


Abstract
Zinc finger protein with KRAB and SCAN domains 3 (ZKSCAN3) has long been known as a master transcriptional repressor of autophagy. Here, we identify a novel role for ZKSCAN3 in alleviating senescence that is independent of its autophagy-related activity. Downregulation of ZKSCAN3 is observed in aged human mesenchymal stem cells (hMSCs) and depletion of ZKSCAN3 accelerates senescence of these cells. Mechanistically, ZKSCAN3 maintains heterochromatin stability via interaction with heterochromatin-associated proteins and nuclear lamina proteins. Further study shows that ZKSCAN3 deficiency results in the detachment of genomic lamina-associated domains (LADs) from the nuclear lamina, loss of heterochromatin, a more accessible chromatin status and consequently, aberrant transcription of repetitive sequences. Overexpression of ZKSCAN3 not only rescues premature senescence phenotypes in ZKSCAN3-deficient hMSCs but also rejuvenates physiologically and pathologically senescent hMSCs. Together, these data reveal for the first time that ZKSCAN3 functions as an epigenetic modulator to maintain heterochromatin organization and thereby attenuate cellular senescence. Our findings establish a new functional link among ZKSCAN3, epigenetic regulation, and stem cell aging.
INTRODUCTION
Zinc finger protein with KRAB and SCAN domains 3 (ZKSCAN3) is a member of the Krüppel-associated box domain zinc finger protein (KRAB-ZFP) family, the largest family of transcription regulators of multiple cellular processes including cell proliferation, apoptosis and neoplastic transformation in higher vertebrates (1,2). There are four subfamilies of KRAB-ZFPs, two with KRAB and C2H2 zinc finger motifs and the other two with an additional SCAN (SCAN-ZFPs) or DUF3669 domain in the N-terminus. The KRAB domain functions to repress transcription, while the C2H2 zinc finger domain binds to specific DNA sequences. The function of the SCAN domain remains poorly understood (1,3). As a member of the SCAN-ZFP subfamily (4), ZKSCAN3 has been reported to promote cancer cell proliferation, migration and invasion (5–9). Emerging evidence has shown that ZKSCAN3 negatively regulates autophagosome formation and lysosomal biogenesis by repressing the expression of autophagy-related genes including MAP1LC3B, ATG5, ATG12 and ATG3 in human cancer cells and mouse neurons (10–12). In contrast, a recent study has reported that Zkscan3-knockout mice exhibit no alterations in the expression of autophagy-related genes (13), raising the intriguing possibility that ZKSCAN3 may function in a species- and/or cell-type-specific manner. Since autophagy is tightly associated with cellular senescence and aging-related human diseases (14,15), whether ZKSCAN3 exerts a geroprotective role needs to be clarified.
Stem cell exhaustion, which diminishes the regenerative potential of various adult tissues, is considered to be a major contributor to progressive impairment of physiological integrity during aging and in aging-associated disorders (16). Functional degeneration and reduction in the number of human mesenchymal stem cells (hMSCs) have been observed in physiologically aged individuals and prematurely aged patients, such as those with Werner Syndrome (17–21). Evidence suggests that stem cell aging and consequent exhaustion are tightly associated with epigenetic alterations (21,22), however, the key epigenetic regulators responsible for human stem cell aging remain largely unidentified.
Heterochromatin is structurally inaccessible and functionally inactive. It typically comprises a high density of repetitive DNA elements, such as satellite sequences (e.g. ALR, Satellite 2 and ACRO1), and transposable elements (TEs) including short interspersed elements (SINEs) (e.g. SVA VNTR, SVA for short), long interspersed elements (LINEs) (e.g. LINE1) and long terminal repeat (LTR)-retrotransposons (e.g. LTR3B) (23–28). Notably, lamina-associated domains (LADs), genomic regions that interact extensively with the nuclear lamina, possess transcriptionally inactive features similar to heterochromatin (29). Previous studies have demonstrated that LADs detach from the nuclear lamina in oncogene-challenged cells (30). Structurally, it has also been reported that chromatin in aged human cells shows more accessible than that in young counterparts (31,32). However, the precise architecture and accessibility alterations of the genome during aging are largely unknown. This is particularly true for the origin of changes in repetitive sequence loci within LADs. Additionally, heterochromatin disorganization has been reported to correlate with abnormal activation of repetitive sequences (such as LINE1) and give rise to genome instability, which is frequently observed in aged tissues and cells (18,19,33–41). In contrast, restoration of condensed heterochromatin delays senescence in human cells (20–22,33), suggesting that the stability of heterochromatin organization could be an effective target for intervention against aging (21,23,42,43). While understanding the mechanisms underlying epigenomic instability is important to the field of geroscience, key stabilizers of heterochromatin that exert a geroprotective effect have not been comprehensively explored.
Here, by combining CRISPR/Cas9-mediated gene knockout and human embryonic stem cell (hESC)-directed differentiation techniques, we generated ZKSCAN3-deficient hMSCs and discovered that the absence of ZKSCAN3 resulted in premature aging in hMSCs. In-depth mechanistic investigation revealed that ZKSCAN3 did not regulate autophagy but functioned as a geroprotector to stabilize heterochromatin and counteract hMSC senescence. This novel function for ZKSCAN3 in the maintenance of heterochromatin stability is independent of its role as an autophagy and lysosome regulator, thus suggesting a new potential role for SCAN-ZFP family proteins in the regulation of stem cell aging.
MATERIALS AND METHODS
Cell culture
ZKSCAN3 +/+ and ZKSCAN3-/- hESCs (derived from Line H9 from WiCell Research Institute) were cultured either on mitomycin C-inactivated mouse embryonic fibroblast (MEF) feeders in hESC medium including DMEM/F12 (Thermo Fisher Scientific), 20% Knockout Serum Replacement (Thermo Fisher Scientific), 1% penicillin/streptomycin (Thermo Fisher Scientific), 2 mM GlutaMAX (Thermo Fisher Scientific), 0.1 mM non-essential amino acids (NEAAs, Thermo Fisher Scientific), 55 μM β-mercaptoethanol (Thermo Fisher Scientific), and 10 ng/ml bFGF (Joint Protein Central, Incheon, Korea) or on Matrigel (BD Biosciences)-coated plates in mTeSR medium (STEMCELL Technologies). hMSCs (hESC-derived or primary hMSCs) were cultured in hMSC medium which contained MEMα basal medium (Thermo Fisher Scientific), 10% fetal bovine serum (FBS, Gemcell), 1% penicillin/streptomycin (Thermo Fisher Scientific), 0.1 mM NEAAs (Thermo Fisher Scientific), and 1 ng/ml bFGF (Joint Protein Central, Incheon, Korea). HEK293T cells were cultured in high glucose DMEM (Hyclone) supplemented with 10% FBS (Gibco). There was no mycoplasma contamination observed during cell culture.
Generation of ZKSCAN3-/- hESCs
CRISPR/Cas9-mediated gene knockout was performed as previously described (44,45). Briefly, guide RNA was designed to target the ATG codon within exon 3 of the ZKSCAN3 gene, and then cloned into pCAG-mCherry-gRNA vector (Addgene #87110). H9 hESCs were cultured on Matrigel-coated plates and treated with ROCK inhibitor Y-27632 (Selleck) for 24 hr before being electroporated by 4D-Nucleofector (Lonza) with both 7 μg gRNA vector and 14 μg pCAG-1BPNLS-Cas9-1BPNLS-2AGFP (Addgene #87109). Subsequently, cells were plated on Matrigel-coated plates in mTeSR medium for 48 hr. GFP/mCherry double-positive cells were then purified by fluorescence activated cell sorting (FACS) and cultured on MEF feeders in hESC culture medium. Guide RNA sequences for targeting ZKSCAN3 locus and primers used for gene targeting and off-target identification are listed in Supplementary Table S1.
Generation and characterization of ZKSCAN3+/+ and ZKSCAN3-/- hMSCs
Differentiation of ZKSCAN3+/+ and ZKSCAN3-/- hMSCs from hESCs was performed as previously described (44,46–52). Briefly, hESCs were cultured on MEF feeders for approximately three days and digested with Dispase (Gibco) to obtain embryoid bodies (EBs) which were plated in differentiation medium for 72 hr. The EBs were transferred to Matrigel-coated plates in hMSC differentiation medium (MEMα (Thermo Fisher Scientific), 10% fetal bovine serum (FBS, Gemcell), 1% penicillin/streptomycin (Thermo Fisher Scientific), 0.1 mM NEAAs (Thermo Fisher Scientific), 1 ng/ml bFGF (Joint Protein Central, Incheon, Korea) and 5 ng/ml TGFβ (Humanzyme)) for approximately 10 days. Cells were then plated on Gelatin (Sigma)-coated plates and maintained in hMSC culture medium until they reached 90% confluence. Next, the differentiated cells were subjected to FACS to purify CD73, CD90, and CD105 (hMSC-specific surface markers)-triple positive hMSCs; hMSC-irrelevant antigens including CD34, CD43, and CD45 were also evaluated by FACS analysis. The following antibodies were used for FACS: anti-CD73-PE (BD Biosciences, 550257), anti-CD90-FITC (BD Biosciences, 555595), anti-CD105-APC (BD Biosciences, 17-1057-42), anti-CD34-PE (BD Biosciences, 555822), anti-CD43-FITC (BD Biosciences, 580198), and anti-CD45-FITC (BD Biosciences, 555482). Anti-IgG-FITC (BD Biosciences, 555748), anti-IgG-PE (BD Biosciences, 555749), and anti-IgG-APC (BD Biosciences, 555751) antibodies were used as isotype controls. Osteoblasts, chondrocytes and adipocytes were differentiated as previously described (48) and characterized by von Kossa staining (Genmed Scientifics, GMS80045.3), Toluidine blue (sigma) staining and Oil red O staining (Sigma) respectively, following the manufacturers’ instructions.
Isolation and culture of primary hMSCs
Primary hMSCs were isolated from the gingiva tissues of different individuals as previously reported, with modification (22,52). Briefly, the tissues were cut into fine-grained chippings and digested in TrypLE™ Express Enzyme (Gibco) plus Dispase (Gibco) at 37°C for 30 min. The digested tissues were collected and fully dissociated by pipetting up and down in hMSC medium. Cell suspension was then collected and centrifuged at 200 g for 5 min at room temperature (RT). Subsequently, the supernatant was removed and the pellet was cultured on Gelatin-coated plates in hMSC culture medium for ∼14 days.
