the strain’s abundance for that sample (Figure S6D).
We developed a tool for normalization that consists of two steps: PreProcessR which processes the input genomes and generates the indices for k-mer normalization, and StrainR which calculates abundances and can be run in parallel on individual samples (Figure S6E). Using StrainR normalization we observed that the skew of the input pool decreased from 28-fold to 1.8-fold between the highest and lowest abundance strains in an even in silico pool (Figure S6CD). Furthermore, StrainR was capable of accurately recapitulating community abundances across a range of sequencing depths and compositions, including detecting the deletion of strains from the pool (Figure S6F).
Having validated our methods for strain quantification from metagenomes, we next sought to establish an experimental system for screening competitive fitness. We constructed a synthetic population of 22 E. lenta strains pooled at equal colony forming units (CFUs). The resulting 22-strain mix was cultured in brain heart infusion (BHI) media supplemented with 1% arginine. The same pool was also used to colonize germ-free Swiss-Webster mice to compare and contrast the outcomes of strain-level competition under laboratory conditions and within the mammalian gastrointestinal tract (Figure 6A). As a positive control, half of our cultures and mice were exposed to tetracycline providing a strong selective pressure for TetR strains with a known genetic determinant (Figure 4).
Figure 6. Mapping determinants of intra-species competition in vitro.
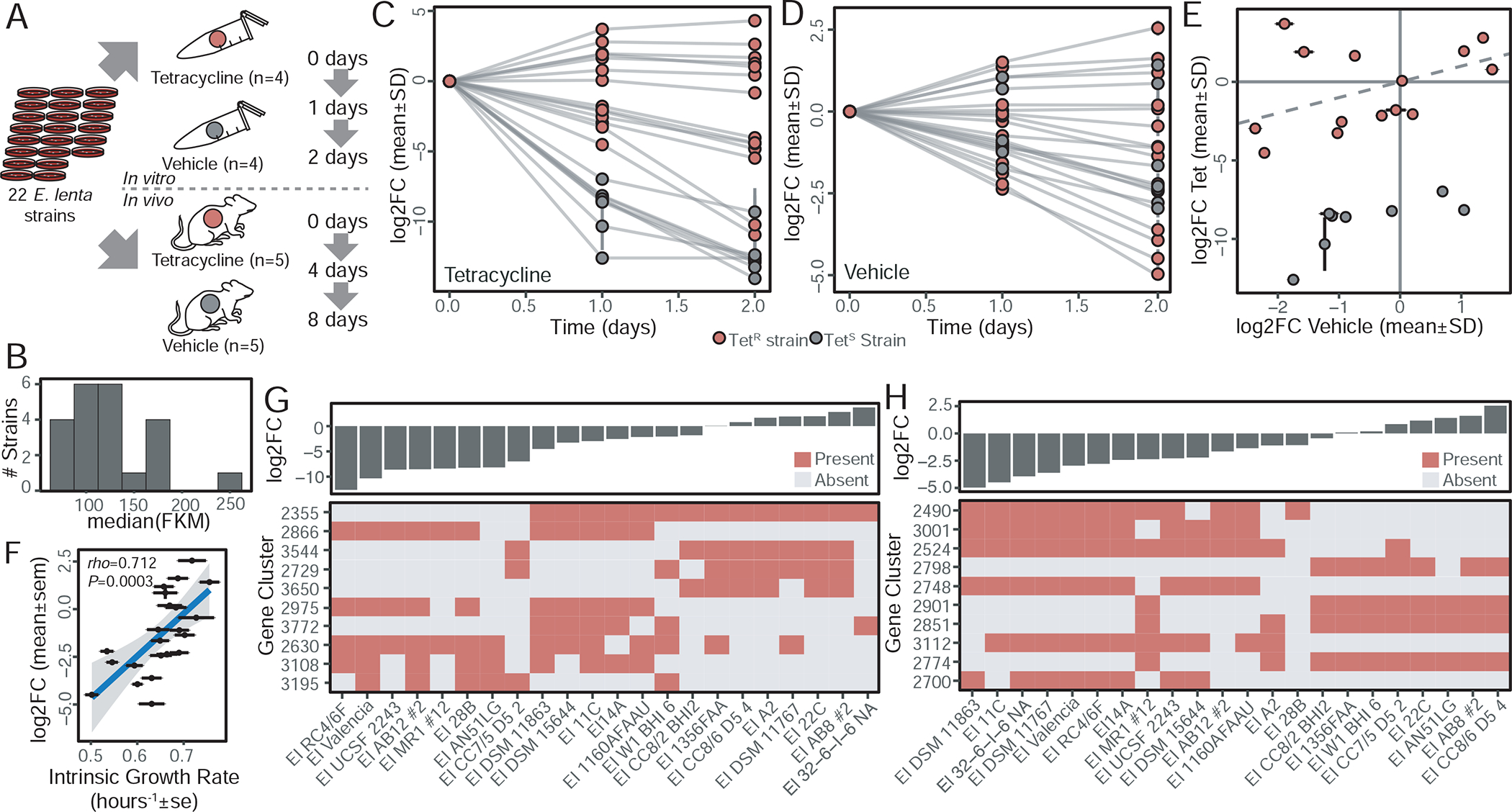
(A) Experimental design: 22 dereplicated E. lenta strains were pooled at equal CFU and passaged in vitro and in vivo with the addition of tetracycline as a known selective pressure. (B) All expected strains were observed in the input pool with concentrations ranging from 74.3±1.7 to 238.5±18.2 fragments per thousand unique k-mers per million reads mapped (FKM; mean±sd). (C) In the presence of tetracycline (12 μg/mL), TetR strains (n=14) are enriched in the output community at the expense of TetS strains (n=8). (D) In the absence of tetracycline, reproducible differences in strain abundances were observed. (E) Correlation of growth in the presence and absence of tetracycline demonstrates consistent outcomes for extremes of TetR strains (dashed line x=y, time=1 day). (F) Bacterial abundance in the absence of tetracycline is correlated with growth rate in mono-culture, but not carrying capacity (rho=0.158, P=0.482). (G) The 10 gene clusters which are most predictive of competitive growth in tetracycline (% Inc MSE, random forest regression) shows the most predictive gene (2355) corresponds to tetW, demonstrating the validity of this method to uncover biologically meaningful predictors. (H) Gene clusters predictive of competitive fitness in the absence of tetracycline reveal signatures for both strong and weak fitness.
The total abundance of E. lenta was unaffected by the inclusion of tetracycline both in vitro and in vivo (Figure S7A), suggesting that any antibiotic-induced differences occur in the relative proportions of strains in the population. After StrainR normalization, we observed all 22 strains present in our input pool with a 3.2-fold maximum difference in strain abundances (Figure 6B, Table S6). Highly reproducible community shifts were observed across replicates (Figure 6CD) with a selection for drug-resistant strains in the presence of tetracycline indicating its utility as a positive control. In the vehicle control, a wide spectrum of abundances was observed but without a complete loss of any of the strains (Figure 6D). Despite the marked effect of tetracycline, there was still a significant correlation between the two treatment groups for TetS strains (rho=0.88, P=0.007) when considered separately (Figure 6E). We also detected a significant correlation between growth rate in isolation and competitive growth in the vehicle control (Figure 6F, Figure S7B–D).
To map the genetic determinants of intra-species competition, we adapted ElenMatchR to accept a continuous outcome variable and applied a Random Forest regression model. As expected, this method identified tetW/tetO as the major determinant of competitive advantage in the presence of tetracycline at both time points (Figure 6G and Table S7). In the absence of tetracycline, multiple genetic signatures of competitive advantage and disadvantage were observed; however, there is no single strong predictor suggesting that multiple mechanisms are involved in the competitive growth phenotype (Figure 6H).
Of the top 20 predictors of competitive growth (Table S7), 7 are putative transcriptional regulators. Two of these regulators are the strongest predictors of a competitive growth advantage (gene clusters 2798 and 2901). Gene cluster 2798 in particular flanks carbamate kinase which catalyzes the final enzymatic step of the L-Arginine degradation V pathway, which fits with the observation that arginine is an essential substrate for the growth of E. lenta (Sperry and Wilkins, 1976). We also identified a component of a chromosomal toxin-antitoxin system which negatively predicted competitive fitness (2748). When found on the chromosome, these systems have been suggested to slow growth and/or initiate cell death in an altruistic manner which may be taken advantage of by toxin-antitoxin system-deficient strains (Yamaguchi and Inouye, 2011).
The outcomes of in vivo competition differed with the near-complete dominance of two TetR strains (22C and 1-3-56FAA) irrespective of the presence of tetracycline (Figure 7AB). Despite these condition-dependent differences in fitness, we were still able to detect a significant correlation between in vitro (24 h) and in vivo (4 days) (rho=0.63, P=0.002, Figure 7C). While the rank order outcomes may be correlated, their magnitude and distribution vary considerably emphasizing the unique selective pressures found within the gastrointestinal tract. Mapping of colonization outcomes at the endpoint samples using ElenMatchR revealed that 19/20 predictors were non-overlapping (Table S7, Figure 7D). The sole exception was the arginine metabolism-associated transcriptional regulator (2798, Table S7), emphasizing the critical role of arginine for E. lenta growth. The strongest predictor of competitive growth during in vivo colonization was a putative surface adhesin enriched in poor colonizers (Figure 7E). This membrane protein has homology to the repeated collagen-binding protein b-type domains of Staphylococcus aureus (Deivanayagam et al., 2000) and the prealbumin-like fold domain of the epithelial-binding SpaA-encoded pilus of Corynebacterium diphtheriae (Kang et al., 2009) (Figure 7F). We also detected a significant correlation (Blomberg et al., 2003) between phylogeny and colonization efficiency (K=0.93, P=0.013), and between phylogeny and the putative adhesin (K=1.05, P=0.004). These results suggest that more recently diverged strains are less fit in vivo (Figure 7G) and highlight the importance of considering physical interactions between E. lenta and host tissues mediating bacterial colonization and long-term fitness.
