


Abstract
Multiplex single-molecule fluorescent in situ hybridization (smFISH) is a powerful method for validating RNA sequencing and emerging spatial transcriptomic data, but quantification remains a computational challenge. We present a framework for generating and analyzing smFISH data in complex tissues while overcoming autofluorescence and increasing multiplexing capacity. We developed dotdotdot (https://github.com/LieberInstitute/dotdotdot) as a corresponding software package to quantify RNA transcripts in single nuclei and perform differential expression analysis. We first demonstrate robustness of our platform in single mouse neurons by quantifying differential expression of activity-regulated genes. We then quantify spatial gene expression in human dorsolateral prefrontal cortex (DLPFC) using spectral imaging and dotdotdot to mask lipofuscin autofluorescence. We lastly apply machine learning to predict cell types and perform downstream cell type-specific expression analysis. In summary, we provide experimental workflows, imaging acquisition and analytic strategies for quantification and biological interpretation of smFISH data in complex tissues.
INTRODUCTION
In the age of rapidly advancing Next Generation Sequencing technologies, such as single cell RNA-sequencing (RNA-seq) and spatial transcriptomics (1,2), single-molecule fluorescent in situ hybridization (smFISH) has emerged as a potential gold standard for validating and extending findings derived from large scale transcriptomic data. The widespread generation of single-cell RNA-seq data sets in the neurosciences has fueled a resurgence of smFISH approaches to validate cell type-specific molecular profiles by visualizing individual transcripts at cellular resolution (3,4). Information from single-cell RNA-seq data has revealed increasingly complex transcriptomic signatures for functionally distinct cell types, including the recently identified Rosehip neurons in cortical layer one (5) and cells with neurogenic potential in the dentate gyrus of the hippocampus (6,7), such that molecular definition of these cells necessitates combinatorial labeling with multiple probes to confirm both presence and absence of specific transcripts within a spatially-defined context.
While chromogenic and fluorescent in situ hybridization methodologies have been utilized for decades (8,9), recent advances in hybridization/probe technologies, imaging techniques, and data analysis tools have streamlined smFISH assays and improved sensitivity and specificity (10–12). Despite these methodological advances, multiplexing in complex tissues with extensive cellular heterogeneity, such as post-mortem human brain, remains a significant challenge. Studying human brain tissue is also complicated by high levels of autofluorescence due to lipofuscin granules (13,14). Indeed, to avoid confounding signals from lipofuscin, the majority of smFISH investigations in post-mortem human brain have been limited to single or duplex chromogenic approaches (6,7,15–17).
While studies have begun to incorporate multiplex fluorescent approaches in post-mortem human brain tissue, no consistent strategy for eliminating, masking, or subtracting lipofuscin autofluorescence has been described (3,5,18–22). Some studies have characterized lipofuscin autofluorescence based on size and intensity or utilized custom filter cubes (3,4,21), but these reports do not document how these approaches impact quantification of fluorescent signals from probe hybridization. An image processing approach in non-human primate brain tissue using spectral imaging and linear unmixing showed promise for characterizing and removing lipofuscin autofluorescence (23). However, this approach has not yet been validated and widely implemented in post-mortem human brain. Furthermore, the four dimensional data sets acquired using multispectral imaging across a tissue depth add additional computational hurdles for automating image analysis and quantifying single transcripts.
Several microscopy-based methodologies for single cell, spatially resolved transcriptomics have been developed (24–28). However, these highly specialized platforms still rely on the availability and accuracy of algorithms for fluorescence segmentation, and often require sophisticated microscopy equipment and reagents that are not readily available to the majority of laboratories. Commercially available smFISH platforms have the capacity for higher-order multiplexing and can, in theory, be used for differential expression analysis within molecularly and spatially defined cell types. However, the downstream computational tools for analyzing these types of data have lagged behind their widespread use, and those tools that have been developed remain largely inaccessible to most neurobiology labs without strong computational expertise. Hence, the majority of current smFISH applications have been qualitative, rather than quantitative, and have therefore not maximized the utility of these potentially rich imaging datasets.
To address this need, we developed an intuitive and adaptable computational workflow called dotdotdot to quantify individual RNA transcripts at single cell resolution in intact tissues and performed differential expression analysis of smFISH data. We validate the accuracy of dotdotdot for quantifying RNA transcripts in both mouse and post-mortem human brain and use differential expression and machine learning approaches, such as K-means clustering and Classification and Regression Trees (CART), to answer biological questions about gene co-expression and molecular cell type based on quantitative analysis of spatial gene expression. In summary, we present an imaging platform coupled with computational tools for smFISH data that can be readily implemented in most laboratories without need for highly specialized expertise or equipment to elevate spatial analyses of gene expression and complement growing single cell and spatial transcriptomic data sets in the field of neuroscience and beyond.
MATERIALS AND METHODS
Animals and electroconvulsive seizure (ECS) treatment
Six-week old male mice (C57BL/6J) were administered either Sham or ECS treatment as previously described (29,30). Briefly, ECS was delivered with an Ugo Basile pulse generator using a corneal electrode fork placed over the frontal cortex (model #57800-001, shock parameters: 100 pulse/s frequency, 0.3 ms pulse width, 1 s shock duration and 50 mA current). The stimulation parameters were chosen because they reliably induced tonic-clonic convulsions. Mice were administered inhaled isoflurane anesthesia prior to ECS sessions, and remained anesthetized for the procedure. Each mouse received a single session of Sham or ECS and was euthanized 90 min following the treatment. All animal experiments were approved by the SoBran Institutional Animal Care and Use Committee.
Post-mortem human tissue samples
Post-mortem human brain tissue from two donors (both male: one 17 years old of African American ancestry and the other 25 years old with European ancestry) was obtained by autopsy primarily from the Offices of the Chief Medical Examiner of the District of Columbia, and of the Commonwealth of Virginia, Northern District, all with informed consent from the legal next of kin (protocol 90-M-0142 approved by the NIMH/NIH Institutional Review Board). Clinical characterization, diagnoses, and macro- and microscopic neuropathological examinations were performed on all samples using a standardized paradigm, and subjects with evidence of macro- or microscopic neuropathology were excluded. Details of tissue acquisition, handling, processing, dissection, clinical characterization, diagnoses, neuropathological examinations, RNA extraction and quality control measures have been described previously (31).
3-Plex smFISH and image acquisition in mouse tissue
Mice (n = 3 Sham and n = 4 ECS-treated) were cervically dislocated and brains were removed from the skull, flash frozen in isopentane, and stored at −80°C. Brain tissue was equilibrated to −20°C in a cryostat (Leica, Wetzlar, Germany) and serial sections of piriform cortex were collected at 16 μm (four sections per slide). Sections were stored at −80°C until completion of the smFISH assay. For mouse studies (Figure 1A), in situ hybridization assays were performed with RNAscope technology utilizing the RNAscope Fluorescent Multiplex Kit V1 (Cat # 320850 Advanced Cell Diagnostics [ACD], Hayward, CA, USA) according to manufacturer's instructions as previously described (32). Briefly, tissue sections were fixed with a 10% neutral buffered formalin solution (Cat # HT501128 Sigma-Aldrich, St. Louis, MO, USA) for 20 min at room temperature (RT), series dehydrated with ethanol, and pretreated with protease IV for 20 min. Sections were incubated with a custom-designed probe for Bdnf exon IV (Cat # 482981-C3, ACD) and commercially available probes for Bdnf exon I and Arc (Cat #457321-C2 and #316911, ACD, Hayward, CA, USA). Probes were fluorescently labeled with orange (excitation 550 nm), green (excitation 488 nm), or far red (excitation 647) fluorophores using the Amp 4 Alt B-FL and stained with DAPI (4′,6-diamidino-2-phenylindole) to demarcate the nucleus. Confocal images were acquired in z-series using a Zeiss LSM700 confocal microscope. For each mouse (biological replicate), two images were randomly captured in the piriform cortex per section (four sections; eight images total).
Figure 1.

Experimental workflows and imaging protocols for smFISH in mouse and human tissues. (A) Brains were extracted from wild-type (WT) mice 90 min following electroconvulsive seizures (ECS) or Sham treatment and sectioned on a cryostat. Gene targets were visualized with the RNAscope Multiplex Fluorescence V1 kit. RNAscope technology uses hybridization of two independent probes (double Z probes), referred to as a ‘ZZ pair,’ that must bind to the target sequence in tandem for signal amplification to proceed via the subsequent binding of preamplifiers, amplifiers, and fluorescent detection molecules. Approximately 5–30 ZZ pairs are designed for each target gene. After completion of the RNAscope V1 assay, slides are imaged in x, y and z-dimensions using confocal microscopy. (B) Fresh frozen post-mortem human tissue was sectioned on a cryostat and gene targets were visualized using the RNAscope Multiplex Fluorescence V2 kit. The V2 assay uses the same RNAscope technology with added TSA technology for customization of dyes/concentrations and the ability to include a fourth gene target. The V2 assay is also better suited for tissues with autofluorescence, such as post-mortem human brain tissue, which contains an abundance of highly autofluorescent lipofuscin granules. Multispectral imaging and linear unmixing were used to separate individual probe signals and lipofuscin autofluorescence. Lipofuscin signals served as a mask during downstream analysis to exclude pixels confounded by autofluorescence.