CNV identification
The genomic DNA from 1 × 106 hESCs or hMSCs was extracted using a DNeasy Blood & Tissue Kit (Qiagen). To obtain DNA fragments of approximately 300 base pairs, the extracted genomic DNA was subjected to ultrasonication by Covaris. Sequencing libraries were constructed with the Next DNA Library Prep Reagent Set for Illumina (NEB). The published R package HMMcopy was used for CNV identification (53). In brief, the genome was divided into continuous 500-kb windows with readCounter, and the absolute number of reads detected in each window was calculated. The copy number with GC and mappability corrections was evaluated with HMMcopy.
Animal experiments
All animal experiments conducted in this study were approved by the Chinese Academy of Science Institutional Animal Care and Use Committee. For the teratoma formation assay, hESCs were cultured on Matrigel-coated plates and collected in a Matrigel/mTeSR (1:4) solution. Subsequently, the mixture was injected into the inguinal region of NOD/SCID mice (male, 4-6 weeks). Teratoma data was collected until the tumours reached a size of ∼10 mm in diameter. The hMSC transplantation assay was carried out as previously described (47). In brief, ∼1 × 106 hMSCs expressing luciferase were injected into the tibialis anterior (TA) muscle of nude mice (male, 6-8 weeks). IVIS spectrum imaging system (XENOGEN, Caliper) was used to detect luciferase activity at 0, 2 and 4 days after injection.
Western blot
Cells were lysed using SDS lysis buffer (containing 4% SDS and 100 mM Tris-HCl (pH=6.8)) and boiled at 105°C on a thermomixer for 10 min. Protein samples were diluted (ranging from 1:10 to 1:20) and protein concentration was measured by BCA kit. About 20 μg protein per sample was subjected to SDS-PAGE and electrotransferred to a PVDF membrane (Millipore). The membrane was blocked with 5% skim milk (powder from BBI Life Sciences) and incubated with primary antibodies for ∼12 hr at 4°C, then with horseradish peroxidase (HRP)-conjugated secondary antibodies. The visualization and data processing were performed by a ChemiDoc XRS system (Bio-Rad). Antibodies used in this study were as follows: anti-ZKSCAN3 (Santa Cruz, sc-515285), anti-HP1α (Cell Signaling Technology, #2616S) and anti-KAP1 (Abcam, Ab22553), anti-Lamin B1 (Abcam, Ab16048), anti-LBR (Abcam, Ab32535), anti-P16 (BD Bioscience, 550834), anti-P21 (Cell Signaling Technology, #2947), anti-β-actin (Santa Cruz, sc-69879), anti-Flag (Sigma, F1804) and anti-GAPDH (Sigma, G8795).
DNA and RNA analyses
A DNA extraction kit (TIANGEN, Beijing, China) was used to extract genomic DNA. PCR on genomic DNA was performed using PrimeSTAR reagents (TAKARA). The primers used for genomic DNA PCR are listed in Supplementary Table S1. For total RNA extraction, cells were collected in TRIzol (Thermo Fisher Scientific) and genomic DNA was removed using a DNA-free kit (Thermo Fisher Scientific). GoScript Reverse Transcription System (Promega) was used to generate cDNA. Quantitative reverse transcription PCR (RT-qPCR) was carried out with the qPCR Mix (TOYOBO) using a CFX384 Real-Time PCR system (Bio-Rad). The primers used for RT-qPCR are listed in Supplementary Table S2. For RNA-seq, 1.5 μg total RNA was constructed into a sequencing library using the Next Ultra RNA Library Prep Kit for Illumina (NEB) following the manufacturer's protocol. The libraries were then sequenced on Illumina Hisses X-Ten platforms with paired-end 150-bp sequencing. Quality control and sequencing were done by NoVo gene Bioinformatics Technology.
RNA-seq data processing
RNA-seq data processing was performed as previously described (50,54,55). The quality of RNA-seq reads was inspected with FASTQC software (v0.11.6). Low quality reads and Illumina adaptors were removed by TrimGalore (v0.4.4_dev) with default parameters. Trimmed reads were mapped to the UCSC human hg19 genome with HISAT2 software (v2.1.0) (56). Transcriptional expression levels of annotated genes were determined using HTSeq (v0.6.1) (57). Differentially expressed genes (DEGs) were identified using DESeq2 (58) with a cut-off Benjamini-Hochberg-adjusted P value < 0.05 and an absolute fold change (FC) of > 1.5 (|log2FC| > 0.58). The Euclidian distance (R) was based on DESeq2 regularized-logarithm (rlog) normalized read counts. Autophagy- and lysosome-related genes were obtained from the KEGG database (https://www.genome.jp/kegg/) and are listed in Supplementary Table S3.
For repetitive sequence analyses (59), trimmed and filtered reads were aligned to the human hg19 genome (RepEnrich2 provided setup) using Bowtie2 (v2.2.9). RepEnrich2 software was used to assign unique and multi-mapped reads to repetitive sequences. Differentially expressed repetitive sequence analysis was performed with DESeq2 (58) on the fractional counts data. Repetitive sequences with an adjusted P value < 0.05 and |log2FC| > 0.3 were considered statistically significant. GO term enrichment analysis was performed using Toppgene (https://toppgene.cchmc.org/) with an adjusted P value < 0.05 (60). The differentially expressed genes and repetitive sequences are listed in Supplementary Table S3.
Immunofluorescence staining
5 × 105 cells were seeded on coverslips (Thermo Fisher Scientific), washed with PBS and fixed with 4% paraformaldehyde (PFA) for 30 min and permeabilized with 0.4% Triton X-100 in PBS for 30 min at RT. After washing with PBS three times, the cells were blocked with 10% donkey serum (Jackson ImmunoResearch) in PBS for 1 hr at RT. The cells were then incubated with primary antibodies in 10% donkey serum at 4°C overnight. Afterwards, cells were washed with PBS and stained with secondary antibodies and Hoechst 33342 (Thermo Scientific) for 1 hr at RT. A Leica SP5 confocal system was used for imaging. Antibodies used in this study were as follows: anti-Ki67 (ZSGB-BIO, ZM0166), anti-53BP1 (Bethyl Laboratories, A300-273A), anti-FOXA2 (Cell Signaling Technology, 8186S), anti-SMA (Sigma, A5228), anti-TuJ1 (Sigma, T2220), anti-H3K9me3 (Abcam, Ab8898), anti-NANOG (Abcam, Ab21624), anti-OCT3/4 (Santa Cruz, sc-5279), anti-SOX2 (Santa Cruz, sc-17320), and anti-LAP2 (BD Bioscience, 611000), anti-HP1α (Cell Signaling Technology, #2616S), and anti-Lamin A/C (Santa Cruz, sc-376248).
SA-β-gal staining
The SA-β-gal staining of hMSCs was conducted as previously described (50).
Clonal expansion assay
2,000 cells were seeded in each well of 12-well culture plates and cultured for ∼2 weeks, after which point cells were fixed with 4% PFA for 30 min and stained with 0.2% crystal violet (Biohao, C0520) for 30 min at RT. The relative colony area was then measured using ImageJ.
Lentivirus packaging
HEK293T cells were co-transfected with lentiviral overexpression plasmids and lentiviral vectors psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259). The viral particles were collected at 48 hr and 72 hr after transfection and concentrated by ultracentrifugation at 19 400 g for 2.5 hr.
Plasmid construction
ZKSCAN3, KAP1, HP1α cDNAs were generated from hMSC cDNA via PCR amplification and then cloned into pLE4 vector that had been pre-cleaved by XhoI and MluI (a kind gift from Dr. Tomoaki Hishida) (22). Cloning primers are listed in Supplementary Table S2.
Cell cycle analysis
hESCs and hMSCs were collected and fixed in 70% ethyl alcohol overnight at −20°C. Cells were then washed with PBS and stained in buffer containing 0.1% Triton X-100, 0.2 mg/ml RNase A and 0.02 mg/ml propidium iodide at 37°C for 30 min. Next, samples were analysed with an LSRFortessa cell analyser (BD), and data were analysed using the ModFit software.
Co-immunoprecipitation (Co-IP)
The Co-IP experiments were performed as previously described (52). Briefly, HEK293T cells were transfected with Flag-Luc and Flag-ZKSCAN3 plasmids, collected and lysed in CHAPS lysis solution (containing 0.3% CHAPS, 40 mM HEPES, 120 mM NaCl, 1 mM EDTA, and complete protease inhibitor cocktail (Roche) at pH 7.5) at 4°C for 2 hr, following which the samples were centrifuged at 12 000 g at 4°C for 30 min. The supernatants were collected and mixed with anti-Flag antibody (Sigma, F1804) coupled with beads (ANTI-FLAG® M2 Affinity Gel), and rotated overnight at 4°C. After centrifugation at 900 g at 4°C for 2 min, the supernatant was discarded and the beads were washed with CHAPS buffer three times. Samples were eluted by Flag peptides (Sigma) at 4°C for 2 hr. After centrifugation, the supernatant was collected, mixed with 5 x SDS loading buffer and denatured at 105°C on a thermomixer for 10 min in preparation for western blot analysis.
LC-MS/MS analysis and protein identification
The eluted proteins from Co-IP were separated on a 10% SDS-PAGE gel and stained with Coomassie brilliant blue. After decolouration, the gel slice containing proteins of interest was cut and subjected to dehydration (in 100% acetonitrile), reduction (with 10 mM DTT in 25 mM NH4HCO3 for 45 min at 56°C) and alkylation (with 40 mM iodoacetamide in 25 mM NH4HCO3 for 45 min at RT in the dark) (22,52). Proteins were then digested into peptides by sequencing grade trypsin (Worthington) overnight at 37°C. The resultant peptides were homogenized in 0.1% formic acid and separated by the online Easy-nLC 1000 system (Thermo Fisher Scientific) with a C18 reverse-phase column. The column was eluted with a linear gradient of 5–30% acetonitrile in 0.2% formic acid at a rate of 300 nl/min for 100 min. The mass spectra was acquired by nanoLC-Q EXACTIVE (Thermo Fisher Scientific) equipped with a nano-ES ion source (Proxeon Biosystems). Full scan spectra (from m/z 300 to 1600) was acquired in the Orbitrap analyzer with a resolution of 60 000 at 400 m/z after the accumulation of 1 000 000 ions. The five most intense ions in each scan were selected for collision-induced dissociation (CID) fragmentation in the linear ion trap after 3000 ions were accumulated. The maximal filling time was set at 500 ms for the full scans and 150 ms for the MS/MS scans. The dynamic exclusion list was defined as a maximum of 500 entries with a maximum retention period of 60 sec and a relative mass window of 10 ppm.
The raw files were processed using MaxQuant software (v1.3.0.5). The generated peak list files were analysed with Thermo Proteome Discoverer (1.4.0.288) based on the UniProt-proteome-human database (update-20160226). The parameters for analysing were set as follows: trypsin enzyme; up to two missed cleavages; alkylated cysteine as fixed modification; oxidized methionine as variable modifications. MS tolerance was 10 ppm while MS/MS tolerance was 0.02 Da. The required false discovery rate (FDR) was set to 1% at peptide and protein levels, and the minimum length for the acquired peptide was set to seven amino acids. At least one unique or razor peptide per protein group was required for protein identification. ZKSCAN3 interaction proteins are listed in Supplementary Table S4.
