


Abstract
The Cys2His2 zinc finger is the most common DNA-binding domain expanding in metazoans since the fungi human split. A proposed catalyst for this expansion is an arms race to silence transposable elements yet it remains poorly understood how this domain is able to evolve the required specificities. Likewise, models of its DNA binding specificity remain error prone due to a lack of understanding of how adjacent fingers influence each other's binding specificity. Here, we use a synthetic approach to exhaustively investigate binding geometry, one of the dominant influences on adjacent finger function. By screening over 28 billion protein–DNA interactions in various geometric contexts we find the plasticity of the most common natural geometry enables more functional amino acid combinations across all targets. Further, residues that define this geometry are enriched in genomes where zinc fingers are prevalent and specificity transitions would be limited in alternative geometries. Finally, these results demonstrate an exhaustive synthetic screen can produce an accurate model of domain function while providing mechanistic insight that may have assisted in the domains expansion.
INTRODUCTION
The Cys2His2 zinc finger (ZF) is the most common DNA-binding domain (DBD) in metazoan genomes yet many influences that govern its base recognition remain poorly understood (1–3). This seemingly simplistic DBD recognizes a 3–4 base target using residues on the amino-terminus of its alpha helix (4) (Figure 1A). The stability of this small domain (typically 23 amino acids) is provided by the two cysteines and two histidines that coordinate a zinc ion as well as a small number of hydrophobic residues that pack into the core of the domain (5). These stabilizing residues can be thought of as structural as mutations at these positions lead to misfolding and loss of function. The remaining positions are somewhat flexible as a wide range of amino acids can be found at the nonstructural positions of the domain in nature (2,6,7). Screens of natural ZFs (7,8), as well as synthetic proteins that only assay residues on the helix (9,10), establish that examples of individual domains exist that are able to recognize any 3-base target (11). These results demonstrate that while any nonstructural residues within a single ZF may have subtle influences on specificity and affinity, recognition of any 3-base sequence can be as simple as 3–6 contacts provided by the residues on the helix. Nevertheless, models of ZF specificity struggle to accurately predict the target preference of ZF transcription factors (TFs), or even which ZFs of the protein engage the DNA. This failure implies that influences beyond the helix have been absent or underappreciated in our understanding of the domain.
Figure 1.

Investigating the influence of boundary residues on adjacent finger function. (A) A single zinc finger structure and the common interactions with a 3–4bp target. Residues –1 through 6 of the alpha helix can be positioned to contact three primary bases labeled B1, B2, and B3, from 5′ to 3′. A fourth contact is sometimes observed through a cross-strand contact between position 2 of the helix and the base immediately 3′ to its primary triplet (shown with dotted arrow). Residues –1, 2, 3 and 6 are the primary contributors to specificity (bold, yellow) and are therefore referred to as the ‘core helical residues’, however positions 1 and 5 can also contribute to base recognition. (B) A structural model of adjacent fingers and their potential interface contacts. The core residues of each helix have been labeled with arrows indicating which base they are likely to specify. Red arrows indicate contacts made by position 6 of the N-terminal finger and position 2 of the C-terminal finger. This base pair is boxed in red and referred to as the overlap position as both fingers may interact with the base pair simultaneously leading to synergy or conflict. Positions 9 and -2 of the N and C –terminal fingers, respectively, are shown in green. These are referred to as the boundary residues, or as a set, the boundary residue pair, and structural evidence indicates their interaction influences the geometry with which adjacent fingers bind the DNA. (C) Structural evidence is limited to 8 unique boundary pairs recovered from 22 adjacent finger structures (left) and models of the boundary residue influence are limited by this small set (middle) while human zinc fingers employ significantly more boundary residue pairs than the models can account for (right). (D) Six libraries of zinc fingers were screened with a bacterial one-hybrid assay to recover amino acid combinations able to bind each of the possible 64 NNN targets. In this assay, only functional zinc finger – DNA interactions will recruit the polymerase to the weak promoter that drives the reporter (HIS3) and lead to survival, allowing for the recovery of potentially rare but functional variants from the library. Each of the libraries employed a different set of boundary residues that represent the six most common adjacent finger geometries (RS, RR, VS, KS, IN and KR) while the six base-specifying residues of the helix (noted in 1A) were fully randomized. Each library was screened in 64 independent experiments, one for each binding site in the ‘NNN’ position of the cartoon. (E) From each combined set of 64 screens, different numbers of unique helices were recovered depending on which boundary pair was employed, with mode 1 showing a roughly 5-fold higher number than mode 5. (F) Not all selections were successful. The number of failed selections by mode is shown.
In human, the ZF is utilized by nearly half of the TFs (12) but unlike other DBDs that are strongly conserved, the ZF appears to be evolving rapidly with the base-specifying residues under positive selection (6,11). PRDM9 is an excellent example as this protein's function to define meiotic recombination hotspots has remained conserved from chicken to human while the base-specifying residues are different (13,14), allowing the protein to evolve and bind new sequences that may provide advantages for the host species. Similarly, the largest class of ZFs in human are the KRAB ZFs that have been proposed to bind and silence transposable elements and, supporting this hypothesis, for the KRAB ZFs where their DNA-binding specificities are known, they are mostly predicted to bind one of these elements (3,15,16). This proposed ‘arms race’ provides a plausible explanation for the necessity for TFs in more complex eukaryotes to be able to quickly adopt new DNA binding preferences. However, while it's clear ZFs have expanded to take on a wide range of specificities, it is not clear mechanistically how the ZF domain provides such plasticity. The modularity of the ZF, which appears to bind DNA in independent 3 bp subunits (17), is likely a contributing factor but numerous attempts to design ZF specificity would argue the contrary. Decades of work focused on the engineering of tandem ZF arrays have repeatedly found that the designed assembly of tandem ZF monomers largely fail to bind the desired target (18). Rather, each ZF monomer needs to be evolved in the neighboring finger context under which it will be utilized (19,20). Therefore, despite the fact that most metazoan genomes contain at least one ZF predicted to bind each of the 64 possible 3 bp targets, while fungi and green plants have much more limited portfolios, it is not clear how or if these adjacent finger influences that challenge engineering have been mitigated in nature.
To better understand how ZF specificity is governed, and ultimately how TFs function across varied genomes, it has been a long-standing goal to provide a predictive model or ‘code’ of ZF specificity. Toward this end, much focus has been placed on characterizing large sets of ZF-TFs (8,16,21). However, DNA-binding specificity of many ZF proteins have proven challenging to characterize leaving a large hole in our understanding of how these TFs function (1). In fact, the DNA-binding preference for ∼40% of the human ZFs have remained undefined though a substantial number have been characterized in the last few years by chromatin immunoprecipitation followed by next generation sequencing (ChIP-seq). Still, these results must be cautioned as the foundation of a code as this technique can be limited by the cell type employed, the cell-type specific fraction of the genome that is accessible for binding, and the size of the genome relative to the potential target length of the protein. In addition, as we now know that each human genome contains millions of variations relative to another (22), each of these variants represent the potential creation or disruption of a TF binding site. It is therefore unclear if ChIP-seq results are universally predictive of the protein's function across the human population or simply an assay of what the protein is capable of in the particular cell-type utilized in that singular genome. However, many other techniques have been applied to characterize the DNA-binding preferences of ZF-TFs. Large scale bacterial-hybrid (8,23), protein-binding microarray (24–26), and high throughput SELEX (21,27–29) assays have focused on ZF specificity as well. Despite these extensive studies, the models that result remain error prone and often fail to accurately predict specificities of known ZF-TF target preferences, much less the consequence of a mutation within the domain. Interestingly, while efforts that characterize large sets of full-length ZF-TF proteins benefit by describing what that TF is capable of binding in vivo, many specificities determined for these proteins cover fewer nucleotides than one would expect based on the number of ZFs they employ. Therefore, it is difficult to determine empirically which ZFs are making the base-specifying contacts, complicating any models derived from the results. Conversely, projects that have exhaustively assayed libraries of single ZFs have produced the most predictive models (7,10). This is likely due to the small collection of amino acid combinations sampled by any set of naturally occurring TFs while the exhaustive screens of single ZFs consider enormous helical diversity in a single, controlled context providing a more comprehensive starting point from which to build a model. However, these investigations have failed to consider the influence of neighboring fingers which ultimately limits their predictive potential.
Currently, the most accurate model of ZF specificity is based on the synthetic screen of a collection of over 47,000 single ZF domains taken from naturally occurring ZF-TFs (7). This collection samples diversity across the domain but the scale of the screen is limited when asking questions that go beyond the base-specifying residues of the helix. In addition, the model is derived from the 8138 ZFs that were enriched in the screens, or just 17.3% of the ZFs sampled, yet the model predicts over 62% of natural ZFs should be functional. Therefore, over 70% of the ZFs predicted to be functional fail this screen presumably due to their expression out of their natural context and the external factors that might influence their engagement with the DNA. These results imply that context is a dominant influence on ZF specificity that has either been overlooked or under sampled in all prior screens. A truly predictive model of ZF specificity will require the exhaustive investigation of each factor that impacts this context and how they influence the ZF-DNA engagement, both individually and combinatorially. For instance, two residues on adjacent ZFs interact with one another and have been proposed to modify the geometry with which the ZFs engage the DNA (positions 9 and –2 of Figure 1B) (30). Models based on ZF structures reveal six distinct geometries (hereafter referred to as modes), each that place the ZF helix at different angles and/or distances from the DNA, modifying the strategies with which it might engage any target. This geometry may have a large impact on ZF function as mutations in position 9 of human ZFs are enriched in cancer samples (31). However, the influence of these residues on binding mode is still somewhat hypothetical, based on a small set of structures and a comprehensive investigation is lacking. In addition to geometry, as ZF-TFs use tandem arrays of closely linked ZFs to recognize their targets, this proximal, linked binding leads to several such factors that contribute to complex interaction networks as contacts made by adjacent ZFs are known to influence one another. The most common example is a cross-strand contact that is sometimes made by position 2 of the helix with the base that precedes the ZF’s primary 3-base target (Figure 1B) (32). This contact is observed in several crystal structures, for example, in the Zif268 structure position 2 aspartic acids of different ZFs make cross-strand contacts with either an adenine or cytosine (4,33). Since the adjacent ZF is also specifying the same base pair, this position of overlap in ZF targets can be a source of synergy or conflict. In addition to the cross-strand contact, all of these interactions with the DNA are made in close proximity and contacts made by helical residue 6 of an N-terminal finger can be within hydrogen bond distance of contacts made by position –1 of the adjacent, C-terminal ZF. As such, the predicted specificity of a single ZF could be ‘overruled’ by incompatibility with the overlap base specified by the adjacent finger or through conflict between the proximal side chains at the ZF’s interface.
