

Abstract
ABCA3 transports phospholipids across lamellar body membranes in pulmonary alveolar type II cells and is required for surfactant assembly. Rare, biallelic, pathogenic ABCA3 variants result in lethal neonatal respiratory distress syndrome and childhood interstitial lung disease. Qualitative functional characterization of ABCA3 missense variants suggests 2 pathogenic classes: disrupted intracellular trafficking (type I mutant) or impaired ATPase-mediated phospholipid transport into the lamellar bodies (type II mutant). We qualitatively compared wild-type (WT-ABCA3) with 4 uncharacterized ABCA3 variants (c.418A>C;p.Asn140His, c.3609_3611delCTT;p.Phe1203del, c.3784A>G;p.Ser1262Gly, c.4195G>A;p.Val1399Met) in A549 cells using protein processing, co-localization with intracellular organelles, lamellar body ultrastructure, and ATPase activity. We quantitatively measured lamellar body-like vesicle diameter and intracellular ABCA3 trafficking using fluorescence-based co-localization. Three ABCA3 variants (p.Asn140His, p.Ser1262Gly, p.Val1399Met) were processed and trafficked normally and demonstrated well-organized lamellar body-like vesicles, but had reduced ATPase activity consistent with type II mutants. P.Phe1203del was processed normally, had reduced ATPase activity, and well-organized lamellar body-like vesicles, but quantitatively co-localized with both endoplasmic reticulum and lysosomal markers, an intermediate phenotype suggesting disruption of both intracellular trafficking and phospholipid transport. All ABCA3 mutants demonstrated mean vesicle diameter smaller than WT-ABCA3. Qualitative and quantitative functional characterization of ABCA3 mutants informs mechanisms of pathogenicity.
Keywords: ABCA3, surfactant, lamellar bodies, respiratory distress syndrome (RDS), childhood interstitial lung disease (chILD)
Introduction
Pulmonary surfactant is a complex mixture of phospholipids and proteins that reduces surface tension at the air-liquid interface and prevents alveolar collapse at end-expiration. Surfactant is assembled and stored in lamellar bodies, lysosome-related organelles in pulmonary alveolar type II (ATII) cells (Ridsdale, Na, Xu, Greis, & Weaver, 2011). ATP-binding cassette transporter A3 (ABCA3) is a large integral transmembrane protein that is highly expressed and developmentally regulated in ATII cells, localizes to the limiting membranes of lamellar bodies, is required for lamellar body biogenesis, and transports phospholipids (specifically phosphatidylcholine and phosphatidylglycerol) from the cytoplasm into lamellar bodies for assembly of pulmonary surfactant (Beers & Mulugeta, 2017; Mulugeta et al., 2002; Shulenin et al., 2004; Tryka, Wert, Mazursky, Arrington, & Nogee, 2000; Whitsett, Wert, & Weaver, 2015).
Rare or private, biallelic, pathogenic variants in ABCA3 (NM_001089.2) are the most frequent monogenic cause of surfactant dysfunction in infants and children. Over 200 disease-associated ABCA3 variants have been identified among term or late preterm infants with progressive neonatal respiratory distress syndrome (RDS) or older infants and children with childhood interstitial lung disease (chILD) (Kroner et al., 2017; Wambach et al., 2014), and fewer than 10% have been functionally characterized in surrogate cell model systems. Variants in ABCA3 encode mutant proteins with disrupted intracellular trafficking (type I mutant) or impaired ATPase-mediated phospholipid transport into the lamellar bodies (type II mutant) (Beers et al., 2013; Cheong et al., 2006; Flamein et al., 2012; Matsumura, Ban, & Inagaki, 2008; Matsumura, Ban, Ueda, & Inagaki, 2006; Tryka et al., 2000; Wambach et al., 2016). Infants with biallelic nonsense or frameshift variants in ABCA3 present with neonatal RDS and die by 1 year of age without lung transplantation (Kroner et al., 2017; Wambach et al., 2014). However, patients with missense, splice-site, and/or in-frame insertions or deletions have variable and unpredictable presentation, progression, and severity of lung disease (Kroner et al., 2017; Wambach et al., 2014). Additionally, variant-encoded pathogenesis mechanisms are difficult to predict based on variant position within the gene or mature protein (Beers & Mulugeta, 2017; Matsumura et al., 2008). Phenotypic variability is likely related to variant-encoded effects on ABCA3 function (type I vs. type II mutant), activation of intracellular stress pathways, or other genetic or environmental modifiers (Beers et al., 2013; Cheong et al., 2006; Flamein et al., 2012; Kroner et al., 2017; Wambach et al., 2016; Weichert et al., 2011). Therapies for infants and children with ABCA3 deficiency remain limited, non-specific (e.g., surfactant, corticosteroids, azithromycin, hydroxychloroquine), and largely ineffective (Kroner et al., 2017). Lung transplantation (5-year survival of ~50%) remains the only available treatment for affected infants and children with progressive respiratory failure (Eldridge et al., 2017). Prior functional characterization studies of disease-associated ABCA3 variants suggest pathogenesis mechanisms similar to those observed in CFTR, another member of the ABC transporter protein superfamily (Denman, 2018; Kinting et al., 2018; Kinting et al., 2019; Lopes-Pacheco, 2016). Current methods of functional characterization include immunoblotting, immunohistochemistry, electron microscopy, assessment vesicular size, ATPase activity, and liposome uptake (Beers et al., 2013; Cheong et al., 2006; Flamein et al., 2012; Hoppner et al., 2017; Kinting et al., 2018; Matsumura et al., 2008; Matsumura et al., 2006; Wittmann et al., 2016). Using previously described methods of functional characterization and quantitative measurement of co-localization using confocal imaging and Pearson’s correlation, we characterized 4 disease-associated ABCA3 variants associated with diverse pulmonary phenotypes.