Transmission electron microscope (TEM)
ZKSCAN3+/+ and ZKSCAN3-/- hMSCs were collected enzymatically by TrypLE (Gibco) and centrifuged at 1500 g for 5 min at RT. The pellets were fixed with 4% glutaraldehyde in PBS at 4°C overnight. Samples were dehydrated in a graded series of ethanol, infiltrated and embedded in Lowicryl resin HM20. Two hundred nanometre sections were obtained and imaged by a Spirit transmission electron microscope (FEI Company) operating at 100 kV.
Telomere length analysis
Detection of telomere length by qPCR and Southern blotting was conducted as previously described (51). The primers used for detection of telomere length are listed in Supplementary Table S2.
Induction of endogenous expression of ZKSCAN3 by using CRISPR-dCas9 transcriptional activation system
The CRISPR/dCas9-mediated gene activation was performed as previously described (61,62). In brief, guide RNA targeting the transcriptional start site (TSS) locus of ZKSCAN3 and two non-targeting controls (NTCs) were constructed into lentiSAM v2 vector (Addgene #75112). For the production of lentivirus particles, HEK293T cells were co-transfected with lentiviral sgRNA plasmids or lentiMPH v2 (Addgene #89308), along with psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259). For the induction of endogenous expression of ZKSCAN3, RS-hMSCs were co-transduced with the produced LentiSAM v2 and LentiMPH v2 as described above to transcriptionally activate the expression of ZKSCAN3 for 48 hr before selected with blasticidin and hygromycin for 7 days. The selected cells were collected for the subsequent analysis of ZKSCAN3 mRNA and protein expression levels and senescence phenotypes.
ELISA analysis of the secretion of MCP1 and IL6
To analyse the protein secretion of MCP1 and IL6 in hMSCs, the supernatants from ZKSCAN3+/+ and ZKSCAN3-/- hMSCs were gathered and centrifuged at 500 g for 5 min at RT. Then the supernatants were incubated with MCP1 or IL6 antibody-coated ELISA plates according to the manufacturer's manual (MCP1 (R&D Systems), IL6 (BioLegend)). Finally, the positive signals were detected at 450 nm by using Synergy H1 (BioTek) and the measurements were normalized by cell numbers for data analysis.
Chromatin immunoprecipitation (ChIP)-qPCR and ChIP-seq
ChIP-qPCR and ChIP-seq were performed according to previous protocols with slight modifications (22,44). Briefly, 1 × 106 hMSCs were crosslinked with 1% (vol/vol) formaldehyde diluted in PBS for 8 min or 12 min. The reaction was stopped by incubation in 0.125 M Glycine for 5 min at RT. After washes with PBS, cells were resuspended in ice-cold lysis buffer (50 mM Tris–HCl, 10 mM EDTA, 1% SDS, pH 8.0) for 5 min. After sonication by a Bioruptor® Plus device (Diagenode), supernatants were incubated overnight at 4°C with Protein A/G dynabeads (Thermo Fisher Scientific, 10004D) conjugated with anti-H3K9me3 (Abcam, Ab8898), anti-ZKSCAN3 (Santa Cruz, sc-515285), or rabbit IgG (Cell Signaling Technology, 2729S)/mouse IgG (Santa Cruz, sc-69786). Subsequently, elution and reverse cross-linking were performed at 68°C for 3 hr on a thermomixer. DNA was then isolated by the phenol–chloroform–isoamylalcohol extraction and ethanol precipitation method, after which purified DNA was subjected to qPCR for evaluation of H3K9me3 or ZKSCAN3 occupation at repetitive sequences. The primers used for ChIP-qPCR are listed in Supplementary Table S2. For H3K9me3 ChIP-seq, the DNA fragments were used to construct the library with the incorporation of spike-in controls via KAPA Hyper Prep Kits with PCR Library Amplification/Illumina series (KK8504) following the manufacturer's instructions for subsequent analyses.
ChIP-seq data processing
First, the genome sequences for human (hg19) and drosophila (dm3) were merged to a combined genome sequence. To avoid confusion caused by use of the same chromosome names, all human chromosomes were identified by a ‘hg19_’ prefix and all drosophila chromosomes by a ‘dm3_’ prefix. A custom Bowtie2 index was built from this mixed genome sequence using the ‘bowtie2-build’ command. After removing low quality reads and Illumina adapters, cleaned reads were mapped to this custom library using Bowtie2 (v2.2.9) with default parameters. The resultant SAM files were split, such that reads mapping to human chromosomes (hg19_) and reads mapping to drosophila chromosomes (dm3_) were placed in two separate files. We used reads aligning to human chromosomes for downstream analysis and reads aligning to drosophila chromosomes to calculate the ChIP-Rx ratio (denoted as Rx) as previously described (63,64):
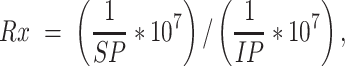 |
in which SP is the number of reads aligning to drosophila chromosomes in the target sample and IP indicates input reads. For downstream analysis, reads from mitochondrial DNA and the Y chromosome, and reads with low mapping quality (MAPQ score < 10) were removed using samtools (v1.9) (65). Duplicated reads were also identified and removed for future analysis using Picard software (v1.113). To visualize the ChIP-seq signals, we calculated the normalized read counts by RPKM (reads per kilobase per million mapped reads) for each 10 bp bin and multiplied by Rx using bamCoverage function in deepTools (v3.3.0) software with parameter ‘-scaleFactor’ (66).
Identification of ‘H3K9me3 mountains’ across genome
First, we called H3K9me3 peaks using SICER (v1.1) with the parameter ‘-w 500 -g 5’ (67), and removed all the peaks with a cut-off FDR (false discovery rate) as more than 1%. Then we calculated H3K9me3 signals (CPM, count per million) for each H3K9me3 peak, ranked H3K9me3 peaks by increasing CPM, and plotted the H3K9me3 occupancy. In these plots, we identified an obvious inflection point, after which the H3K9me3 signals increases dramatically; inflection points in these curves were calculated using R package inflection (v1.3.5). We further defined H3K9me3 peaks above the inflection point to be ‘H3K9me3 mountains’. The locations of ‘H3K9me3 mountains’ are listed in Supplementary Table S5.
ATAC-seq
A total of 50,000 cells of ZKSCAN3+/+ and ZKSCAN3-/- hMSCs were washed twice with 500 μl cold PBS and dissociated in 50 μl lysis buffer (10 mM Tris–HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% (v/v) Nonidet P-40 Substitute). The sample was then centrifuged at 500 g for 10 min at 4°C, followed by incubation at 37°C for 30 min supplemented with 50 μl transposition reaction mix (10 μl 5× TTBL buffer, 4 μl TTE mix and 36 μl nuclease-free H2O) from the TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme Biotech). TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme Biotech) was used to amplify and purify the library. Library quality was checked via Fragment Analyzer. Finally, 150-bp paired-end sequencing was performed on an Illumina HiSeq X-10.
ATAC-seq data processing
For ATAC-seq data analysis, low quality reads and Illumina adapters were removed by TrimGalore (v0.4.4_dev). The remaining clean reads were mapped to the UCSC human hg19 genome using Bowtie2 (v2.2.9) with default parameters. To avoid the effect of sequencing bias and depth to the best extent possible, we merged all replicates for each sample and randomly sampled the same number (56 million) of high-quality reads for each cell type. Mapped reads from mitochondrial DNA and the Y chromosome, and reads with low mapping quality (MAPQ score < 10) were filtered using samtools (v1.9) (65). Duplicate reads were removed using Picard software (v1.113) for future analysis. We extended each read by 250 bp and normalized read counts by RPKM for each 10 bp using the bamCoverage function in deepTools (v3.3.0) software (66).
Peak calling was performed with MACS2 (v2.1.2) after exclusion of blacklisted regions (with parameters ‘-nomodel -shift 0 -extsize 250’ (68)). Genome annotation was performed with HOMER using the ‘annotatePeaks’ function (69). To identify consensus peaks, we obtained a set of all open chromatin peaks that were present in ZKSCAN+/+ and ZKSCAN3-/-hMSCs, and identified the overlapping peaks using Diffbind (70). We then analyzed the differential ATAC-seq peaks between ZKSCAN3+/+ and ZKSCAN3-/- hMSCs using DiffBind defined by abs (log2FC) > 1 and BH-adjusted FDR < 0.05. The locations of ATAC-seq peaks are listed in Supplementary Table S5.
DamID-seq
pLgw V5-EcoDam and pLgw EcoDam-V5-EMD were kind gifts from Prof. Bas van Steensel, NKI. DamID-seq was performed as previously described with minor modifications (71). In brief, Dam and Dam-EMD lentiviruses were concentrated by ultracentrifugation at 19 400 g for 2.5 hr and then resuspended in PBS. 2 × 105ZKSCAN3+/+ or ZKSCAN3-/- hMSCs were plated in each well of a six-well plate. After 24 hr, culture medium was substituted with fresh culture medium containing either Dam or Dam-EMD lentivirus. Cells were collected 72 hr after transduction and genomic DNA was isolated using a DNeasy Blood & Tissue Kit (Qiagen). Genomic DNA was subjected to DpnI digestion, adaptor ligation, DpnII digestion, PCR amplification and purification as previously described (71). The amplified DNA was then sonicated and digested with AlwI (New England Biolabs) to remove the adaptors. The DNA library was constructed using a NEBNext ultra DNA library prep kit for Illumina (New England Biolabs, E7370S). The libraries were pooled and sequenced by 150-bp paired-end sequencing on an Illumina NovaSeq sequencer.
DamID data processing
For DamID-seq data analysis, raw reads of Dam and Dam-EMD data were trimmed by TrimGalore (v0.4.4_dev) software. The remaining clean reads were mapped to the UCSC human hg19 genome using bowtie2 (v2.2.9) with default parameters. Mapped reads from mitochondrial DNA and the Y chromosome, and reads with low mapping quality (MAPQ score < 10) were filtered using samtools (v1.9) (65). Duplicate reads were also removed using Picard and saved for future analysis. To avoid the effect of sequencing bias and depth to the best extent possible, we merged all replicates for each sample and randomly sampled the same number (105 million) of high-quality reads for each cell type. LAD calling was performed based on sampled bam files using ChromHMM (v1.20) (72). Only the medians of DamID signals of LADs with a cut-off of 0 or better were kept as LADs. To visualize the DamID signals, we calculated the log2 ratio of Dam-EMD and Dam signals [log2(Dam-EMD versus Dam)] for each 10-bp bin using deepTools (v3.3.0) software (66). We calculated the difference of DamID signals at LADs between ZKSCAN3+/+ and ZKSCAN3-/- hMSCs with a cutoff of 0.3. LADs exceeding this cutoff were considered to have changed. The locations of LADs are listed in Supplementary Table S5.