Figure 7. Mapping determinants of in vivo fitness.
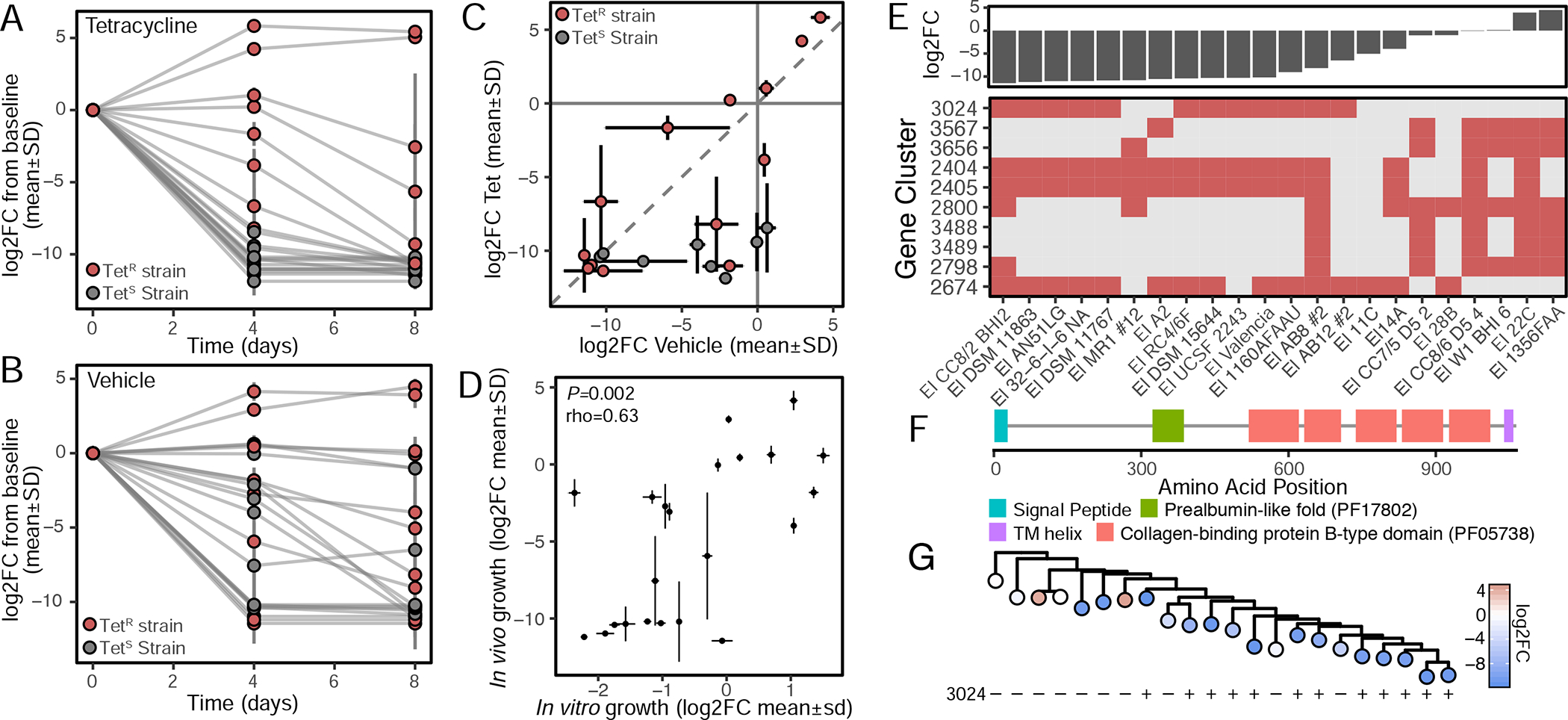
(A) While TetR strains have a selective advantage at 4-days post inoculation in the presence of tetracycline, (B) a subset of TetR strains are strongly favored irrespective of tetracycline, particularly E. lenta 22C and 1-3-56 FAA. (C) Correlation of community shifts at 4-days post inoculation suggests the selective pressure induced by tetracycline is less than differences in in vivo competitive advantage (dashed line x=y). (D) Competition outcomes in vivo are correlated with in vitro outcomes in the absence of tetracycline (Spearman correlation, 4 days in vivo, 1 day in vitro). (E) Top 10 gene clusters which are predictive of outcomes in competition in vehicle control (%Inc MSE) reveals signatures of both strong and weak competitive fitness. (F) A putative membrane-anchored adhesion protein is negatively correlated with fitness (gene cluster 3024). (G) More recently diverged E. lenta strains are less fit during competition with evidence of more recent acquisition(s) of the putative adhesin protein 3024 (phylogenetic tree of core SNPs [Figure 1B]).
DISCUSSION
Our investigation of the Coriobacteriia demonstrates the utility, generalizability, and recyclability of comparative genomics for conducting mechanistic studies on intractable taxa. Additionally, the identified and validated resistance markers combined with the plasmids and bacteriophage we have identified provide a first step towards the development of tools for genetic engineering of E. lenta. Our sensitive assay for the detection and quantification of E. lenta and a key gene of interest (cgr2) will provide a useful tool in the context of human cohort and pharmacokinetic studies. More broadly, the additional genomes generated in this analysis will aid future metagenomic studies by providing a more comprehensive representation of the E. lenta pangenome and the taxonomy of the Coriobacteriia. The importance of strain-resolved studies is also reinforced by our finding of extensive within-species variability in E. lenta metabolite production. This observation is consistent with previous work showing that metabolic divergence of gut microbes is associated with phylogeny on a large scale but not at close range (Bauer et al., 2016; Plata et al., 2015).
To date, there are limited large-scale experimental studies on bacterial intraspecies (intraspecific) competition in members of the human gut microbiome. Much of this work has focused on the role of Type VI secretion systems among gram-negative microbes (Hecht et al., 2016), but additional mechanisms that drive this phenomenon need further study. Previous work has demonstrated both lottery-like and non-random assembly at the species level in public datasets (Verster and Borenstein, 2018); however, experimental systems to understand the dynamics of colonization and competition within species are needed given the increasing shift towards strain-resolved microbiome studies (Ferretti et al., 2018; Garud et al., 2019; Goltsman et al., 2018; Lee et al., 2013).
Our observations that the outcomes of competition in vitro are correlated with growth rate rather than a potential mechanism of competitive interference and/or antagonism are of note given conventional ecological theory suggesting that interference is often highest among related organisms sharing an overlapping niche (Connell, 1983). Given that these experiments were performed in rich media and the cultures were kept in a near-continuous state of exponential growth, perhaps these mechanisms never manifested. Alternatively, our in vivo model may provide a more plausible biological system wherein clear exclusion took place. This experiment also supports the importance of host interactions in shaping bacterial fitness, due to the decreased growth of strains containing a putative host adhesin. This result is surprising given that host adhesion is typically considered to promote colonization (Kankainen et al., 2009); however, our results may suggest that adhesion could be detrimental because of immune interactions, reduced growth, enhanced clearance, or other mechanisms. The appearance of this trait later in the evolutionary history of E. lenta may indicate a more specialized role in the tropism of E. lenta for the human and perhaps a function in persistence rather than colonization. Experiments exploring longer-term colonization, higher density sampling, and intraspecies competition in the context of a more complete microbiota will help establish the significance of this finding. A key caveat of these results is that the analysis of our competition experiment does not address recombination between strains and treats each strain as a static unit. However, our method of quantification would not be affected by this phenomenon given its use of multiple genome segments for quantification. Re-analysis of communities with long-read approaches would help to detect these events (Douglas and Langille, 2019); however, the non-stochastic outcomes across multiple isolated communities would suggest these low-frequency occurrences are likely not a major driving force of outcomes. A second caveat is that the determinants of fitness in the human and mouse GI tracts may differ; however, this in and of itself creates opportunities to better understand the nature of host-microbe interactions for this understudied taxon.
Together, these results highlight that moving beyond shallow coverage of diversity in gut microbiota isolates will improve our collective understanding of the role of the microbiome in health and disease. Our data emphasize that single type strains and their genomes do not accurately represent the genotypic and phenotypic diversity within a given clade. In some cases, even single nucleotide polymorphisms can have phenotypic consequences, necessitating genome-wide nucleotide. Mechanistic studies into the basic biology of the Coriobacteriia coupled to translational studies in preclinical models and human cohorts are critical to gaining a greater understanding of the role of these intestinal symbionts in human health and disease. The tools and resources described in this manuscript have been developed for the greater scientific community including our open source ElenMatchR and StrainR approaches. Newly isolated strains have been deposited to the German Collection of Microorganisms and Cell Cultures (DSMZ) facilitating additional phenotype-genotype matching experiments by interested groups. Furthermore, the use of naturally occurring genetic and phenotypic variation within as-yet genetically intractable species to uncover effector genes is generalizable and will prove valuable in the study of other poorly characterized members of the human microbiome.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact Peter Turnbaugh (Peter.Turnbaugh@ucsf.edu).