Methods
Mutation Selection
We selected 4 previously uncharacterized ABCA3 variants associated with diverse pulmonary phenotypes for functional study (Supplemental Fig. S1) Three ABCA3 variants were identified among neonates with RDS or infants and children with chILD. The c.418A>C;p.Asn140His variant (minor allele frequency (MAF) 1.4x10−4, https://gnomad.broadinstitute.org/ accessed March 2020 (Karczewski KJ, 2019)) was identified in an infant with chILD.6 The c.4195G>A;p.Val1399Met variant (MAF 8.0x10−6) was identified in 3 unrelated infants with severe neonatal RDS who died during infancy or underwent lung transplantation (Wambach et al., 2014). The c.3609_3611delCTT;p.Phe1203del variant (MAF 7.4x10−5) was identified in 7 unrelated individuals with phenotypes of RDS or chILD (Wambach et al., 2014). The c.3784A>G;p.Ser1262Gly variant is present in ~1/1000 individuals (MAF 1.0x10−3), did not segregate with pulmonary fibrosis in families with surfactant protein C variants(van Moorsel et al., 2010), and has conflicting predictions of pathogenicity (Adzhubei, Jordan, & Sunyaev, 2013; Ng & Henikoff, 2003; Rentzsch, Witten, Cooper, Shendure, & Kircher, 2019; Schwarz, Cooper, Schuelke, & Seelow, 2014). We used 2 previously characterized ABCA3 variants as positive controls: c.302T>C;p.Leu101Pro (type I, intracellular trafficking mutant) and c.875A>T;p.Glu292Val (type II, impaired ATP-mediated phospholipid transport mutant). This study was reviewed and approved by the Washington University School of Medicine Human Research Protection Office.
shRNA/ABCA3/GFP Constructs
We generated adenoviral vectors that expressed GFP-tagged wild-type ABCA3 (WT-ABCA3-GFP, C-terminus tagged) or cDNAs from the 4 selected variants (c.418A>C;p.Asn140His; c.3609_3611delCTT;p.Phe1203del; c.3784A>G;p.Ser1262Gly; c.4195G>A;p.Val1399Met) or the 2 positive controls (c.302T>C;p.Leu101Pro and c.875A>T;p.Glu292Val) as previously described (Wambach et al., 2016).
Transduction of A549 Cells
We transduced A549 cells with adenovirus that express WT-ABCA3 or ABCA3 variant constructs as previously described (Wambach et al., 2016). We performed immunohistochemistry, membrane or total protein extraction, and fixation for electron microscopy 48 hours after viral transduction.
Extraction of Membrane Protein
To extract membrane protein, we resuspended cell pellets in hypotonic buffer (10mM Tris-HCl pH 7.5, 0.5mM MgCl2, 1mM EDTA) with protease inhibitors [Roche Applied Science®, Indianapolis, IN]) on ice for 15 minutes, sonicated for 30 seconds in solution M (10mM Tris-HCl pH 7.5, 0.5M sucrose, 0.3M KCl, 6mM β-mercaptoethanol, 40μM CaCl2), and centrifuged (8000 xg at 4°C) for 20 minutes. We isolated microsomal pellets by centrifugation of the supernatant for 1 hour (100,000 xg at 4°C) and resuspended pellets in solution M1 (10mM Tris-HCl pH 7.5, 0.25M sucrose, 0.15M KCl, 3mM β-mercaptoethanol, 20μM CaCl2). We quantified membrane protein using the bicinchoninic acid (BCA) assay (Pierce Biotechnology®, Rockford, IL).
Immunoblotting
We separated total or membrane proteins by gel electrophoresis (3-8% tris-acetate or 12% bis-tris NuPage gel [Thermo-Fisher Scientific®, Waltham, MA]), transferred to nitrocellulose membrane, probed with full-length polyclonal anti-GFP (Takara Bio®, Mountain View, CA) or monoclonal anti-β-actin (MilliporeSigma®, Burlington, MA) followed by horseradish peroxidase-conjugated secondary antibody (GE Healthcare®, Pittsburgh, PA). After chemiluminescent labelling (Amersham ECL Detection Reagent, GE Healthcare®), we captured images by film exposure.
Confocal Microscopy Imaging of Immunofluorescence-Tagged Intracellular Markers
We fixed GFP tagged WT-ABCA3 and mutant ABCA3 (Ex 488nm/Em 500-540nm) transduced A549 cells with 4% paraformaldehyde and permeabilized (0.1% Triton X-100) prior to incubation with anti-CD63 (lysosome-related organelle marker, Thermo-Fisher Scientific®) or anti-SelectFX_ER (endoplasmic reticulum marker, Thermo-Fisher Scientific®). We detected the CD63 antibody and ER marker with Alexa Fluor 568 anti-mouse IgG (Ex 568nm/Em 580-620nm, Thermo-Fisher Scientific®). Images were acquired using a Leica SP8X laser scanning confocal microscope fitted with a white light laser and HyD detectors using a 63x Apochromat oil objective (N.A. 1.4) and LASX software (v3.6.0; Leica Microsystems). A single x-y plane that contained the most anti-CD63 positive vesicles was selected for analysis of co-localization and vesicle size.
Quantitative Co-Localization of GFP-tagged ABCA3 with Intracellular Markers
We used Volocity quantification (v6.3.3, PerkinElmer®, Waltham, MA) to determine the Pearson’s correlation coefficient (PCC) (Manders, Stap, Brakenhoff, van Driel, & Aten, 1992) of the fluorescence co-localization of GFP-tagged WT-ABCA3 or mutant ABCA3 proteins with anti-CD63 or anti-SelectFX_ER for 15 cells per construct. We compared ABCA3 mutant protein co-localization results to WT-ABCA3 using paired Student’s t-test.
Quantitative Vesicle Diameter of GFP-tagged ABCA3 Positive Vesicles
We used ImageJ® with Bioformats for manual measurement of 20 vesicles positive for GFP-tagged WT-ABCA3 or individual mutant ABCA3 proteins from 10 individual cells (n=200 vesicles per construct) and compared vesicle diameter using paired Student’s t-test.