To fully understand these adjacent finger influences and provide mechanistic insight will require that we go beyond the relatively small set of ZF helices that have evolved in nature and provide comprehensive studies directly focused on these parameters. Here we have exhaustively screened large synthetic ZF libraries that systematically investigate one adjacent finger influence, geometry, by independently considering pairs of residues at positions 9 and –2 that are predicted to result in the six most common geometric modes. While we should note that we cannot be certain that the amino acids chosen to represent each binding mode will universally place each zinc finger in the exact, predicted geometric arrangement, this exhaustive approach has uncovered several consequences related to the chosen residues and therefore their presumed geometric relationship. For example, each mode has a different code of specificity though ZFs often engage targets with similar amino acid strategies. In fact, a minor fraction of helices are unique to a mode but the binding activity of more general helices can vary greatly from one mode to the next. Interestingly, the most common mode found in human ZF-TFs appears the most flexible with regards to its specificity as functional ZFs were recovered to bind more 3 bp targets than any other mode and with more helical diversity. Moreover, this plasticity could provide a mechanistic explanation for the ease of this domain's expansion as ZFs utilize this mode with greater frequency in organisms where ZFs represent their most common DNA-binding domain. We also demonstrate with a pair of human TFs that their flexibility to transition specificity would be restricted if their ZFs utilized alternative modes. In addition, Molecular Dynamic (MD) simulations indicate that this plasticity may be influenced by a more prevalent hydrogen bond between positions 9 and –2 as well as additional contacts that are possible between position 9 and the phosphate backbone of the DNA. Finally, by using a convolutional neural network we produce a model that predicts natural TF-ZF specificity as accurately as the prior model that was based on natural ZFs that offered diversity throughout the domain. Conversely, our model is based on proteins that are 90% identical (78/86 amino acids) in every assay employed demonstrating that a synthetic approach can provide mechanistic insight without the loss of accuracy. In sum, by using a completely synthetic but exhaustive approach we demonstrate the importance of the influence between adjacent ZFs and the necessity for any model that hopes to accurately predict the functional consequence of a mutation to reflect a full appreciation of these influences.
MATERIALS AND METHODS
Bacterial one-hybrid selections to survey the influence of mode geometry
In general, bacterial one-hybrid selections were carried out as previously described (34). Below we first detail the approach and any modification to the prior protocols that we have used here and provide pertinent details that explain why and how we have carried out the experiments in these particular ways. We also include a general protocol for B1H selections. However, details such as protocols for the minimal media used in our B1H assays or the construction and prep of the cell line are best found in those prior works (10,23). Also, these protocols are available upon request.
To survey the influence of neighboring ZF geometry on the DNA-binding landscape of the ZF domain, we used site-directed mutagenesis to assemble diverse ZF coding libraries as guided by the original description of ZF geometry (30), and our previously published work (a description of the library building process is listed below). Our libraries use an NNS coding scheme at each randomized position which provides at least one codon for each of the 20 amino acids and one stop codon. Positions –1, 1, 2, 3, 5 and 6 of the α-helix of the C-terminal ZF (F3) of a model Zif268-based system were randomized. The constant fingers at positions 1 and 2 of the 3-fingered protein are listed below as well as the sequences they interact with. For each mode library, the same randomization scheme was employed with the same constant fingers, however, the amino acids at helical position 9 of finger 2 and position –2 of finger 3 were chosen to represent each mode so they are the critical difference from one mode library to another. We refer to positions (9 and –2) as the boundary residues (BRs) or as a pair, the boundary residue pair (BRP). In each case, the libraries are expressed with an N-terminal omega fusion from a strong promoter (LppC).
For each library, a comprehensive set of protein selections were performed in an attempt to recover amino acid combinations able to bind each of the 64 possible 3 bp DNA targets. As depicted in the cartoon of Figure 1D, the two constant fingers function as anchors as they will bind to the sequence they are known to specify and these target sequences are placed adjacent to the 3 bp test target specific to the selection. The anchor finger binding then places the random finger in a position to interact with the 3 bp target unique to each selection. Only helices able to interact with that sequence will have sufficient affinity to recruit polymerase to a weak promoter that drives the reporter gene, HIS3. As these cells are grown on media that lacks histidine, only cells that harbor a ZF with a helix that can interact with the desired sequence will activate the reporter and survive the selection. In addition, the stringency of the selection can be controlled by the addition of 3-amino triazole (3-AT, a competitive inhibitor of HIS3) in the selection media. However, to maintain a low stringency and recover as many functional helices as possible, all of our protein selections were performed with 2-mM 3-AT which we have previously shown is the minimal stringency that will still eliminate background, false positive survival. To screen the libraries we plated a minimum of 5 × 108 cells on 2 mM 3AT plates, though the great majority of screens considered over 1 × 109 cells. Below are the protein sequences used for each library screened and the general target sequences for each reporter. BRPs are listed position 9 followed by position –2.
Mode 1 – BRP = RS
GTERPYACPVESCDRRFSRSDELTRHIRIHTGQK PFQCRICMRNFSRSDNLRAHIRTHTGEKPFACDICGRKFSXXXXLXXHTKIHLRGS
Mode 1 – BRP = RA
GTERPYACPVESCDRRFSRSDELTRHIRIHTGQK PFQCRICMRNFSRSDNLRAHIRTHTGEKPFACDICGRKFAXXXXLXXHTKIHLRGS
Mode 2 – BRP = RR
GTERPYACPVESCDRRFSRSDELTRHIRIHTGQK PFQCRICMRNFSRSDNLRAHIRTHTGEKPFACDICGRKFRXXXXLXXHTKIHLRGS
Mode 3 – BRP = VS
GTERPYACPVESCDRRFSRSDELTRHIRIHTGQK PFQCRICMRNFSRSDNLRAHIVTHTGEKPFACDICGRKFSXXXXLXXHTKIHLRGS
Mode 4 – BRP = KS
GTERPYACPVESCDRRFSRSDELTRHIRIHTGQK PFQCRICMRNFSRSDNLRAHIKTHTGEKPFACDICGRKFSXXXXLXXHTKIHLRGS
Mode 5 – BRP = IN
GTERPYACPVESCDRRFSRSDELTRHIRIHTGQK PFQCRICMRNFSRSDNLRAHIITHTGEKPFACDICGRKFNXXXXLXXHTKIHLRGS
Mode 6 – BRP = KR
GTERPYACPVESCDRRFSRSDELTRHIRIHTGQK PFQCRICMRNFSRSDNLRAHIKTHTGEKPFACDICGRKFRXXXXLXXHTKIHLRGS
In each protein above, omega is present as an N-terminal fusion to these proteins. The base specifying residues at positions –1 through 6 are bold and underlined for each finger. italic letters represent BRs while ‘X’s’ are the positions in each library that were randomized. The libraries were cloned into our expression vector using the restriction sites Kpn1 and Xba1 as shown below:
ggtaccGAACGCCCATATGCTTGCCCTGTCGAGTC CTGCGATCGCCGCTTTTCTCGCTCGGATGAGCTTACCCGCCATATCCGCATCCATACCGGTCAGAAGCCCTTCCAGTGTCGAATCTGCATGCGTAACTTCAGTCGTTCTGATAACCTTCGCGCCCACATCXXXACCCACACCGGCGAGAAGCCTTTTGCCTGTGACATTTGTGGGAGGAAGTTTXXXNNSNNSNNSNNSCTGNNSNNSCATACCAAAATCCATTTACGTCAGAAGGACGTCTAAGtctaga
Binding sites were cloned into the GHUC vector (35) between Not1 and EcoRI allowing for the regulation of HIS3 or GFP from the same target. The targets between those restriction sites followed the format below. A set of 64 reporters were constructed, one for each 3 bp target represented as ‘NNN’:
All selections: gcggccgcAANNNAAGGCGTAACgaattc
Characterizing the specificity of zinc finger by selecting interacting sequences from random DNA libraries
The original application of the omega-based bacterial one hybrid system was to characterize the DNA-binding preference of transcription factors (36). For this purpose, the technique has been successfully applied to factors from many model organisms with similar predictive value to any other common method. To characterize the full-length specificity (all KLF6 and Snai2 proteins) we used the 28 bp random library employed in those published screens (23). To characterize individual fingers recovered in this paper, we created a new library in the pH3U3 vector that places a 4 bp region of random DNA sequences in the same position that the targets are placed in each of the individual mode screens (see cartoon in Figure 2C). However, because this library offers relatively low complexity (256 members) it is possible that a false positive could easily dominate a selection. To avoid this, we also installed a 20 bp barcode in the vector 400 bp upstream of the reporter. By doing so we can determine if a low number of very similar 4 bp sequences are actually selected for as they will be paired with many different barcodes. Conversely, if a false positive cell escaped selective pressure, only one sequence would be represented both at the target and barcode. Therefore, we first filtered by only considering unique barcodes once per selection and then using those 4 bp targets to create our logos that represent the factor's specificity. As above, the sequence between the Not1 and EcoR1 sites of the reporter plasmid is listed below for the 28 bp library and between Age1 and EcoRI for the 4 bp library indicating placement of the barcode:
Figure 2.