Materials Availability
Bacterial Strains generated as part of this study are available from the German collection of Microorganisms and Cell Cultures (DSMZ) and will be made available directly from the authors upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Studies
Human samples were collected and analyzed as part of a diet-intervention study whose analysis is described elsewhere (clinicaltrials.gov registration: NCT01105143). Human experiments were approved by the ethics committee of the Charité-Universitätsmedizin Berlin.
Mouse Husbandry and Experiments
All mouse experiments were approved by the University of California San Francisco Institutional Animal Care and Use Committee. For the quantification of E. lenta in gnotobiotic mice, germ-free Swiss Webster mice were obtained from the UCSF Gnotobiotics core facility (gnotobiotics.ucsf.edu) and housed in gnotobiotic isolators (Class Biologically Clean). For the mono-colonization experiment, each of 10 male Swiss Webster 10–15 week old mice were orally gavaged with an inoculum of 1E8 CFUs of E. lenta DSM 2243 (prepared in an anaerobic environment and suspended in 200 μL PBS, which contained 0.05% L-cysteine-HCl). Mice were allowed to feed ad libitum on one of two isocaloric, isonitrogenous semi-purified diets composed of 10% sesame seeds (Teklad custom research diet). One diet was supplemented with 0.5% L-arginine (TD.150470) and provided to 5 mice; the other diet contained no supplemented L-arginine (TD.150471) and was provided to the other 5 mice. At 14 days following colonization, mice were euthanized, and the contents from each segment of the GI tract were collected. Arginine levels were not found to impact colonization (P>0.05, linear mixed effects model). Mice in all subsequent experiments were fed LabDiet 5021 ad libitum. For the 10-member mock community, 1E8 CFUs of each member (Table S5) was combined and 1E8 CFUs (in 200 μL saline + 0.5% cysteine) was administered to 7 germ-free 10 week old Swiss Webster mice (3 males and 4 females). After 18 days, mice were sacrificed and contents collected from each segment of the GI tract. For the competition experiments, the pooled strains were administered via oral gavage to 10 11-week old female Swiss Webster germ-free mice housed individually in duplex cages which were sacrificed after 8 days.
METHOD DETAILS
Strain Isolation and Culture.
Strains were isolated in multiple medias. Briefly, this includes Eubacterial minimal media supplemented with 1% w/v arginine, 50 μg/mL hygromycin, and 25 μg/mL nalidixic acid; brain heart infusion agar (BHI) supplemented with 1% arginine; fastidious anaerobe agar (FAA); and nutrient agar. All strains were isolated under anaerobic conditions at 37°C. Routine culturing was carried out under anaerobic conditions (Coy Lab Products) using the appropriate media listed in Table S1. Media used for routine experimentation were BHI+ (BHI with 1% arginine), BHI++ (BHI with 1% arginine, 0.05% L-cysteine-HCl, 1 μg/mL vitamin K, 5 μg/mL hemin, and 0.0001% w/v resazurin), or TSA blood + (tryptic soy agar with 0.5% arginine and 5% sheep blood).
Library preparation and sequencing.
Genomic DNA was prepared from anaerobic 24 h liquid cultures using the media described above before extraction using the Powersoil DNA extraction kit (MoBio), PureLink genomic DNA minikit (ThermoFisher) or DNeasy UltraClean Microbial kit (Qiagen). Libraries were prepared with either an Apollo 324 instrument, the Nextera XT kit (Illumina), or TruSeq PCR-free kit (Illumina). Libraries were sequenced according to the platform and chemistry listed in Table S1.
Genome assembly and annotation.
Demultiplexed sequences were first stripped of PhiX using bbduk version 37.97 before quality filtering using fastp version 0.20.0 (Chen et al., 2018) with the following parameters: --detect_adapter_for_pe --trim_poly_g --cut_front --cut_tail --cut_windowsize 5 --cut_mean_quality 20 --length_required 70. Resulting reads were overlapped using vsearch 2.13.4 (Rognes et al., 2016) with both paired and overlapped reads being used for assembly with SPAdes 3.13.1 (Bankevich et al., 2012). Coverage was estimated using bbmap version 37.97. Prokka version 1.13.3 was used to annotate genomes with the following parameters --kingdom Bacteria --gram pos --mincontiglen 200. Genome completeness and contamination was determined using CheckM (Parks et al., 2015). Assembly statistics were generated using Quast version 5.0.2 with --min-contig 200 (Gurevich et al., 2013). The 16S rRNA phylogenetic tree was determined by extracting the longest predicted 16S rRNA from genome assemblies with Bifidobacterium animalis subsp. lactis DSM 10140 as an outgroup (accession GCA_000022965.1). The tree was generated using FastTree (Price et al., 2009) from alignments created using DECIPHER (Wright et al., 2016). Phylogenetic trees were created from 400 universal conserved proteins (PhyloPhlAn (Segata et al., 2013)) and a subset of 195 core Eggerthella genes determined using Roary (Page et al., 2015) with Fast Tree. Whole genome average nucleotide identity was calculated by the Pyani package (widdowquinn.github.io/pyani/). When necessary, coding sequences were manually annotated using InterProScan5 (Jones et al., 2014).
Comparative genomic analysis.
Clustering of gene orthologs were carried out using ProteinOrtho6 (Lechner et al., 2011) across variable coverage and identity settings using Diamond for alignment (Buchfink et al., 2015). Determination of core and accessory gene counts were conducted using the output of ProteinOrtho6 at 60% identity and 80% coverage cutoffs using 42 samplings of strain combinations at each step of genomes analyzed. Pangenome calculations were benchmarked against Roary (Page et al., 2015) and LS BSR (Sahl et al., 2014) with default parameters providing consistent results (spearman correlations of binary distance based on gene content rho>0.941, P<2.2e-16). KEGG orthologies were determined using GhostKoala (Kanehisa et al., 2016). The circular BLAST comparison was generated by splitting the E. lenta DSM 2243 type strain genome into successive 1000 bp ranges and aligning these against all strains (BLAST). These were then plotted in a circular representation colored by nucleotide identity. Genetic variation between DSM 2243 isolates was determined by using the tool Snippy (github.com/tseemann/snippyv) after downsampling to an even 7.2 million reads and was verified using manual inspection of read pileups and breseq (Barrick et al., 2014). >99.83% of reads mapped to the reference assembly (CP001726.1) and assembly of the unmapped portion yielded single contigs with 100% coverage and 99.92% identity to PhiX174.
Identification of biosynthetic gene clusters.
AntiSMASH 5.0 was used to initially identify gene clusters in the E. lenta genomes (Blin et al., 2019). Where gene clusters were split across multiple contigs, tBLASTn was used to identify any contigs not identified by antiSMASH. PRISM (Skinnider et al., 2017), InterProScan (Jones et al., 2014), HMMScan (Potter et al., 2018), PKS/NRPS (Bachmann and Ravel, 2009), and RODEO (Tietz et al., 2017) were used to further investigate or confirm the identification of specific genes and their encoded domains. Manual annotation was used to confirm RiPP precursor peptide identification. To investigate the relationship between the RiPP-modifying radical SAM and potentially related enzymes, the top 500 IMG (Chen et al., 2019) BLAST results for representative enzymes (the E. lenta enzyme, TrnC, HuaB, SkfB, AlbA, SboB, ThnB, CteB, PapB, TTE1186, QhpD, and NxxcB) were pooled and submitted to the EFI-EST web tool (Zallot et al., 2019) in order to construct a sequence similarity network. The final network used a 95% identity cutoff for representative node condensation and a 1e-60 alignment score cutoff for clustering. Nodes were colored according to identifiable membership in a specific RiPP biosynthetic enzyme family.
ElenMatchR.
A user provided xlsx file is uploaded containing assigned categorical phenotypes. If running on genes, user defined thresholds for clustering identity and coverage are used to recall a table of gene content which is subsequently binarized and features which occur in all or no strains are removed. If running on k-mers, a sparse matrix of k-mer occurrence which has been dereplicated into co-occurring clusters is loaded and subset as above. The feature table is then fed to the randomForest function (Random Forest 4.16–4, R) which is repeated by a user defined parameter. Importance values (mean decrease GINI) are subsequently extracted and summary statistics reported. Features are subsequently ranked by their average importance for display. Visual outputs are generated using ggplot2 (Wickham, 2016) using user defined parameters for number of features and reference genomes, and a fasta file is generated for top features containing either each coding sequence or individual k-mer within a given dereplicated k-mer cluster. For demonstration cases, clonal-isolates have been dereplicated, i.e. represented as a single strain. Source code and databases are available at github.com/turnbaughlab/ElenMatchR. ElenMatchR was benchmarked against LS BSR (compare_bsr.py) and Scoary (Brynildsrud et al., 2016), outperforming both tools in the identification of TetO/TetW homologs.
Metabolomic analysis.