Electron Microscopy
After fixation (2% glutaraldehyde at 4°C) for 4 hours, cells were post-fixed (2% osmium tetroxide for 1 hour) and en bloc stained (2% aqueous uranyl acetate) for 30 minutes, dehydrated, and embedded in PolyBed 812 (Polysciences, Hatfield, PA). Sections (90 nm) were post-stained (Veneable’s lead citrate) and viewed with a JEOL model 1200EX electron microscope (JEOL®, Toyko, Japan). We acquired digital images using an AMT Advantage HR (Advanced Microscopy Technology®, Danvers, MA) high definition CCD, 1.3 megapixel TEM camera. We reviewed electron microscopy images for each construct with a pediatric pulmonary pathologist (F.V.W).
ATPase Assay
Using a colorimetric ATPase assay (Expedon®, San Diego, CA) and triplicate measurements (15 μg membrane protein for each construct), we assessed ATP hydrolysis activity by measuring free phosphate (Pi) release as previously described (Wambach et al., 2016). We normalized Pi release from each sample with densitometric measurement of ABCA3-GFP tagged protein, expressed activity as a percentage of WT-ABCA3 Pi release, and compared WT-ABCA3 and mutant protein ATPase activities using paired Student’s t-test. A previously characterized type II, impaired ATP-mediated phospholipid transport mutant p.Glu292Val was included as a positive control. These experiments were independently performed three times.
Results
Protein Processing
The GFP-tagged ABCA3 primary translation product (220 kD) typically undergoes processing (residue p.Lys174) to a 180 kD peptide in post-Golgi, acidic, LAMP-3-positive vesicles, specifically multivesicular bodies and lamellar bodies (Beers & Mulugeta, 2017; Cheong et al., 2006; Engelbrecht, Kaltenborn, Griese, & Kern, 2010; Hofmann et al., 2016; Matsumura et al., 2008; Matsumura et al., 2006). Type I ABCA3-GFP mutant proteins (190 kD ABCA3+ 30 kD GFP tag) are retained in the endoplasmic reticulum (ER), do not undergo processing, and only a single incleaved band (220 kD) is detected by immunoblotting (Beers et al., 2013; Flamein et al., 2012; Matsumura et al., 2006; Wambach et al., 2016; Weichert et al., 2011). We found that mutant ABCA3 proteins p.Asn140His, p.Phe1203del, p.Ser1262Gly, and p.Val1399Met undergo protein processing similar to WT-ABCA3 [Figure 1].
Figure 1. Immunoblotting analysis of wild-type ABCA3 and mutant proteins.
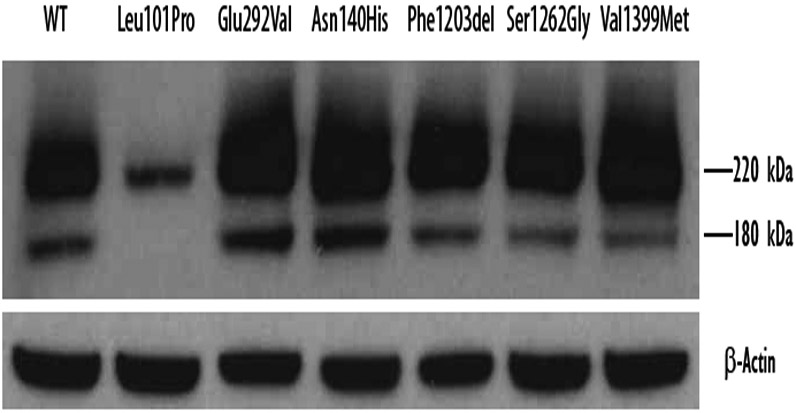
Total protein was extracted from A549 cells transduced with adenovirus expressing WT or ABCA3 variant constructs encoding p.Asn140His, p.Phe1203del, p.Ser1262Gly, p.Val1399Met, p.Leu101Pro, or p.Glu292Val. ABCA3-GFP protein was detected using anti-GFP antibody and normalized to β-actin. Except for the previously characterized type I trafficking mutant p.Leu101Pro, all mutant ABCA3 proteins demonstrated immunoblotting patterns similar to WT-ABCA3.
Qualitative Co-Localization of Mutant ABCA3 Proteins with Intracellular Markers
We observed that mutant ABCA3 proteins p.Asn140His, p.Phe1203del, p.Ser1262Gly, and p.Val1399Met co-localize to CD63 positive subcellular organelles, suggesting these mutants traffic to lysosome-related, lamellar body-like vesicles (anti-CD63+) and demonstrate cellular distribution (dotted, ring-like pattern) similar to WT-ABCA3 [Figure 2]. We confirmed previous reports that mutant protein p.Leu101Pro co-localizes with ER markers (anti-SelectFX_ER+) and that p.Glu292Val co-localizes similarly to WT-ABCA3 (Matsumura et al., 2006; Wambach et al., 2016) [Figure 2].
Figure 2. Qualitative co-localization of wild-type ABCA3 and mutant proteins.

A549 cells transduced with adenovirus expressing WT-ABCA3 or ABCA3 variant constructs encoding p.Asn140His, p.Phe1203del, p.Ser1262Gly, p.Val1399Met, p.Leu101Pro were analyzed using confocal microscopy. Anti-CD63 and anti-SelectFX_ER were used to stain lysosome-related, lamellar body-like vesicles and the ER, respectively. Except for the previously characterized type I trafficking mutant p.Leu101Pro, all mutant ABCA3 proteins demonstrated qualitative co-localization patterns similar to WT-ABCA3. Scale bar = 10 mm
Quantitative Co-Localization of ABCA3 Mutant Proteins with Intracellular Markers
In addition to qualitative assessment of co-localization patterns, we performed quantitative co-localization using Pearson’s correlation coefficient (PCC) (Manders et al., 1992) of GFP-tagged mutant ABCA3 proteins with cellular markers for lysosome-related, lamellar body-like vesicles (anti-CD63+) or ER (SelectFX_ER+). We found that p.Asn140His (PCC mean ±s.d.= 0.79±0.09), p.Ser1262Gly (0.81±0.07), and p.Val1399Met (0.77±0.08) quantitatively co-localized with lysosomal marker anti-CD63 similarly to WT-ABCA3 (mean PCC=0.84± 0.06) [Figure 3]. In contrast to qualitative co-localization, quantitative co-localization demonstrated that p.Phe1203del co-localized with both the lysosomal marker anti-CD63 (0.77±0.08) and with the ER marker SelectFX_ER (0.33± 0.10), p<0.001 [Figure 3]. Similar to previous reports, the type I mistrafficking mutant p.Leu101Pro co-localized with the ER marker (p<0.001) and p.Glu292Val co-localized similarly to WT-ABCA3 [Figure 3].