4-Plex smFISH and image acquisition in post-mortem human tissue
Two blocks of fresh frozen dorsolateral prefrontal cortex (DLPFC) from neurotypical control individuals ages 24 and 17 were sectioned at 10μm and stored at −80°C. RNA integrity numbers (RINS) were 8.4 and 8.8, respectively. For post-mortem human studies (Figure 1B), in situ hybridization assays were performed with RNAscope technology utilizing the RNAscope Fluorescent Multiplex Kit V2 and 4-plex Ancillary Kit (Cat # 323100, 323120 ACD, Hayward, CA, USA) according to manufacturer's instructions. Briefly, tissue sections were fixed with a 10% neutral buffered formalin solution (Cat # HT501128 Sigma-Aldrich, St. Louis, MO, USA) for 30 min at RT, series dehydrated in ethanol, pretreated with hydrogen peroxide for 10 min at RT, and treated with protease IV for 30 min. Sections were incubated with probes for SNAP25, SLC17A7, GAD1, and MBP (Cat #518851, 415611-C2, 573061-C3, 573051-C4, ACD, Hayward, CA, USA) and stored overnight in 4x SSC (saline-sodium citrate) buffer. Probes were fluorescently labeled with Opal Dyes (Perkin Elmer, Waltham, MA; Opal690 diluted at 1:1000 and assigned to SNAP25; Opal570 diluted at 1:1500 and assigned to SLC17A7; Opal620 diluted at 1:500 and assigned to GAD1; Opal520 diluted at 1:1500 and assigned to MBP) and stained with DAPI (4′,6-diamidino-2-phenylindole) to label the nucleus.
For experiments with SNX19, sections were incubated with probes for SLC17A7, GAD1 and SNX19 (Cat #415611-C3, Cat #404031, Cat #518861-C2, ACD, Hayward, CA) and stored overnight in 4× SSC buffer. Probes were fluorescently labeled with Opal Dyes (Opal690 diluted at 1:1500 and assigned to SLC17A7; Opal570 diluted at 1:500 and assigned to SNX19; Opal520 diluted at 1:1000 and assigned to GAD1) and stained with DAPI to label the nucleus
Lambda stacks were acquired in z-series using a Zeiss LSM780 confocal microscope equipped with 20× 1.4 NA and 63× 1.4NA objectives, a GaAsP spectral detector, and 405, 488, 555 and 647 lasers. All lambda stacks were acquired with the same imaging settings and laser power intensities. For each subject, two cortical strips were tile imaged at 20× to capture layers I to VI (Figure 5). Layer II/II and layer VI were identified by measuring 20–30% and 80–90% of the cortical layer thickness, respectively. This strategy reliability delineated layer II/III and VI across 10 individuals and cortical strips with varying absolute thicknesses. After demarcation of cortical layers, the positions feature in Zen software was used to randomly select six fields per layer per strip (n = 12 layer II/III and n = 12 layer VI in two different cortical strips per subject) for high magnification imaging at 63×. Following image acquisition, lambda stacks in z-series were linearly unmixed in Zen software (weighted; no autoscale) using reference emission spectral profiles previously created in Zen (see below) and saved as Carl Zeiss Image ‘.czi’ files.
Figure 5.

Layer-specific cell type analysis in post-mortem human DLPFC demonstrates sensitivity and specificity of dotdotdot for transcript quantification. (A–G) Maximum z-projection of unmixed and stitched lambda stacks acquired across a single cortical strip (layer I to VI) of post-mortem human DLPFC depicting expression of DAPI (A), SNAP25 (B), SLC17A7 (C), GAD1 (D), MBP (E), lipofuscin (F) and merged (G). High magnification (63×) images used for analysis were randomly acquired within a strip in layer II/III (80–90% of strip thickness; n = 24 images total) or layer VI (20–30% of strip thickness; n = 24 images total) from 2 individuals from 2 different strips (see Figure 3 for representative 63x image). (H) Validation of cell type calling using CART analysis demonstrating an enrichment of SNAP25 transcripts in predicted excitatory (SLC17A7) and inhibitory (GAD1) neurons compared to oligodendrocytes (MBP) and SLC17A7-/GAD1-/MBP- cells (likely astrocytes and microglia). Color represents different subjects and shape represents different layers (circle = layer II/III and triangle = layer VI). (I) Proportion of cortical cells expressing markers for neurons, excitatory neurons, inhibitory neurons, and oligodendrocytes in layer II/III versus layer VI of DLPFC (n = 519 ROIs layer II/III and n = 442 ROIs layer VI). Scale bar is 200 um.
Generation of reference emission spectral profiles
Reference emission spectral profiles, or ‘fingerprints,’ were created for each Opal dye in Zen software. Briefly, four single positive slides were generated in mouse tissue using the RNAscope Fluorescent Multiplex Kit V2 and 4-plex Ancillary Kit (Cat # 323100, 323120 ACD, Hayward, California) and a control probe against the housekeeping gene POLR2A according to manufacturer's instructions as described above (Supplementary Figure S6). Mouse tissue was used in place of human tissue due to lower tissue autofluorescence (i.e. the absence of confounding lipofuscin signals). For each single positive slide, POLR2A was labeled with either Opal520, Opal570, Opal620 or Opal690 dye. A single positive slide was generated for DAPI using the same pretreatment conditions, but omission of probe hybridization steps. To generate a reference emission spectral profile for lipofuscin autofluorescence, a negative control slide was generated in post-mortem DLPFC tissue using a 4-plex negative control probe against 4 bacterial genes (Cat #321831, ACD, Hayward, CA) in which all Opal dyes were applied, but no probe signal was amplified.
Automated imaging analysis
dotdotdot is a MATLAB-based command line toolbox for automated nuclei and transcript segmentation and quantification. Confocal images are processed in MATLAB, but downstream data analyses can be performed in R (as done here), MATLAB, python or any statistical software. Briefly, the processing pipeline involves smoothing/filtering raw images, thresholding, watershed segmentation, autofluorescence masking and extracting dot metrics. The analysis pipeline involves k-means clustering for classifying nuclei expression (low, medium and high) and CART (Classification and Regression Trees) for classifying cell types (astrocytes, oligodendrocytes, GABAergic or glutamatergic neurons). Bio-formats toolbox ‘bfmatlab’ is used to read the image data into a MATLAB structure with fields containing gene data, DAPI and lipofuscin. Processing techniques for human nuclei, mouse nuclei and transcript channels are different, as described below.
Mouse nuclei segmentation
Processing and segmentation of mouse nuclei is performed using the MATLAB toolbox called ‘CellSegm’ (33). The toolbox provides the user with several input options for smoothing (coherence enhancing diffusion, edge enhancing diffusion, gaussian) and thresholding (iterative thresholding, adaptive thresholding, gradient thresholding, ridge enhancement). A 2D planewise gaussian smoothing with default settings (filter size = [5 5], standard deviation = 2) was used to filter the raw images (X = Y = 512 pixels) (Supplementary Figure S1.A1). Adaptive thresholding with an average filter (size = [42 42]) followed by several morphological operations (like imopen, imerode, imfill) were performed on the gaussian smoothed images (Supplementary Figure S1.B1) to obtain the binary image (Supplementary Figure S1.C1). The irregularly large objects in the binary image are then split into smaller segments using watershed segmentation based on local maxima and the euclidean distances (Supplementary Figure S1.D1).
Human nuclei segmentation
A 3D median filter (size = [19 19 3]) is used to smooth (Supplementary Figure S2.B1) the intensity irregularities in the raw image (X = Y = 1024 pixels) (Supplementary Figure S2.A1) that are produced from heterogeneously-stained nuclei. An intensity threshold from the image histogram is then used to segment the DAPI stained nuclei from the background. A technique called ‘minima imposition’ is applied to the binary image (Supplementary Figure S2.C1) before watershed transform to filter the tiny local minima that might cause over-splitting of large segmented nuclei blobs. A modified distance transform of the binary image is then computed for the watershed segmentation on the maximum z projection (Supplementary Figure S2.D1).
Transcript segmentation and lipofuscin masking
Background noise (i.e. potential bleed-through from adjacent wavelengths) in the gene channels is eliminated using the function ‘imhmin’ (34). Here all the minima in the grayscale image whose depth is less than the standard deviation of the image is suppressed (Supplementary Figures S1.B2, S2.B2). A histogram-based intensity threshold is used to segment the RNA signal (Supplementary Figures S1.C2, S2.C2). Watershed segmentation based on the minima of the image is then performed to split the detected pixel clusters in each channel into identified transcripts (Supplementary Figures S1.D2, S2.D2). Lipofuscin segmentation is similar, except it does not include the background suppression step (Supplementary Figure S2.Lipofuscin channel).
Extract nuclei and transcript metrics
Custom MATLAB functions (regionprops3 function in Image Processing toolbox) were then used to calculate relevant metrics (count, size, location, intensity) of detected nuclei and transcripts (Supplementary Figures S1.E(1,2), S2.E(1,2)). For human data, before transcript quantification, the segmented RNA channels (Supplementary Figure S2.D2) are masked (Supplementary Figure S2.E3) with the segmented lipofuscin channel (Supplementary Figure S2.C3). Nuclei and transcript colocalization data (Supplementary Figure S1.Data analysis.1, Supplementary Figure S2.Data analysis.1) are then obtained by assigning each transcript to a cell based on its position in three dimensions. For gene expression analysis (mouse data) a dot is assigned to a nucleus if its center falls within the boundary of the nucleus and for cell type classification (human data) each segmented gene pixel is considered as a transcript and is assigned to nucleus if it is within the boundary of the nucleus.
Downstream data analysis
(a) Gene expression analysis (Supplementary Figure S1.F): all the nuclei are clustered into low, medium and high expressers for each gene type by the k-means clustering method (Supplementary Figure S1.F.2) based on the total transcript count and the average transcript size per nuclei. The choice of k=3 here was biologically motivated, but can be a flexible parameter dependent on the underlying study design and research question. Nuclei with at least one transcript are recruited for k-means clustering, and the nuclei with zero transcripts are explicitly labeled as low expressers. (b) Cell type classification (Supplementary Figure S2.F): the proportion of each type of transcript in individual nuclei is used by Classification and Regression Trees (CART) to predict the underlying cell type (Supplementary Figure S2.F.3). The initial CART model was built on a test and train dataset created from 89 manually annotated nuclei (60 random ROIs were used to train the model and the rest were used to test) from five random images (Supplementary Figure S2.F.2) from the whole dataset. The predictions from this model were used to classify the rest of the data into predefined categories. This strategy can be used to develop other analogous classification models for other cell and tissue types.