Strains were inoculated into 5 mL ISP-2 media (4 g/L yeast extract, 10 g/L meat extract, 4 g/L dextrose, pH=7.1) supplemented with 1% arginine and grown 48 h. The cultures were then subcultured at 1% v/v in triplicate and allowed to grow for 72 h. Cells were removed via centrifugation at 5000 g for 20 min at 4°C and solid phase extracted with a Waters Oasis HLB 96-well plate (WAT058951). The untargeted metabolomics analysis using high-performance LC-MS/MS (HPLC-MS) was carried out as described previously (McDonald et al., 2018). The chromatography was performed on a Dionex UltiMate 3000 Thermo Fisher Scientific high-performance liquid chromatography system (Thermo Fisher Scientific, Waltham, MA) coupled to a Bruker Impact HD quadrupole time of flight (qTOF) mass spectrometer. The chromatographic separation was carried out on a reverse phase (RP) Kinetex C18 1.7-μm, 100-Å ultrahigh-performance liquid chromatography (UHPLC) column (50 mm by 2.1 mm) (Phenomenex, Torrance, CA), held at 40°C during analysis. A total of 5 μl of each sample was injected. Mobile phase A was water, and mobile phase B was acetonitrile, both with added 0.1% (vol/vol) formic acid. The solvent gradient table was set as follows: initial mobile phase composition was 5% B for 1 min, increased to 40% B over 1 min and then to 100% B over 6 min, held at 100% B for 1 min, and decreased back to 5% B in 0.1 min, followed by a washout cycle and equilibration for a total analysis time of 13 min. The scanned m/z range was 80 to 2,000, the capillary voltage was 4,500 V, the nebulizer gas pressure was 2 × 105 Pa, the drying gas flow rate was 9 liters/min, and the temperature was 200°C. Each full MS scan was followed by tandem MS (MS/MS) using collision-induced dissociation (CID) fragmentation of the seven most abundant ions in the spectrum. For MS/MS, the collision cell collision energy was set at 3 eV and the collision energy was stepped 50%, 75%, 150%, and 200% to obtain optimal fragmentation for differentially sized ions. The scan rate was 3 Hz. An HP-921 lock mass compound was infused during the analysis to carry out postprocessing mass correction. All of the raw data are publicly available at the UCSD Center for Computational Mass Spectrometry (massive.ucsd.edu dataset ID MSV000083734). The collected data were processed as previously described (Dührkop et al., 2019). The feature tables were obtained using MZmine2 (Pluskal et al., 2010). The collected HPLC-MS raw data were converted from Bruker’s .d to .mzXML format. The data were then batch-processed with the following settings for each step: mass detection (noise level 1000), chromatogram builder (minimum time span 0.01 min, minimum peak height 3000, m/z tolerance 0.1 m/z or 20 ppm), chromatogram deconvolution - baseline cutoff (minimum peak height 3000, peak duration range 0.01–3.00 min, baseline level 300), deisotopisation - isotopic peak grouper (m/z tolerance 0.1 m/z or 20 ppm, RT tolerance 0.1 min, maximum charge 4), peak alignment - join aligner (m/z tolerance 0.1 m/z or 20 ppm, weight for m/z 75, weight for RT 25, RT tolerance 0.1 min), peak filtering - peak list raw filter (minimum peak in a row 3, minimum peak in an isotope pattern 2).
Initial pre-processing produced a dataset of 860 mass spectrometry features. Missing values in the feature table were imputed to 75% of the minimum measured peak area. One replicate sample from E. lenta W1BHI6 had a high share of missing and low feature abundances and clustered with the quality control samples and was therefore removed from the dataset. Features with more than 75% missing values were also removed, as were features that were inconsistently detected across 3 or more replicate sets. We noticed the feature abundance table displayed strong correlations with both sample run order and column placement in the initial plate, so the parametric ComBat method implemented in the R package sva (Leek et al., 2019) was used to normalize for both of these factors simultaneously. Although our samples were not fully randomized across these variables, applying this correction substantially increased the correlation between metabolomic and phylogenetic distance and the profile similarity between the type strain isolates DSM 2243D, UCSF 2243, and ATCC 25559, which supported the value of the correction. These isolates were then treated as separate strains in downstream analysis in order to maintain equal numbers of replicates for each strain and to evaluate potential metabolic differences between the three isolates. Finally, to focus on the most informative features, downstream analysis was restricted to features with high (>0.4) repeatability (i.e. high variance across replicate groups relative to within-group variance), following (Wehrens et al., 2016). The resulting dataset consisted of 173 features across 89 strain samples and 6 media controls, of which 31 features were identified based on spectral match. To obtain additional putative metabolite identifications, the CEU Mass Mediator v3.0 (Gil-de-la-Fuente et al., 2019) was used to perform a feature search based on m/z and retention time, with an error tolerance of 5ppm and standard positive ionization adducts. The resulting potential identifications were evaluated using MAGI (Erbilgin et al., 2019). All potential metabolites from the feature search were provided to the program along with annotated gene sequences for all strains.
Differentially abundant features in strain cultures compared to media controls were identified based on t-tests comparing normalized feature abundances in samples versus controls, using a Benjamini-Hochberg false discovery rate correction and a corrected p-value cutoff of 0.05. Euclidean distances between strain metabolite profiles were calculated between the log2 median fold change of the abundances compared with media controls as these quantities are approximately normally distributed for each metabolite feature. Phylogenetic distances were obtained from the phylogeny constructed with PhyloPhlan (see above), using the cophenetic.phylo function in the R package ape (Paradis and Schliep, 2018). To confirm that data type and distance metric did not significantly affect our conclusions, we also calculated the Manhattan distance between strains on the basis of categorical profiles of whether each metabolite is enriched, depleted, or neither (1, −1, or 0) by each strain which produced similar significant correlations (Mantel Spearman correlation). To identify putative gene-metabolite links, we searched for metabolite shifts that corresponded precisely with the presence or absence of a variable gene in the E. lenta pan-genome. Specifically, we evaluated every unique co-occurrence pattern of variable genes within E. lenta as identified by ProteinOrtho with 30% identity and 80% coverage thresholds (see above). We removed variable gene clusters with a KO annotation identical to a complementary gene cluster annotated with the same KO, inferring that these likely represent somewhat more diverged but non-variable gene families. We then assessed whether any metabolite features were strongly differentially abundant and separable between strains with and without the gene(s) in question, identifying promising links as those with a Benjamini-Hochberg-adjusted p-value less than 0.01, and for which at least 30% of samples from strains with the gene set and 30% from strains without the gene set could be perfectly separated by the abundance of the metabolite feature. We filtered the identified gene-feature links to the gene set with the highest separability of abundances for each feature (smallest number of overlapping samples). We compared the resulting pairs with the set of gene-feature links with a non-zero homology score in the MAGI analysis described above. Gene clusters were categorized as having an annotation of enzymatic function if annotated with a KEGG Ortholog linked to an Enzyme Commission number.
The annotations and visualizations of chemical distributions were explored on GNPS using molecular networking (Wang et al., 2016) as follows. MS/MS spectra were window filtered by choosing only the top 6 peaks in the 50-Da window throughout the spectrum. The MS spectra were then clustered with a parent mass tolerance of 0.02 Da and an MS/MS fragment ion tolerance of 0.02; consensus spectra that contained fewer than 4 spectra were discarded. Network was created where with edges filtered to have a cosine score above 0.65 and more than 5 matched peaks. The edges between two nodes are kept in the network if and only if each of the nodes appeared in each other’s respective top 10 most similar nodes. The required library matches were set to have a score above 0.7 and at least 6 matched peaks when searched the spectra in the network against GNPS spectral libraries. All resulting annotations are at level 2/3 according to the proposed minimum standards in metabolomics (Sumner et al., 2007). The GNPS results are located at https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=85c9922a8b8548e3a537dda24301673f Feature-based molecular network (Nothias et al., 2019) results can be found at https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=e3ae9e9add4d484ea3715aa45dee8447. The raw data are available at the MassIVE repository (massive.ucsd.edu) under dataset ID: MSV000083734.
Antibiotic resistance screening.
Screening was carried out using Epsilometer (Etest) strips (bioMerieux) or broth dilution minimum inhibitory concentration (MIC) assay. For Etest strips, a 24 h broth culture was diluted to an OD600 of 0.1 before 500 μL was spread on 135 mm plates with 4 strips per plate. The assays used were: CL 256, TC 256, KM 256, AM 256, VA 256, MX 32, PG 32, and MZ 256. Resistance was determined based on EuCAST breakpoint tables (version 8.0 2018) where available, or by MIC distribution. For the broth MIC assay, a 24h broth culture was inoculated at 1% v/v in a 96-well plate and incubated in anaerobic conditions at 37°C for 48h with OD600 recorded by an Eon microplate reader (BioTek). Strains were assayed twice and the mean value reported. Resistance was determined based on the bimodal distribution of MICs. Antibiotic resistance determinants were predicted using Abricate version 0.9.8 using default parameters (https://github.com/tseemann/abricate).
Heterologous expression.
The sequences of a putative tetW and aminoglycoside 3’-phosphotransferase were amplified from genomic DNA of E. lenta DSM 11767 with Q5 polymerase (NEB) using for 30 cycles using 500nM primers (ACTGATCATATGAAAATAATCAATATTGGAATTC, AGCTATGGATCCTTACATTATCTTCTGAAACATATAG, ACTGATCATATGGCTAAAATGAGAATATCA, GCTATGGATCCCTAAAACAATTCATCCAGTAAA, where bold sites represent restriction enzymes sites). pET-19bTEV (a derivative of pET-19b with a tobacco etch virus [TEV] cut site replacing the enterokinase cut site) was used as the expression vector. Both inserts and plasmid were cut with NdeI and BamHI. The plasmid backbone was treated with recombinant shrimp alkaline phosphatase (rSAP) before both plasmid and inserts were gel purified (Qiaquick gel purification kit). Ligation was carried out with T4 ligase. All enzymes for cloning were purchased from New England Biolabs and used according to the manufacturer’s instructions. Ligation reactions were heat inactivated and transformed into E. coli DH5α and selected using LB media with 100μg/mL ampicillin. Plasmids were extracted from transformants and confirmed by sanger sequencing (GeneWiz) from the T7 promoter and terminator. Verified plasmids were then transformed into E. coli Rosetta and selected on LB with 50 μg/mL carbenicillin (for pET-19bTEV) and 30 μg/mL chloramphenicol (for selection of pRARE plasmid carrying rare codons). Verified transformants were then grown in LB broth for 8h with appropriate selection before being inoculated at 0.5% v/v across a range of tetracycline, kanamycin, and IPTG concentrations in 96 well plates. Plates were incubated for 16 h at 37°C and read at OD600 using an Eon microplate reader (BioTek).