Figure 3. Quantitative co-localization of GFP-tagged wild-type ABCA3 and mutant proteins with intracellular markers using Pearson’s correlation coefficient (PCC).
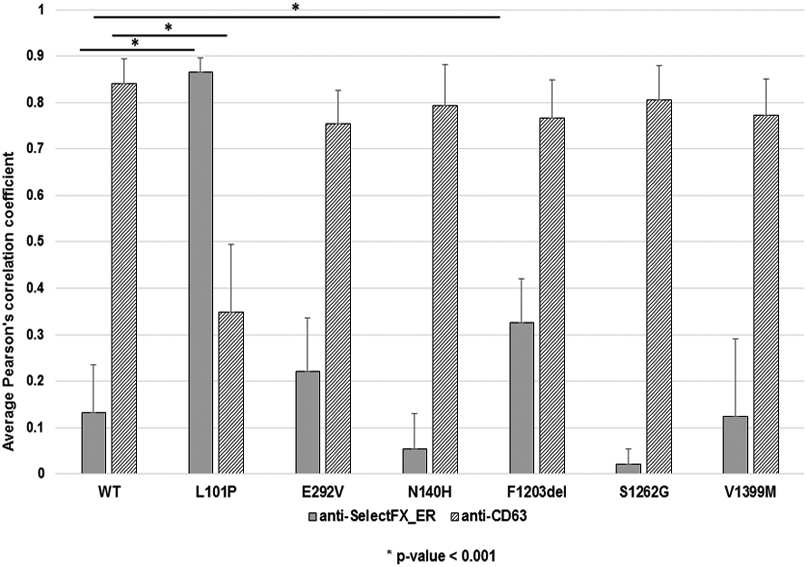
A549 cells transduced with adenovirus expressing GFP-tagged WT-ABCA3 or ABCA3 variant constructs encoding p.Asn140His, p.Phe1203del, p.Ser1262Gly, p.Val1399Met, p.Leu101Pro, or p.Glu292Val were analyzed using confocal microscopy. Anti-CD63 and anti-SelectFX_ER were used to stain lysosome-related, lamellar body-like vesicles and the ER, respectively. Fifteen cells were selected from each construct, co-localization was determined by Pearson’s correlation coefficient (PCC) measured using Volocity Quantification software for each condition. Similar to results from qualitative co-localization, previously uncharacterized mutant ABCA3 proteins p.Asn140His, p.Ser1262Gly, and p.Val1399Met quantitatively co-localized similar to WT-ABCA3, whereas mutant protein p.Phe1203del quantitatively co-localized with both ER and lysosomal markers, suggesting an intermediate phenotype. The quantitative co-localization patterns of p.Leu101Pro and p.Glu292Val were similar to previous qualitative co-localization studies. *p<0.001
Qualitative Vesicle Ultrastructure
A549 cells transduced with p.Asn140His, p.Phe1203del, p.Ser1262Gly, and p.Val1399Met had multiple well-organized lamellar body-like vesicles (white arrows) similar to A549 cells transduced with WT-ABCA3 [Figure 4]. Similar to previously reported findings, A549 cells expressing p.Leu101Pro (previously characterized type I, trafficking mutant) demonstrated frequent abnormal appearing lamellar body-like vesicles with dense inclusions (black arrows), and A549 cells expressing p.Glu292Val (previously characterized type II, impaired phospholipid transport mutant) demonstrated well-organized lamellar body-like vesicles (white arrows) [Figure 4].
Figure 4. Qualitative vesicle ultrastructure of A549 cells transduced with wild-type or mutant ABCA3 constructs.
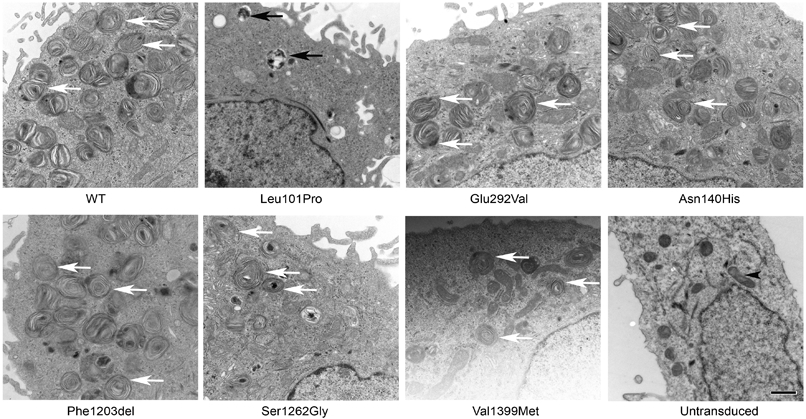
A549 cells transduced with adenovirus expressing WT-ABCA3 or ABCA3 variant constructs encoding p.Asn140His, p.Phe1203del, p.Ser1262Gly, p.Val1399Met, or p.Leu101Pro were fixed for assessment of lamellar body-like vesicle ultrastructure. White arrows indicate well-organized lamellar body-like vesicles, black arrows indicate abnormal appearing lamellar body-like vesicles. A549 cells expressing variants p.Asn140His, p.Phe1203del, p.Ser1262Gly, and p.Val1399Met had multiple well-organized lamellar body-like vesicles similar to WT-ABCA3. Similar to previous results, A549 cells expressing p.Leu101Pro had frequent abnormal appearing lamellar body-like vesicles with dense inclusions, whereas A549 cells expressing p.Glu292Val had well organized appearing lamellar body-like vesicles. Black arrowheads (untransduced A549 cells) indicate mitochondria. Scale bar = 0.8 mm
Quantitative Vesicle Diameter of GFP-tagged ABCA3 Positive Vesicles
When compared to GFP-tagged ABCA3+ vesicles in A549 cells that express WT-ABCA3, we observed smaller mean vesicle diameters in A549 cells that express individual mutant proteins p.Asn140His, p.Phe1203del, p.Ser1262Gly, and p.Val1399Met as well as p.Glu292Val (previously characterized type II, impaired phospholipid transport mutant) (p<0.001) [Figure 5]. Reliable quantification of vesicle diameter of p.Leu101Pro expressing A549 cells was difficult due to a diffuse staining pattern consistent with mistrafficking of the mutant protein [Figure 2].