Statistics
All statistical analyses were performed in R. For mouse data analyses, we used linear mixed effects modeling with the lmerTest package to analyze nuclei size, total number of nuclei in an image, gene expression (Bdnf Ex1, Bdnf Ex4, Arc) differences for each group (low, medium and high) as a function of the treatment main effect (Sham versus ECS) and three nested groups as random intercepts: animal ID, brain section and the image/scan ID. For human data analysis, we also used linear mixed effects modeling for the analysis of nuclei size, total number of nuclei in image, proportions of cell types in an image as a function of brain layer (layer II/III versus layer VI) as a main effect and three nested random intercepts of: brain donor, strip number, and the image ID. For the validation of SNAP25 enrichment in GAD1 and SLC17A7 positive cells, GAD1 and SLC17A7 cells were combined into one group and compared to the MBP and negative labelled cells. The analysis was performed using the linear mixed effects model with main effects as the predicted cell labels and random intercepts being the same variables as above.
RESULTS
The dotdotdot framework for image acquisition and data analysis
We first introduce the dotdotdot framework, which involves (i) image acquisition using confocal microscopy or spectral imaging/linear unmixing, (ii) image processing to extract nuclei/regions of interest (ROIs) and quantitative transcript abundances and (iii) transcript colocalization analysis to classify cell types and (iv) differential expression analysis. To localize and quantify single transcripts in individual nuclei, we developed parallel gene-labeling, fluorescence microscopy, and image analysis workflows for mouse and human brain tissues. The general workflows for mouse (Figure 1A) and human (Figure 1B) tissues are similar, but they include optimized conditions for sample preparation (i.e. section thickness, fixation, protease treatment), smFISH labeling (i.e. V1 versus V2 RNAscope Multiplex Fluorescence Technology, number of gene targets, fluorophores), fluorescent imaging (i.e. confocal microscopy vs. multispectral imaging/linear unmixing), and image analysis (segmentation, dot/transcript detection). Differences in image processing and data analysis workflows in mouse (Supplementary Figure S1) and human (Supplementary Figure S2) tissues arose from the need to address a challenge specifically associated with fluorescent imaging in post-mortem human brain tissue—lipofuscin autofluorescence. In addition, because post-mortem human brain tissue is a highly limited resource, we sought to maximize multiplexing capabilities by utilizing V2 4-plex RNAscope technology, which allowed for visualization of an additional gene target compared to the V1 3-plex technology used for mouse tissues. For the human workflow (Figure 1B), we used four different fluorophores (Opal520, Opal570, Opal620 and Opal690) to label four distinct gene targets. Importantly, the number associated with each Opal dye corresponds to its maximum emission wavelength ([520 nm] green, [570 nm] orange, [620 nm] red, [690 nm] far red, respectively). Following image acquisition, raw fluorescent data are processed in MATLAB using dotdotdot. This toolbox and example vignettes are available at: https://github.com/LieberInstitute/dotdotdot. We demonstrate the utility of this framework (Figure 2) and software using several experimental examples across diverse applications in mouse and human tissues.
Figure 2.
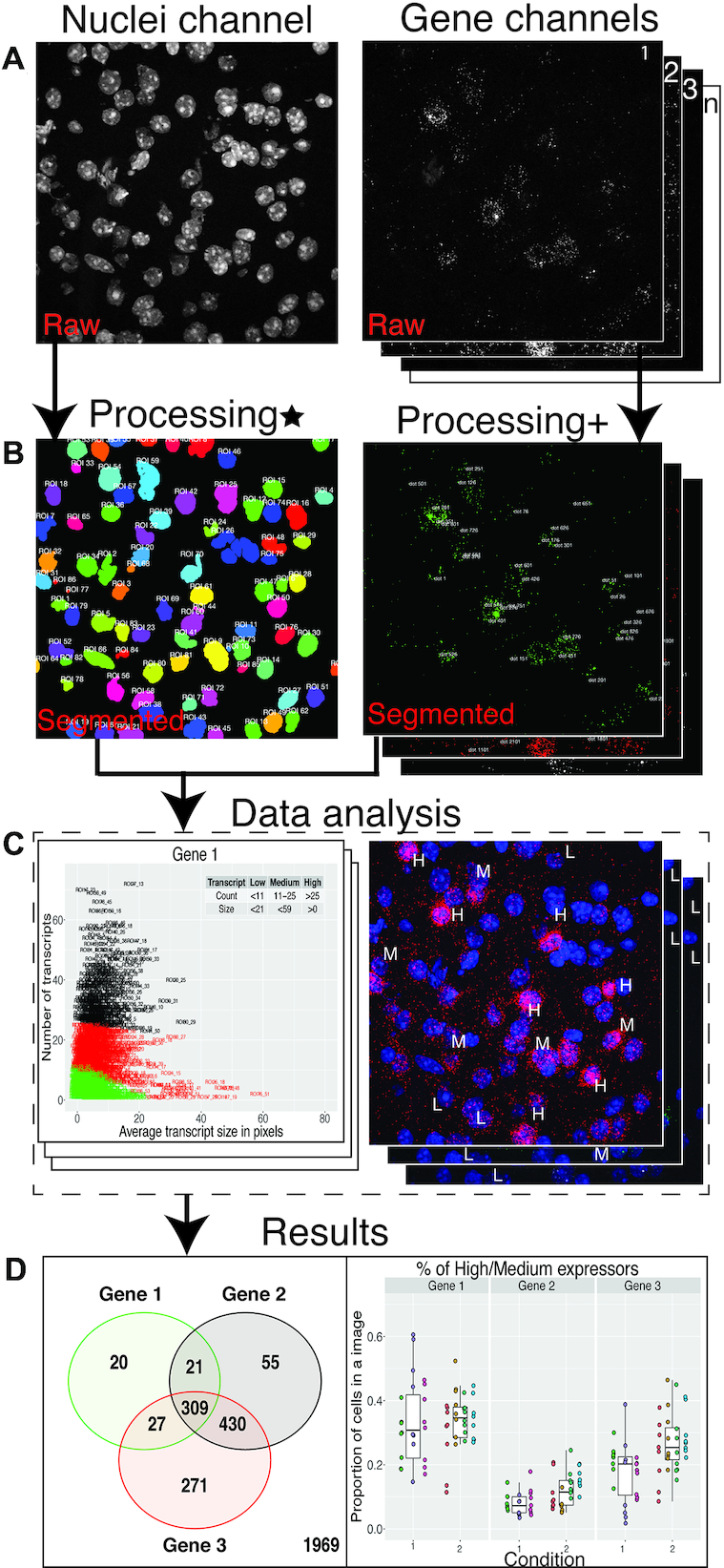
Dotdotdot image processing and data analysis workflow: (A) Raw ‘.czi’ images of nuclei and gene channels. (B) Final segmented images of nuclei and gene channels (Processing* involves gaussian smoothening, adaptive thresholding and watershed segmentation of nuclei channel, Processing+ involves background filtering, histogram-based thresholding and watershed segmentation of transcript channel). (C) Data analysis steps (for example, K-means) that are executed based on metrics from segmented images. (D) Predictions from data analysis steps are used to produce final results.
dotdotdot quantifies dynamics of two activity-regulated genes at cellular resolution following induction of widespread neural activity
We first conducted a proof-of-concept experiment in mouse tissue examining the expression of two well-established activity-regulated genes in piriform cortex following brain stimulation to establish and validate the robustness of dotdotdot for quantifying smFISH data acquired with RNAscope technology. Mice were administered electroconvulsive seizures (ECS) to induce widespread neural activity and subsequent upregulation of activity-regulated genes (35). After 90 min, brains from ECS- and Sham-treated animals were collected, snap frozen, and processed through our smFISH workflow for mouse tissue to visualize individual transcripts for activity regulated cytoskeleton associated protein (Arc) and brain-derived neurotrophic factor (Bdnf) splice variants (Figure 1A). Bdnf transcription is initiated from one of nine promoters upstream of individual 5′-untranslated regions that are spliced to a common coding exon (36,37). We previously designed and validated RNAscope probes for Bdnf splice variants containing untranslated exons 1 (Ex1) and 4 (Ex4), which are strongly induced by neural activity (32,38,39). As expected, confocal images show a qualitative upregulation of Arc, Bdnf Ex1 and Bdnf Ex4 transcripts following induction of neural activity (Figure 3A and B).
Figure 3.

Image analysis with dotdotdot captures differential expression of two activity-regulated gene (ARG) transcripts in single cells of mouse cortex following activity induction. (A) Maximum intensity confocal projections of piriform cortex depicting expression of transcripts for Arc (A), Bdnf Ex1, (A’) Bdnf Ex4 (A’'), and merged (A’'') from a mouse receiving Sham treatment. (B) Maximum intensity confocal projections of piriform cortex depicting expression of transcripts for Arc (B), Bdnf Ex1, (B’) Bdnf Ex4 (B’'), and merged (B’'') 90 min after an acute ECS treatment. (C–E) Proportion of cortical cells expressing low, medium and high levels of Arc (C), Bdnf Ex1 (D) and Bdnf Ex4 (E) in Sham vs. ECS treatment (n = 3 mice, 24 images, 2543 nuclei and n = 4 mice, 32 images and 3102 nuclei, respectively). (F) Proportion of cortical cells expressing medium/high levels of Arc, Bdnf Ex1, Bdnf Ex4 following ECS treatment. (G) Venn diagrams showing co-expression of different ARGs in high/medium expressers following Sham or ECS (n = 867 high/medium ROIs out of 2543 total ROIs with n = 1676 low expressers excluded for Sham, n = 1133 high/medium ROIs out of 3102 total ROIs for ECS with n = 1969 low expressers). Yellow arrows highlight cells preferentially expressing Bdnf Ex4 compared to Bdnf Ex1. Cyan arrows highlight cells expressing Arc, but not Bdnf. Pink arrows highlight cells enriched in Bdnf Ex1, which often co-express Bdnf Ex4 and Arc. Scale bar is 20um.