Identification of LAD-localized repetitive elements
First, repetitive sequence annotations were downloaded using ‘configureHomer.pl -install hg19’ in HOMER software (v4.9.1) and LAD calling was performed as described above. Next, we extracted the repetitive sequences in LADs using the GenomicRanges Bioconductor R package (v1.36.0) and calculated the DamID signals in these repetitive sequences using deepTools (v3.3.0) (66). Only the medians of DamID signals of repetitive sequences with a cut-off of 0 or better were kept as LAD-localized repetitive sequences. LAD-random sequences were generated using the ‘shuffle’ function in bedtools (v2.25) (73).
Statistical analyses
All the data are presented as the mean ± SEMs. To compare the differences between treatment and control groups, we assumed equal variance and performed statistical analyses using a two-tailed Student's t-test in PRISM software (GraphPad 8 Software). P values < 0.05 , < 0.01 and < 0.001 are considered statistically significant and indicated using *, ** and ***, respectively.
RESULTS
Downregulation of ZKSCAN3 in senescent hMSCs
To examine whether the expression of ZKSCAN3 changes during stem cell aging, we first investigated the ZKSCAN3 level in replicative-senescent (RS) hMSCs and premature-senescent hMSCs, including Hutchinson-Gilford Progeria Syndrome hMSCs (HGPS-hMSC, LMNAG608G/+ and LMNAG608G/G608G) and Werner Syndrome hMSCs (WS-hMSC, WRN-/-) (19,21,22,50,52). The results showed decreased ZKSCAN3 protein levels in these senescent hMSCs compared to those in the corresponding control lines (Figure 1A–C). Consistently, ZKSCAN3 was also downregulated in human primary hMSCs derived from aged individuals compared to those derived from young individuals (Figure 1D) (21,22). The downregulation of ZKSCAN3 in a variety of senescent hMSCs suggests its potential as a regulator of hMSC aging.
Figure 1.

Absence of ZKSCAN3 results in premature cellular senescence in hMSCs. (A) Western blot analysis of ZKSCAN3 protein in early-, middle- and late-passage (P3, P8, P13) hMSCs. Representative western blot images are shown on the left, and the statistical analysis of relative protein expression levels of ZKSCAN3 is on the right. GAPDH was used as the loading control. Data are presented as the mean ± SEMs, n = 3. *P < 0.05 (two tailed t-test). The asterisk indicates the band of ZKSCAN3 and P indicates passage. (B) Western blot analysis of ZKSCAN3 protein in LMNA+/+, LMNAG608G/+ and LMNAG608G/G608G hMSCs (P7). Representative western blot images are shown on the left, and the statistical analysis of the relative protein expression levels of ZKSCAN3 is on the right. β-actin was used as the loading control. Data are presented as the mean ± SEMs, n = 3. *P < 0.05, **P < 0.01 (two tailed t-test). (C) Western blot analysis of ZKSCAN3 protein in WRN+/+ and WRN-/- hMSCs (P7). Representative western blot images are shown to the left, and the statistical analysis of the relative protein expression levels of ZKSCAN3 is on the right. β-actin was used as the loading control. Data are presented as the mean ± SEMs, n = 3. **P < 0.01 (two tailed t-test). (D) Western blot analysis of ZKSCAN3 protein in young and physiologically aged hMSCs (P7). Representative western blot images are shown on the left, and the statistical analysis of the relative protein expression levels of ZKSCAN3 is on the right. GAPDH was used as the loading control. Data are presented as the mean ± SEMs, n = 4. **P < 0.01 (two tailed t-test). (E) Schematic of hMSC differentiation. (F) FACS analysis of hMSC surface markers CD73, CD90 and CD105 in ZKSCAN3+/+ and ZKSCAN3-/- hMSCs. (G) Western blot analysis of ZKSCAN3 protein in ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P4). GAPDH was used as the loading control. (H) Clonal expansion ability analysis of ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10). Data are presented as the mean ± SEMs, n = 3. ***P < 0.001 (two tailed t-test). (I) SA-β-gal staining of ZKSCAN3+/+ and ZKSCAN3-/-hMSCs (P10). Scale bar, 100 μm. Data are presented as the mean ± SEMs, n = 3. ***P < 0.001 (two tailed t-test). (J) Staining of cell nuclei by Hoechst 33342 in ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10). The relative nuclear size is shown on the right. Scale bar, 10 μm. Data are presented as the mean ± SEMs, n = 300. ***P < 0.001 (two tailed t-test). (K) Telomere length analysis of ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10) by qPCR. Data are presented as the mean ± SEMs, n = 4. ***P < 0.001 (two tailed t-test). (L) ELISA analysis of the secretion of MCP1 and IL6 in ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10). Data are presented as the mean ± SEMs, n = 5 for MCP1 detection, n = 3 for IL6 detection. *P < 0.05, ***P < 0.001 (two tailed t-test). (M) Western blot analysis of P16, P21 in ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10). GAPDH was used as the loading control. (N) ZKSCAN3+/+ and ZKSCAN3-/- hMSCs infected with a lentivirus expressing luciferase were implanted into the tibialis anterior (TA) muscle of nude mice. Luciferase activities were detected at day D0, D2 and D4 after injection by an in vivo imaging system (IVIS). Data are presented as the mean ± SEMs, n = 5. **P < 0.01, ***P < 0.001 (two tailed t-test).
Generation of ZKSCAN3-deficient hMSCs
To further dissect the role of ZKSCAN3 in hMSC aging, we generated ZKSCAN3-knockout human embryonic stem cells (hESCs) via CRISPR/Cas9-mediated gene knockout technology (Supplementary Figure S1A) (22,44). Genomic polymerase chain reaction (PCR) and DNA sequencing verified the successful targeting of the ZKSCAN3 locus (Supplementary Figure S1A). Western blot analysis further confirmed the loss of ZKSCAN3 protein (Supplementary Figure S1B). ZKSCAN3-/- hESCs maintained normal morphology and expressed pluripotency markers NANOG, SOX2 and OCT4, as did ZKSCAN3+/+ hESCs (Supplementary Figure S1C). ZKSCAN3-/- hESCs maintained the capacity to differentiate into three germ layer lineages in vivo as indicated by the teratoma formation assay (Supplementary Figure S1D). No off-target cleavage was observed upon gene editing (Supplementary Figure S1E). In addition, ZKSCAN3-/- hESCs maintained genomic integrity after more than 30 passages, which was verified by karyotype analysis and genome-wide copy number variation (CNV) analysis (Supplementary Figure S1F and G). ZKSCAN3-/- hESCs also exhibited comparable growth kinetics to ZKSCAN3+/+ hESCs (Supplementary Figure S1H and I). Overall, these data show that ZKSCAN3-/-hESCs manifest normal pluripotent characteristics.
To elucidate the function of ZKSCAN3 in hMSC aging, we next differentiated ZKSCAN3+/+ and ZKSCAN3-/- hESCs into hMSCs (Figure 1E). Both ZKSCAN3+/+ and ZKSCAN3-/- hMSCs expressed comparable levels of mesenchymal progenitor markers CD73, CD90, and CD105 (Figure 1F), and did not express hMSC-irrelevant antigens CD34, CD43 and CD45 (Supplementary Figure S2A). In addition, both ZKSCAN3+/+ and ZKSCAN3-/- hMSCs maintained chromosomal integrity as indicated by genome-wide CNV analysis (Supplementary Figure S2B), and were able to differentiate towards osteoblasts, chondrocytes and adipocytes, in spite of the knockout hMSCs having partially compromised chondrogenesis potential (Supplementary Figure S2C). The absence of ZKSCAN3 protein in ZKSCAN3-/- hMSCs was confirmed by western blot (Figure 1G).
ZKSCAN3 deficiency accelerates hMSC senescence
Further analysis of the cellular characteristics of ZKSCAN3+/+ and ZKSCAN3-/- hMSCs revealed that ZKSCAN3 deficiency accelerated hMSC senescence, as evidenced by early-onset growth arrest, impaired clonal expansion ability, reduced proportion of Ki67-positive cells upon serial passaging, and decreased percentage of cells in synthesis (S) phase (Figure 1H and Supplementary Figure S2D–F). Additionally, increased senescence-associated (SA)-β-galactosidase (SA-β-gal) activity and increased nuclear size were observed in ZKSCAN3-/- hMSCs (Figure 1I and J). Typical indicators of senescence including shortened telomere, increased DNA damage, downregulation of DNA damage response-related genes and chromosome segregation-related genes were detected in ZKSCAN3-/- hMSCs (Figure 1K and Supplementary Figure S2G–L). In addition, ZKSCAN3-deficiency activated senescence-associated secretory phenotype (SASP) and upregulated the expression of senescence markers in hMSCs (Figure 1L and M) (74). Consistent with the in vitro observations, ZKSCAN3-/- hMSCs exhibited an accelerated decay in vivo when implanted into muscles of nude mice (Figure 1N). Taken together, these results indicate that deficiency of ZKSCAN3 promotes premature senescence and functional attrition of hMSCs.
ZKSCAN3 acts as an epigenetic regulator to maintain heterochromatin organization in hMSCs
Given that ZKSCAN3 is a known transcriptional repressor of autophagy in human cancer cells (11), we next explored if depletion of ZKSCAN3 would induce the expression of autophagy- and lysosome-associated genes in hMSCs. However, RNA-seq and RT-qPCR data revealed no obvious changes in the expression of such genes (including MAP1LC3B, ATG5 and ULK1) upon ZKSCAN3 depletion (Supplementary Figure S2M and N). This implies that the accelerated senescence caused by ZKSCAN3 deficiency is unrelated to the transcriptional regulation of autophagy by ZKSCAN3, at least in hMSCs.
To elucidate the underlying mechanisms by which ZKSCAN3 accelerates hMSC senescence, we searched for interaction partners of ZKSCAN3 by expressing Flag-tagged ZKSCAN3 in HEK293T cells and performing co-immunoprecipitation (Co-IP) followed by mass spectrometry (MS) (Figure 2A). Many proteins were identified in this process, including those related to heterochromatin, chromosome and nucleus (e.g. nuclear envelope) (Figure 2B). Among these, we confirmed the interaction of ZKSCAN3 with two nuclear lamina proteins, Lamin B1 and Lamin B receptor (LBR) (Figure 2C and Supplementary Figure S3A). Given the limited resolution of Co-IP-MS in characterizing trypsin-insensitive or low abundance proteins, we also performed a Co-IP assay and identified additional heterochromatin components, including KAP1, HP1α as interacting partners of ZKSCAN3 (Figure 2C).