Figure 5. Quantitative diameter of wild-type and mutant ABCA3-containing vesicles.
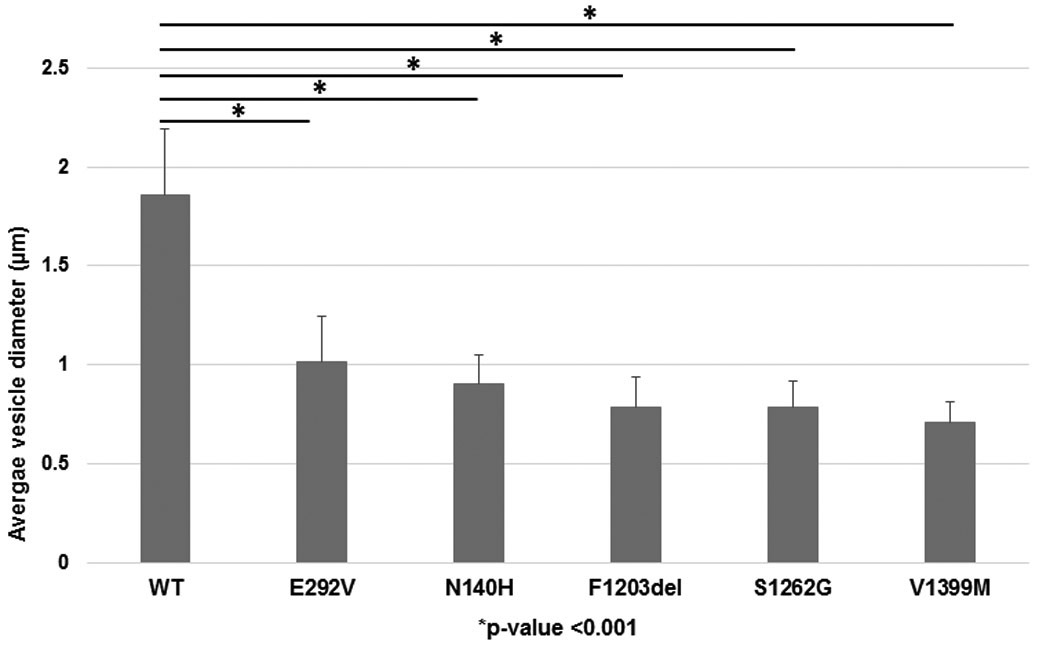
A549 cells transduced with adenovirus expressing WT-ABCA3 or ABCA3 variant constructs encoding p.Asn140His, p.Phe1203del, p.Ser1262Gly, p.Val1399Met, or p.Glu292Val were analyzed using confocal microscopy. Ten cells were selected from each condition and 20 vesicles from individual cells were selected (n=200) for manual diameter measurements using ImageJ. The ABCA3 positive vesicle diameters of A549 cells expressing p.Asn140His, p.Phe1203del, p.Ser1262Gly, p.Val1399Met, or p.Glu292Val were smaller as compared to WT-ABCA3. 1 pixel=0.11 μm. *p<0.001
ATPase Assay
Using membrane protein isolated from A549 cells transduced with adenovirus that expresses individual ABCA3 variants or WT-ABCA3, we found decreased ATP hydrolysis activity of mutant proteins p.Asn140His, p.Phe1203del, p.Ser1262Gly, and p.Val1399Met compared to WT-ABCA3 (p<0.001) [Figure 6]. We confirmed previous results of significantly decreased ATP hydrolysis activity for p.Glu292Val (type II, impaired phospholipid transport mutant) (Wambach et al., 2016).
Figure 6. ATPase activity of wild-type ABCA3 and mutant proteins.