To quantitatively analyze increases in these activity-regulated gene transcripts (n = 3 Sham mice, 24 images, 2543 nuclei; n = 4 ECS mice, 32 images and 3102 nuclei), dotdotdot first uses nuclear segmentation in x, y and z-dimensions to define nuclei regions of interest (ROIs) based on DAPI staining (Supplementary Figures S1, S3A–C). Here, we focus on nuclear mRNA expression due to the ambiguity of defining cell boundaries in brain tissue based solely on RNA expression. While RNA expression is lower in the nucleus than the cytoplasm, several studies have shown that these measures are highly correlated (40,41). As expected, quantification of nuclei/ROI number and size reveals similar metrics between Sham and ECS images (nuclei size: P = 0.7, total nuclei: P = 0.47; Supplementary Figure S3D-E), demonstrating accurate and effective automated three-dimensional nuclear segmentation. After defining ROIs, dotdotdot next performs transcript segmentation for each gene in x, y, and z-dimensions (Supplementary Figures S1, S4A–L). Metrics such as dot location, size, fluorescence intensity and total number are extracted per image for each transcript channel. Using dot number metrics, analysis of total, nuclear (co-localizing with DAPI), and non-nuclear (not co-localizing with DAPI) Arc, Bdnf Ex1, and Bdnf Ex4 transcripts per image reveals increases in these activity-regulated genes following electroconvulsive seizures (Supplementary Figure S4M-O). Quantification showed significant increases in Bdnf Ex1 and Ex4 transcripts in both nuclear and non-nuclear (cytoplasmic) compartments (Bdnf Ex1 nuc: P = 5.05e–4, cyt: P = 9.53e–4; Bdnf Ex4 nuc: P = 1.63e–4, cyt: P = 0.022) following ECS administration. Interestingly, we see specific increases in cytoplasmic, but not nuclear, Arc transcripts following activity induction, which is consistent with the rapid transportation of newly synthesized Arc mRNA to dendrites and recently activated synapses (42–44) (nuc: P = 0.782, cyt: P = 0.0658).
While increases in these activity-regulated genes following induction of widespread neural activity have been appreciated for decades, two questions have remained outstanding in the field. First, are global increases in activity-induced gene transcription mediated by small increases in transcript abundance across many cells or large increases in transcript abundance in a sub-population of ‘expresser’ cells? Second, do individual neurons differentially express and utilize distinct Bdnf splice variants as their source of activity-dependent BDNF? Definitively answering these questions requires quantifying transcript levels at single cell resolution. Rather than arbitrarily thresholding signal from each gene, we instead performed k-means clustering using both dot count and size metrics. We selected 3 groups/clusters a priori and the algorithm used the underlying dot-level metrics to classify each nucleus into one of three groups, corresponding approximately to low, medium, and high expressers for each activity-regulated gene (Supplementary Figure S5). Expression cutoffs for each gene were therefore based on minimizing the distance between dot-level metrics between all ROIs in the same cluster, and maximizing distance between ROIs in different clusters. For example, Bdnf Ex1 low expressers had <11 dots (transcripts) with an average dot size of less than 17 pixels, while Bdnf Ex1 high expressers had >25 dots. We then examined the proportion of low, medium, and high expressers between Sham and electroconvulsive seizure treatment (Figure 3C-E) using linear mixed effects modeling to account for high similarity between ROIs measured in the same image from the same tissue sections and animals. For both activity-regulated genes, we saw shifts in the proportions of low to medium and high expressers following activity-induction. This was especially true for Bdnf Ex 4 (low: P = 0.00717, medium P = 0.0135, high P = 0.00984, Figure 3E). Pooling the proportion of high and medium expressers further demonstrated increased Bdnf transcription in single cells following electroconvulsive seizures (Figure 3F, Bdnf Ex1: P = 0.0122, Bdnf Ex4 P = 0.00717). Surprisingly, co-expression analysis of these activity-regulated genes in high and medium expressers following Sham or ECS showed that Arc and Bdnf splice variants are differentially expressed in single cells at baseline and following activity (Figure 3G). While there is extensive overlap among ROIs expressing Arc, Bdnf Ex1 and Bdnf Ex4, there are several ROIs that express only one or both transcripts suggesting that these activity-regulated genes can be dynamically regulated in single cells. These data validate the robustness of dotdotdot for transcript segmentation and quantification, and illustrate its utility to provide novel biological insights by analyzing data at cellular resolution.
Visualization and quantification of single transcripts in post-mortem human brain tissue using spectral imaging, linear unmixing and dotdotdot
Extensive efforts are underway to more fully characterize the human brain transcriptome within and across cell types to better understand changes in RNA expression associated with brain development and aging, developmental or psychiatric brain disorders, and local genetic variation. Many of these studies incorporate smFISH in post-mortem human brain to validate RNA-seq findings. However, post-mortem human brain tissue contains abundant lipofuscin, a highly autofluorescent product of lysosomal digestion that confounds quantification of smFISH signals (13,14,23). To address this problem, we employed multispectral imaging and linear unmixing to isolate and exclude lipofuscin autofluorescence from analysis (Figure 4A–C). In addition to allowing for isolation of lipofuscin autofluorescence, this strategy also allows precise separation of spectrally overlapping fluorophores (i.e. orange [Opal570] and red [Opal620]), which is necessary for utilizing 4-plex technology.
Figure 4.

Visualization and quantification of single transcripts in post-mortem human brain tissue using spectral imaging/linear unmixing and dotdotdot. (A) Matrix of raw confocal images acquired during spectral imaging in z-series of a single field of post-mortem human cortex at 63x magnification. A lambda stack is captured at each z-plane detecting expression of SNAP25, SLC17A7, GAD1, MBP (labeled using Opal690 [emission maximum at 690nm], Opal570 [emission maximum at 570 nm], Opal620 [emission maximum at 620 nm], and Opal 520 [emission maximum at 520 nm] dyes, respectively), and lipofuscin autofluorescence. (B) Representative lambda stack depicting a single z-plane acquired at different wavelength bands, each spanning a limited spectral region (∼8.7 nm). (C) Combined emission signals across the lambda stack in each z-plane are linearly unmixed using reference emission spectral profiles from each Opal dye and lipofuscin to separate the contribution of individual fluorescent gene probes. Unmixed data is then projected across the z-axis. Single transcripts for SNAP25 (C), SLC17A7 (C’), GAD1 (C’’) and MBP (C''') (canonical markers for neurons, excitatory neurons, inhibitory neurons, and oligodendrocytes, respectively) can be separated from each other and from lipofuscin autofluorescence (C’'''). (D) Segmentation of unmixed fluorescent signals using dotdotdot. (E) Nuclear segmentation overlaid with transcript segmentation. (F) Masking with lipofuscin signal removes pixels confounded by autofluorescence from analysis. Green arrows highlight MBP+/SNAP25-/SLC17A7-/GAD1- oligodendrocytes. Red arrows highlight MBP-/SNAP25+/SLC17A7+/GAD1- excitatory neurons. Pink arrows highlight MBP-/SNAP25+/SLC17A7-/GAD1+ inhibitory neurons. Cyan double arrows highlight lipofuscin. Scale bar is 20um.
To validate spectral imaging and linear unmixing parameters in post-mortem human brain, we used probes targeting canonical cell type markers in dorsolateral prefrontal cortex (DLPFC), including synaptosome associated protein 5 (SNAP25), solute carrier family 17 member 7 (SLC17A7), glutamate decarboxylase 1 (GAD1) and myelin basic protein (MBP), which identify neurons, excitatory neurons, inhibitory neurons, and oligodendrocytes, respectively (3) (Figure 4). For fluorescence visualization, we assigned Opal690 to SNAP25, Opal570 to SLC17A7, Opal620 to GAD1 and Opal520 to MBP and co-labeled samples with DAPI (maximum emission wavelength at 461 nm). We then performed spectral imaging across multiple z planes to generate a matrix of mixed fluorescent signals spanning the tissue depth (Figure 4A). For a given z-plane, we captured a spectral image stack, or a lambda stack, which is a collection of images of the same field of view (x, y) captured at different wavelengths (Figure 4B). This four-dimensional matrix of mixed fluorescent signals (x, y, z, lambda; Figure 4A) was decoded, or ‘unmixed,’ (Figure 4C) using a linear unmixing algorithm in Zen software, which separates signals from individual probes and lipofuscin autofluorescence using reference emission spectral profiles, or emission ‘fingerprints,’ for each fluorophore (Supplementary Figure S6) and lipofuscin (Supplementary Figure S7). A single Opal dye, regardless of its degree of spectral overlap with other Opal dyes and DAPI, has a unique spectral signature that can be cataloged and used to assign the spatial contribution of that fluorophore to individual pixels in a lambda stack during linear unmixing.
Given that reference emission spectral profiles are critical for accurate unmixing, we carefully generated and validated fingerprints for DAPI (Supplementary Figure S6A), Opal520 (Supplementary Figure S6B), Opal570, (Supplementary Figure S6C), Opal620 (Supplementary Figure S6D), and Opal690 (Supplementary Figure S6E) in mouse tissue, which lacks lipofuscin autofluorescence. Fingerprints were created in Zen software for each of the Opal fluorophores using a series of 4 ‘single positive’ slides of mouse brain tissue hybridized with a positive control probe against the ‘house-keeping’ gene, POLR2A. For the DAPI fingerprint, mouse brain tissue was subjected to pretreatment conditions, but no additional probe labeling before incubation with DAPI. Linear unmixing of single positive slides with all fingerprints (DAPI, Opal520, Opal570, Opal620 and Opal690) verified that reference emission spectral profiles are highly specific for the targeted fluorophore. For example, when POLR2A is labeled with the Opal570 fluorophore (Supplementary Figure S6C), unmixing with the Opal570 fingerprint captures Opal570 fluorescence, while no fluorescent signals are captured with other fingerprints, including the spectrally overlapping Opal620 fingerprint. Transcript segmentation with dotdotdot similarly captures fluorescent signals in the appropriate spectral range for each single positive slide. Quantification of dot count, intensity, and size further demonstrates the specificity of reference emission spectral profiles used for linear unmixing of lambda stacks (Supplementary Figure S6).