E. lenta quantification in mouse and human samples.
The mock community was profiled using 515F/806R golay-barcodes (Caporaso et al., 2012) and denoised using DADA2 (Callahan et al., 2016). DNA was extracted from 100 mg aliquots of material using the ZymoBIOMICS 96 MagBead DNA Kit (Zymo D4302) according to the manufacturer’s protocol with an additional 10 min incubation after mechanical cell disruption at 65°C. Disruption was carried out using a BioSpec Mini-Beadbeater-96 for 5 min. qPCR was carried out using Taq Universal Probes Supermix (BioRad 1725131), in a CFX384 thermocycler (BioRad) in triplicate 10 μL reactions with all oligos present at 200 nM final concentration and 4 μL of purified gDNA. The following oligos were used elnmrk1_for: GTACAACATGCTCCTTGCGG, elnmrk1_rev CGAACAGAGGATCGGGATGG, elnmrk1_probe [6FAM]TTCTGGCTGCACCGTTCGCGGTCCA[BHQ1], cgr2_for GAGGCCGTCGATTGGATGAT, cgr2_rev ACCGTAGGCATTGTGGTTGT, and cgr2_probe [HEX]CGACACGGAGGCCGATGTCG[BHQ1]. DNA was amplified using 40 cycles of 5 s at 95°C and 30 s at 60°C after 5 min initial denaturation at 95°C. Absolute copies were determined by comparison against a standard curve of E. lenta DSM2243 genomic DNA. The primer efficiencies of the elnmrk1 and cgr2 assays are 105% and 103.5% respectively. A practical limit of quantification of 1400 genome copies/mL was determined as it was the last dilution in the series fitting the linear trend-line with concordant detection in all replicate wells. The primer/probe sets were designed by first focusing on highly conserved genes within E. lenta that were absent in all other members of the Coriobacteriia. Primers were designed with the aid of Primer-BLAST checking for specificity against the NCBI non-redundant database.
StrainR.
The initial preprocessing step (PreProcessR) prepares an index for subsequent per-sample analysis. To provide multiple estimates of genome abundance, and to account for varied states of assembly quality, genomes are first fragmented to 50kbp with fragments smaller than 10kbp discarded. Next canonical k-mers (the lexicographically first of the forward and reverse complement) are generated using Jellyfish 2 (Marcais and Kingsford, 2012) and a dereplicated list is generated and used to build a sparse matrix. Finally, a bbmap index is prepared for all fragments. The quantification step (StrainR) first filters and trims reads using strict user-modifiable parameters before mapping reads using bbmap with the following parameters: perfectmode=t, local=f, ambiguous=toss, pairedonly=t. For each genome subfragment, the mapped fragments per thousand unique k-mers per million reads mapped in sample is calculated (FKM). This data is provided on a per-sample basis to the user and the median value across all subfragments taken forward preventing undue bias due to library preparation, multicopy elements (ie plasmids), and other unobserved factors. StrainR has been tested on Mac OS × Mojave, Ubuntu 14.04.5 and CentOS 7. It requires BBmap >=37.97, Jellyfish 2, and Samtools >=1.9 installed and the following R packages: tidyverse, Biostrings, doParallel, foreach, data.table, Matrix, Matrix.utils, vegan, openssl, dada2, and ShortRead.
To generate mock data for validation and testing, an actual Illumina Nova-seq containing a pilot run of input pool was used to generate error profiles for InSilicoSeq (Gourlé et al., 2019). Next, InSilicoSeq was used to generate variable input communities containing even, skewed, random abundances, and a data set missing strains across variable sequencing depths. These profiles were then compared back to expected profiles to determine accuracy.
Competition Experiments.
A set of 22 non-clonal strains (Table S6) was selected for competition experiments including 8 tetracycline sensitive and 14 resistant strains. Strains were grown on BHI+ before drop-plate CFU estimation and preservation with 10% glycerol. Strains were subsequently thawed and pooled based on CFU for in vitro and in vivo competitions. For in vitro experiments, the pool was subcultured at 0.5% v/v into 50 mL BHI+ in 4 Erlenmeyer flasks and incubated for 24h at 37°C. After 24 h, subculturing again took place into fresh media and the resulting cultures were incubated for an additional 24 h. In the tetracycline group, 12 μg/mL tetracycline was added to media. Drop-plates were used to determine CFU after both passages demonstrating that tetracycline did not lead to a loss in total cell count. For the in vivo experiment, 100μL of the pooled strain collection (7e8CFU/mL) was administered via oral gavage to 10 11-week old female Swiss Webster germ-free mice housed individually in duplex cages. Five mice received 32 μg/mL tetracycline in their drinking water, corresponding to a final fecal concentration of 12 μg/g (Corpet et al., 1989). Fecal pellets were collected for analysis after 4 and 8 days with cages changed at 2 day intervals to prevent re-inoculation of the seed community due to coprophagy. For both experiments, DNA was extracted from fecal and cell pellets using protocol Q from the International Human Microbiome Standards Consortium (IHMS_SOP 06 V1). Libraries were prepared using the Nextera XT library protocol and sequenced via an Illumina Nova-Seq with 2×141 chemistry. The median number of reads obtained after quality filtering was 20,972,910 (range 2,041,797 – 126,953,787). Strain abundances in pools were determined using StrainR. Per-strain growth parameters were generated by inoculating 1.5 μL of 48 h broth cultures at OD600=0.2 into 150ul of BHI+ in a 96-well microplate and measuring OD600 every 20 min for 48 h at 37°C under anaerobic conditions in an Eon Microplate Spectrophotometer (BioTek) and estimated using GrowthCurveR (Sprouffske and Wagner, 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise specified, statistical analysis was carried out in R 3.6.1 using appropriate base functions. Individual datapoints have been shown where possible but are otherwise represented as the mean ± standard error unless otherwise stated. Significance was determined as P<0.05 unless otherwise stated using the appropriately identified test.
DATA AND CODE AVAILABILITY
Genome accessions are listed in Table S1 and newly sequenced genomes are available as part of BioProject PRJNA412637. Raw reads for assembly and community competition were deposited under PRJNA578765. Source code for ElenMatchR and StrainR are available at github.com/turnbaughlab/ElenMatchR and github.com/turnbaughlab/StrainR. Raw metabolomic data is available at massive.ucsd.edu under accession MSV000083734.
Supplementary Material
Table S1. Genome metadata relating to Figure 1.
Table S2. Genomic variants in resequenced type strain isolates versus reference assembly (CP001726.1), related to Figure 1.
Table S3. Minimum inhibitory concentration (MIC) testing in Coriobacteriia, related to Figure 4.
Table S4. Associations between gene clusters and metabolite features, related to Figure 2.