Figure 2.

ZKSCAN3 forms a complex with heterochromatin proteins and nuclear envelope proteins in hMSCs. (A) Schematic representation of Co-IP followed by LC-MS/MS. (B) Gene Ontology Cellular Component (GO-CC) enrichment analysis of ZKSCAN3 interaction proteins identified by Co-IP/MS. (C) Co-IP analysis showing that exogenous ZKSCAN3 interacts with Lamin B1, LBR, KAP1 and HP1α proteins in HEK293T cells. The asterisks represent the bands of indicated proteins. (D) Enrichment of ZKSCAN3 within the regions of repetitive sequences (LINE1, SVA, ALR, Satellite 2) in ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P4) as measured by ChIP-qPCR. Data are presented as the mean ± SEMs, n = 4. ***P < 0.001 (two tailed t-test). (E) Representative TEM images for ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10) showing reduced heterochromatin at the nuclear periphery in ZKSCAN3-/- hMSCs. The percentages of cells with heterochromatin loss at the nuclear periphery are shown on the bottom. Scale bar, 1 μm. (F) Immunostaining of H3K9me3 and Lamin A/C in ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10). Scale bar, 5 μm. The mean intensity of H3K9me3 was measured with ImageJ and the calculated data are shown as the mean ± SEMs, n = 300. ***P < 0.001 (two tailed t-test). (G) Immunostaining of LAP2 in ZKSCAN3+/+ and ZKSCAN3-/-hMSCs (P10). White arrows represent the cells with decreased LAP2 expression. Scale bar, 25 μm. The mean intensity of LAP2 was measured with ImageJ and the calculated data are shown as the mean ± SEMs, n = 150. ***P < 0.001 (two tailed t-test). (H) Western blot analysis of the expression levels of Lamin B1, LBR, KAP1 and HP1α proteins in ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10). GAPDH was used as the loading control.
Based on these data, we hypothesized that ZKSCAN3 may function as a potential component of heterochromatin to modulate genomic organization and cellular senescence. As heterochromatin is characterized by the enrichment of repetitive sequences that are often transcriptionally derepressed in senescent cells (23,29), we explored whether ZKSCAN3 regulates these repetitive sequences. Chromatin immunoprecipitation-qPCR (ChIP-qPCR) analysis identified specific occupation of ZKSCAN3 at a number of genomic repetitive sequences including LINE1, SVA, ALR and Satellite 2 in hMSCs (Figure 2D). Consistent with the previous findings that Lamin B1, LBR and lamina-associated protein LAP2 physically interact with heterochromatin proteins to maintain heterochromatin stability and genomic LAD organization at the nuclear periphery (75,76), we observed that ZKSCAN3 deficiency caused a general loss of heterochromatin, especially beneath the nuclear membrane (Figure 2E and F). We further noted a decrease in protein level of nuclear lamina-associated proteins LAP2, Lamin B1 and LBR, and heterochromatin-associated proteins KAP1 and HP1α in ZKSCAN3-/- hMSCs (Figure 2G and H). The enrichment of KAP1 and HP1α at genomic repetitive sequences was also reduced in ZKSCAN3-/- hMSCs (Supplementary Figure S3B and C). Together, these findings demonstrate that ZKSCAN3 functions as a direct epigenetic regulator to maintain heterochromatin stability.
Epigenetic derepression of LAD-localized repetitive sequences in ZKSCAN3-deficient hMSCs
To characterize the genome-wide epigenetic alterations caused by ZKSCAN3 deficiency, we performed DNA adenine methyltransferase identification following next-generation sequencing (DamID-seq) and H3K9me3 chromatin immunoprecipitation sequencing (ChIP-seq). DamID was performed by introducing DNA adenine methyltransferase (Dam) fused to Emerin (EMD, an inner nuclear membrane-associated protein), or Dam protein alone in hMSCs (Figure 3A) (77,78). We observed a decrease in DamID signals across annotated LADs in ZKSCAN3-/- hMSCs compared to those in ZKSCAN3+/+ hMSCs (Figure 3B and Supplementary Figure S3D and E), indicating that LADs had become unanchored from the nuclear lamina in the absence of ZKSCAN3. Specifically, LAD-localized repetitive sequences (LINE, SINE and Satellite elements) showed the most significant reduction in DamID signals (Figure 3C and Supplementary Figure S3F and G). This is consistent with the reduction of heterochromatin marker H3K9me3 at the nuclear periphery of ZKSCAN3-deficient hMSCs observed by immunofluorescence staining (Figure 2F). Consistently, ChIP-seq analysis indicated a global loss of H3K9me3 occupancy in ZKSCAN3-/- hMSCs compared to that in ZKSCAN3+/+ hMSCs, especially within LADs and heterochromatic regions across the genome (Figure 3D and E and Supplementary Figure S3H-L) (21,22). The enrichment of H3K9me3 on LAD-localized repetitive sequences (LINE, SINE and Satellite elements) was also decreased in ZKSCAN3-/- hMSCs (Figure 3F and Supplementary Figure S3M), which was confirmed by ChIP-qPCR (Figure 3G).
Figure 3.

ZKSCAN3 regulates heterochromatin architecture and represses repetitive sequence expression in hMSCs. (A) Schematic representation of DamID-seq strategy. (B) Reduced DamID signals [log2 (Dam-EMD vs. Dam)] at LAD regions in ZKSCAN3-/- hMSCs (P10) compared to ZKSCAN3+/+ hMSCs (P10), shown by both violin plot (left) and heatmaps (right). (C) Line plots showing DamID signals [log2 (Dam-EMD vs. Dam)] at LAD-localized repetitive sequence loci in ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10). (D) Sketch map showing the loss of ‘H3K9me3 mountain’ distribution over 23 chromosomes in ZKSCAN3-/- hMSCs (P10) compared to ZKSCAN3+/+ hMSCs (P10). (E) Violin plots showing the loss of H3K9me3 signals in H3K9me3 mountains, LAD and heterochromatin (HC) regions in ZKSCAN3-/- hMSCs (P10) compared to ZKSCAN3+/+ hMSCs (P10). (F) Violin plots showing the loss of H3K9me3 signals at LAD-localized repetitive sequence regions (LINE, SINE, Satellite family) in ZKSCAN3-/- hMSCs (P10) compared to ZKSCAN3+/+ hMSCs (P10). (G) Enrichment of H3K9me3 within the regions of repetitive sequences in ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10) as measured by ChIP-qPCR. Data are presented as the mean ± SEMs, n = 4. *P < 0.05, ***P < 0.001 (two tailed t-test). (H) Violin plot showing the increase of ATAC signals at ATAC-seq peaks within LADs in ZKSCAN3-/- hMSCs (P10) compared to ZKSCAN3+/+ hMSCs (P10). (I) The distribution feature of ATAC-seq peaks annotated to different repetitive sequence regions in ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10). (J) Violin plots showing ATAC signals of LAD-localized repetitive sequence regions in ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10). (K) Representative tracks of DamID-seq, H3K9me3 ChIP-seq and ATAC-seq analyses showing decreased DamID signal, reduced H3K9me3 signal coupled with increased ATAC signal in ZKSCAN3-/- hMSCs (P10) compared to ZKSCAN3+/+ hMSCs (P10). (L) RNA-seq analysis showing the increased mRNA expression levels of repetitive sequences in ZKSCAN3-/- hMSCs (P10) compared to ZKSCAN3+/+ hMSCs (P10). (M) Heatmap showing the mRNA expression levels of repetitive sequences in the ZKSCAN3+/+ and ZKSCAN3-/- hMSCs (P10) by RT-qPCR.
To further detail the changes in chromatin accessibility on a genome-wide scale and identify regions with more accessibility (and hence higher activity), we performed transposase-accessible chromatin sequencing (ATAC-seq) (79,80). The ATAC signals were increased across the genome, including within the heterochromatin-enriched LADs and LAD-localized repetitive sequences in ZKSCAN3-/- hMSCs (Figure 3H–J and Supplementary Figure S4A-H), implying that the condensed heterochromatin underneath the nuclear lamina was more accessible in ZKSCAN3-deficient hMSCs. Notably, the global ATAC peak signals were negatively correlated to those of DamID and H3K9me3 (Figure 3K and Supplementary Figure S4I). Consistent with these epigenomic changes, RNA-seq and RT-qPCR analyses revealed increased transcript levels of these repetitive sequences in ZKSCAN3-/- hMSCs compared to those in their ZKSCAN3+/+ counterparts (Figure 3L and M). Taken together, these findings suggest that ZKSCAN3 deficiency in hMSCs triggers a series of epigenetic changes that include detachment of LADs from the nuclear lamina, loss of constitutive heterochromatin and increase of chromatin accessibility at the nuclear periphery. Collectively, these changes result in the transcriptional induction of repetitive genomic sequences.
Overexpression of ZKSCAN3 attenuates hMSC senescence
To further define the role of ZKSCAN3 in defying hMSC aging, we transduced ZKSCAN3-/- hMSCs with lentiviral vector encoding ZKSCAN3, and found that the senescence-associated phenotypes were ameliorated, demonstrated by increased Ki67-positive cells and clonal expansion ability, decreased SA-β-gal-positive cells, reduced nuclear size and downregulated P16 and IL6 (Figure 4A–E and Supplementary Figure S5A). Likewise, ZKSCAN3-/- hMSCs supplemented with ZKSCAN3 showed recovery in the levels of heterochromatin markers H3K9me3, HP1α and LAP2 (Figure 4F–H). Notably, overexpression of heterochromatin-associated proteins KAP1 or HP1α, the newly identified partners of ZKSCAN3, reversed cellular senescence in ZKSCAN3-/- hMSCs (Supplementary Figure S5A–F). We further confirmed that the exogenously introduced ZKSCAN3 bound to genomic repetitive sequences, increased enrichment of H3K9me3 at these elements and thus re-silenced their transcription (Figure 4I–K). Likewise, ectopic expression of KAP1 or HP1α in ZKSCAN3-/- hMSCs also restored the enrichment of H3K9me3 on repetitive sequences and repressed their expression (Supplementary Figure S5G and H). Altogether, these observations indicate that the re-introduction of ZKSCAN3 in ZKSCAN3-/-hMSCs alleviates hMSC aging via the restoration of heterochromatin.
Figure 4.