In addition to validating robust emission fingerprints for DAPI and each Opal dye, we also generated and validated a spectral signature for lipofuscin autofluorescence in post-mortem human dorsolateral prefrontal cortex (Supplementary Figure S7A, B). Here, we hybridized tissue from a representative subject with a negative control probe against the bacterial gene dapB. As there was no probe binding, fluorescent signals were attributed exclusively to lipofuscin autofluorescence, and a lipofuscin fingerprint was created in Zen software. Using fingerprints for DAPI, Opal520, Opal570, Opal690 and lipofuscin, we performed linear unmixing of eight lambda stacks acquired from negative control slides from four different subjects (Supplementary Figure S7C). The lipofuscin fingerprint was equally effective in detecting lipofuscin autofluorescence across subjects. For all images, segregated lipofuscin signals were used to successfully mask and excludes pixels confounded by autofluorescence across the electromagnetic spectrum (Supplementary Figure S7C).
After linear unmixing with our validated reference emission spectra, we qualitatively observed co-localization of SLC17A7 (excitatory neurons) and GAD1 (inhibitory neurons) with SNAP25 (pan-neuronal) as expected. Similarly, we saw no overlap of MBP (oligodendrocytes) with SNAP25 (neurons), and exclusive expression of either SLC17A7 (excitatory) or GAD1 (inhibitory) in SNAP25+ neurons (Figure 4C). Nuclear and transcript segmentation with dodotdot faithfully represented fluorescence signals (Figure 4D-E), and masking with lipofuscin signals removed background autofluorescence (Figure 4F). For quantitative analysis of unmixed spectral data using dotdotdot, we acquired a set of images at 63× magnification in layers II/III and VI of post-mortem dorsolateral prefrontal cortex (n = 2 subjects, n = 2 cortical strips per subject, n = 6 images per layer per strip, Figures 4C, 5A–G). Given differences in the density and arrangement of nuclei in mouse and human tissue, we first optimized nuclear segmentation for human tissue (Supplementary Figure S8A–C), and demonstrated successful identification of nuclei/ROIs. We observed a similar number and size of nuclei in layer II/III versus layer VI (Supplementary Figure S8D-E, nuclei number P = 0.376, nuclei size P = 0.592).
Following transcript segmentation and lipofuscin masking with dotdotdot, we quantified the number, size, and fluorescence intensity of SNAP25, MBP, SLC17A7 and GAD1 dots and used these data to predict the cell type of each nucleus/ROI using a classification and regression tree (CART) model (Supplementary Figure S9). Given the close spatial proximity of nuclei/ROIs that may represent different cell types (i.e. MBP+ oligodendrocytes situated next to SLC17A7+ neurons), this type of model accommodates potentially conflicting signals (i.e. cytoplasm of SLC17A7+ neuron overlapping the nuclear ROI of MBP+ oligodendrocyte) allowing accurate cell type calling. We gave the model 60 random nuclei/ROIs from 11 manually annotated images as training data and built a classification tree for defining SLC17A7+, MBP+, GAD1+ or triple negative ROIs (Supplementary Figure S9A). The confusion matrix for all 201 manually annotated ROIs demonstrates high accuracy between predicted and actual (manual) cell type calling (182 correctly predicted out of 201; Supplementary Figure S9B). For example, the classifier identifies 31 MBP+ ROIs out of 35 actual MBP+ ROIs. Of these 31 identified MBP+ ROIs, 30 are correctly predicted and 1 is mislabeled as negative. Plotting the manual and predicted cell type for each ROI against the proportion of the ROI positive for GAD1, SLC17A7, and MBP demonstrates the high sensitivity, specificity, and precision of the classifier, which has a 90.5% accuracy (Supplementary Figure S9C). Furthermore, for the 961 ROIs identified in layer II/III and layer VI, we confirmed that cells predicted to be excitatory and inhibitory neurons (SLC17A7+ and GAD1+, respectively) were highly enriched for the pan-neuronal marker SNAP25 (SLC17A7 and GAD1 versus Neg, P = <2e–16; SLC17A7 and GAD1 versus MBP, P = <2e–16, Figure 5H). Consistent with a predicted glial cell type, ROIs classified as oligodendrocytes (MBP+) or negative for MBP, SLC17A7 and GAD1 (likely astrocyte and microglia populations) showed low or zero levels of SNAP25. Further supporting the accuracy of transcript quantification with dotdotdot and subsequent cell type calling with CART analysis, we observed a higher proportion of excitatory neurons compared to inhibitory neurons in both layers of dorsolateral prefrontal cortex and a higher proportion of oligodendrocytes in layer VI compared to layer II/III (Figure 5I, MBP: P = 3.97e–05, SLC17A7: P = 0.029, GAD1: P = 0.0493, Neg: P = 0.859874). Taken together, we show robust analysis of gene expression in post-mortem human dorsolateral prefrontal cortex using 4-plex RNAscope technology and spectral imaging/linear unmixing in combination with dotdotdot and machine learning.
dotdotdot accommodates hi-plex smFISH data acquired with alternative imaging acquisition parameters and saved in diverse file formats
To demonstrate the utility of our image acquisition and analysis workflow, we enabled flexible solutions for users who may be unable to image lipofuscin autofluorescence with spectral imaging or who may acquire images in alternative file formats. First, we evaluated whether a narrower spectral range, such as the range detected by the Opal520 fingerprint, could be used for lipofuscin detection and masking. This would allow users to sacrifice data collection for one gene target in order to capture lipofuscin autofluorescence in the available spectral range (i.e. use the spectral range of one fluorophore to capture lipofuscin instead of a gene target).
To evaluate this possibility, we hybridized post-mortem human dorsolateral prefrontal cortex with probes targeting SLC17A7 (Opal690), GAD1 (Opal620) and SNX19 (sorting nexin 19, Opal570), a gene associated with genetic risk for schizophrenia (45) (Supplementary Figure S10). As the Opal520 fluorophore was not used to detect a probe target, signals unmixed with the Opal520 fingerprint represented autofluorescence due to lipofuscin (Supplementary Figure S10A). Unmixing with the Opal520 fingerprint as a proxy for the lipofuscin fingerprint captured the majority of autofluorescence. However, unmixing the same lambda stack with the custom lipofuscin fingerprint was more comprehensive and captured a larger number of autofluorescent pixels (Supplementary Figure S10B). Quantification of fluorescent pixels assigned to each gene demonstrated that more confounding autofluorescent pixels were removed from gene channels when unmixing with the lipofuscin compared to Opal520 fingerprint (Supplementary Figure S10C). However, masking with either Opal520 or lipofuscin unmixed signals removed a substantial portion of background autofluorescence (Supplementary Figure S10A, B), suggesting that a more narrow spectral range can be used to detect and monitor lipofuscin autofluorescence if imaging capabilities are limited.
Next, given the diversity of imaging file formats for smFISH data, we evaluated the performance of dotdotdot on images acquired and saved using a different microscope system and customized file format (Nikon ‘.nd2’ files versus Zeiss ‘.czi’ files). For three distinct combinations of probe targets hybridized to post-mortem human cortical tissue (images courtesy of Jennie Close and Ed Lein at the Allen Brain Institute), we show accurate segmentation of fluorescent RNAscope signals (Supplementary Figure S11). dotdotdot is compatible with several additional file formats, including those supported by Zeiss, Leica, Nikon, Olympus and MetaMorph systems.
Finally, as chemistry for higher order multiplexing is rapidly coming online, we evaluated the performance of dotdotdot on higher order multiplexed images acquired using the RNAscope Hi-plex assay, which visualizes up to 12 gene targets in a single tissue slice with several rounds of hybridization and stripping (Supplementary Figure S12). dotdotdot faithfully segments fluorescent signals from 12 positive control probes against different 'house-keeping' genes in the same brain section of mouse tissue (image courtesy of Advanced Cell Diagnostics).
DISCUSSION
This manuscript reports a robust, flexible, and automated approach for quantification of multiplex single molecule fluorescent in situ hybridization (smFISH) images in complex tissues, including post-mortem human brain. For smFISH analysis, we present and validate dotdotdot, a versatile computational tool for quantitative analysis of smFISH data. In combination with spectral imaging workflows and machine learning strategies, we demonstrate robustness of this software for quantifying gene expression in single cells in mouse brain as well as post-mortem human brain. Importantly, we develop imaging acquisition and analysis strategies for detecting and removing tissue autofluorescence in smFISH data to improve the accuracy of transcript quantification. Furthermore, we demonstrate that quantitative segmentation data can be used in combination with machine learning approaches to define cell types in a systematic and unbiased manner. Finally, we demonstrate the flexibility of dotdotdot for diverse data file formats, imaging acquisition parameters, and multiplexed experiments thereby increasing its utility for different types of smFISH data.
dotdotdot confers several advantages for quantitative analysis of smFISH data
We developed dotdotdot to address several challenges currently facing users attempting to quantify smFISH data acquired in complex tissues using RNAscope. A major goal of our project was to design user-friendly software that can process multidimensional images at high throughput, and provide several unbiased quantitative measures of smFISH data while controlling for tissue autofluorescence.
A major roadblock hindering the ability of standard biological laboratories to effectively quantify smFISH data is the lack of accessible and intuitive software for image segmentation and quantification. While a number of commercial platforms are available (e.g. HALO; Indica Labs and Aperio; Leica), for smFISH data analysis, these softwares are only accessible through purchase and utilize proprietary algorithms that offer less flexibility for segmentation, customization, and autofluorescence masking. To provide a more accessible alternative (Supplementary Table S1), our platform, dotdotdot is an open source tool that can be implemented and customized by any MATLAB user. We have provided code, tutorials, and example images to make our software user-friendly and adaptable for different applications, images, and tissues.