Mode Similarity and Predicted Function. (A) Principal component analysis to compare the complete data sets provided by each mode library screen of 64 targets. (B) Comparison of helices predicted to have altered function between modes (See also Supplementary Figure S1). The tested helices are examples that were recovered in one mode selection (left), but not another (right), for the same target. In each case, the mode tested is listed after the helical residues used at positions –1 through 6 for each test construct. These helices were expressed with the BRPs for either mode tested and then challenged to activate a GFP reporter. The mean fluorescence of three replicates is shown (error bars = S.E.M.). The dotted red line indicates mean background GFP activation from a negative control. (C) A cartoon representing the 4bp reporter library created to determine the DNA-binding preference of helices recovered in our screens. In each case a unique helix is expressed as the third finger of a 3-fingered protein. The first two fingers are common to all proteins in our assay. A functional interaction between the test helix and a sequence in our library is required for activation of the HIS3 reporter and survival on minimal media. At least 10 helices were tested for each mode. (D–G) Examples of predicted and determined specificities for helices recovered in our screens. For each comparison a logo on the left has been generated from the frequency with which that helix was recovered across all binding sites of a mode library screen (See also Supplementary Figure S2). On the right is the determined specificity of that helix by selecting sequences from our random 4bp reporter library. (D) An example comparison of predicted and determined specificities where the preferred base is accurately predicted at all three positions. The percent of helices tested that fall in this category is noted to the left. (E) An example comparison of predicted and determined specificities where the preferred base is accurately predicted at two of three positions and the predicted base at the third position is one of the top 2 recovered. The percent of helices tested that fall in this category is noted to the left. (F) An example comparison of predicted and determined specificities where the preferred base is accurately predicted at two of three positions, and at the unmatched position the predicted base does not appear to be selected for. The percent of helices tested that fall in this category is noted to the left. (G) Comparison of predicted and determined specificities where the preferred base is accurately predicted at 1 of 3 positions. The percent of helices tested that fall in this category is noted to the left.
28 bp library:
gcggccgcNNNNNNNNNNNNNNNNNNNNNNNNNNNNCgaattc
4 bp library:
accggtAGNNNNNNNNNNNNNNNNNNNN(barcode)GCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCCTGAGTAGGACAAATCCGCCGGGAGCGGATTTGAACGTTGCGAAGCAACGGCCCGGAGGGTGGCGGGCAGGACGCCCGCCATAAACTGCCAGGCATCAAATTAAGCAGAAGGCCATCCTGACGGATGGCCTTTTTGCGTTTCTACAAACTCTTTTGTTTATTTTTCTAAATACATTCAAATATGTATCCGCTCATGACATTAACCTATAAAAATAGGCGTATCACGAGGCCCTTTCGTCTTCAAACGCGTGTACACCCGGGCGGCCGCAANNNNAAGGCGTAACgaattc
Binding site selections were carried out as described above, in minimal media challenged by 3-AT. However, binding site selections often require higher stringencies to avoid self-activating sequences. Therefore, these selections were carried out at 5 mM 3AT. In addition, the library complexity is much smaller than our protein selections. For the 28 bp library we routinely plate 5 × 107 cells which covers the diversity required for nearly any 12 bp target. With the 4 bp library, we plated ∼5 × 104, a 195-fold over sampling of the library space, but simple to screen in our system.
Defining boundary residue pairs by B1H selection in mode exclusive contexts
To define what BRPs would be functional in each mode we searched for helices that uniquely represented each mode. To do so, we analyzed our mode selection data to find helices found only in a single mode for a given target, or at least highly enriched in a single mode (see Supplemental Table S3). We think of these helices as mode exclusive as their activity on that particular target appears to be dependent on the mode employed. We can then think of these helix–target pairs as representative of the parameters particular to that mode. However, it is possible that a rare but unique helix would be recovered in a single mode by chance. Therefore, to reduce the impact of potential sampling error, we reduced or analysis to consider only core helices as there are potentially 400 versions of each protein that has the same residues at the four core helical positions. As a result, it would be incredibly unlikely that a core helix is found in only one mode by chance. An example is the core helix DRCR. We recovered over 5000 reads for this core helix in the CCC target selection in mode 1 but not in any other mode.
To define the BRPs that offer features exclusive to each mode we created libraries that randomized the BR positions with the set of mode exclusive helices (Supplemental Table S3). We then screened these libraries with the complementary targets (listed in Supplemental Table S3) to select BRPs that are functional with each mode exclusive helix (see cartoon, Figure 3A). In each case, the library diversity consists of 400 amino acid combinations, however, we plated 50 000 cells in each selection, a >100-fold over sampling of each library. Below is a general template of the protein sequences for the mode exclusive libraries:
Figure 3.
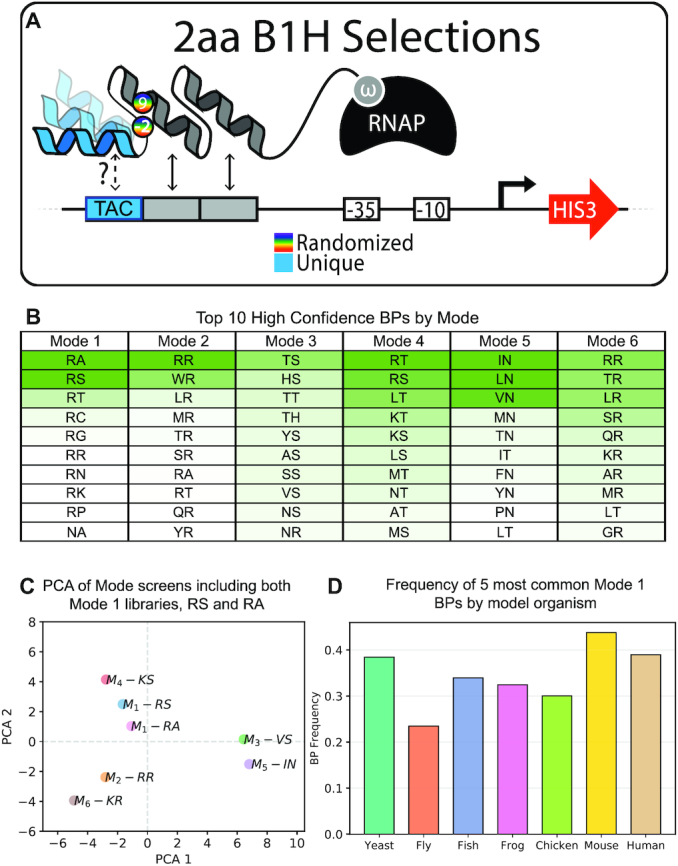
Defining the sets of boundary pairs included in each mode. (A) Cartoon of B1H selections to define functional BRPs within each mode. For each library, a helix was chosen that was enriched in a single mode-target pair across all of our screens (see Supplemental Table S3). These were considered ‘mode exclusive’ helices (in the example the blue helix interacts with TAC exclusively in a single mode). Mode exclusive libraries were generated by randomizing the BRs between the common second finger of the 3-fingered protein and each mode-exclusive helix in the third finger position. Only functional BRPs were able to activate the HIS3 reporter and survive the selective conditions. (B) Table of the top 10 BRPs recovered in the mode exclusive screens. This table is displayed as a heat plot to indicate the relative enrichments of each BRP per mode (see Also Supplementary Figure S3 and Supplemental Tables S4–S10). (C) Principal component analysis that includes a seventh mode screen across all 64 targets that uses the BRP ‘RA’. This second screen allows us to demonstrate the reproducibility of screens that use BRPs determined to be within the same mode. Here the two mode 1 screens tested used BRPs RS and RA and they are most similar to one another (See also Supplementary Figure S4). (D) The frequency with which the five most common mode 1 BRPs are present in model organisms that use ZFs as their most common DNA-binding domain (See also Supplementary Figure S5).
Mode exclusive library sequence: GTERPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDNLRAHIXTHTGEKPFACDICGRKFXDPRCLSRHTKIHLRGS
Above is an example sequence for the mode exclusive helix DPRCLSR. The bold and underlined ‘X’s’ at the BR positions are randomized and this protein library would then be challenged to activate the CCC reporter. For each mode-exclusive helix listed in Supplemental Table S3, the helix residues listed would be coded in place of the blue residues above and the library screened with the listed, complementary 3 bp target.
Building the libraries
While all libraries were built using the same approach, below we detail how the approach using the original mode libraries as the example.
For each library, the vector DNA (LPPC-omega with a Kanamycin insert between Kpn1 and Xba1) was prepped by maxiprep (Qiagen:12963) of 1L overnight culture. The recovered DNA was digestion with kpnI-HF (NEB:R3142L) and XbaI (NEB:R0145L), followed by gel isolation using minelute gel extraction kits (QIAGEN:28606). Library inserts were amplified using degenerate primers with high fidelity Expand polymerase. The DNA was purified by PCR purification (Qiagen) then digested with Kpn1 and Xba1 before gel isolation. For each library, 15 ug of LppC-omega backbone was ligated with 5 ug insert using T4 DNA Ligase (NEB:B0202S) in a 150 ul reaction overnight at 16°C.
All mode libraries were built in a similar fashion: To confirm the success of the library build, 1 μl of ethanol precipitated library ligation was electroporated into 85 ul of our ΔrpoZE. coli strain and recovered for 1 h in 10 ml SOB (Difco:244310) + 0.5% glucose (SOC). The culture was serially diluted onto carbenicillin (100 μg/ml) plates, kanamycin (50 μg/ml) plates, and plates containing both antibiotics. If the CFU on the carb plate was 100× the CFU on the dual antibiotic plate and kan plate, the library build was considered a success. Note that we have previously shown that by using the Kanamycin cassette as the fragment removed from the parent vector we are able to quantify ligation background to true insertion by comparing the fraction of the ligated material that still contains the Kan cassette versus those that only contain that ampicillin marker, expressed elsewhere on the plasmid. If the ligation was successful, the remaining library DNA was electroporated into electrocompetent cells (85 μl cells per 1 ul DNA), and recovered by shaking at 37°C in 1 l SOC. After 1 h, a small amount of culture was serially diluted and plated on carbenicillin, the CFU on this plate represents the total library size (each library was built to over 1 × 109 complexity). After dilution, carbenicillin was added at 100 μg/ml to the 1 l culture and grown until the OD was between 0.5 and 0.7 compared to a negative control. The DNA was then isolated from the pellet by maxiprep (Qiagen:12963). The resulting DNA is used for B1H selections.
B1H ZF selections
ZF selections were performed as previously described (10). Briefly, libraries were built in a vector that will express the ZF-omega fusion using a strong promoter (LppC). The binding site reporters were built by placing the binding site of interest 10 BRP upstream of the −35 box of the promoter that drives HIS3 and GFP expression in the previously described GHUC vector (35). For example, for the ‘AAA’ selections, a reporter with the binding site 5′ AAA-AAG-GCG 3′ was placed 10BRP upstream of the promoter, and so on. These sites were cloned between the Not1 and EcoRI sites of the reporter plasmid.