Table S5. Mock community composition, related to Figure 5.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Asaccharobacter celatus AP38TSA | This Paper | NA |
| Asaccharobacter celatus OB21 GAM11 | This Paper | NA |
| Collinsella aerofaciens ATCC 25986 | This Paper | NA |
| Collinsella aerofaciens CaUC5 | This Paper | NA |
| Eggerthella lenta FAA 1-1-60A | This Paper | NA |
| Eggerthella lenta 11C | This Paper | NA |
| Eggerthella lenta FAA 1 −3–56 | This Paper | NA |
| Eggerthella lenta 14A | This Paper | NA |
| Eggerthella lenta 16A | This Paper | NA |
| Eggerthella lenta 22C | This Paper | NA |
| Eggerthella lenta 28B | This Paper | NA |
| Eggerthella lenta 32–6-I-6 NA | This Paper | NA |
| Eggerthella lenta A2 | This Paper | NA |
| Eggerthella lenta AB12 #2 | This Paper | NA |
| Eggerthella lenta AB8 #2 | This Paper | NA |
| Eggerthella lenta AN51 LG | This Paper | NA |
| Eggerthella lenta ATCC 25559 | ATCC | 25559 |
| Eggerthella lenta CC7/5 D5 2 | This Paper | NA |
| Eggerthella lenta CC8/2 BHI2 | This Paper | NA |
| Eggerthella lenta CC8/6 D5 4 | This Paper | NA |
| Eggerthella lenta RJX1626 | This Paper | NA |
| Eggerthella lenta RJX1627 | This Paper | NA |
| Eggerthella lenta DSM 11767 | DSMZ | 11767 |
| Eggerthella lenta DSM 11863 | DSMZ | 11863 |
| Eggerthella lenta DSM 15644 | DSMZ | 15644 |
| Eggerthella lenta DSM 2243D | DSMZ | 2243 |
| Eggerthella lenta ElUC2 | This Paper | NA |
| Eggerthella lenta ElUC5 | This Paper | NA |
| Eggerthella lenta MR1 #12 | This Paper | NA |
| Eggerthella lenta RC4/6F | This Paper | NA |
| Eggerthella lenta SECO-Mt75m2 | This Paper | NA |
| Eggerthella lenta UCSF 2243 | This Paper | NA |
| Eggerthella lenta Valencia | This Paper | NA |
| Eggerthella lenta W1 BHI 6 | This Paper | NA |
| Eggerthella sinensis DSM 16107 | DSMZ | 16107 |
| Gordonibacter pamelaeae 3C | This Paper | NA |
| Gordonibacter pamelaeae DSM 19378 | DSMZ | 19378 |
| Gordonibacter species 28C | This Paper | NA |
| Gordonibacter urolithinfaciens DSM 27213 | DSMZ | 27213 |
| Paraeggerthella hongkongensis CD11 | This Paper | NA |
| Paraeggerthella hongkongensis RC2/2 A | This Paper | NA |
| Senegalimassilia anaerobia AP69FAA | This Paper | NA |
| Slackia exigua UCSF 15923 | This Paper | NA |
| Slackia heliotrinireducens DSM 20476 | DSMZ | 20476 |
| Slackia isoflavoniconvertens OB21 GAM31 | This Paper | NA |
| Bacteroides thetaiotaomicron | DSMZ | 2079 |
| Bacteroides uniformis | DSMZ | 6597 |
| Clostridium spiroforme | DSMZ | 1552 |
| Collinsella aerofaciens | DSMZ | 3979 |
| Akkermansia muciniphila | DSMZ | 22959 |
| Clostridium scindens | DSMZ | 5676 |
| Dorea longicatena | DSMZ | 13814 |
| Prevotella copri | DSMZ | 18205 |
| E. coli DH5a | NEB | C2989K |
| E. coli Rosetta | Millipore Sigma | 70953–3 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Brain Heart Infusion | BD | 237500 |
| Arginine | Sigma Aldrich | W381918 |
| Tryptic Soy Broth | Millipore | 1054590500 |
| Gifu Anaerobic Media | HiMedia | M1801 |
| rSAP | NEB | M0371 |
| NdeI | NEB | R0111 |
| BamHI | NEB | R0136 |
| T4 Ligase | NEB | M0202 |
| ZymoBIOMICS 96 Well DNA kit | Zymo Research | D4302 |
| iTaq Universal Probes Supermix | BioRad | 1725131 |
| Tetracycline (USP) | Sigma Aldrich | T4062 |
| Critical Commercial Assays | ||
| Purelink genomic DNA Mini Kit | ThermoFisher | K182002 |
| DNeasy UltraClean Microbial kit | Qiagen | 12224–250 |
| Nextera XT kit | Illumina | FC-131–1024 |
| Nextera XT Index Kit | Illumina | FC-131–1001 |
| Epsilometer Strips | bioMerieux | CL 256, TC 256, KM 256, AM 256, VA 256, MX 32, PG 32, MZ 256 |
| Deposited Data | ||
| Competition Experiment Reads | Sequence Read Archive | PRJNA578765 |
| Genomes Sequenced in this Manuscript | NCBI Genbank | PRJNA412637 |
| Metabolomics Data | massive.ucsd.edu | MSV000083734 |
| Adlercreutzia caecimuris DSM 21839 | NCBI Genbank | GCA_000403355.2 |
| Adlercreutzia equolifaciens DSM 19450 | NCBI Genbank | GCA_000478885.1 |
| Adlercreutzia caecicola DSM 22242 | NCBI Genbank | GCA_003725335. 1 |
| Adlercreutzia mucosicola DSM 19490 | NCBI Genbank | GCA_000422625. 1 |
| Arabia massiliensis Marseille-P3078 | NCBI Genbank | GCA_900169505.1 |
| Asaccharobacter celatus DSM 18785 | NCBI Genbank | GCA_003726015.1 |
| Collinsella aerofaciens ATCC 25986 | NCBI Genbank | GCA_000169035.1 |
| Coriobacterium glomerans DSM20642 | NCBI Genbank | GCA_000195315.1 |
| Cryptobacterium curtum DSM 15641 | NCBI Genbank | GCA_000023845. 1 |
| Denitrobacterium detoxificans DSM 21843 | NCBI Genbank | GCA_900110565.1 |
| Eggerthella lenta FAA 1-1-60ABroad | NCBI Genbank | GCA_000763035. 1 |
| Eggerthella lenta FAA 1-3-56 | NCBI Genbank | GCA_000185625.1 |
| Eggerthella lenta C592 | NCBI Genbank | GCA_002148255.1 |
| Eggerthella lenta HGA1 | NCBI Genbank | GCA_000191845.2 |
| Eggerthella lenta TF05–26B-4 | NCBI Genbank | GCA_003438525. 1 |
| Eggerthella sp. YY7918 | NCBI Genbank | GCA_000270285. 1 |
| Eggerthella timonensis Marseille-P3135 | NCBI Genbank | GCA_900184265.1 |
| Enorma massiliensis DSM25476 | NCBI Genbank | GCA_000311845.1 |
| Enteroscipio rubneri DSM105130 | NCBI Genbank | GCA_002899715.1 |
| Gordonibacter faecihominis JCM 16058 | NCBI Genbank | GCA_003788985.1 |
| Gordonibacter massiliensis Marseille-P2775 | NCBI Genbank | GCA_900170005.1 |
| Gordonibacter pamelaeae DSM 19378 | NCBI Genbank | GCA_000210055. 1 |
| Gordonibacter urolithinfaciens DSM 27213 | NCBI Genbank | GCA_003788975.1 |
| Atopobium rimae ATCC 49626 | NCBI Genbank | GCA_000174015.1 |
| Libanicoccus massiliensis Marseille-P3237 | NCBI Genbank | GCA_900143685.1 |
| Olegusella massiliensis DSM 101849 | NCBI Genbank | GCA_900078545.1 |
| Olsenella uli DSM 7084 | NCBI Genbank | GCA_000143845.1 |
| Paraeggerthella hongkongensis DSM 16106 | NCBI Genbank | GCA_003726035. 1 |
| Parolsenella catena DSM 105194 | NCBI Genbank | GCA_003966955. 1 |
| Phoenicibacter congonensis Marseille-P3241 | NCBI Genbank | GCA_900169485.1 |
| Raoultibacter massiliensis Marseille-P2849 | NCBI Genbank | GCA_900199545.1 |
| Raoultibacter timonensis Marseille-P3277 | NCBI Genbank | GCA_900240215.1 |
| Rubneribacter badeniensis DSM 105129 | NCBI Genbank | GCA_002899695.1 |
| Senegalimassilia anaerobia JC110 | NCBI Genbank | GCA_000236865. 1 |
| Slackia equolifaciens DSM 24851 | NCBI Genbank | GCA_003725995. 1 |
| Slackia exigua DSM 15923 | NCBI Genbank | GCA_000162875.1 |
| Slackia faecicanis DSM 17537 | NCBI Genbank | GCA_003725295. 1 |
| Slackia heliotrinireducens DSM 20476 | NCBI Genbank | GCA_000023885. 1 |
| Slackia isoflavoniconvertens DSM 22006 | NCBI Genbank | GCA_003725955. 1 |
| Slackia piriformis DSM 22477 | NCBI Genbank | GCA_000296445. 1 |
| Experimental Models: Organisms/Strains | ||
| Germ-free Swiss Webster Mice | UCSF Gnotobiotics Core | NA |
| Oligonucleotides | ||
| Tetw_FACTGATCATATGAAAATAATCAATATTGGAATTC | This paper | NA |
| Tetw RAGCTATGGATCCTTACATTATCTTCTGAAACATATAG | This paper | NA |
| Apt FACTGATCATATGGCTAAAATG | This paper | NA |
| AGAATATCA | ||
| Apt RGCTATGGATCCCTAAAACAAT TCATCCAGTAAA | This paper | NA |
| Elenmkr1_FGT ACAACAT G CT CCTT GCGG | This paper | NA |
| Elenmkr1_RCGAACAGAGGATCGGGATGG | This paper | NA |
| Elenmkr1_P[6FAM]TTCTGGCTGCACCGTT CGCGGTCCA[BHQ1] | This paper | NA |
| Cgr2FGAGGCCGTCGATTGGATGAT | This paper | NA |
| Cgr2RACCGTAGGCATTGTGGTTGT | This paper | NA |
| Cgr2P[HEX]CGACACGGAGGCCGATGTCG[BHQ1] | This paper | NA |
| Software and Algorithms | ||
| BBtools | jgi.doe.gov/data-and-tools/bbtools/ | v37.97 |
| fastp | Chen et al. (2018) | v0.20.0 |
| vsearch | Rognes et al. (2016) | v2.13.4 |
| SPAdes | Bankevich et al. (2012) | v3.13.1 |
| Prokka | github.com/tseemann/prokka | v1.13.3 |
| CheckM | Parks et al. (2015) | v1.0.12 |
| Quast | Gurevich et al. (2013) | v5.0.2 |
| FastT ree | Price et al. (2009) | 2.1.10 |
| DECIPHER | Wright et al. (2016) | v2.12.0 |
| PhyloPhlAn | Segata et al. (2013) | v0.99 |
| Roary | Page et al. (2015) | v3.12.0 |
| Pyani | github.com/widdowquinn/pyani | v0.2.9 |
| Interproscan | Jones et al. (2014) | v5 |
| ProteinOrtho | Lechner et al. (2011) | v6 |
| DIAMOND | Buchfink et al. (2015) | v0.9.14.115 |
| GhostKoala | Kanehisa et al. (2016) | v2.2 |
| Snippy | github.com/tseemann/snippy | v4.4.3 |
| MZmine2 | Pluskal et al. (2010) | NA |
| Abricate | github.com/tseemann/abricate | v0.9.8 |
| Jellyfish 2 | Marcais and Kingsford (2012) | v2.2.10 |
| Samtools | http://www.htslib.org/ | v1.9 |
| InSilicoSeq | Gourle et al. (2019) | v1.4.3 |
| GNPS Molecular Networking | Wang et al. (2016) | v1.2.3 |
| ComBat | Leek et al. (2019) | 3.32.1 |
| CEU Mass Mediator | Gil-de-la-Fuente et al. (2019) | 3.0 |
| MAGI | Erbilgin et al. (2019) | Docker image 1804676bbef1 |
| Other | ||
| HLB 96-well plate | Waters Oasis | WAT058951 |
| C18 column | Kinetix | 1.7μM 100-A 50×2.1mm |
Highlights:
Curated analysis of paired genome and isolate collection for the study of Coriobacteriia
Eggerthella lenta is genotypically and phenotypically diverse
Development of tools for discovering and validating effector genes in E. lenta
Intra-species competition in E. lenta is correlated with a putative host adhesion
ACKNOWLEDGEMENTS
We are indebted to the UCSF Gnotobiotic Core Facility, Michelle Daigneault (UoG), Stephen Nayfach (UCSF), and Peter Spanogiannopoulos, An Luong, Niki Arab, and the other members of the Turnbaugh lab for technical assistance. We also thank the labs of Ramnik Xavier and Michael Blaut for sharing isolates. Support is acknowledged from the National Institutes of Health (R01HL122593; R21CA227232; R01AR074500; 2T32AI060537–16), Searle Scholars Program (SSP-2016–1352), Burroughs Wellcome Fund, Chan Zuckerberg Biohub, Damon Runyon Cancer Research Foundation (DRR-42–16, DRG-2369–19), Natural Sciences and Engineering Research Council of Canada and the Canadian Institutes of Health Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
PJT is on the scientific advisory boards for Kaleido, Pendulum, Seres, and SNIPRbiome. EAV is co-founder and CSO of NuBiyota. All other authors have no relevant declarations.