The multidimensionality associated with smFISH data is an additional, significant challenge for quantifying images. Typical smFISH experiments include multiple genes/probes/channels, image planes (i.e. z-stacks), objects (nuclei and transcript dots), and parameters for analysis (dot size, intensity, and number). We designed dotdotdot to accommodate all of these dimensions simultaneously, allowing users more flexibility in their data analysis. dotdotdot is compatible with data acquired from several smFISH workflows, including RNAscope Multiplex Fluorescent V1, V2, and Hi-plex assays (Figure 1). We demonstrate analysis of six separate fluorescent signals using spectral imaging and linear unmixing (Figure 5). However, there is no limit to the number of gene targets or fluorescent signals (i.e. DAPI or lipofuscin) that dotdotdot can process in parallel. Indeed, using RNAscope Hi-plex data, we demonstrate the utility of dotdotdot for accurately segmenting 12 different probes in the same tissue slice (Supplementary Figure S12).
Importantly, dotdotdot readily accommodates z-stacks and retains unique z-information in the same x, y coordinate for images with densely packed transcripts or cells. dotdotdot can also handle tiled z-stacks containing multiple fields of view over the tissue depth. This is in contrast to other commercially available softwares and open-source platforms, such as FISH-quant (12), which perform nuclear segmentation on two-dimensional images. Moreover, dotdotdot provides several quantitative metrics, including dot size, number and intensity, that can be incorporated into computational data analysis strategies such as K-means clustering and Classification and Regression Trees (CART). This is an advantage to other programs, such as Count Nuclear Foci-ImageJ (46), that are tailored to one metric (i.e. nuclei number) that may not fully capture the dimensionality of the dataset.
Finally, it is important to note that other open-source platforms require manual adjustment of several parameters per image or have limited documentation to implement the software, which makes it challenging to automate for batch processing or utilize for multidimensional RNAscope data (46–49). While other high throughput, programs/pipelines for cell segmentation and spot counting are in various stages of development, including Allen Cell Structure Segmenter (50) and Starfish (51), these softwares still have limitations for rapid analysis of RNAscope data, including requirements of human selected ground truth images for iterative deep learning or reformatting of microscopy data to complex SpaceTx format. We developed dotdotdot to meet the needs of the standard biological laboratory aiming to quantitatively analyze multidimensional smFISH data with a rapid and unbiased approach to answer hypothesis-driven questions about gene expression and co-localization.
Quantifying spatial gene expression at single cell resolution with dotdotdot delivers novel biological insights
We confirmed the accuracy of dotdotdot by quantifying expression of the activity-regulated genes Arc and Bdnf, which are expected to increase following brain stimulation with electroconvulsive seizures (35,52–55). While induction of Arc and Bdnf in response to electroconvulsive seizures has long been appreciated (30), it has remained unclear whether activity-induced increases are driven by increased transcription in a stable population of cell ‘expressers,’ recruitment of additional neurons that become transcriptionally active, or a combination of both. Furthermore, whether multiple Bdnf isoforms can be induced within an individual BDNF-expressing cell in vivo has been difficult to demonstrate. While the necessary spatial and cellular resolution to answer these questions is lost in bulk homogenate analysis of gene expression, it can be resolved with quantitative analysis of smFISH data.
Using dotdotdot, we identify distinct ensembles of neurons expressing activity-regulated genes following electroconvulsive seizures. While the majority of cells upregulate Arc and Bdnf upon activity, we identify some cells that appear to selectively upregulate Arc, and some that preferentially upregulate specific Bdnf isoforms. The dynamics in regulation of these activity-regulated genes following activity have important implications for studies using activity-induced promoters (e.g. Arc and Fos) to tag ensembles of activated neurons (56,57). These data suggest that ‘activated’ ensembles may not be a homogenous population as some activity-tagged cells may have the capacity to release BDNF and engage downstream plasticity cascades, while others may not. Similarly, our results suggest that activity-induced, BDNF-expressing neurons are likely not a uniform population as some BDNF-expressing neurons may preferentially utilize one isoform over another. Because Bdnf isoforms show distinct expression kinetics and subcellular targeting (58–60), it is likely that differences in upregulation between single cells impact neuron structure and function. Future studies should aim to link differences in activity-regulated gene expression that occur in response to induction of neural activity or behavioral experiences to correlates of cell function, including morphology and activity, to better understand how dynamic, activity-dependent gene expression impacts network function.
dodotdot overcomes tissue autofluorescence to accurately profile post-mortem human brain tissue
Post-mortem human brain poses several intrinsic and extrinsic challenges for quantifying smFISH data, including extensive cellular heterogeneity (61), high lipofuscin autofluorescence (13), and RNA degradation due to the interval between death and brain extraction and processing. To overcome these challenges, we carefully optimized the RNAscope assay as well as imaging acquisition and analysis parameters. We found that RNAscope Multiplex Fluorescence V2 reagents afforded higher signal-to-noise in post-mortem human brain tissue, especially for moderate or low expressing gene targets, such as SNX19 (Supplementary Figure S10). Flexibility to dilute fluorophore concentrations for higher expressing gene targets, such as SLC17A7 (Figure 4), also facilitated more accurate target labeling and quantification. While we utilized RNAscope technology for fluorescent probe labeling, non-commercial smFISH technologies have also proven successful in post-mortem human brain tissue (4). However, RNAscope offers a rapid, robust, and universal approach that has now been replicated by several groups for qualitative validation of RNA-seq data in post-mortem human brain tissue (3–5,18).
For quantitative analysis of smFISH signals in post-mortem human brain tissue, it is necessary to identify and remove fluorescent pixels derived from lipofuscin autofluorescence. There are several lipophilic reagents marketed to quench tissue autofluorescence that can be incorporated into the RNAscope assay (23,62). However, dye-based approaches proved ineffective in mitigating lipofuscin signals in our hands. While we did not explore optical tissue clearing methods to remove lipofuscin granules, several approaches, such as CLARITY, have been utilized in conjunction with smFISH (28,63,64) and may be compatible with the RNAScope assay.
Given the persistence of lipofuscin in our human brain samples, we employed a spectral imaging/linear unmixing approach to isolate and mask autofluorescent pixels attributed to lipofuscin and other tissue artifacts (23). This approach also allowed us to conduct 4-plex labeling as we were able to separate signals arising from spectrally overlapping fluorophores (Opal570 and Opal620). We carefully validated reference emission profiles used for linear unmixing of fluorescent probe signals (Supplementary Figure S6) and lipofuscin autofluorescence (Supplementary Figure S7). Importantly, we demonstrated that the spectral quality of lipofuscin autofluorescence is comparable among subjects close in age and a common lipofuscin fingerprint can be employed for multiple subjects. However, studies examining gene expression across development may require age-specific lipofuscin fingerprints as biochemical and autofluorescent properties of lipofuscin are dynamic across the lifespan (14). Although spectral imaging or a custom lipofuscin filter cube are superior imaging strategies for detecting and masking lipofuscin autofluorescence (21,23), we also demonstrate the feasibility of using a narrower spectral range, such as that detected by Opal520 (or a standard FITC filter), to detect and mask lipofuscin fluorescence with dotdotdot. Given that neurons are more profoundly affected by lipofuscin masking than glia (13), it is important to consider whether removal of lipofuscin pixels biases quantification of gene expression in particular cell types. Our data suggest that neurons are not adversely affected by lipofuscin masking as we detect the expected proportions of neuronal and glial cell types in dorsolateral prefrontal cortex (65) (Figure 5).
dotdotdot complements growing computational approaches for spatial analysis of genome-wide expression
We developed dotdotdot to faithfully segment fluorescent probe signals and provide quantitative information on transcript/dot size, number, and fluorescence intensity. While this quantitative output of dotdotdot can stand alone to answer many biological questions (66), we provide examples of how further computational approaches can be utilized to answer more complex questions about gene expression. First, in mouse tissue, we used K-means clustering (67) to identify groups of cells defined as high, medium, and low expressers for individual genes based on transcript dot size and number (Figure 3). Second, using thresholds established with the K-means approach, we examined co-localization of different activity-regulated genes to understand co-regulation of activity-induced transcripts. A key advantage of the K-means approach is that several features of transcript segmentation, such as dot size and number, are provided to offer a more comprehensive and unbiased interpretation of gene expression. Finally, given the diversity and intermingled spatial position of different cell types in post-mortem human brain tissue, we used a machine learning approach—CART, (68) to systematically assign cell types to individual ROIs with 91% accuracy (Figure 5 and Supplementary Figure S9). This model accommodated potentially conflicting signals from mutually exclusive canonical cell type markers (i.e. MBP and SLC17A7) to assign the most likely cell type for each ROI. As molecular profiles of cell types become more complex, machine learning approaches such as CART may become increasingly necessary to interpret overlapping patterns of gene expression (69). Regardless of the classification approach, we strongly suggest using linear mixed effects models to analyze smFISH data that account for the nested nature of these data when combining across many images and ROIs. Regular linear regression will likely result in overly-liberal p-values for differential expression since it ignores the high similarity between ROIs/cells in the same image.
Spatial analysis of genome-wide expression is a rapidly emerging field (2,70–72). With the advent of spatial transcriptomics (73) and Slide-seq (74), smFISH will continue to be a gold standard for validating spatial RNA-seq approaches. dotdotdot is an intuitive computational tool that can add quantitative dimensions to traditionally qualitative smFISH data. Furthermore, as tools for integrating smFISH and single cell RNA-seq data continue to develop (75,76), dotdotdot can augment these approaches by extracting quantitative information from existing datasets for integration with spatial gene expression databases.
In summary, we present a computational tool for smFISH data that can be readily implemented in wet-bench laboratories to elevate spatial analyses of gene expression and complement growing single cell and spatial transcriptomic data sets in the field of neuroscience (77) and beyond (78).