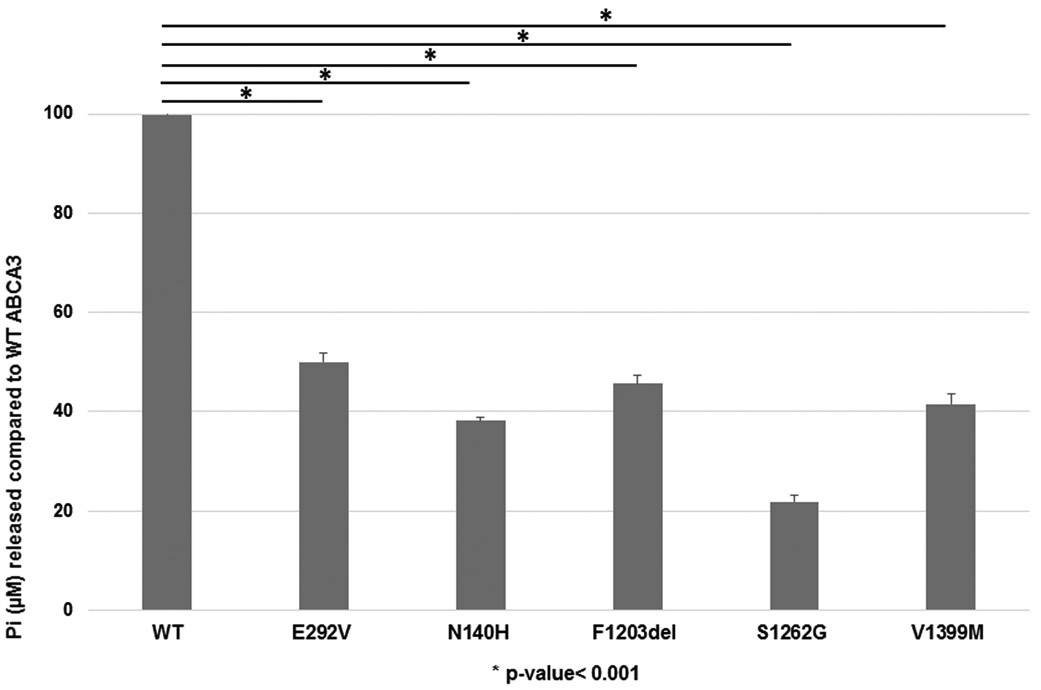
Membrane protein was extracted from A549 cells transduced with adenoviral vectors expressing WT-ABCA3 or ABCA3 variant constructs encoding p.Asn140His, p.Phe1203del, p.Ser1262Gly, p.Val1399Met, or p.Glu292Val (previously characterized type II mutant). ATPase activity was measured as free phosphate (Pi) released as percentage of WT activity and normalized to Western blot with anti-GFP antibody. Mutant proteins p.Asn140His, p.Phe1203del, p.Ser1262Gly, and p.Val1399Met had decreased ATPase activity as compared to WT-ABCA3. Similar to previous results, p.Glu292Val had reduced ATPase activity as compared to WT-ABCA3.*p<0.001
Discussion
Based on our functional characterization, the previously uncharacterized ABCA3 variants c.418A>C;p.Asn140His, c.3609_3611delCTT;p.Phe1203del, c.3784A>G;p.Ser1262Gly, and c.4195G>A;p.Val1399Met are type II mutants. Despite ABCA3 mutant protein processing similar to WT-ABCA3 and well-organized lamellar body-like vesicle ultrastructure in cells transduced with each of these ABCA3 variants, variant pathogenicity is suggested by the quantitatively smaller ABCA3+ vesicle diameter and reduced ATPase activity compared to WT-ABCA3. Quantitative co-localization of fluorescence markers using confocal microscopy also confirmed and extended prior methods for functional characterization of ABCA3 mutants (Beers et al., 2013; Cheong et al., 2006; Flamein et al., 2012; Kinting et al., 2018; Matsumura et al., 2008; Matsumura et al., 2006; Wambach et al., 2016; Weichert et al., 2011; Wittmann et al., 2016).
Amino acid residue N140 is located within the first luminal loop of ABCA3 and is glycosylated (Beers et al., 2013). Substitution of glutamine for asparagine at residue 140 (p.Asn140Gln) results in normal intracellular trafficking (similar to our result for p.Asn140His), whereas the double mutant p.Asn124Gln/p.Asn140Gln is partially retained in the ER, and both p.Asn140Gln and p.Asn124Gln/p.Asn140Gln result in decreased ABCA3 expression (Beers et al., 2013). Restored ABCA3 expression for both the single p.Asn140Gln and the double p.Asn124Gln/p.Asn140Gln mutants in the presence of the proteasome inhibitor MG132 demonstrates that these mutant proteins undergo intracellular degradation via the ubiquitin-proteasome mediated pathway (Beers et al., 2013) and suggests that activation of intracellular protein degradation pathways may contribute to disease pathogenesis.
Quantitative co-localization of p.Phe1203del with both lysosome-related, ABCA3+ lamellar body-like vesicles and ER markers suggests some ABCA3 mutant proteins may have intermediate phenotypes not previously identified with qualitative co-localization studies. Of the 185 ABCA3 deficient reported individuals with biallelic ABCA3 variants and either severe neonatal RDS or chILD, 7 unrelated individuals were compound heterozygous for p.Phe1203del and another ABCA3 variant (Wambach et al., 2014). Five of these individuals were alive without lung transplant at last contact including an adult (age 26 years); one underwent lung transplantation at 5 months of age and one died at 4 months of age (Wambach et al., 2014). The clinical courses of these individuals in combination with the data from functional characterization studies suggest that p.Phe1203del is more likely to be associated with chILD than the neonatal RDS phenotype similar to p.Glu292Val. Activation of cellular stress pathways rather than direct disruption of surfactant metabolism by ABCA3 mutant proteins may contribute to chILD pathogenesis as has been described for SFTPC mutant proteins (Katzen et al., 2019; Nureki et al., 2018). Such pathogenic mechanisms may provide targets for new therapeutic strategies.
Recently, Kinting and colleagues demonstrated increased lipid transport activity as measured by increased portion of labelled phosphatidylcholine (TopF-PC) filled vesicles and increased fluorescence intensity of filled vesicles when A549 cells expressing the type II mutant c.1702A>G;p.N568D were exposed to CFTR potentiators ivacaftor and genistein (Kinting et al., 2019). While not all type II mutants studied demonstrated increased lipid transport activity in response to ivacaftor or genistein, these data demonstrate proof of concept for development of pharmacologic strategies to correct ABCA3 function among symptomatic infants and children (Kinting et al., 2019). Quantitative differences in mean vesicle diameter and immunoflourescent co-localization may be useful in pathogenicity screening of uncharacterized ABCA3 variants. Future automation of these measurements could permit high-throughput screening of pharmacologic correctors similar to strategies used to identify compounds that rescue CFTR function (Merkert et al., 2019; Pedemonte et al., 2005; Veit et al., 2018).
Limitations of our study include the use of an immortalized, human pulmonary epithelial cell line (A549) which lacks patient-specific genetic backgrounds and spatial and paracrine cellular topography (Matsumura, Sakai, Sasaki, Ban, & Inagaki, 2007). However, similar ABCA3 protein processing and co-localization in A549 cells and primary AT2 cells suggests biologic relevance of the A549 cell line (Beers et al., 2013; Cheong et al., 2006). In addition, lamellar body phenotypes from patients with ABCA3 deficiency are consistent with our measurements of smaller lamellar body diameter (Citti et al., 2013; Doan et al., 2008; Jackson et al., 2015; Shulenin et al., 2004; Wert, Whitsett, & Nogee, 2009). Finally, quantitative measurements using this A549 cell-based model permit functional characterization of uncharacterized variants and may be a useful model for drug discovery.