For selection, the ΔrpoZ selection strain was transformed with one of the ZF libraries and one of the reporter plasmids by electroporation. The cells were expanded in 10 ml SOC for 1 h at 37°C with rotation, recovered and resuspended in minimal media supplemented with histidine and grown with rotation for an additional hour at 37°C. Finally, cells were washing in minimal media that lacks histidine, recovered in 1 ml of this media, and 20 μl plated in serial dilution on rich plates containing Kanamycin and Carbenicillin to quantify double transformants. This plate was grown at 37°C overnight while the remaining 980 μl of the transformed cells was stored at 4°C. Once grown, the serial dilutions were counted and a volume containing a minimum of 5 × 108 cells were taken from the transformants stored at 4°C and plated on selective media. These plates contain 3-AT concentrations best suited for the experiment (protein selections for mode were done at low stringency, 2 mM, while binding site selections from the 28BRP or 4BRP library were done at 5 mM. In previous work we have shown 10 mM is a suitable high stringency while 20 mM is helpful only in extreme cases of very high affinity proteins). Cells were grown on the selection plates for 36–48 h at 37°C. Colonies were counted and cells were pooled by scraping everything from these plates and harvesting the DNA. This DNA was used as the template for Illumina sequencing. All selections resulted in hundreds to thousands of surviving colonies while reporters combined with a negative control result in zero to single digit surviving colonies.
GFP expression assays
In the GHUC vector, a GFP cassette follows HIS3 after an internal ribosome entry site (Shine-Dalgarno sequence) that separates the two coding sequences. In this way, the same sequence that can drive HIS3 in our selections can also activate GFP allowing for a visualization and quantification of activation driven by a unique protein-DNA interaction. To do so, two plasmids, one containing the unique omega-ZF construct to be tested and one containing a unique, corresponding binding site upstream of the HIS3/GFP cassette, were transformed into our ΔrpoZ E. coli strain via heat shock, recovered for 1 h in SOC, and plates on dual antibiotic rich media (2XYT) plates. The following day, single colonies were picked in biological triplicate and incubated for 8hr (or until OD ∼ 0.6) in 2XYT media + 2% glucose. 5 ul of each culture was then used to inoculate 5 ml of NM media supplemented with histidine, kanamycin, carbenicillin and IPTG. These new NM cultures are incubated overnight with rotation at 37C (∼18 h). The next day, 25 μl of culture was resuspended in 500 ul PBS + 0.5% FBS and analyzed by fluorescent activated cell sorting (FACS), where mean GFP expression levels (AU) were recorded for each sample.
Illumina prep
Helix selections, boundary residue selections, and 4 bp selections were prepped for Illumina sequencing as follows. Cells were scraped from selection plates and plasmid DNA was recovered with Qiagen miniprep kits. The resulting DNA was used as the template for PCR in order to attach a barcode to each sample. A series of sixty four 8 bp barcodes were designed to minimize similarity between each 8 bp sequence and avoid falsely including a variant in the wrong bin because of mutations that occur in the PCR or illumina reactions. Below, general templates for the oligonucleotides used are provided with the barcode “N" regions and the region that anneals to our template underlined.
Barcodes used for helix selections.
Forward: AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACGCTCTTCCGATCTGACATTTGTGGGAGGAAGTTT
Reverse: CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTTCTGTCTTAAATGGATTTTGGT
Barcodes used to sequence selected BRPs in mode exclusive selections.
Forward: AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACGCTCTTCCGATCTGTCGTTCTGATAACCTTCGC
Reverse: CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTGACGTAAATGGATTTTGGTATG
Barcodes used binding site selections.
Forward: AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACGCTCTTCCGATCTCAGCTGGCAATTCCGACGT
Reverse: CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCGAGCCGGAAGCATAAAGTGTA
Amplification from recovered templates were done in 96-well formats according to the manufacturer's suggested reaction conditions using 15 cycles of amplification. After PCR, each reaction was run on a 1.0% agarose gel to confirm the PCR reaction worked. If successful, 5 ul of each reaction was pooled, purified, and run out on a 1.0% agarose gel and recovered using a Qiagen gel extraction kit. The DNA was eluted from the Qiagen minelute column in 25 μl of elution buffer. The product concentration was measured (Thermo scientific, Nanodrop 2000c) and diluted to 10 nM and sent for Illumina sequencing at the core facility, the NYU Genome Technology Center.
ORF ZF sequences used (modified BRPs bold and underlined) for ZF-TF GFP and specificity experiments
EGR1
WT (RA): GTERPYACPVESCDRRFSRSDE LTRHIRIHTGQKPFQCRICMRNFSRSDHLTTHIRTHTGEKPFACDICGRKFARSDERKRHTKIHLRQKD*
RR: GTERPYACPVESCDRRFSRSDELTRHIRIHT GQKPFQCRICMRNFSRSDHLTTHIRTHTGEKPFACDICGRKFRRSDERKRHTKIHLRQKD*
KA: GTERPYACPVESCDRRFSRSDELTRHIRIHT GQKPFQCRICMRNFSRSDHLTTHIKTHTGEKPFACDICGRKFARSDERKRHTKIHLRQKD*
VA: GTERPYACPVESCDRRFSRSDELTRHIRIHT GQKPFQCRICMRNFSRSDHLTTHIVTHTGEKPFACDICGRKFARSDERKRHTKIHLRQKD*
VS: GTERPYACPVESCDRRFSRSDELTRHIRIHT GQKPFQCRICMRNFSRSDHLTTHIVTHTGEKPFACDICGRKFSRSDERKRHTKIHLRQKD*
KLF6
WT (RS): GTGRRRVHRCHFNGCRKVYTKS SHLKAHQRTHTGEKPYRCSWEGCEWRFARSDELTRHFRKHTGAKPFKCSHCDRCFSRSDHLALHMKRHL*
Mode 4 KS: GTGRRRVHRCHFNGCRKVYTKSSHLKAHQRTHTGEKPYRCSWEGCEWRFARSDELTRHFKKHTGAKPFKCSHCDRCFSRSDHLALHMKRHL*
Mode 3 VS: GTGRRRVHRCHFNGCRKVYTKSSHLKAHQRTHTGEKPYRCSWEGCEWRFARSDELTRHFVKHTGAKPFKCSHCDRCFSRSDHLALHMKRHL*
Mode 5 IN: GTGRRRVHRCHFNGCRKVYTKSSHLKAHQRTHTGEKPYRCSWEGCEWRFARSDELTRHFIKHTGAKPFKCSHCDRCFNRSDHLALHMKRHL*
WT (RS), position 3Tyr: GTGRRRVHRCHFNGCRKVYTKSSHLKAHQRTHTGEKPYRCSWEGCEWRFARSDELTRHFRKHTGAKPFKCSHCDRCFSRSDYLALHMKRHL*
WT (RS), position 3Asn: GTGRRRVHRCHFNGCRKVYTKSSHLKAHQRTHTGEKPYRCSWEGCEWRFARSDELTRHFRKHTGAKPFKCSHCDRCFSRSDNLALHMKRHL*
WT (RS), position 3Asp: GTGRRRVHRCHFNGCRKVYTKSSHLKAHQRTHTGEKPYRCSWEGCEWRFARSDELTRHFRKHTGAKPFKCSHCDRCFSRSDDLALHMKRHL*
WT (RS), position 3Thr: GTGRRRVHRCHFNGCRKVYTKSSHLKAHQRTHTGEKPYRCSWEGCEWRFARSDELTRHFRKHTGAKPFKCSHCDRCFSRSDTLALHMKRHL*
WT (RS), position 3Ser: GTGRRRVHRCHFNGCRKVYTKSSHLKAHQRTHTGEKPYRCSWEGCEWRFARSDELTRHFRKHTGAKPFKCSHCDRCFSRSDSLALHMKRHL*
*shown are the position 3 mutants in mode 1, the equivalent mutations were also made in each of the mode templates above.
Snail
WT (RA):
GTQSRKSFSCKYCDKEYVSLGALKMHIRTHTL PCVCKICGKAFSRPWLLQGHIRTHTGEKPFSCPHCNRAFADRSNLRAHLQTHSDVKKYQCKNCSKTFSRMSLLHKHEESGCCVAH
Mode 2 (RR):
GTQSRKSFSCKYCDKEYVSLGALKMHIRTHTL PCVCKICGKAFSRPWLLQGHIRTHTGEKPFSCPHCNRAFRDRSNLRAHLQTHSDVKKYQCKNCSKTFSRMSLLHKHEESGCCVAH
Mode 3 (VS):
GTQSRKSFSCKYCDKEYVSLGALKMHIRTHTL PCVCKICGKAFSRPWLLQGHIVTHTGEKPFSCPHCNRAFSDRSNLRAHLQTHSDVKKYQCKNCSKTFSRMSLLHKHEESGCCVAH
Mode 4 (KS):
GTQSRKSFSCKYCDKEYVSLGALKMHIRTHTL PCVCKICGKAFSRPWLLQGHIKTHTGEKPFSCPHCNRAFSDRSNLRAHLQTHSDVKKYQCKNCSKTFSRMSLLHKHEESGCCVAH
Mode 5 (IN):
GTQSRKSFSCKYCDKEYVSLGALKMHIRTHTL PCVCKICGKAFSRPWLLQGHIITHTGEKPFSCPHCNRAFNDRSNLRAHLQTHSDVKKYQCKNCSKTFSRMSLLHKHEESGCCVAH
WT (RA), position 6 His:
GTQSRKSFSCKYCDKEYVSLGALKMHIRTHTL PCVCKICGKAFSRPWLLQGHIRTHTGEKPFSCPHCNRAFADRSNLRHHLQTHSDVKKYQCKNCSKTFSRMSLLHKHEESGCCVAH
WT (RA), position 6 Arg:
GTQSRKSFSCKYCDKEYVSLGALKMHIRTHTL PCVCKICGKAFSRPWLLQGHIRTHTGEKPFSCPHCNRAFADRSNLRRHLQTHSDVKKYQCKNCSKTFSRMSLLHKHEESGCCVAH
WT (RA), position 6 Lys:
GTQSRKSFSCKYCDKEYVSLGALKMHIRTHTL PCVCKICGKAFSRPWLLQGHIRTHTGEKPFSCPHCNRAFADRSNLRKHLQTHSDVKKYQCKNCSKTFSRMSLLHKHEESGCCVAH
*shown are the position 6 mutants in mode 1, the equivalent mutations were also made in each of the mode templates above.