DATA AVAILABILITY
The source code for dotdotdot is available together with test data and detailed tutorials at https://github.com/LieberInstitute/dotdotdot. Raw confocal images are available at https://www.doi.org/10.6084/m9.figshare.12104082 (human data) and https://www.doi.org/10.6084/m9.figshare.12118830 (mouse data).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful for the contributions of the Clinical Brain Disorders Branch of the National Institute of Mental Health in assisting the Lieber Institute for Brain Development in the acquisition and curation of brain tissue donations for this study. We thank Ed Lein and Jennie Close at the Allen Brain Institute and Li Wang, Mohammed Qutaish, Bingqing Zhang and Xiao-Jun Ma at Advanced Cell Diagnostics for generously sharing RNAscope image data. We thank Daniel Weinberger and Stephanie Page for comments on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Lieber Institute for Brain Development; National Institutes of Mental Health [R21MH118725 to K.M.]. Funding for open access charge: Lieber Institute for Brain Development.
Conflict of interest statement. None declared.
REFERENCES
- 1. Poulin J.-F., Tasic B., Hjerling-Leffler J., Trimarchi J.M., Awatramani R.. Disentangling neural cell diversity using single-cell transcriptomics. Nat. Neurosci. 2016; 19:1131–1141. [DOI] [PubMed] [Google Scholar]
- 2. Lein E., Borm L.E., Linnarsson S.. The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science. 2017; 358:64–69. [DOI] [PubMed] [Google Scholar]
- 3. Lake B.B., Ai R., Kaeser G.E., Salathia N.S., Yung Y.C., Liu R., Wildberg A., Gao D., Fung H.-L., Chen S. et al.. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016; 352:1586–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hodge R.D., Bakken T.E., Miller J.A., Smith K.A., Barkan E.R., Graybuck L.T., Close J.L., Long B., Johansen N., Penn O. et al.. Conserved cell types with divergent features in human versus mouse cortex. Nature. 2019; 573:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boldog E., Bakken T.E., Hodge R.D., Novotny M., Aevermann B.D., Baka J., Bordé S., Close J.L., Diez-Fuertes F., Ding S.-L. et al.. Transcriptomic and morphophysiological evidence for a specialized human cortical GABAergic cell type. Nat. Neurosci. 2018; 21:1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tartt A.N., Fulmore C.A., Liu Y., Rosoklija G.B., Dwork A.J., Arango V., Hen R., Mann J.J., Boldrini M.. Considerations for assessing the extent of hippocampal neurogenesis in the adult and aging human brain. Cell Stem Cell. 2018; 23:782–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sorrells S.F., Paredes M.F., Cebrian-Silla A., Sandoval K., Qi D., Kelley K.W., James D., Mayer S., Chang J., Auguste K.I. et al.. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature. 2018; 555:377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McNicol A.M., Farquharson M.A.. In situ hybridization and its diagnostic applications in pathology. J. Pathol. 1997; 182:250–261. [DOI] [PubMed] [Google Scholar]
- 9. Weickert C.S., Rothmond D.A., Purves-Tyson T.D.. Considerations for optimal use of post-mortem human brains for molecular psychiatry: lessons from schizophrenia. Handb. Clin. Neurol. 2018; 150:221–235. [DOI] [PubMed] [Google Scholar]
- 10. Wang F., Flanagan J., Su N., Wang L.-C., Bui S., Nielson A., Wu X., Vo H.-T., Ma X.-J., Luo Y.. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012; 14:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Z., Portier B.P., Gruver A.M., Bui S., Wang H., Su N., Vo H.-T., Ma X.-J., Luo Y., Budd G.T. et al.. Automated quantitative RNA in situ hybridization for resolution of equivocal and heterogeneous ERBB2 (HER2) status in invasive breast carcinoma. J. Mol. Diagn. 2013; 15:210–219. [DOI] [PubMed] [Google Scholar]
- 12. Tsanov N., Samacoits A., Chouaib R., Traboulsi A.-M., Gostan T., Weber C., Zimmer C., Zibara K., Walter T., Peter M. et al.. smiFISH and FISH-quant - a flexible single RNA detection approach with super-resolution capability. Nucleic Acids Res. 2016; 44:e165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dowson J.H., Harris S.J.. Quantitative studies of the autofluorescence derived from neuronal lipofuscin. J. Microsc. 1981; 123:249–258. [DOI] [PubMed] [Google Scholar]
- 14. Benavides S.H., Monserrat A.J., Fariña S., Porta E.A.. Sequential histochemical studies of neuronal lipofuscin in human cerebral cortex from the first to the ninth decade of life. Arch. Gerontol. Geriatr. 2002; 34:219–231. [DOI] [PubMed] [Google Scholar]
- 15. Baleriola J., Walker C.A., Jean Y.Y., Crary J.F., Troy C.M., Nagy P.L., Hengst U.. Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell. 2014; 158:1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bissel S.J., Auer R.N., Chiang C.-H., Kofler J., Murdoch G.H., Nix W.A., Painter M., Richer M., Sartelet H., Wang G. et al.. Human parechovirus 3 meningitis and fatal leukoencephalopathy. J. Neuropathol. Exp. Neurol. 2015; 74:767–777. [DOI] [PubMed] [Google Scholar]
- 17. Bialas A.R., Presumey J., Das A., van der Poel C.E., Lapchak P.H., Mesin L., Victora G., Tsokos G.C., Mawrin C., Herbst R. et al.. Microglia-dependent synapse loss in type I interferon-mediated lupus. Nature. 2017; 546:539–543. [DOI] [PubMed] [Google Scholar]
- 18. Gandal M.J., Zhang P., Hadjimichael E., Walker R.L., Chen C., Liu S., Won H., van Bakel H., Varghese M., Wang Y. et al.. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 2018; 362:eaat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith R.S., Kenny C.J., Ganesh V., Jang A., Borges-Monroy R., Partlow J.N., Hill R.S., Shin T., Chen A.Y., Doan R.N. et al.. Sodium channel SCN3A (nav1.3) regulation of human cerebral cortical folding and oral motor development. Neuron. 2018; 99:905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fish K.N., Rocco B.R., Lewis D.A.. Laminar distribution of subsets of gabaergic axon terminals in human prefrontal cortex. Front. Neuroanat. 2018; 12:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rocco B.R., Oh H., Shukla R., Mechawar N., Sibille E.. Fluorescence-based cell-specific detection for laser-capture microdissection in human brain. Sci. Rep. 2017; 7:14213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jolly S., Lang V., Koelzer V.H., Sala Frigerio C., Magno L., Salinas P.C., Whiting P., Palomer E.. Single-Cell quantification of mRNA expression in the human brain. Sci. Rep. 2019; 9:12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pyon W.S., Gray D.T., Barnes C.A.. An alternative to Dye-Based approaches to remove background autofluorescence from primate brain tissue. Front. Neuroanat. 2019; 13:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shah S., Lubeck E., Zhou W., Cai L.. seqFISH accurately detects transcripts in single cells and reveals robust spatial organization in the hippocampus. Neuron. 2017; 94:752–758. [DOI] [PubMed] [Google Scholar]
- 25. Chen K.H., Boettiger A.N., Moffitt J.R., Wang S., Zhuang X.. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015; 348:aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ke R., Mignardi M., Pacureanu A., Svedlund J., Botling J., Wählby C., Nilsson M.. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods. 2013; 10:857–860. [DOI] [PubMed] [Google Scholar]
- 27. Lee J.H., Daugharthy E.R., Scheiman J., Kalhor R., Yang J.L., Ferrante T.C., Terry R., Jeanty S.S.F., Li C., Amamoto R. et al.. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014; 343:1360–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang X., Allen W.E., Wright M.A., Sylwestrak E.L., Samusik N., Vesuna S., Evans K., Liu C., Ramakrishnan C., Liu J. et al.. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 2018; 361:eaat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schloesser R.J., Orvoen S., Jimenez D.V., Hardy N.F., Maynard K.R., Sukumar M., Manji H.K., Gardier A.M., David D.J., Martinowich K.. Antidepressant-like effects of electroconvulsive seizures require adult neurogenesis in a neuroendocrine model of depression. Brain Stimulat. 2015; 8:862–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maynard K.R., Hobbs J.W., Rajpurohit S.K., Martinowich K.. Electroconvulsive seizures influence dendritic spine morphology and BDNF expression in a neuroendocrine model of depression. Brain Stimulat. 2018; 11:856–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lipska B.K., Deep-Soboslay A., Weickert C.S., Hyde T.M., Martin C.E., Herman M.M., Kleinman J.E.. Critical factors in gene expression in postmortem human brain: Focus on studies in schizophrenia. Biol. Psychiatry. 2006; 60:650–658. [DOI] [PubMed] [Google Scholar]
- 32. Colliva A., Maynard K.R., Martinowich K., Tongiorgi E.. Duarte C.B., Tongiorgi E.. Detecting single and multiple BDNF transcripts by in situ hybridization in neuronal cultures and brain sections. Brain-Derived Neurotrophic Factor (BDNF). 2019; 143:NY: Neuromethods. Springer; 27–53. [Google Scholar]
- 33. Hodneland E., Kögel T., Frei D.M., Gerdes H.-H., Lundervold A.. CellSegm - a MATLAB toolbox for high-throughput 3D cell segmentation. Source Code Biol. Med. 2013; 8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ram S., Rodríguez J.J., Bosco G.. Segmentation and detection of fluorescent 3D spots. Cytometry A. 2012; 81:198–212. [DOI] [PubMed] [Google Scholar]
- 35. Altar C.A., Laeng P., Jurata L.W., Brockman J.A., Lemire A., Bullard J., Bukhman Y.V., Young T.A., Charles V., Palfreyman M.G.. Electroconvulsive seizures regulate gene expression of distinct neurotrophic signaling pathways. J. Neurosci. 2004; 24:2667–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Timmusk T., Palm K., Metsis M., Reintam T., Paalme V., Saarma M., Persson H.. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993; 10:475–489. [DOI] [PubMed] [Google Scholar]