Overexpression of ZKSCAN3 delays ZKSCAN3-/- hMSC senescence. (A) Immunostaining of Ki67 in ZKSCAN3-/- hMSCs (P8) transduced with lentiviruses expressing luciferase (Luc) or ZKSCAN3. Scale bar, 25 μm. White arrows represent Ki67-positive cells. Data are presented as the mean ± SEMs, n = 3. **P < 0.01 (two tailed t-test). (B) Clonal expansion ability analysis of ZKSCAN3-/- hMSCs (P8) transduced with lentiviruses expressing Luc or ZKSCAN3. Data are presented as the mean ± SEMs, n = 3. **P < 0.01 (two tailed t-test). (C) SA-β-gal staining of ZKSCAN3-/- hMSCs (P8) transduced with lentiviruses expressing Luc or ZKSCAN3. Data are presented as the mean ± SEMs, n = 3. **P < 0.01 (two tailed t-test). (D) Staining of cell nuclei by Hoechst 33342 in ZKSCAN3-/- hMSCs (P8) transduced with lentiviruses expressing Luc or ZKSCAN3. The nuclear size was measured with ImageJ and the data are presented as the mean ± SEMs, n = 150. ***P < 0.001 (two tailed t-test). (E) Heatmap showing the mRNA expression levels of P16 and IL6 in ZKSCAN3-/- hMSCs (P8) transduced with lentiviruses expressing Luc or ZKSCAN3 by RT-qPCR. (F) Immunostaining of H3K9me3 and Lamin A/C in ZKSCAN3-/- hMSCs (P8) transduced with lentiviruses expressing Luc or ZKSCAN3. Scale bar, 5 μm. The mean intensity of H3K9me3 was measured with ImageJ and the data are presented as the mean ± SEMs, n = 150. ***P < 0.001 (two tailed t-test). (G) Immunostaining of HP1α in ZKSCAN3-/- hMSCs (P8) transduced with lentiviruses expressing Luc or ZKSCAN3. Scale bar, 25 μm. White arrows represent cells with decreased HP1α expression. The mean intensity of HP1α was measured with ImageJ and the data are presented as the mean ± SEMs, n = 300. ***P < 0.001 (two tailed t-test). (H) Immunostaining of LAP2 in ZKSCAN3-/- hMSCs (P8) transduced with lentiviruses expressing Luc or ZKSCAN3. Scale bar, 25 μm. White arrows represent the cells with decreased LAP2 expression. The mean intensity of LAP2 was measured with ImageJ and the data are presented as the mean ± SEMs, n = 300. ***P < 0.001 (two tailed t-test). (I) Enrichment of ZKSCAN3 within the regions of repetitive sequences in ZKSCAN3-/- hMSCs (P8) transduced with lentiviruses expressing Luc or ZKSCAN3 as measured by ChIP-qPCR. Data are presented as the mean ± SEMs, n = 4. **P < 0.01, ***P < 0.001 (two tailed t-test). (J) Enrichment of H3K9me3 within the regions of repetitive sequence in ZKSCAN3-/- hMSCs (P8) transduced with lentiviruses expressing Luc or ZKSCAN3 as measured by ChIP-qPCR. Data are presented as the mean ± SEMs, n = 4. ***P < 0.001 (two tailed t-test). (K) Heatmap showing the mRNA expression levels of repetitive sequences in ZKSCAN3-/- hMSCs (P8) transduced with lentiviruses expressing Luc or ZKSCAN3 by RT-qPCR.
Finally, we tested whether the overexpression of ZKSCAN3 would rejuvenate replicatively, pathologically or physiologically aged hMSCs. ZKSCAN3 was introduced via lentivirus into RS-hMSCs, premature senescent HGPS-hMSCs and WS-hMSCs, and primary hMSCs from healthy elderly individuals (Supplementary Figure S6A). ZKSCAN3 overexpression rescued the senescence-associated phenotypes in all senescent hMSCs tested (Figure 5A–F and Supplementary Figure S6B–F), accompanied by an increase in H3K9me3 levels and a decrease in transcript levels of repetitive sequences (Figures 5G and H and Supplementary Figure S6G and H). Furthermore, we induced the endogenous expression of ZKSCAN3 in RS-hMSCs by using CRISPR-dCas9 transcriptional activation system (Supplementary Figure S7A-C) (61,62). We found the activation of endogenous ZKSCAN3 rescued the senescence-associated phenotypes in RS hMSCs (Supplementary Figure S7D-J). Altogether, these results indicate that ZKSCAN3 overexpression exerts a geroprotective role in aged hMSCs, at least partially through heterochromatin stabilization.
Figure 5.

ZKSCAN3 overexpression alleviates hMSC aging. (A) Immunostaining of Ki67 in RS (P10) (upper), LMNAG608G/+ (P6) (middle) and an aged individual-derived primary (P8) (lower) hMSCs transduced with lentiviruses expressing Luc or ZKSCAN3, respectively. Scale bar, 25 μm. White arrows represent Ki67-positive cells. Data are presented as the mean ± SEMs, n = 3. *P < 0.05, **P < 0.01 (two tailed t-test). (B) Clonal expansion ability analysis of RS (P10) (upper), LMNAG608G/+ (P6) (middle) and an aged individual-derived primary (P8) (lower) hMSCs transduced with lentiviruses expressing Luc or ZKSCAN3. Data are presented as the mean ± SEMs, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001 (two tailed t-test). (C) SA-β-gal staining of RS (P10) (upper), LMNAG608G/+ (P6) (middle) and an aged individual-derived primary (P8) (lower) hMSCs transduced with lentiviruses expressing Luc or ZKSCAN3. Data are presented as the mean ± SEMs, n = 3. *P < 0.05, **P < 0.01 (two tailed t-test). (D) Relative nuclear size analysis in RS (P10) (upper), LMNAG608G/+ (P6) (middle) and an aged individual-derived primary (P8) (lower) hMSCs transduced with lentiviruses expressing Luc or ZKSCAN3. Data are presented as the mean ± SEMs, n = 200. *P < 0.05, **P < 0.01 (two tailed t-test). (E) Heatmaps showing the mRNA expression levels of P16 and IL6 in RS (P10) (upper), LMNAG608G/+ (P6) (middle) and an aged individual-derived primary (P8) (lower) hMSCs transduced with lentiviruses expressing Luc or ZKSCAN3 by RT-qPCR. (F) Immunostaining of HP1α in RS (P10) (left), LMNAG608G/+ (P6) (middle) and an aged individual-derived primary (P8) (right) hMSCs transduced with lentiviruses expressing Luc or ZKSCAN3. Scale bar, 25 μm. Data are presented as the mean ± SEMs, n = 300. ***P < 0.001 (two tailed t-test). (G) Enrichment of H3K9me3 within the regions of repetitive sequences in RS (P10) (left), LMNAG608G/+ (P6) (middle) and an aged individual-derived primary (P8) (right) hMSCs transduced with lentiviruses expressing Luc or ZKSCAN3 as measured by ChIP-qPCR. Data are presented as the mean ± SEMs. n = 4. **P < 0.01, ***P < 0.001 (two tailed t-test). (H) Heatmaps showing the mRNA expression levels of repetitive sequences in RS (P10) (left), LMNAG608G/+ (P6) (middle) and an aged individual-derived primary (P8) (right) hMSCs transduced with lentiviruses expressing Luc or ZKSCAN3 by RT-qPCR.
DISCUSSION
We have demonstrated a novel role for ZKSCAN3 in safeguarding hMSCs from senescence. Highlights of this study include the following: (i) ZKSCAN3-knockout hESCs and hMSCs were generated for the first time by combining CRISPR/Cas9 gene editing and stem cell directed differentiation. (ii) The level of ZKSCAN3 was consistently decreased in replicatively, prematurely, and physiologically senescent hMSCs. (iii) ZKSCAN3 deficiency accelerated cellular senescence in hMSCs. (iv) ZKSCAN3 maintained hMSC heterochromatin stability as an epigenetic regulator by tethering heterochromatin components to nuclear lamina-associated proteins, thus anchoring the LADs to the nuclear periphery and stabilizing repetitive genomic sequences (Figure 6). (v) Exogenous overexpression of ZKSCAN3 stabilized heterochromatin and rejuvenated aged hMSCs.
Figure 6.

ZKSCAN3 protects hMSC from aging via the maintenance of heterochromatin stability. A model showing that ZKSCAN3 safeguards hMSCs from senescence by stabilizing heterochromatin. In young hMSCs, ZKSCAN3 couples with the heterochromatin-associated proteins KAP1 and HP1α as well as nuclear lamina proteins Lamin B1 and LBR. The complex tethers H3K9me3-enriched heterochromatin to the nuclear periphery and represses transcription of repetitive sequences. In aged hMSCs, downregulation of ZKSCAN3 results in heterochromatin instability, upregulation of repetitive sequences, and cellular senescence.
In contrast to the abundant studies on other members of the KRAB-ZFP family, the function of human ZKSCAN3 was largely unknown, partially due to a lack of appropriate research models; most of the existing studies on ZKSCAN3 were conducted in human cancer cell lines (6–9,11) or murine models (13). For the first time, we generated the ZKSCAN3-deficient hESCs and their hMSC derivatives and provided a valuable experimental platform for studying the role of human ZKSCAN3. Consistent with prior studies that identified ZKSCAN3 as a promoter of cancer cell proliferation (6,9,11), we showed that ZKSCAN3 deficiency retarded cell proliferation in hMSCs and was associated with early-onset senescence. Yet, different from its known role as a transcription repressor that regulates autophagosome formation and lysosomal biogenesis in cancer cells and neurons (11,12), neither our study nor another recently published study revealed any change in the expression of autophagy-related genes in ZKSCAN3-deficient hMSCs or mice (13), respectively. These differences may be due to the distinct cellular features between human transformed tumour cell lines, human diploid stem cells, and murine cells. Instead, we found that ZKSCAN3 acts as an epigenetic regulator to maintain heterochromatin organization and repress LAD-localized repetitive sequences in hMSCs. Thus, we have uncovered a novel function of ZKSACN3 in the regulation of homeostasis and aging in human adult stem cells, which contributes to an unprecedented understanding of SCAN-ZFP proteins.
As far as we know, this is the first evidence that ZKSCAN3 functions as a heterochromatin stabilizer by interacting with the nuclear lamina components and heterochromatin-related proteins (required for directing heterochromatin-enriched LADs to the nuclear envelope). It should be noted that some other (non-SCAN) KRAB-ZFPs have been reported to interact with KAP1 as a scaffold to recruit DNA methyltransferases (DNMTs) and histone methyltransferases, and form a complex that represses the expression of transposon elements (TEs) (1,3,81). For example, ZFP809 associates with KAP1 to repress the expression of exogenous murine leukemia virus (MLV) in mouse embryonic stem cells (82). Derepression of TEs has been implicated in disruption of early embryogenesis and diverse pathological processes including cancers, muscular dystrophy and many other congenital or acquired human diseases (83–87). In contrast, our data indicate that ZKSCAN3, a member of the SCAN-ZFP subfamily, is dispensable for maintenance of hESC pluripotency but counteracts senescence in hMSCs via the repression of TEs and other repetitive sequences, a finding that broadens the scope of biological roles of SCAN-ZFPs.