REFERENCES
- Bachmann BO, and Ravel J (2009). In silico prediction of microbial secondary metabolic pathways from DNA sequence data. Methods Enzymol 458, 181–217. [DOI] [PubMed] [Google Scholar]
- Bai X, Fu S, Zhang J, Fan R, Xu Y, Sun H, He X, Xu J, and Xiong Y (2018). Identification and pathogenomic analysis of an Escherichia coli strain producing a novel Shiga toxin 2 subtype. Sci. Rep 8, 6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Colburn G, Deatherage DE, Traverse CC, Strand MD, Borges JJ, Knoester DB, Reba A, and Meyer AG (2014). Identifying structural variation in haploid microbial genomes from short-read resequencing data using breseq. BMC Genom 15, 1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer E, Laczny CC, Magnusdottir S, Wilmes P, and Thiele I (2016). Phenotypic differentiation of gastrointestinal microbes is reflected in their encoded metabolic repertoires. Microbiome 4, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayjanov JR, Molenaar D, Tzeneva V, Siezen RJ, and van Hijum SA (2012). PhenoLink-a web-tool for linking phenotype to omics data for bacteria: application to gene-trait matching for Lactobacillus plantarum strains. BMC Genom 13, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bess EN, Bisanz JE, Yarza F, Bustion A, Rich BE, Li X, Kitamura S, Waligurski E, Ang QY, Alba DL, et al. (2020). Genetic basis for the cooperative bioactivation of plant lignans by Eggerthella lenta and other human gut bacteria. Nat Microbiol 5, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K, Shaw S, Steinke K, Villebro R, Ziemert N, Lee SY, Medema MH, and Weber T (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res 47, W81–W87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg SP, Garland T Jr, and Ives AR (2003). Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57, 717–745. [DOI] [PubMed] [Google Scholar]
- Breiman L (2001). Random Forests. Mach. Learn 45, 5–32. [Google Scholar]
- Brynildsrud O, Bohlin J, Scheffer L, and Eldholm V (2016). Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol 17, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, and Huson DH (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, Kanner R, Bencosme Y, Lee YK, Hauser SL, et al. (2017). Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. U. S. A 114, 10713–10718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I-MA, Chu K, Palaniappan K, Pillay M, Ratner A, Huang J, Huntemann M, Varghese N, White JR, Seshadri R, et al. (2019). IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res 47, D666–D677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, Nelson H, Matteson EL, and Taneja V (2016). An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med 8, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, and Gu J (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung L, Orberg ET, Geis AL, Chan JL, Fu K, DeStefano Shields CE, Dejea CM, Fathi P, Chen J, Finard BB, et al. (2018). Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 23, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell JH (1983). On the Prevalence and Relative Importance of Interspecific Competition: Evidence from Field Experiments. Am. Nat 122, 661–696. [Google Scholar]
- Corpet DE, Lumeau S, and Corpet F (1989). Minimum antibiotic levels for selecting a resistance plasmid in a gnotobiotic animal model. Antimicrob. Agents Chemother 33, 535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deivanayagam CC, Rich RL, Carson M, Owens RT, Danthuluri S, Bice T, Höök M, and Narayana SV (2000). Novel fold and assembly of the repetitive B region of the Staphylococcus aureus collagen-binding surface protein. Structure 8, 67–78. [DOI] [PubMed] [Google Scholar]
- Devlin AS, Sloan Devlin A, and Fischbach MA (2015). A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat. Chem. Biol 11, 685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas GM, and Langille MGI (2019). Current and Promising Approaches to Identify Horizontal Gene Transfer Events in Metagenomes. Genome Biol. Evol 11, 2750–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dührkop K, Fleischauer M, Ludwig M, Aksenov AA, Melnik AV, Meusel M, Dorrestein PC, Rousu J, and Böcker S (2019). SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 16, 299–302. [DOI] [PubMed] [Google Scholar]
- Erbilgin O, Rübel O, Louie KB, Trinh M, Raad M. de, Wildish T, Udwary D, Hoover C, Deutsch S, Northen TR, et al. (2019). MAGI: A Method for Metabolite Annotation and Gene Integration. ACS Chem. Biol 14, 704–714. [DOI] [PubMed] [Google Scholar]
- Ferretti P, Pasolli E, Tett A, Asnicar F, Gorfer V, Fedi S, Armanini F, Truong DT, Manara S, Zolfo M, et al. (2018). Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe 24, 133–145.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner BJ, Tai AY, Kotsanas D, Francis MJ, Roberts SA, Ballard SA, Junckerstorff RK, and Korman TM (2015). Clinical and microbiological characteristics of Eggerthella lenta bacteremia. J. Clin. Microbiol 53, 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garud NR, Good BH, Hallatschek O, and Pollard KS (2019). Evolutionary dynamics of bacteria in the gut microbiome within and across hosts. PLoS Biol 17, e3000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giessen TW, and Silver PA (2017). Widespread distribution of encapsulin nanocompartments reveals functional diversity. Nat. Microbiol 2, 17029. [DOI] [PubMed] [Google Scholar]
- Gil-de-la-Fuente A, Godzien J, Saugar S, Garcia-Carmona R, Badran H, Wishart DS, Barbas C, and Otero A (2019). CEU Mass Mediator 3.0: A Metabolite Annotation Tool. J. Proteome Res 18, 797–802. [DOI] [PubMed] [Google Scholar]
- Goltsman DSA, Aliaga Goltsman DS, Sun CL, Proctor DM, DiGiulio DB, Robaczewska A, Thomas BC, Shaw GM, Stevenson DK, Holmes SP, et al. (2018). Metagenomic analysis with strain-level resolution reveals fine-scale variation in the human pregnancy microbiome. Genome Res 28, 1467–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourlé H, Karlsson-Lindsjö O, Hayer J, and Bongcam-Rudloff E (2019). Simulating Illumina metagenomic data with InSilicoSeq. Bioinformatics 35, 521–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblum S, Carr R, and Borenstein E (2015). Extensive strain-level copy-number variation across human gut microbiome species. Cell 160, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, and Tesler G (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, and Turnbaugh PJ (2013). Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 341, 295–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SC, Devendran S, Méndez-García C, Mythen SM, Wright CL, Fields CJ, Hernandez AG, Cann I, Hylemon PB, and Ridlon JM (2018). Bile acid oxidation by Eggerthella lenta strains C592 and DSM 2243T. Gut Microbes 9, 523–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Håvarstein LS, Diep DB, and Nes IF (1995). A family of bacteriocin ABC transporters carry out proteolytic processing of their substrates concomitant with export. Mol. Microbiol 16, 229–240. [DOI] [PubMed] [Google Scholar]
- Hecht AL, Casterline BW, Earley ZM, Goo YA, Goodlett DR, and Bubeck Wardenburg J (2016). Strain competition restricts colonization of an enteric pathogen and prevents colitis. EMBO Rep 17, 1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson GA, Burkhart BJ, DiCaprio AJ, Schwalen CJ, Kille B, Pogorelov TV, and Mitchell DA (2019). Bioinformatic Mapping of Radical S-Adenosylmethionine-Dependent Ribosomally Synthesized and Post-Translationally Modified Peptides Identifies New Cα, Cβ, and Cγ-Linked Thioether-Containing Peptides. J. Am. Chem. Soc 141, 8228–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, and Morishima K (2016). BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol 428, 726–731. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Paterson NG, Gaspar AH, Ton-That H, and Baker EN (2009). The Corynebacterium diphtheriae shaft pilin SpaA is built of tandem Ig-like modules with stabilizing isopeptide and disulfide bonds. Proc. Natl. Acad. Sci. U. S. A 106, 16967–16971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankainen M, Paulin L, Tynkkynen S, von Ossowski I, Reunanen J, Partanen P, Satokari R, Vesterlund S, Hendrickx APA, Lebeer S, et al. (2009). Comparative genomic analysis of Lactobacillus rhamnosus GG reveals pili containing a human- mucus binding protein. Proc. Natl. Acad. Sci. U. S. A 106, 17193–17198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel N, Bisanz JE, Pandelia M-E, Turnbaugh PJ, and Balskus EP (2018). Discovery and characterization of a prevalent human gut bacterial enzyme sufficient for the inactivation of a family of plant toxins. eLife 7, e33953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier J-C, Khelaifia S, Alou MT, Ndongo S, Dione N, Hugon P, Caputo A, Cadoret F, Traore SI, Seck EH, et al. (2016). Culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol 1, 16203. [DOI] [PubMed] [Google Scholar]