ZNF713 (WT)
TGEKPYKCDECGKRFSQRIHLIQH QRIHTGEKPFICNGCGKAFRQHSSFTQHLRIHTGEKPYKCNQCGKAFSRITSLTEHHRLHTGEKPYECGFCGKAFSQRTHLNQHERTHTGEKPYKCNECGKAFSQSAHLNQHRKIHTREK
ZNF713 (F1,2 BRP mutation)
TGEKPYKCDECGKRFSQRIHLIQHQIIHTGEKP FICNGCGKAFRQHSSFTQHLRIHTGEKPYKCNQCGKAFSRITSLTEHHRLHTGEKPYECGFCGKAFSQRTHLNQHERTHTGEKPYKCNECGKAFSQSAHLNQHRKIHTREK
ZNF713 (F2,3 BRP mutation)
TGEKPYKCDECGKRFSQRIHLIQHQRIHTGEKP FICNGCGKAFRQHSSFTQHLIIHTGEKPYKCNQCGKAFSRITSLTEHHRLHTGEKPYECGFCGKAFSQRTHLNQHERTHTGEKPYKCNECGKAFSQSAHLNQHRKIHTREK
ZNF713 (F3,4 BRP mutation)
TGEKPYKCDECGKRFSQRIHLIQHQRIHTGEKP FICNGCGKAFRQHSSFTQHLRIHTGEKPYKCNQCGKAFSRITSLTEHHILHTGEKPYECGFCGKAFSQRTHLNQHERTHTGEKPYKCNECGKAFSQSAHLNQHRKIHTREK
ZNF713 (F4,5 BRP mutation)
TGEKPYKCDECGKRFSQRIHLIQHQRIHTGEKP FICNGCGKAFRQHSSFTQHLRIHTGEKPYKCNQCGKAFSRITSLTEHHRLHTGEKPYECGFCGKAFSQRTHLNQHEITHTGEKPYKCNECGKAFSQSAHLNQHRKIHTREK
QUANTIFICATION AND STATISTICAL ANALYSIS
Recovery of B1H selection data by next generation sequencing
Illumina fastq files were demultiplexed by the Genome Technology Center at NYU Langone Health. For the mode library screens, the randomized region corresponds to 21 nt which were trimmed from all amplicons and translated with transeq (EMBOSS).
Filtering used to determine success of selections and recover positive helices
Since the functional selection works at the protein level, and only in extreme cases is there only one coding scheme for a helix in our library, a protein that is being selected for should be recovered with more than one coding strategy. Therefore, we used both reads and the presence of multiple coding schemes to validate a recovered sequence as a true or false positive. First, only protein sequences with >10 sequencing reads were considered. Second, protein sequences coded by a single unique 21-nt sequence were classified as false positives and eliminated as it is extremely unlikely that the protein would be selected for by only one coding variant of that protein. In addition, a false positive that has escaped selective pressure might be missed by this filter as the PCR step and the sequencing step can often lead to single nt mutations, which would then indicate that the protein was actually coded by more than one sequence. However, what we find is that in these cases there are many low read count single nt mutations that occur similar to one parent sequence with significantly more reads. To account for this, these sequences are removed if for a protein sequence, we found more than 1 log difference between the 21-nt with the highest read count and the 21-nt with the second highest read count.
Screening data preprocessing
A similar approach was used to filter data using Shannon entropy as previously described (10). Both filtering approaches produce similar results. Here, results were demultiplexed using custom python tools (37). After demultiplexing, sequences with insertions, deletions or mutations in constant regions were discarded. Next, the encoding diversity of each helix was evaluated through their entropy using the Shannon entropy equation normalized by the number of potential ways to code the peptide sequence using NNS codons. In the library, helices are represented by multiple nucleotide sequences. However, selection occurs at the protein level. Consequently, helices represented by a single DNA sequence are likely to be spontaneous growing colonies. Finally, helices represented by less than ten reads or a normalized entropy <0.07 were dropped.
Motif derivation from selections
The DNA specificity motif of each helix was constructed using the frequency of reads of each nucleotide at each position. Next, the six DNA-specifying amino acids of each helix were concatenated to their corresponding boundary pair. Finally, the eight amino acid sequence was one-hot encoded to be used as input.
Selections analysis and comparison
After filtering, the helices were aggregated by adding reads of helices with the same core specificity residues. Within each mode, a graph was built considering two helices connected if the hamming distance was equal to one. Network plotting and properties were calculated using the Network X package (38).
A position frequency matrix with the helices recovered for each mode and 3 bp target was built. The principal component analysis of the position frequency matrix was computed using the Scikit-learn package in Python (39). For a more granular comparison, the standardized euclidian distance of the position frequency matrix of the same 3 bp target selection between modes was calculated using Scipy python package (40).
Convolutional neural network
The model was implemented using Keras with Tensorflow backend. Before training, any sequence with specificity residues present in the natural repertoire of natural ZF collected by Najafabadi et al. and the 100 helices selected for additional characterization, and a random selection of 4000 were filtered out of the training set. The random search strategy was used for hyperparameters and architecture optimization. A total of five hundred trials allow us to explore models with one to three convolutional layers with a wide variety on the numbers and sizes of filters. The models were ranked based on the performance of 10-fold cross-validations on an independent dataset of D. melanogaster ZFs (8). The architecture with the best performance has one convolutional layer and two fully connected hidden layers. The output layer is a 12-position vector representing the DNA binding motif of the ZF for three bases. Relu and Sigmoid activation functions were used for our hidden layers and output layer, respectively. The input as the amino acid sequences were represented as a binary matrix by one-hot encoding and is first transformed by a 1D-convolutional layer, which computes the activations for 128 convolutional filters with a stride and a size of 2 positions. We train the model by minimizing the mean square error of the DNA motif. Before training, all the helices present Any helix in the training set present in any of the validation or test sets were removed to avoid data leaking. RMS prop optimized the loss function with a learning rate of 0.0001 and a batch size of 512. Learning was terminated if the validation loss did not improve over five consecutive epochs (early stopping). The neural network output is normalized before comparing with the experimental value.
Correlation score, validations and reference database
To measure the correlation score of a pair of motifs, we measured the Pearson correlation of their affinity scores across 50 000 random sequences of length 100 bp with GC content regulated to be within a reasonable range, and the affinity scores calculated as described previously (7). Following the method described by Najafabadi, for comparison of the predicted with experimental motifs, we measured the correlation score of all the possible alignments and the alignment with the maximum score per position with six or more aligned positions was selected.
Six different datasets were used to benchmark the models; the ZifRC training dataset with 8112 natural zinc fingers characterized by B1H screening (7). Najafabadi's Golden standard and Human datasets, a curated selection across organisms of transcription factors, and selection of representative human transcription factors, respectively. Narasimhan C.elegans dataset (41), which motifs were characterized by PBM. Lastly, 100 validated motifs from the selection extensively characterized (Supplementary Figure S2) and a subset of 4000 ZFs removed from the training set.
The full set of zinc fingers was downloaded from cis-BP build 2.0 database (1). For each kingdom, a set of the most common reference organisms was selected. For green plants, A. thaliana, S. moellendorffii, B. distachyon,P. trichocarpa, O. sativa, B. stricta, L. japonicus and P. patens. For Fungi, N. crassa, S. commune, C. cinerea, S. pombe, A. nidulans, S. cerevisiae, A. gossypii,and U. maydis. Finally, for Metazoa, C. elegans, D. melanogaster, D. rerio, X. tropicalis, G. gallus, M. musculus and H.sapiens.
The linker between two zinc finger need to be seven or less amino acids long between the last His of the N-terminal finger and the first Cys of the C-terminal finger to consider them part of the same array.
All related algorithms were implemented in Python, and Spearman correlations, Fisher exact test, t-test and ANOVA one-way associated P values were calculated using SciPy (40).
Molecular modelling
Structural models were generated with TLEAP in AMBER16 (42) using PDB file 1AAY as the template. Zinc 2+ ions were parametrized using ZAFF, the zinc AMBER force field (43). All models' protonation states were identified using the WHATIF (44). They were then explicitly solvated in a 15 nm3 box of TIP3P water, and Sodium counter-ions were added for overall charge neutrality, and periodic boundary conditions were applied. Bonds to hydrogen were constrained using SHAKE (45), and the particle mesh Ewald (46) algorithm was used to treat long-range electrostatic interactions. The non-bonded cut-off was set at 12.0 Å. Systems were energy minimized using a combination of steepest descent and conjugate gradient methods. The system was thermalized and equilibrated for 3 ns using a multistage protocol. The first step was a 500 ps gradual heating from 0 to 300 K, followed by 250 ps of density equilibration, and positional restraints were gradually removed. The next step was 500 ps of constant pressure equilibration at 300 K. Berendsen thermostat, and barostat was used throughout for both temperature and pressure regulation (47). The final phase of equilibration for a total of 2 ns. Due to the sensitivity of the system, the time step for the MD equilibrations was 1 fs and 2fs for the simulations. MD calculations were carried out with the GPU-accelerated AMBER16 code in conjunction with the FF99 Barcelona forcefield (48). During calculations, a snapshot was saved every 2 ps. Root mean square deviation (RMSD) was evaluated to assess the equilibration of each run. Three independent MD simulations were carried out per model producing a total of 620 ns of simulation time. The RMSD, RMSF, distances, hydrogen bonds and clustering analysis of the trajectories were obtained using the corresponding commands of the CPPTRAJ module (49).