Consistent with this, activation of repetitive sequences has been implicated in senescence and in aging-related disorders in human cellular models and other mammalian models (87–89). We noticed that ZKSCAN3 can interact with lamina-associated proteins and heterochromatin proteins (like some other KRAB-ZFPs do). The interaction of ZKSCAN3 with nuclear lamina proteins may confer its ability to regulate the stability of heterochromatin within LADs that are normally retained at the nuclear periphery. Thus, ZKSCAN3 may regulate heterochromatin condensation not only by stabilizing the nuclear lamina-heterochromatin complex, but also by participating in its spatial organization, particularly within LADs. Through an integrated analysis of the genome-wide nuclear envelope-chromatin interaction (DamID profile), heterochromatin organization (H3K9me3 ChIP-seq), and chromatin accessibility (ATAC-seq), we demonstrated the predominant delocalization of LADs from the nuclear lamina and a more open chromatin status at LAD-localized repetitive sequences in ZKSCAN3-deficient hMSCs. In summary, we identified ZKSCAN3 as a novel epigenetic regulator that alleviates hMSC senescence by maintaining heterochromatin stability. This deepens our understanding of the epigenetic regulation of heterochromatin during human stem cell aging.
DATA AVAILABILITY
RNA-seq, whole genome DNA sequencing, DamID-seq, H3K9me3 ChIP-seq and ATAC-seq data generated in this study are available at GEO Accession Number GSE146387.
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge L. Bai, R. Bai, Q. Chu, J. Lu, S. Ma and Y. Yang for administrative assistance; Y. Hao and J. Zhou for their help in gene-editing; J. Jia (IBP, CAS) and S. Sun (IBP, CAS) for their assistance in the FACS experiments; J Wang for his help in LC–MS/MS experiments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program of China [2018YFA0107203]; Strategic Priority Research Program of the Chinese Academy of Sciences [XDA16010000]; National Key Research and Development Program of China [2018YFC2000100, 2017YFA0102802, 2017YFA0103304, 2019YFA0110100, 2019YFA0802202]; National Natural Science Foundation of China [81701388, 81625009, 81330008, 91749202, 81861168034, 81921006, 91749123, 31671429, 81671377, 81771515, 31601158, 81701388, 81601233, 31601109, 81822018, 81870228, 81801399, 31801010, 81801370, 81861168034, 81870228, 81922027, 31900524, 31970597]; Program of the Beijing Municipal Science and Technology Commission [Z191100001519005]; Beijing Natural Science Foundation [Z190019]; Beijing Municipal Commission of Health and Family Planning [PXM2018_026283_000002]; Advanced Innovation Center for Human Brain Protection [3500-1192012]; Key Research Program of the Chinese Academy of Sciences [KFZD-SW-221]; K.C. Wong Education Foundation [GJTD-2019-06, GJTD-2019-08]; Young Elite Scientists Sponsorship Program by CAST, Youth Innovation Promotion Association of CAS, the State Key Laboratory of Stem Cell and Reproductive Biology and the State Key Laboratory of Membrane Biology. Funding for open access charge: National Key Research and Development Program of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Urrutia R. KRAB-containing zinc-finger repressor proteins. Genome Biol. 2003; 4:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ecco G., Imbeault M., Trono D.. KRAB zinc finger proteins. Development. 2017; 144:2719–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Witzgall R., O’Leary E., Leaf A., Onaldi D., Bonventre J.V.. The Kruppel-associated box-A (KRAB-A) domain of zinc finger proteins mediates transcriptional repression. Proc. Natl Acad. Sci. U.S.A. 1994; 91:4514–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ma X., Wang X., Gao X., Wang L., Lu Y., Gao P., Deng W., Yu P., Ma J., Guo J. et al.. Identification of five human novel genes associated with cell proliferation by cell-based screening from an expressed cDNA ORF library. Life Sci. 2007; 81:1141–1151. [DOI] [PubMed] [Google Scholar]
- 5. Kim C.W., Roh S.A., Tak K.H., Koh B.M., Ha Y.J., Cho D.H., Kim S.Y., Kim Y.S., Kim J.C.. ZKSCAN3 facilitates liver metastasis of colorectal cancer associated with CEA-expressing tumor. Anticancer Res. 2016; 36:2397–2406. [PubMed] [Google Scholar]
- 6. Zhang X., Jing Y., Qin Y., Hunsucker S., Meng H., Sui J., Jiang Y., Gao L., An G., Yang N. et al.. The zinc finger transcription factor ZKSCAN3 promotes prostate cancer cell migration. Int. J. Biochem. Cell Biol. 2012; 44:1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang L., Hamilton S.R., Sood A., Kuwai T., Ellis L., Sanguino A., Lopez-Berestein G., Boyd D.D.. The previously undescribed ZKSCAN3 (ZNF306) is a novel “driver” of colorectal cancer progression. Cancer Res. 2008; 68:4321–4330. [DOI] [PubMed] [Google Scholar]
- 8. Yang L., Wang H., Kornblau S.M., Graber D.A., Zhang N., Matthews J.A., Wang M., Weber D.M., Thomas S.K., Shah J.J. et al.. Evidence of a role for the novel zinc-finger transcription factor ZKSCAN3 in modulating Cyclin D2 expression in multiple myeloma. Oncogene. 2011; 30:1329–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawahara T., Inoue S., Ide H., Kashiwagi E., Ohtake S., Mizushima T., Li P., Li Y., Zheng Y., Uemura H. et al.. ZKSCAN3 promotes bladder cancer cell proliferation, migration, and invasion. Oncotarget. 2016; 7:53599–53610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Settembre C., Di Malta C., Polito V.A., Arencibia M.G., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P. et al.. TFEB links autophagy to lysosomal biogenesis. Science. 2011; 332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chauhan S., Goodwin J.G., Chauhan S., Manyam G., Wang J., Kamat A.M., Boyd D.D.. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol. Cell. 2013; 50:16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rusmini P., Cortese K., Crippa V., Cristofani R., Cicardi M.E., Ferrari V., Vezzoli G., Tedesco B., Meroni M., Messi E. et al.. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy. 2018; 15:631–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pan H., Yan Y., Liu C., Finkel T.. The role of ZKSCAN3 in the transcriptional regulation of autophagy. Autophagy. 2017; 13:1235–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gewirtz D.A. Autophagy and senescence: a partnership in search of definition. Autophagy. 2013; 9:808–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheon S.Y., Kim H., Rubinsztein D.C., Lee J.E.. Autophagy, cellular aging and Age-related human diseases. Exp Neurobiol. 2019; 28:643–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. López-Otín C., Blasco M.A., Partridge L., Serrano M., Kroemer G.. The hallmarks of aging. Cell. 2013; 153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang J., Lian Q., Zhu G., Zhou F., Sui L., Tan C., Mutalif R.A., Navasankari R., Zhang Y., Tse H.F. et al.. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011; 8:31–45. [DOI] [PubMed] [Google Scholar]
- 18. Kubben N., Zhang W., Wang L., Voss T.C., Yang J., Qu J., Liu G.H., Misteli T.. Repression of the antioxidant NRF2 pathway in premature aging. Cell. 2016; 165:1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu Z., Zhang W., Song M., Wang W., Wei G., Li W., Lei J., Huang Y., Sang Y., Chan P. et al.. Differential stem cell aging kinetics in Hutchinson-Gilford progeria syndrome and Werner syndrome. Protein Cell. 2018; 9:333–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Y., Zhang W., Chang L., Han Y., Sun L., Gong X., Tang H., Liu Z., Deng H., Ye Y. et al.. Vitamin C alleviates aging defects in a stem cell model for Werner syndrome. Protein Cell. 2016; 7:478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang W., Li J., Suzuki K., Qu J., Wang P., Zhou J., Liu X., Ren R., Xu X., Ocampo A. et al.. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015; 348:1160–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deng L., Ren R., Liu Z., Song M., Li J., Wu Z., Ren X., Fu L., Li W., Zhang W. et al.. Stabilizing heterochromatin by DGCR8 alleviates senescence and osteoarthritis. Nat. Commun. 2019; 10:3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang W., Qu J., Liu G.H., Belmonte J.C.I.. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020; 21:137–150. [DOI] [PubMed] [Google Scholar]
- 24. Pheasant M., Mattick J.S.. Raising the estimate of functional human sequences. Genome Res. 2007; 17:1245–1253. [DOI] [PubMed] [Google Scholar]
- 25. Mills R.E., Bennett E.A., Iskow R.C., Devine S.E.. Which transposable elements are active in the human genome. Trends Genet. 2007; 23:183–191. [DOI] [PubMed] [Google Scholar]
- 26. Deininger P.L., Moran J.V., Batzer M.A., Kazazian H.H. Jr. Mobile elements and mammalian genome evolution. Curr. Opin. Genet. Dev. 2003; 13:651–658. [DOI] [PubMed] [Google Scholar]
- 27. De Cecco M., Criscione S.W., Peckham E.J., Hillenmeyer S., Hamm E.A., Manivannan J., Peterson A.L., Kreiling J.A., Neretti N., Sedivy J.M.. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell. 2013; 12:247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nishibuchi G., Dejardin J.. The molecular basis of the organization of repetitive DNA-containing constitutive heterochromatin in mammals. Chromosome Res. 2017; 25:77–87. [DOI] [PubMed] [Google Scholar]
- 29. van Steensel B., Belmont A.S.. Lamina-Associated Domains: Links with chromosome architecture, heterochromatin, and gene repression. Cell. 2017; 169:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lenain C., de Graaf C.A., Pagie L., Visser N.L., de Haas M., de Vries S.S., Peric-Hupkes D., van Steensel B., Peeper D.S.. Massive reshaping of genome-nuclear lamina interactions during oncogene-induced senescence. Genome Res. 2017; 27:1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moskowitz D.M., Zhang D.W., Hu B., Le Saux S., Yanes R.E., Ye Z., Buenrostro J.D., Weyand C.M., Greenleaf W.J., Goronzy J.J.. Epigenomics of human CD8 T cell differentiation and aging. Sci. Immunol. 2017; 2:eaag0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ucar D., Márquez E.J., Chung C.-H., Marches R., Rossi R.J., Uyar A., Wu T.-C., George J., Stitzel M.L., Palucka A.K. et al.. The chromatin accessibility signature of human immune aging stems from CD8+ T cells. J. Exp. Med. 2017; 214:3123–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ren R., Ocampo A., Liu G.H., Izpisua Belmonte J.C.. Regulation of stem cell aging by metabolism and epigenetics. Cell Metab. 2017; 26:460–474. [DOI] [PubMed] [Google Scholar]
- 34. Park M., Min B., Jeon K., Cho S., Park J.S., Kim J., Jeon J., Song J., Kim S., Jeong S. et al.. Age-associated chromatin relaxation is enhanced in Huntington's disease mice. Aging (Albany NY). 2017; 9:803–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Villeponteau B. The heterochromatin loss model of aging. Exp. Gerontol. 1997; 32:383–394. [DOI] [PubMed] [Google Scholar]