- Lechner M, Findeiß S, Steiner L, Marz M, Stadler PF, and Prohaska SJ (2011). Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinform 12, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Donaldson GP, Mikulski Z, Boyajian S, Ley K, and Mazmanian SK (2013). Bacterial colonization factors control specificity and stability of the gut microbiota. Nature 501, 426–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD (2019). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcais G, and Kingsford C (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthies A, Loh G, Blaut M, and Braune A (2012). Daidzein and genistein are converted to equol and 5-hydroxy-equol by human intestinal Slackia isoflavoniconvertens in gnotobiotic rats. J. Nutr 142, 40–46. [DOI] [PubMed] [Google Scholar]
- McDonald D, Hyde E, Debelius JW, Morton JT, Gonzalez A, Ackermann G, Aksenov AA, Behsaz B, Brennan C, Chen Y, et al. (2018). American Gut: an Open Platform for Citizen Science Microbiome Research. mSystems 3, e00031–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S, Shi ZJ, Seshadri R, Pollard KS, and Kyrpides NC (2019). New insights from uncultivated genomes of the global human gut microbiome. Nature 568, 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothias LF, Petras D, Schmid R, Dührkop K, Rainer J, Sarvepalli A, Protsyuk I, Ernst M, Tsugawa H, Fleischauer M, et al. (2019). Feature-based Molecular Networking in the GNPS Analysis Environment. bioRxiv, doi.org/10.1101/812404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouioui I, Carro L, García-López M, Meier-Kolthoff JP, Woyke T, Kyrpides NC, Pukall R, Klenk H-P, Goodfellow M, and Göker M (2018). Genome-Based Taxonomic Classification of the Phylum Actinobacteria. Frontiers Microbiol 9, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paauw A, Leverstein-van Hall MA, van Kessel KPM, Verhoef J, and Fluit AC (2009). Yersiniabactin Reduces the Respiratory Oxidative Stress Response of Innate Immune Cells. PLoS ONE 4, e8240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, Fookes M, Falush D, Keane JA, and Parkhill J (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paniagua-Michel J, Olmos-Soto J, and Ruiz MA (2012). Pathways of carotenoid biosynthesis in bacteria and microalgae. Methods Mol. Biol 892, 1–12. [DOI] [PubMed] [Google Scholar]
- Paradis E, and Schliep K (2018). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. [DOI] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, and Tyson GW (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25, 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnode ML, Beller ZW, Han ND, Cheng J, Peters SL, Terrapon N, Henrissat B, Le Gall S, Saulnier L, Hayashi DK, et al. (2019). Interspecies Competition Impacts Targeted Manipulation of Human Gut Bacteria by Fiber-Derived Glycans. Cell 179, 59–73.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plata G, Henry CS, and Vitkup D (2015). Long-term phenotypic evolution of bacteria. Nature 517, 369–372. [DOI] [PubMed] [Google Scholar]
- Pluskal T, Castillo S, Villar-Briones A, and Oresic M (2010). MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, and Finn RD (2018). HMMER web server: 2018 update. Nucleic Acids Res 46, W200–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyet M, Groussin M, Gibbons SM, Avila-Pacheco J, Jiang X, Kearney SM, Perrotta AR, Berdy B, Zhao S, Lieberman TD, et al. (2019). A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat. Med 25, 1442–1452. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol 26, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekdal VM, Bess EN, Bisanz JE, Turnbaugh PJ, and Balskus EP (2019). Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364, eaau6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson AE, Lowe JE, Koh E-I, and Henderson JP (2018). Uropathogenic enterobacteria use the yersiniabactin metallophore system to acquire nickel. J. Biol. Chem 293, 14953–14961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognes T, Flouri T, Nichols B, Quince C, and Mahé F (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahl JW, Caporaso JG, Rasko DA, and Keim P (2014). The large-scale blast score ratio (LS-BSR) pipeline: a method to rapidly comapre genetic content between bacterial genomes. PeerJ 2, e332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders E, Pukall R, Abt B, Lapidus A, Del Rio TG, Copeland A, Tice H, Cheng J-F, Lucas S, Chen F, et al. (2009). Complete genome sequence of Eggerthella lenta type strain (VPI 0255 T). Stand. Genomic Sci 1, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Börnigen D, Morgan XC, and Huttenhower C (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun 4, 2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seipke RF, Song L, Bicz J, Laskaris P, Yaxley AM, Challis GL, and Loria R (2011). The plant pathogen Streptomyces scabies 87–22 has a functional pyochelin biosynthetic pathway that is regulated by TetR- and AfsR-family proteins. Microbiology 157, 2681–2693. [DOI] [PubMed] [Google Scholar]
- Skinnider MA, Merwin NJ, Johnston CW, and Magarvey NA (2017). PRISM 3: expanded prediction of natural product chemical structures from microbial genomes. Nucleic Acids Res 45, W49–W54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Pan Y, Chen S, and Zhang X (2012). Structural characteristics of genomic islands associated with GMP synthases as integration hotspot among sequenced microbial genomes. Comput. Biol. Chem 36, 62–70. [DOI] [PubMed] [Google Scholar]
- Sperry JF, and Wilkins TD (1976). Arginine, a growth-limiting factor for Eubacterium lentum. J. Bacteriol 127, 780–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprouffske K, and Wagner A (2016). Growthcurver: an R package for obtaining interpretable metrics from microbial growth curves. BMC Bioinform 17, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TW-M, Fiehn O, Goodacre R, Griffin JL, et al. (2007). Proposed minimum reporting standards for chemical analysis. Metabolomics 3, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sybesma W, Molenaar D, van IJcken W, Venema K, and Kort R (2013). Genome instability in Lactobacillus rhamnosus GG. Appl. Environ. Microbiol 79, 2233–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot CM, and Young VB (2015). Interactions Between the Gastrointestinal Microbiome and Clostridium difficile. Annu. Rev. Microbiol 69, 445–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietz JI, Schwalen CJ, Patel PS, Maxson T, Blair PM, Tai H-C, Zakai UI, and Mitchell DA (2017). A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat. Chem. Biol 13, 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, and Segata N (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903. [DOI] [PubMed] [Google Scholar]
- Verster AJ, and Borenstein E (2018). Competitive lottery-based assembly of selected clades in the human gut microbiome. Microbiome 6, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M, Hesselman MC, Te Beek TA, van Passel MWJ, and Eyre-Walker A (2015). Rates of Lateral Gene Transfer in Prokaryotes: High but Why? Trends Microbiol 23, 598–605. [DOI] [PubMed] [Google Scholar]
- Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, et al. (2016). Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol 34, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens R, Hageman JA, van Eeuwijk F, Kooke R, Flood PJ, Wijnker E, Keurentjes JJB, Lommen A, van Eekelen HDLM, Hall RD, et al. (2016). Improved batch correction in untargeted MS-based metabolomics. Metabolomics 12, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis (Springer; ). [Google Scholar]
- Wright E., Erik, and Wright S (2016). Using DECIPHER v2.0 to Analyze Big Biological Sequence Data in R. R J 8, 352. [Google Scholar]
- Yamaguchi Y, and Inouye M (2011). Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat. Rev. Microbiol 9, 779–790. [DOI] [PubMed] [Google Scholar]
- Zallot R, Oberg N, and Gerlt JA (2019). The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 58, 4169–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevi D, Korem T, Godneva A, Bar N, Kurilshikov A, Lotan-Pompan M, Weinberger A, Fu J, Wijmenga C, Zhernakova A, et al. (2019). Structural variation in the gut microbiome associates with host health. Nature 568, 43–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genome metadata relating to Figure 1.
Table S2. Genomic variants in resequenced type strain isolates versus reference assembly (CP001726.1), related to Figure 1.
Table S3. Minimum inhibitory concentration (MIC) testing in Coriobacteriia, related to Figure 4.
Table S4. Associations between gene clusters and metabolite features, related to Figure 2.
Table S5. Mock community composition, related to Figure 5.
Data Availability Statement
Genome accessions are listed in Table S1 and newly sequenced genomes are available as part of BioProject PRJNA412637. Raw reads for assembly and community competition were deposited under PRJNA578765. Source code for ElenMatchR and StrainR are available at github.com/turnbaughlab/ElenMatchR and github.com/turnbaughlab/StrainR. Raw metabolomic data is available at massive.ucsd.edu under accession MSV000083734.