Data and code availability
Preprocessed sequencing and code are available upon request. The model of specificity can be applied to ZFs using the utility ‘ZFPred’ found at repository: www.gitlab.com/kimlab/zfgeomodes
RESULTS
Boundary residues influence zinc finger function
Many TF DBDs bind to a limited set of targets (1,50) while ZFs are the exception to the rule offering flexibility in their interactions that include multiple amino acid strategies to bind any 3 bp target. Part of this plasticity may be due to the ZF’s ability to engage the DNA with an assortment of geometries that could influence and expand the number of functional strategies (5,30). Conversely, other common DBDs such as basic helix-loop-helix, leucine zippers, homeodomains, forkhead and ETS domains can bind the DNA as either monomers, dimers or both (51). The structural constraints of the protein-protein interactions required for dimerization may restrict the flexibility of these domains and limit their sampling of novel specificities. For ZFs, the assortment of functional geometries with which the domain might engage the DNA has been suggested to be controlled by helical positions 9 and −2 of adjacent ZF helices (30), henceforth referred to as the boundary residues (BRs) or together, boundary residue pairs (BRPs) (Figure 1B). However, the analysis of these ZF interactions is based on a small collection of X-ray crystal structures resulting in a set of 8 adjacent ZF BRPs collected from 22 adjacent finger structures (Figure 1C, left). From such a small catalog, not only is it difficult to say if all ZFs with the same BRPs will engage the DNA in the same way, it is impossible to gauge how these geometries influence function across all possible targets or all possible helices. Moreover, many naturally occurring ZF-TFs in human contain BRPs that are not found in the structures nor are they recovered in a model derived from these data (30) (Figure 1C, middle and right). Therefore, we first set to exhaustively address how these modes influence ZF function and DNA target preference by investigating whether a disparate set of BRPs predicted to represent each mode allows the domain to interact with any target with an expanded set of amino acid combinations. To investigate this question we applied a bacterial hybrid assay to screen ZF libraries where the 6 base-specifying residues of the third ZF helix of a three-fingered protein had been fully randomized, presenting 64 million amino acid combinations in each screen (Figure 1D). This approach has previously been applied on smaller scales to successfully investigate ZF function (10,52). Six libraries were constructed, each representing one of the six common mode BRPs modeled from structure. Each library was screened in 64 independent selections to recover helices able to interact with each of the 64 possible 3 bp targets. In total, we performed 384 selections that theoretically assayed over 24 billion unique protein-DNA interactions. From these selections we uncovered between 24 and 120 thousand functional helices depending on which mode was employed (Figure 1E and Supplemental Table S1). However, not all target selections enriched for functional helices. Mode 1 was the most successful screen with only four of the 64 selections failing to enrich functional helices (Figure 1F and Supplemental Table S2). Conversely, Mode 4 failed to enrich helices in 23 of the 64 selections despite the fact that the mode 1 and mode 4 BRPs only differ from RS to KS, respectively.
Analysis of the 384 selections reveals that mode has a large impact on ZF function. For example, a principal component analysis (PCA) demonstrates that modes with similar geometries produce more similar data (Figure 2A), while substantial differences in function remain. For example, while modes 1 and 4 are the most similar both in structure and specificity (according to the PCA), as mentioned above, mode 4 failed to enrich for helices in 19 of the target screens that were successful in mode 1 (Figure 1F). To confirm these results are a true reflection of function and not a sampling issue, we tested a series of helices based on their enrichment in one mode, but absence in an alternative mode, for the same binding site selections. These ZFs were then challenged for their ability to activate a GFP reporter driven by the complementary binding site (Figure 2B, Supplementary Figure S1). In all examples, the helix activated GFP significantly stronger in the mode it was recovered in compared to the alternative, with four of the five alternatives producing fluorescence levels similar to a negative control. We also confirmed that recovery in screens of two different modes demonstrates that the given helices have positive function in both modes (Supplementary Figure S1). In this case, all helices strongly activated the GFP reporter. These results demonstrate that the presence or absence of a helix in the proteins recovered from our selection screens is a reasonable approximation of function in that mode. Therefore, our results demonstrated the profound influence that mode can have on function where, depending on mode, ∼6–38% of the 3 bp targets are unable to be specified by any ZF amino acid strategy. In addition, as recovery of a helix within a selection is an approximate measure of function, we used the recovery frequency of each helix in these selections to predict helix specificity. In particular, summing the recovery frequency across all 64 binding sites (treated as nucleotide sequences) yields a target specificity. We then experimentally tested the specificity of over 100 helices, at least 10 helices representing each mode, by selecting their interaction partners from a random DNA library (Figure 2C). We find that by taking this approach we accurately predict the preferred base at all three positions of the binding site over 55% of the time and two of three preferred bases over 93% of the time (Figure 2D–G, Supplementary Figure S2). These results indicate that recovery of any ZF across modes and targets is a reasonable reflection of ZF function and specificity. Interestingly, in 23% of the ZFs tested we find evidence of extended specificity 5′ to the core 3 bp target (see Figure 2G and Supplementary Figure S2), however with our limited set of specificities we were unable to find a trend in helical composition or BRPs that would explain the specificity extension. Still, considering this extension and the potential for a cross-strand contact 3′ to the core target, it is possible that some ZFs influence base preference at 5 bp not the 3–4 commonly noted.
Expanding the boundary residue pair definition
Many naturally occurring ZF pairs do not use the BRPs employed in our screens making it difficult to determine if the results are generalizable. In fact, we still cannot predict the mode that any untested BRP is best represented by de novo. To provide a more detailed view of the BRP-mode landscape we analyzed our mode selections for helix–target pairs that are indicators of mode-exclusive function. We searched for helix-target pairs that were recovered in a single mode, or significantly more represented in one mode than any others, and used these as examples of ZF function that is dependent on, and therefore representative of, that mode's engagement with the DNA (Supplemental Table S3). Next, we created libraries of fully randomized residues at the two BRs in scaffolds that employed these mode-exclusive helices (Figure 3A). We screened 2–3 helices for each mode, totaling 13 mode-exclusive screens. From these results we find that the BRPs that we used in our original mode selections are recovered in the top 2 pairs for three out of the six modes (Modes 1, 2 and 5) while the BRPs used by the other three mode screens were recovered in the top 8 (Figure 3B). Interestingly there appears to be overlap between many of the modes where the same BRPs appear in more than one mode, implying that our definition of mode-exclusive should be thought of as a fluid approximation. Moreover, there are inconsistencies between our selected mode BRPs and the previously reported model (30) that might be explained by the small number of structures used for that model, the internal placement of some ZFs in these structures that place them under the influence of two BRPs simultaneously, or the BRPs that we chose for our primary libraries that dictated the design of our mode-exclusive screens. Nevertheless, since we cannot definitively say whether BRPs recovered in the same screen actually engage the DNA in the same way without structures, high frequency of recovery for any BRP in any of our screens should be thought of more as an indicator of function than an absolute definition of geometry. That said, the overlap between common BRPs often occurs in the most similar modes. For example, RS and RT are both recovered in the top 3 for the similar modes 1 and 4 while 6 of the top 10 BRPs found in mode 2 are also found in mode 6. In addition, with the exception of mode 3, the results of each independent mode-exclusive screen are more correlated with other screens of the same or similar modes (e.g. 1 with 4 and 2 with 6) than with more disparate modes (Supplementary Figure S3). These redundancies make sense as they accommodate general trends observed for each mode. For example, mode 1 appears to be defined by an Arginine at position 9 and a small amino acid at position −2 while mode 4 appears to be strongly biased towards a Serine or Threonine at position −2 but tolerates a more diverse set of amino acids with longer side chains at position 9. Thus, the RS and RT BRPs satisfy both of the mode 1 and mode 4 requirements. Similarly, both modes 2 and 6 can be defined by a preference for a basic residue at the −2 position with the flexibility to accommodate a diverse set of amino acids at position 9. To test the consistency of these results we made substitutions in the mode-exclusive helices that would be consistent, or inconsistent, with our BRP definition of mode and tested the ZFs ability to activate a GFP reporter. In all cases, the BRPs predicted to remain in the proper mode significantly outperform the out of mode substitutions (Supplementary Figure S4). Finally, to test how representative these results are of the mode influences, we built a second mode 1 library to represent the most common BRP recovered in our mode 1-exclusive screens, RA. The library consisted of 64 million amino acid combinations and was screened across all 64 targets, consistent with the previous mode library screens. We find by PCA that both mode 1 screens (BRPs RA and RS) are more similar to one another than they are to any other mode (Figure 3C) and are both functional across more targets (95% and 93% successful) than any other mode. These results demonstrate consistent, mode-related results across the mode defining BRPs that we have determined here.
Boundary residue pairs in nature
Since our BRP selections appear a reasonable proxy of mode activity, we next set to use these results to better define the BRPs in nature. To do so we considered that BRP substitutions resulted in reduced activity for the mode-exclusive ZFs and this reduction is related to the frequency with which the BRP was recovered in the screens (Supplementary Figure S4). Therefore, BRPs recovered with either high (at least 1% of recovered sequences), moderate (within the 95th percentile), and low frequency (the remainder) can be binned into groups of high confidence, low confidence and nonfunctional BRPs, respectively (Supplemental Tables S4–S9). While the relationship between the frequency of recovery and activity is not linear, the general inclusion in these groups does appear to be predictive of strong, weak, or non-functional BRPs. However, since the set BRPs within each confidence group is different depending on mode, a BRP might be high confidence in one mode but predicted to be nonfunctional in an alternative. This simply implies that ZF function is both dependent on the compatibility of the helix with its target and whether that helix is functional in the mode it is presented in. Therefore, to predict ZF function based on mode, we must first ask if the BRP is likely to be functional at all and next whether the helix is likely to be functional in the mode presented. With this in mind we note that across all 13 mode-exclusive screens we recovered a total of 62 high confidence BRPs that can be further divided by mode designation. An additional 85 BRPs fall into our low confidence category (Supplemental Table S10). Finally, we believe the remaining 253 BRPs are unlikely to be functional or they require very specific contexts that enables engagement with the DNA. Interestingly, a survey of the human ZF-TFs demonstrates that over 74% of human ZF pairs use BRPs that fall in our high confidence category and 91% fall into either the high or low confidence groups (Supplemental Table S11). While these results indicate that most ZFs present a functional geometry, this is likely a high-end estimate as we have already shown that a helix across different geometries can present significantly different levels of function.