- 36. Protasova M.S., Gusev F.E., Grigorenko A.P., Kuznetsova I.L., Rogaev E.I., Andreeva T.V.. Quantitative analysis of L1-Retrotransposons in Alzheimer's disease and aging. Biochemistry (Mosc.). 2017; 82:962–971. [DOI] [PubMed] [Google Scholar]
- 37. Simon M., Van Meter M., Ablaeva J., Ke Z., Gonzalez R.S., Taguchi T., De Cecco M., Leonova K.I., Kogan V., Helfand S.L. et al.. LINE1 derepression in aged Wild-Type and SIRT6-Deficient mice drives inflammation. Cell Metab. 2019; 29:871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De Cecco M., Criscione S.W., Peterson A.L., Neretti N., Sedivy J.M., Kreiling J.A.. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging (Albany NY). 2013; 5:867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. De Cecco M., Ito T., Petrashen A.P., Elias A.E., Skvir N.J., Criscione S.W., Caligiana A., Brocculi G., Adney E.M., Boeke J.D. et al.. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. 2019; 566:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma S., Sun S., Geng L., Song M., Wang W., Ye Y., Ji Q., Zou Z., Wang S., He X. et al.. Caloric restriction reprograms the Single-Cell transcriptional landscape of rattus norvegicus aging. Cell. 2020; 180:984–1001. [DOI] [PubMed] [Google Scholar]
- 41. Geng L., Liu Z., Wang S., Sun S., Ma S., Liu X., Chan P., Sun L., Song M., Zhang W. et al.. Low-dose quercetin positively regulates mouse healthspan. Protein Cell. 2019; 10:770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Larson K., Yan S.J., Tsurumi A., Liu J., Zhou J., Gaur K., Guo D., Eickbush T.H., Li W.X.. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLos Genet. 2012; 8:e1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsumagari K., Baribault C., Terragni J., Varley K.E., Gertz J., Pradhan S., Badoo M., Crain C.M., Song L., Crawford G.E. et al.. Early de novo DNA methylation and prolonged demethylation in the muscle lineage. Epigenetics. 2013; 8:317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheng F., Wang S., Song M., Liu Z., Liu P., Wang L., Wang Y., Zhao Q., Yan K., Chan P. et al.. DJ-1 is dispensable for human stem cell homeostasis. Protein Cell. 2019; 10:846–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Suzuki K., Tsunekawa Y., Hernandez-Benitez R., Wu J., Zhu J., Kim E.J., Hatanaka F., Yamamoto M., Araoka T., Li Z. et al.. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016; 540:144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang S., Min Z., Ji Q., Geng L., Su Y., Liu Z., Hu H., Wang L., Zhang W., Suzuiki K. et al.. Rescue of premature aging defects in Cockayne syndrome stem cells by CRISPR/Cas9-mediated gene correction. Protein Cell. 2019; 11:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang S., Hu B., Ding Z., Dang Y., Wu J., Li D., Liu X., Xiao B., Zhang W., Ren R. et al.. ATF6 safeguards organelle homeostasis and cellular aging in human mesenchymal stem cells. Cell Discov. 2018; 4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang P., Liu Z., Zhang X., Li J., Sun L., Ju Z., Li J., Chan P., Liu G.-H., Zhang W. et al.. CRISPR/Cas9-mediated gene knockout reveals a guardian role of NF-κB/RelA in maintaining the homeostasis of human vascular cells. Protein Cell. 2018; 9:945–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pan H., Guan D., Liu X., Li J., Wang L., Wu J., Zhou J., Zhang W., Ren R., Zhang W. et al.. SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res. 2016; 26:190–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fu L., Hu Y., Song M., Liu Z., Zhang W., Yu F.X., Wu J., Wang S., Izpisua Belmonte J.C., Chan P. et al.. Up-regulation of FOXD1 by YAP alleviates senescence and osteoarthritis. PLoS Biol. 2019; 17:e3000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang X., Liu Z., Liu X., Wang S., Zhang Y., He X., Sun S., Ma S., Shyh-Chang N., Liu F. et al.. Telomere-dependent and telomere-independent roles of RAP1 in regulating human stem cell homeostasis. Protein Cell. 2019; 10:649–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ren X., Hu B., Song M., Ding Z., Dang Y., Liu Z., Zhang W., Ji Q., Ren R., Ding J. et al.. Maintenance of nucleolar homeostasis by CBX4 alleviates senescence and osteoarthritis. Cell Rep. 2019; 26:3643–3656. [DOI] [PubMed] [Google Scholar]
- 53. Ha G., Roth A., Lai D., Bashashati A., Ding J., Goya R., Giuliany R., Rosner J., Oloumi A., Shumansky K. et al.. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res. 2012; 22:1995–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang J., Li J., Suzuki K., Liu X., Wu J., Zhang W., Ren R., Zhang W., Chan P., Izpisua Belmonte J.C. et al.. Genetic enhancement in cultured human adult stem cells conferred by a single nucleotide recoding. Cell Res. 2017; 27:1178–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yan P., Li Q., Wang L., Lu P., Suzuki K., Liu Z., Lei J., Li W., He X., Wang S. et al.. FOXO3-Engineered human ESC-Derived vascular cells promote vascular protection and regeneration. Cell Stem Cell. 2019; 24:447–461. [DOI] [PubMed] [Google Scholar]
- 56. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Anders S., Pyl P.T., Huber W.. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang Y.J., Guo L., Gonzales P.K., Gendron T.F., Wu Y., Jansen-West K., O’Raw A.D., Pickles S.R., Prudencio M., Carlomagno Y. et al.. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science. 2019; 363:eaav2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen J., Bardes E.E., Aronow B.J., Jegga A.G.. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009; 37:W305–W311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Joung J., Konermann S., Gootenberg J.S., Abudayyeh O.O., Platt R.J., Brigham M.D., Sanjana N.E., Zhang F.. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017; 12:828–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H. et al.. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015; 517:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Orlando D.A., Chen M.W., Brown V.E., Solanki S., Choi Y.J., Olson E.R., Fritz C.C., Bradner J.E., Guenther M.G.. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 2014; 9:1163–1170. [DOI] [PubMed] [Google Scholar]
- 64. Weinberg D.N., Papillon-Cavanagh S., Chen H., Yue Y., Chen X., Rajagopalan K.N., Horth C., McGuire J.T., Xu X., Nikbakht H. et al.. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature. 2019; 573:281–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome Project Data Processing, S . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zang C., Schones D.E., Zeng C., Cui K., Zhao K., Peng W.. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009; 25:1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ross-Innes C.S., Stark R., Teschendorff A.E., Holmes K.A., Ali H.R., Dunning M.J., Brown G.D., Gojis O., Ellis I.O., Green A.R. et al.. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012; 481:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vogel M.J., Peric-Hupkes D., van Steensel B.. Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat. Protoc. 2007; 2:1467–1478. [DOI] [PubMed] [Google Scholar]
- 72. Ernst J., Kellis M.. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012; 9:215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Watanabe S., Kawamoto S., Ohtani N., Hara E.. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017; 108:563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Brachner A., Foisner R.. Cancer Biology and the Nuclear Envelope. 2014; 143–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Solovei I., Wang A.S., Thanisch K., Schmidt C.S., Krebs S., Zwerger M., Cohen T.V., Devys D., Foisner R., Peichl L. et al.. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell. 2013; 152:584–598. [DOI] [PubMed] [Google Scholar]
- 77. Wang C.-Y., Jégu T., Chu H.-P., Oh H.J., Lee J.T.. SMCHD1 merges chromosome compartments and assists formation of super-Structures on the inactive X. Cell. 2018; 174:406–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Amendola M., van Steensel B.. Nuclear lamins are not required for lamina-associated domain organization in mouse embryonic stem cells. EMBO Rep. 2015; 16:610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Britton E., Rogerson C., Mehta S., Li Y., Li X., consortium O., Fitzgerald R.C., Ang Y.S., Sharrocks A.D.. Open chromatin profiling identifies AP1 as a transcriptional regulator in oesophageal adenocarcinoma. PLos Genet. 2017; 13:e1006879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yang S.H., Andrabi M., Biss R., Murtuza Baker S., Iqbal M., Sharrocks A.D.. ZIC3 controls the transition from naive to primed pluripotency. Cell Rep. 2019; 27:3215–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jacobs F.M., Greenberg D., Nguyen N., Haeussler M., Ewing A.D., Katzman S., Paten B., Salama S.R., Haussler D.. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature. 2014; 516:242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wolf G., Macfarlan T.S.. Revealing the complexity of retroviral repression. Cell. 2015; 163:30–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ecco G., Cassano M., Kauzlaric A., Duc J., Coluccio A., Offner S., Imbeault M., Rowe H.M., Turelli P., Trono D.. Transposable elements and their KRAB-ZFP controllers regulate gene expression in adult tissues. Dev. Cell. 2016; 36:611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Coluccio A., Ecco G., Duc J., Offner S., Turelli P., Trono D.. Individual retrotransposon integrants are differentially controlled by KZFP/KAP1-dependent histone methylation, DNA methylation and TET-mediated hydroxymethylation in naïve embryonic stem cells. Epigenet. Chromatin. 2018; 11:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hancks D.C., Kazazian H.H.. Roles for retrotransposon insertions in human disease. Mobile DNA. 2016; 7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ayarpadikannan S., Lee H.-E., Han K., Kim H.-S.. Transposable element-driven transcript diversification and its relevance to genetic disorders. Gene. 2015; 558:187–194. [DOI] [PubMed] [Google Scholar]
- 87. Mager D.L., Stoye J.P.. Mammalian Endogenous Retroviruses. Microbiol. Spectr. 2015; 3:doi:10.1128/microbiolspec.MDNA3-0009-2014. [DOI] [PubMed] [Google Scholar]
- 88. Gorbunova V., Boeke J.D., Helfand S.L., Sedivy J.M.. Human Genomics. Sleeping dogs of the genome. Science. 2014; 346:1187–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Guo C., Jeong H.-H., Hsieh Y.-C., Klein H.-U., Bennett D.A., De Jager P.L., Liu Z., Shulman J.M.. Tau activates transposable elements in Alzheimer's disease. Cell Rep. 2018; 23:2874–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq, whole genome DNA sequencing, DamID-seq, H3K9me3 ChIP-seq and ATAC-seq data generated in this study are available at GEO Accession Number GSE146387.