Supplementary Material
Supplemental Figure S1: Location of mutants within ABCA3 protein Four ABCA3 mutants identified among infants and children with diverse respiratory phenotypes were selected for functional characterization. Pictured here are the location of the mutants within the ABCA3 protein. Figure modified from Wert SE et al. Genetic Disorders of Surfactant Dysfunction. Pediatr Dev Path. 2009 Jul - Aug;12(4):253-74.
Acknowledgements:
This work was supported by grants from the National Institutes of Health (K08 HL105891 [JAW], R33 HL120760 [FSC], U01 HL134745 [JAW, FSC]), R01DK114047 [GAS, CJL], R01DK104946 [GAS], American Thoracic Society (JAW), from the Children’s Discovery Institute (FSC, CJL), and the Saigh Foundation (FSC). None of the authors have conflicts of interest to declare. The data that support the findings of this study are available from the corresponding author upon reasonable request.
References:
- Adzhubei I, Jordan DM, & Sunyaev SR (2013). Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet, Chapter 7, Unit7 20. doi: 10.1002/0471142905.hg0720s76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers MF, & Mulugeta S (2017). The biology of the ABCA3 lipid transporter in lung health and disease. Cell Tissue Res, 367(3), 481–493. doi: 10.1007/s00441-016-2554-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers MF, Zhao M, Tomer Y, Russo SJ, Zhang P, Gonzales LW, . . . Mulugeta S (2013). Disruption of N-linked glycosylation promotes proteasomal degradation of the human ATP-binding cassette transporter ABCA3. Am J Physiol Lung Cell Mol Physiol, 305(12), L970–980. doi: 10.1152/ajplung.00184.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong N, Madesh M, Gonzales LW, Zhao M, Yu K, Ballard PL, & Shuman H (2006). Functional and trafficking defects in ATP binding cassette A3 mutants associated with respiratory distress syndrome. J Biol Chem, 281(14), 9791–9800. doi: 10.1074/jbc.M507515200 [DOI] [PubMed] [Google Scholar]
- Citti A, Peca D, Petrini S, Cutrera R, Biban P, Haass C, . . . Danhaive O (2013). Ultrastructural characterization of genetic diffuse lung diseases in infants and children: a cohort study and review. Ultrastruct Pathol, 37(5), 356–365. doi: 10.3109/01913123.2013.811454 [DOI] [PubMed] [Google Scholar]
- Denman L (2018). The classification of ATP-binding cassette subfamily A member 3 mutations using the cystic fibrosis transmembrane conductance regulator classification system. Pediatr Invest, 2(1), 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan ML, Guillerman RP, Dishop MK, Nogee LM, Langston C, Mallory GB, . . . Fan LL (2008). Clinical, radiological and pathological features of ABCA3 mutations in children. Thorax, 63(4), 366–373. doi: 10.1136/thx.2007.083766 [DOI] [PubMed] [Google Scholar]
- Eldridge WB, Zhang Q, Faro A, Sweet SC, Eghtesady P, Hamvas A, . . . Wambach JA (2017). Outcomes of Lung Transplantation for Infants and Children with Genetic Disorders of Surfactant Metabolism. J Pediatr, 184, 157–164 e152. doi: 10.1016/j.jpeds.2017.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelbrecht S, Kaltenborn E, Griese M, & Kern S (2010). The surfactant lipid transporter ABCA3 is N-terminally cleaved inside LAMP3-positive vesicles. FEBS Lett, 584(20), 4306–4312. doi: 10.1016/j.febslet.2010.09.026 [DOI] [PubMed] [Google Scholar]
- Flamein F, Riffault L, Muselet-Charlier C, Pernelle J, Feldmann D, Jonard L, . . . Guillot L (2012). Molecular and cellular characteristics of ABCA3 mutations associated with diffuse parenchymal lung diseases in children. Hum Mol Genet, 21(4), 765–775. doi: 10.1093/hmg/ddr508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann N, Galetskiy D, Rauch D, Wittmann T, Marquardt A, Griese M, & Zarbock R (2016). Analysis of the Proteolytic Processing of ABCA3: Identification of Cleavage Site and Involved Proteases. PLoS One, 11(3), e0152594. doi: 10.1371/journal.pone.0152594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppner S, Kinting S, Torrano AA, Schindlbeck U, Brauchle C, Zarbock R, . . . Griese M (2017). Quantification of volume and lipid filling of intracellular vesicles carrying the ABCA3 transporter. Biochim Biophys Acta Mol Cell Res, 1864(12), 2330–2335. doi: 10.1016/j.bbamcr.2017.08.013 [DOI] [PubMed] [Google Scholar]
- Jackson T, Wegner DJ, White FV, Hamvas A, Cole FS, & Wambach JA (2015). Respiratory failure in a term infant with cis and trans mutations in ABCA3. J Perinatol, 35(3), 231–232. doi: 10.1038/jp.2014.236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, F. L, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd N, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, The Genome Aggregation Database Consortium, Neale BM, Daly MJ, MacArthur DG. . (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv. [Google Scholar]
- Katzen J, Wagner BD, Venosa A, Kopp M, Tomer Y, Russo SJ, . . . Beers MF (2019). An SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis. JCI Insight, 4(6). doi: 10.1172/jci.insight.126125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinting S, Hoppner S, Schindlbeck U, Forstner ME, Harfst J, Wittmann T, & Griese M (2018). Functional rescue of misfolding ABCA3 mutations by small molecular correctors. Hum Mol Genet, 27(6), 943–953. doi: 10.1093/hmg/ddy011 [DOI] [PubMed] [Google Scholar]
- Kinting S, Li Y, Forstner M, Delhommel F, Sattler M, & Griese M (2019). Potentiation of ABCA3 lipid transport function by ivacaftor and genistein. J Cell Mol Med, 23(8), 5225–5234. doi: 10.1111/jcmm.14397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroner C, Wittmann T, Reu S, Teusch V, Klemme M, Rauch D, . . . Griese M (2017). Lung disease caused by ABCA3 mutations. Thorax, 72(3), 213–220. doi: 10.1136/thoraxjnl-2016-208649 [DOI] [PubMed] [Google Scholar]