To provide a more general view of BRPs across Kingdoms, we compared the distant Metazoan, Fungal and Green Plant ZF-TF BRPs and find that the most common Mode 1 BRPs from our screens (RA/RT/RS) are common in Metazoan and Fungal ZFs while the more distant Green Plants have instead enriched for mode 2 BRPs (QK, RK, QR and RR) (Supplementary Figure S5). As Fungi have a closer last common ancestor to human and have been shown to have ZFs that can explore a large diversity of binding specificities (11), it is interesting that the frequency of these Mode 1 BRPs have also been enriched in human concurrently with the expansion of the total number of ZF-TFs. Our results demonstrate the diverse functionality of this mode that supports more helical binding strategies across more functional targets. At the same time, ZFs in Green Plants frequently utilize less prolific, non-mode 1 BRPs and their ZF-TFs have not expanded at the same level representing just 4% of the TFs in Arabidopsis thaliana compared to the ∼50% of human factors. In fact, we find the five most common mode 1 BRPs are represented in high frequency (23–43%) for several model organisms where ZFs represent their most common DBD (Figure 3D). What's more, when we consider the 47,000 natural fingers investigated previously by synthetic screen (7), all fingers were presented adjacent to a common, fixed finger that displays an Arg at position 9. This means the ZFs tested may have been sampled in and out of their natural mode depending on what amino acid is present at position 9 in the finger that is naturally adjacent to it. While only 17% of the ZFs were functional in that screen, our results would predict that over 73% of those functional fingers were presented in mode 1. In addition, only ZFs in that screen that present the common mode 1 BRPs RS, RA, RT and RN were more likely to be recovered in the functional group than the non-functional group (Supplemental Table S12). These results indicated that natural fingers from ZF-TFs, regardless of their natural mode, are more likely to be functional when presented in mode 1.
Zinc finger promiscuity
Since mode can have influence on function across targets, it is possible that the mode might influence function within a target. We next considered whether some modes provide more strategies than others to bind the same sequence. To avoid the influence of potential sampling error for any unique 6 amino acid helix, we grouped helices with the same residues at the core binding positions −1, 2, 3, and 6, referring to these as ‘core helices’. This allows for diversity at positions 1 and 5, providing 400 version of every core helix and significantly reducing the likelihood that a core helix selected in one mode would be absent in another due to sampling error alone. This approach has been used before to approximate helical strategies as positions 1 and 5 are typically minor contributors to specificity (10,53,54). We found that mode 1 can utilize more unique core helices across all targets and, on average, significantly more core helices per 3 bp target (Figure 4A, B, and Supplemental Table S13). In addition, over 84% of the core helices used to bind a given target in a non-mode 1 selection are also found in at least one of the mode 1 screens. These results indicate that the flexibility provided across multiple mode geometries do expand the number of functional helices but most solutions are enabled by mode 1. Nevertheless, 16% of the helices do appear to depend on a geometry outside of mode 1, or 1634 of the 10 193 core helices recovered across all of our screens.
Figure 4.
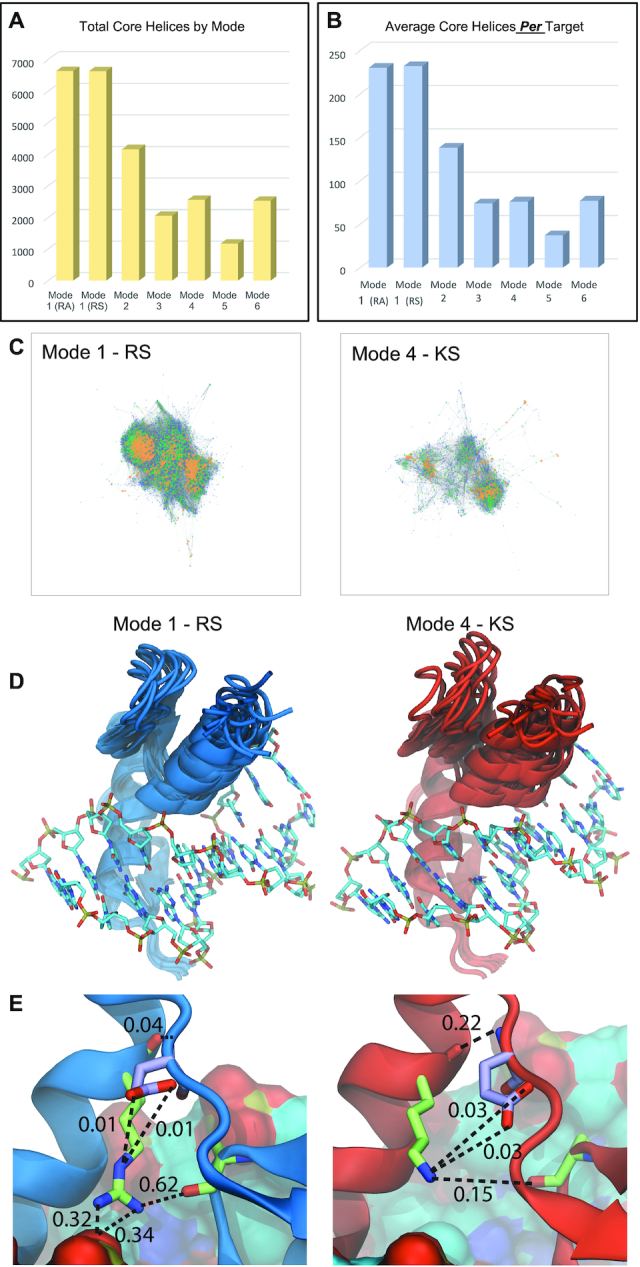
Helical plasticity across modes. (A) The number of unique core helices (positions −1, 2, 3, and 6) recovered across mode screens. (B) The average number of core helices per target by mode. (C) Connectivity plot comparison of Mode 1 and Mode 4. These modes use the BRPs RS and KS, respectively. Node color represents the number of target selections the helix was recovered in. Blue = 1, Green = 2–4, Yellow = 5 or more. (See also Supplementary Figure S7). (D) Top 10 conformational clusters identified on the molecular dynamic simulations of zif268 in mode 1 and mode 4. (E) Detail of the hydrogen bonds network of the BRs over the most frequent conformational cluster for mode 1 and mode 4. The numbers indicate the fraction of time a hydrogen bond (dash lines) was observed during the simulations. The Arg at position 9 (green licorice) of mode 1 is found within hydrogen bond distance of the backbone-carbonyl of Ser at position −2 (green licorice) four times more frequently than the Lys (green licorice) at position 9 of mode 4. The glutamic acid at position 51 (purple licorice) of the finger linker demonstrates an alternative hydrogen bond pattern in mode 4 respective to mode 1.
Since there are more mode 1 helices that can bind a given target, we next asked the reverse question, are there more mode 1 helices that offer the flexibility to bind more than one target? When we again consider core helices, we find that on average mode 1 core helices are recovered in more target selections than for any of the other modes, indicating that in general, mode 1 helices can be more promiscuous (Supplementary Figure S6 and Supplemental Table S13). While this increased promiscuity is slight, it is significant (ANOVA P < 0.00001) and increased flexibility could enable a ZF-TF to sample a new target with a single-base substitution that may provide an evolutionary advantage while still binding its natural target. This flexibility would certainly contribute to the expansion of a domain. Therefore, we considered how promiscuity might be related to helix similarity. We created connectivity plots for each mode screen using the recovered core helices across all selections. Each helix that differs by a single core amino acid is represented by a node while edges represent a single amino acid change between core helices. The size and color of the node indicates the number of target sequences the helix was able to bind (Figure 4C and Supplementary Figure S7). While both mode 1 plots are densely connected with the most promiscuous helices clustered in the center, non-mode 1 plot are sparse with distinct but separate clusters. For instance, the average node degree in mode 1 is 23 versus less than 20 for other modes (meaning a higher connectedness of the graph), while betweenness centrality is 0.0004 in mode 1 versus 0.0007 and higher in other modes (meaning less clustering of the graph, t-test P < 0.00001, see Supplementary Figure S7) (55). The more promiscuous helices are again at the center of these clusters, having both a higher average degree and betweenness centrality (see Supplementary Figure S7). These results imply that promiscuous helices within a mode are often related to large groups of helices that are also functional in that mode. This may provide an advantage as a promiscuous finger might sample multiple sequences and if one provided an evolutionary advantage, a secondary mutation that locked in that specificity would likely still be functional.
Hydrogen bond stability may contribute to Mode 1 plasticity
Our results demonstrate mode 1 is able to functionally employ a more diverse set of amino acids and interact with more targets successful. These results are surprising especially when we consider the similarity of many of the BRPs employed. The BRPs of mode 1 (RS) and mode 4 (KS) both present a basic residue at position 9 paired with a serine at position −2 yet nearly 5-fold more target selections failed in the mode 4 selections (Figure 1F) and on average, more than 3 times as many core helices are recovered per target in mode 1 relative to mode 4 (Figure 4B). Hence, we sought a structural explanation for the advantage provided by the Arg at position 9 and we carried out molecular dynamic simulations (MD) for ZFs modeled after zif268 that represent mode 1 (position 9 = Arg) or mode 4 (position 9 = Lys) geometries between fingers 2 and 3 of the protein (Figure 4D). We find that the position of finger 3 in the mode 4 MD fluctuates more when compared to finger 3 of mode 1 (Figure 4D, Supplementary Figure S8, and Supplemental Table S14), consistent with lower binding affinity. In addition, we find that the hydrogen bond between the backbone-carbonyl of Ser −2 and the side chains of the BRs in mode 1 occurs in 62% of the MD trajectory while only 15% in mode 4 (Figure 4E and Supplemental Table S15). Further, the Arg at position 9 in mode 1 makes additional hydrogen bonds with the phosphate backbone of the DNA that are not observed in the mode 4 MD. While additional contacts are possible with residues in the linker, in the MD these appear to make a minor contribution in mode 1 with a more substantial contact made between the backbone in mode 4 (Supplemental Table S16). Still, while possible, linker contacts will be difficult to predict because the substantial diversity in ZF linkers will impact the feasibility and strength of these additional interactions (Supplemental Table S11). Nevertheless, the hydrogen bonds more frequently observed between the BRs and the DNA in the mode 1 MD could increase the baseline affinity of the ZFs, which might explain the increased plasticity of mode 1 relative to mode 4 as an increased baseline affinity could allow more low affinity helices to survive our selections. Considering these potential differences, we tested a series of three ZF-TFs that in multiple modes, mutating their wildtype mode 1 BRs, for their ability to activate GFP and find in all cases mode 1 (wt) produces the strongest output consistent with it offering the highest affinity to DNA (Supplementary Figure S9).