- 37. Aid T., Kazantseva A., Piirsoo M., Palm K., Timmusk T.. Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 2007; 85:525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maynard K.R., Hill J.L., Calcaterra N.E., Palko M.E., Kardian A., Paredes D., Sukumar M., Adler B.D., Jimenez D.V., Schloesser R.J. et al.. Functional role of BDNF production from unique promoters in aggression and serotonin signaling. Neuropsychopharmacology. 2016; 41:1943–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hong E.J., McCord A.E., Greenberg M.E.. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008; 60:610–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bakken T.E., Hodge R.D., Miller J.A., Yao Z., Nguyen T.N., Aevermann B., Barkan E., Bertagnolli D., Casper T., Dee N. et al.. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One. 2018; 13:e0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Price A.J., Hwang T., Tao R., Burke E.E., Rajpurohit A., Shin J.H., Hyde T.M., Kleinman J.E., Jaffe A.E., Weinberger D.R.. Characterizing the nuclear and cytoplasmic transcriptomes in developing and mature human cortex uncovers new insight into psychiatric disease gene regulation. Genome Res. 2020; 30:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steward O., Farris S., Pirbhoy P.S., Darnell J., Driesche S.J.V.. Localization and local translation of Arc/Arg3.1 mRNA at synapses: some observations and paradoxes. Front. Mol. Neurosci. 2014; 7:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lyford G.L., Yamagata K., Kaufmann W.E., Barnes C.A., Sanders L.K., Copeland N.G., Gilbert D.J., Jenkins N.A., Lanahan A.A., Worley P.F.. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron. 1995; 14:433–445. [DOI] [PubMed] [Google Scholar]
- 44. Link W., Konietzko U., Kauselmann G., Krug M., Schwanke B., Frey U., Kuhl D.. Somatodendritic expression of an immediate early gene is regulated by synaptic activity. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:5734–5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ma L., Semick S.A., Chen Q., Li C., Tao R., Price A.J., Shin J.H., Jia Y. BrainSeq Consortium BrainSeq Consortium Brandon N.J. et al.. Schizophrenia risk variants influence multiple classes of transcripts of sorting nexin 19 (SNX19). Mol. Psychiatry. 2019; 5:831–843. [DOI] [PubMed] [Google Scholar]
- 46. Abramoff M., Magalhães P., Ram S.J.. Image Processing with ImageJ. Biophoton. Int. 2003; 11:36–42. [Google Scholar]
- 47. Xie F., Timme K.A., Wood J.R.. Using Single Molecule mRNA Fluorescent in Situ Hybridization (RNA-FISH) to Quantify mRNAs in Individual Murine Oocytes and Embryos. Sci. Rep. 2018; 8:7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Buxbaum A.R., Wu B., Singer R.H.. Single β-actin mRNA detection in neurons reveals a mechanism for regulating its translatability. Science. 2014; 343:419–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thompson R.E., Larson D.R., Webb W.W.. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002; 82:2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jianxu C., Ding L., Viana M.P., Hendershott M.C., Yang R., Mueller I.A., Rafelski S.M.. The Allen CellStructure Segmenter: a new open source toolkit for segmenting 3D intracellularstructures in fluorescence microscopy images. 2018; bioRxiv doi:08 December 2018, preprint: not peer reviewed 10.1101/491035. [DOI]
- 51. Perkel J.M. Starfish enterprise: finding RNA patterns in single cells. Nature. 2019; 572:549–551. [DOI] [PubMed] [Google Scholar]
- 52. Larsen M.H., Olesen M., Woldbye D.P.D., Hay-Schmidt A., Hansen H.H., Rønn L.C.B., Mikkelsen J.D.. Regulation of activity-regulated cytoskeleton protein (Arc) mRNA after acute and chronic electroconvulsive stimulation in the rat. Brain Res. 2005; 1064:161–165. [DOI] [PubMed] [Google Scholar]
- 53. Vismer M.S., Forcelli P.A., Skopin M.D., Gale K., Koubeissi M.Z.. The piriform, perirhinal, and entorhinal cortex in seizure generation. Front. Neural Circuits. 2015; 9:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Revuelta M., Castaño A., Venero J.L., Machado A., Cano J.. Long-lasting induction of brain-derived neurotrophic factor is restricted to resistant cell populations in an animal model of status epilepticus. Neuroscience. 2001; 103:955–969. [DOI] [PubMed] [Google Scholar]
- 55. Sun W., Park K.W., Choe J., Rhyu I.J., Kim I.H., Park S.K., Choi B., Choi S.-H., Park S.H., Kim H.. Identification of novel electroconvulsive shock-induced and activity-dependent genes in the rat brain. Biochem. Biophys. Res. Commun. 2005; 327:848–856. [DOI] [PubMed] [Google Scholar]
- 56. DeNardo L.A., Liu C.D., Allen W.E., Adams E.L., Friedmann D., Fu L., Guenthner C.J., Tessier-Lavigne M., Luo L.. Temporal evolution of cortical ensembles promoting remote memory retrieval. Nat. Neurosci. 2019; 22:460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Denny C.A., Kheirbek M.A., Alba E.L., Tanaka K.F., Brachman R.A., Laughman K.B., Tomm N.K., Turi G.F., Losonczy A., Hen R.. Hippocampal memory traces are differentially modulated by experience, time, and adult neurogenesis. Neuron. 2014; 83:189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. West A.E., Pruunsild P., Timmusk T.. Neurotrophins: transcription and translation. Handb. Exp. Pharmacol. 2014; 220:67–100. [DOI] [PubMed] [Google Scholar]
- 59. Baj G., Leone E., Chao M.V., Tongiorgi E.. Spatial segregation of BDNF transcripts enables BDNF to differentially shape distinct dendritic compartments. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:16813–16818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pattabiraman P.P., Tropea D., Chiaruttini C., Tongiorgi E., Cattaneo A., Domenici L.. Neuronal activity regulates the developmental expression and subcellular localization of cortical BDNF mRNA isoforms in vivo. Mol. Cell. Neurosci. 2005; 28:556–570. [DOI] [PubMed] [Google Scholar]
- 61. Molyneaux B.J., Arlotta P., Menezes J.R.L., Macklis J.D.. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 2007; 8:427–437. [DOI] [PubMed] [Google Scholar]
- 62. Sun Y., Ip P., Chakrabartty A.. Simple Elimination of Background Fluorescence in Formalin-Fixed Human Brain Tissue for Immunofluorescence Microscopy. J. Vis. Exp. 2017; 3:56188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Moffitt J.R., Hao J., Bambah-Mukku D., Lu T., Dulac C., Zhuang X.. High-performance multiplexed fluorescence in situ hybridization in culture and tissue with matrix imprinting and clearing. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:14456–14461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sylwestrak E.L., Rajasethupathy P., Wright M.A., Jaffe A., Deisseroth K.. Multiplexed Intact-Tissue Transcriptional Analysis at Cellular Resolution. Cell. 2016; 164:792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guintivano J., Aryee M.J., Kaminsky Z.A.. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013; 8:290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hallock H.L., Quillian H.M., Mai Y., Maynard K.R., Hill J.L., Martinowich K.. Manipulation of a genetically and spatially defined sub-population of BDNF-expressing neurons potentiates learned fear and decreases hippocampal-prefrontal synchrony in mice. Neuropsychopharmacology. 2019; 44:2239–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. MacQueen J. Some methods for classification and analysis of multivariate observations. Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability, Vol. 1: Statistics. 1967; Berkeley: University of California Press; 281–297. [Google Scholar]
- 68. Breiman L., Friedman J.H., Olshen R.A., Stone C.J.. Classification and Regression Trees Wadsworth & Brooks/Cole Advanced Books & Software. 1984; Monterey. [Google Scholar]
- 69. Alegro M., Theofilas P., Nguy A., Castruita P.A., Seeley W., Heinsen H., Ushizima D.M., Grinberg L.T.. Automating cell detection and classification in human brain fluorescent microscopy images using dictionary learning and sparse coding. J. Neurosci. Methods. 2017; 282:20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Strell C., Hilscher M.M., Laxman N., Svedlund J., Wu C., Yokota C., Nilsson M.. Placing RNA in context and space - methods for spatially resolved transcriptomics. FEBS J. 2019; 286:1468–1481. [DOI] [PubMed] [Google Scholar]
- 71. Burgess D.J. Spatial transcriptomics coming of age. Nat. Rev. Genet. 2019; 20:317. [DOI] [PubMed] [Google Scholar]
- 72. Maynard K.R., Jaffe A.E., Martinowich K.. Spatial transcriptomics: putting genome-wide expression on the map. Neuropsychopharmacology. 2020; 45:232–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ståhl P.L., Salmén F., Vickovic S., Lundmark A., Navarro J.F., Magnusson J., Giacomello S., Asp M., Westholm J.O., Huss M. et al.. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016; 353:78–82. [DOI] [PubMed] [Google Scholar]
- 74. Rodriques S.G., Stickels R.R., Goeva A., Martin C.A., Murray E., Vanderburg C.R., Welch J., Chen L.M., Chen F., Macosko E.Z.. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019; 363:1463–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Svensson V., Teichmann S.A., Stegle O.. SpatialDE: identification of spatially variable genes. Nat. Methods. 2018; 15:343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Edsgärd D., Johnsson P., Sandberg R.. Identification of spatial expression trends in single-cell gene expression data. Nat. Methods. 2018; 15:339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Maniatis S., Äijö T., Vickovic S., Braine C., Kang K., Mollbrink A., Fagegaltier D., Andrusivová Ž., Saarenpää S., Saiz-Castro G. et al.. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science. 2019; 364:89–93. [DOI] [PubMed] [Google Scholar]
- 78. Moncada R., Barkley D., Wagner F., Chiodin M., Devlin J.C., Baron M., Hajdu C.H., Simeone D.M., Yanai I.. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020; 38:333–342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code for dotdotdot is available together with test data and detailed tutorials at https://github.com/LieberInstitute/dotdotdot. Raw confocal images are available at https://www.doi.org/10.6084/m9.figshare.12104082 (human data) and https://www.doi.org/10.6084/m9.figshare.12118830 (mouse data).