- Lopes-Pacheco M (2016). CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front Pharmacol, 7, 275. doi: 10.3389/fphar.2016.00275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EM, Stap J, Brakenhoff GJ, van Driel R, & Aten JA (1992). Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci, 103 (Pt 3), 857–862. [DOI] [PubMed] [Google Scholar]
- Matsumura Y, Ban N, & Inagaki N (2008). Aberrant catalytic cycle and impaired lipid transport into intracellular vesicles in ABCA3 mutants associated with nonfatal pediatric interstitial lung disease. Am J Physiol Lung Cell Mol Physiol, 295(4), L698–707. doi: 10.1152/ajplung.90352.2008 [DOI] [PubMed] [Google Scholar]
- Matsumura Y, Ban N, Ueda K, & Inagaki N (2006). Characterization and classification of ATP-binding cassette transporter ABCA3 mutants in fatal surfactant deficiency. J Biol Chem, 281(45), 34503–34514. doi: 10.1074/jbc.M600071200 [DOI] [PubMed] [Google Scholar]
- Matsumura Y, Sakai H, Sasaki M, Ban N, & Inagaki N (2007). ABCA3-mediated choline-phospholipids uptake into intracellular vesicles in A549 cells. FEBS Lett, 581(17), 3139–3144. doi: 10.1016/j.febslet.2007.05.078 [DOI] [PubMed] [Google Scholar]
- Merkert S, Schubert M, Olmer R, Engels L, Radetzki S, Veltman M, . . . Martin U (2019). High-Throughput Screening for Modulators of CFTR Activity Based on Genetically Engineered Cystic Fibrosis Disease-Specific iPSCs. Stem Cell Reports, 12(6), 1389–1403. doi: 10.1016/j.stemcr.2019.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulugeta S, Gray JM, Notarfrancesco KL, Gonzales LW, Koval M, Feinstein SI, . . . Shuman H (2002). Identification of LBM180, a lamellar body limiting membrane protein of alveolar type II cells, as the ABC transporter protein ABCA3. J Biol Chem, 277(25), 22147–22155. doi: 10.1074/jbc.M201812200 [DOI] [PubMed] [Google Scholar]
- Ng PC, & Henikoff S (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res, 31(13), 3812–3814. doi: 10.1093/nar/gkg509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nureki SI, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, . . . Beers MF (2018). Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest, 128(9), 4008–4024. doi: 10.1172/JCI99287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, & Verkman AS (2005). Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest, 115(9), 2564–2571. doi: 10.1172/JCI24898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, & Kircher M (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res, 47(D1), D886–D894. doi: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridsdale R, Na CL, Xu Y, Greis KD, & Weaver T (2011). Comparative proteomic analysis of lung lamellar bodies and lysosome-related organelles. PLoS One, 6(1), e16482. doi: 10.1371/journal.pone.0016482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, & Seelow D (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods, 11(4), 361–362. doi: 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, & Dean M (2004). ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med, 350(13), 1296–1303. doi: 10.1056/NEJMoa032178 [DOI] [PubMed] [Google Scholar]
- Tryka AF, Wert SE, Mazursky JE, Arrington RW, & Nogee LM (2000). Absence of lamellar bodies with accumulation of dense bodies characterizes a novel form of congenital surfactant defect. Pediatr Dev Pathol, 3(4), 335–345. [DOI] [PubMed] [Google Scholar]
- van Moorsel CH, van Oosterhout MF, Barlo NP, de Jong PA, van der Vis JJ, Ruven HJ, . . . Grutters JC (2010). Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med, 182(11), 1419–1425. doi: 10.1164/rccm.200906-0953OC [DOI] [PubMed] [Google Scholar]
- Veit G, Xu H, Dreano E, Avramescu RG, Bagdany M, Beitel LK, . . . Lukacs GL (2018). Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat Med, 24(11), 1732–1742. doi: 10.1038/s41591-018-0200-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wambach JA, Casey AM, Fishman MP, Wegner DJ, Wert SE, Cole FS, . . . Nogee LM (2014). Genotype-phenotype correlations for infants and children with ABCA3 deficiency. Am J Respir Crit Care Med, 189(12), 1538–1543. doi: 10.1164/rccm.201402-0342OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wambach JA, Yang P, Wegner DJ, Heins HB, Kaliberova LN, Kaliberov SA, . . . Cole FS (2016). Functional Characterization of ATP-Binding Cassette Transporter A3 Mutations from Infants with Respiratory Distress Syndrome. Am J Respir Cell Mol Biol, 55(5), 716–721. doi: 10.1165/rcmb.2016-0008OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichert N, Kaltenborn E, Hector A, Woischnik M, Schams A, Holzinger A, . . . Griese M (2011). Some ABCA3 mutations elevate ER stress and initiate apoptosis of lung epithelial cells. Respir Res, 12, 4. doi: 10.1186/1465-9921-12-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wert SE, Whitsett JA, & Nogee LM (2009). Genetic disorders of surfactant dysfunction. Pediatr Dev Pathol, 12(4), 253–274. doi: 10.2350/09-01-0586.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitsett JA, Wert SE, & Weaver TE (2015). Diseases of pulmonary surfactant homeostasis. Annu Rev Pathol, 10, 371–393. doi: 10.1146/annurev-pathol-012513-104644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann T, Schindlbeck U, Hoppner S, Kinting S, Frixel S, Kroner C, . . . Griese M (2016). Tools to explore ABCA3 mutations causing interstitial lung disease. Pediatr Pulmonol, 51(12), 1284–1294. doi: 10.1002/ppul.23471 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1: Location of mutants within ABCA3 protein Four ABCA3 mutants identified among infants and children with diverse respiratory phenotypes were selected for functional characterization. Pictured here are the location of the mutants within the ABCA3 protein. Figure modified from Wert SE et al. Genetic Disorders of Surfactant Dysfunction. Pediatr Dev Path. 2009 Jul - Aug;12(4):253-74.