Modes restrict zinc finger plasticity of transcription factors
To further test if mode 1 flexibility influences the ability of ZF-TFs to more easily sample new specificities we assayed a series of mutations in the 3rd ZFs of the human transcription factors KLF6 and Snai2. KLF6 is a member of a family of 18 orthologous human ZF-TFs that all bind similar targets using conserved residues at the core helical positions of its three ZF domains. KLF6 has core helical residues that match the consensus for all 18 family members, making it an excellent example of the entire KLF family (Supplementary Figure S10). Based on our selections across modes we identified single substitutions at position 3 of the KLF finger 3 helix that could transition its specificity from TGG to TAG or TCG, in the context of the core residues naturally expressed by finger 3 (Figure 5). We tested these substitution in 4 different mode contexts by also varying the BRPs and assaying the target specificity of each construct using a similar bacterial hybrid screen previously used to characterize hundreds of transcription factors across multiple model organisms (36,56,57). We find that while the mode 1 versions of the proteins are all functional and each specify the predicted targets, the other modes are less flexible. In general, mode 3 and mode 5 are less functional with significantly fewer colonies surviving selective pressure for all mutants and with multiple examples where the selections failed to provide any enrichment over background. The Mode 4 variants are more successful than modes 3 and 5, enriching for colonies above background in all but 1 selection. However, in all cases Mode 4 appears less successful than mode 1, producing far less colonies above background. In addition, while the two variants tested to modify the KLF finger 3 target preference from TGG to TAG are both functional in mode 1, mode 4 is only functional in one of the two (Figure 5, right). These results demonstrate that mode 1 would enable variants that provide TAG specificity more successfully and with more strategies. Interestingly, a common strategy selected to bind TCG uses Asp at the 3rd position of the helix (Figure 5, left). While this mutation is functional in modes 1 and 4, but not 3 and 5, it is the weakest of all mode 1 variants tested producing only 3-fold more colonies compared to a negative control. Therefore, we next tested substitutions of Ser and Thr at position 3 that are also commonly enriched in this context to bind TCG. However, these substitutions would require two mutations at the DNA level starting from the natural CAC codon of position 3. Again, we find modes 1 and 4 are functional, modes 3 and 5 are not, and mode 1 out performs mode 4 in each case. Therefore, substitutions at position 3 of finger 3 in KLF factors could transition the specificity in modes 1 and 4 but not 3 and 5. Further, based on these results, the most functional transition to TCG specificity would go through an intermediate where Tyr is at position 3 allowing for recognition of A or C at the middle base. If C were favored, a second mutation could switch the Tyr to a Ser and improve overall activity on TCG targets. However, as the Tyr substitution is only functional in mode 1, both of these specificity trajectories would be dependent on the plasticity of mode 1.
Figure 5.

Transcription factor specificity transitions are restricted by mode. (Top, center). Finger 3 of the KLF transcription factor family use a His at position 3 of the helix (green) to contact a guanine at the middle base (B2) of its target, tGG. Four versions of KLF6, a representative member of the KLF family, were constructed (see Supplementary Figure S10). Each version modified the BRPs between fingers 2 and 3 to create Mode 1, 3, 4, and 5 versions of the protein. Sequences that the KLF6 variants were able to interact with were selected from a 28bp random library by B1H selection. The fold increase in the number of colonies that survived those selections relative to a negative control, are shown in addition to the specificity for finger 3 of KLF6 in mode 1. (Right panel) Transitioning specificity to tAG. Substitutions at position 3 of the helix from His to Asn or Tyr are predicted to provide Adenine specificity at the middle base. These substitutions were made in the KLF6 variants as Modes 1, 3, 4 and 5 and subjected to B1H selection. The fold increase in the number of colonies that survived those selections are shown in addition to the specificity for finger 3 of KLF6 in mode 1. (Left panel) Transitioning specificity to tCG. Substitutions at position 3 of the helix from His to Asp, Thr, or Ser are predicted to provide cytosine specificity at the middle base. These substitutions were made in the KLF6 variants as Modes 1, 3, 4 and 5 and subjected to B1H selection. The fold increase in the number of colonies that survived those selections are shown in addition to the specificity for finger 3 of KLF6 in mode 1.
Snai2, the human homolog of Slug (58), is a 4-fingered transcription factor but only fingers 2 and 3 appear to specify its target. This makes Snai2 an excellent test case as function should be completely dependent on the single BRP between these two fingers. From our selection data it appears modification of the finger 3 core helix (DSNA) at helical position 6 could lead to tolerance of alternative bases at the first base of its target, CAC. Our data would predict functional substitutions in this core helix from Ala at position 6 to His, Arg or Lys with varying levels of mode-dependent success. These mutations could modify binding preference from CAC to at least tolerate AAC, GAC and TAC, allowing altered target preference to any NAC target with a single amino acid substitution. Therefore, we again screened a series of Snai2 variants in five modes to test the TFs ability to transition specificity in each mode. Again, we find that mode 1 is the most functional and able to transition to the predicted binding tolerance for each substitution (Supplementary Figure S11). Mode 2 is functional, to a lesser degree, with two of the three substitutions while each other mode is restricted to weak function with an Arg at position 6 or no function in all cases. These results and those above demonstrate that in KLF and Snai2, mode 1 enables the flexibility for the ZFs to easily transition to new target specificities that would be restricted in any other mode.
An improved specificity model based on BRP influences and deep neural networks
Previous studies have used a number of different machine learning techniques to build predictive models of DNA binding specificity for ZFs including those based on k-nearest neighbors (59), neural networks (60), support vector machines (61) and random forests (7). Although these different approaches have proven to be useful tools, the increment in computational power has made feasible the application of more complex and powerful techniques. Therefore, to take advantage of the wealth of data provided here, and in particular the fact that it should take into account the neighbor-effects on specificity, we implemented a new prediction model based on convolutional neural networks. Such methods have recently been shown to be powerful when coupled with large amounts of data (62). In particular, we implemented a one-dimensional convolutional neural network (1D-CNN) using Keras (https://github.com/fchollet/keras), where the input layer is the one-hot encoded amino acid sequence of the helix in addition to the BRP and the output layer is a 3 × 4 matrix representing the 3-base binding motif of a zinc finger (See Figure 6A). The hyperparameters and the architecture of the network where determined by random search using as evolution the two independent data sets generated by B1H, including the previous data from our lab (10) and Hughes et al. (63), as well as the new data presented here. Finally, for testing and performance comparison, we used a number of different curated datasets from other methods, including Chip-Seq (1,5,8,41,63). These present a variety of natural fingers from various species, including human, fly and worm.
Figure 6.

Validation of the synthetic model of zinc finger specificity. (A) Schema of the network architecture trained with the mode selection screens. (B, C) Effect of the consecutive expansion of the training set with new modes subsets on the performance of the network. Network performance was measured as the mean square error of the predicted motif to the experimental D. melanogaster ZF-TFs and ZifRC datasets. Error estimations from independent trainings over 10-fold cross-validations. (D−F) Comparison of performance of ZifRC and neural network across a series of single-finger datasets measured by correlation score (described in the methods section) over different datasets: ZifRC training dataset with 8,112 motifs (D), a subset of 4000 motifs removed from the neural network training set (E), 100 validated motifs from the selection (Supplementary Figure S2) also absent in the training set (F). (G−I) Comparison of performance of ZifRC and neural network on a series of characterized ZF-TFs including 129 C. elegans factors (G) and two sets curated by Najafadabi et al., 2015, the gold standard set (H) and a set of 39 human ZF-TFs (I).
When comparing model performance with incremental addition of more mode-specific training data, we observe a steady increase in accuracy for each mode added, in line with the intuition that our mode-specific data encodes some of the neighbor-effects on specificity (see Figure 6B, C). We also find that when compared to a previous model, our new specificity predictor outperforms it in most datasets, while performing similarly in others (see Figure 6 D–I). A utility based on this model for ZF predictions, ZFPred, can be found here, www.gitlab.com/kimlab/zfgeomodes.
DISCUSSION
The Cys2His2 zinc finger domain is the most common DNA-binding in most metazoan genomes and despite its seemingly simplistic interaction with the DNA, efforts to provide predictive rules that define this domain have fallen short. We applied the largest synthetic screen of any protein domain to better understand how ZFs engage their DNA targets. Seven libraries, each consisting of 64 million amino acid combinations, were screened in 448 selections to uncover ZFs able to bind each of the 64 possible 3 bp targets. The theoretical complexity sampled here, over 28 billion unique protein–DNA interactions, eclipses the diversity experimentally sampled in any prior work. These screens and the models derived from them include several orders of magnitude more ZF-target diversity than screens of naturally occurring ZFs (∼16,000-fold) and 10X more diversity than our original synthetic screen of zif268’s third finger (7,10). Here, we demonstrate two benefits to this exhaustive approach. First, by systematically changing a single variable that influence adjacent finger function and then comprehensively screening the consequence on base recognition, we are able to provide mechanistic insight into domain function that would be extremely difficult to derive from the limited complexity that has evolved naturally. Second, we are able to use this purely synthetic data to create a model of ZF-TF specificity as accurate as any previously reported. The implications of this synthetic model are important to note. Other models include data produced for naturally occurring ZFs and ZF-TFs that offer diversity through the domain and between adjacent fingers and then use these data to predict other ZF-TFs. Conversely, all proteins expressed in our screens are 90% identical and yet we are able to predict the specificity of diverse ZF-TFs with the same level of accuracy.
Our results also allow a preliminary prediction of ZF function based on the BRPs employed. One benefit of predicting BRP functionality is that it allows us to begin to investigate which fingers of large ZF-TFs actually engage the DNA. CTCF is an excellent example as only five of its 11 ZFs, fingers 3–7, are responsible for its target recognition. Interestingly, our data predicts that the BRPs that surround these five functional ZFs are low probability BRPs. Between fingers 2 and 3 the BRP (NV) is predicted to be nonfunctional and the BRP between fingers 7 and 8 (RH) is predicted to be low confidence, representing less than 0.5% of the sequences recovered in the mode 6 screens alone. What's more, in the CTCF structure while fingers 3–7 are engaged with the DNA with canonical angles and distances, fingers 2 and 8 appear removed from the major groove and interact with the phosphate backbone (64). This offers at least a preliminary example of predicted BRP function that is supported by structure though we caution that many more variables will ultimately impact adjacent finger activity and our prediction of BRP functionality should be thought of as an approximation. For example, the position within the ZF array may influence the functionality of a given BRP as the function of some ZFs are known to be dependent on position. However, we chose the C-terminal position for our libraries as it reduces the influence on the library selection to a single BRP and we have previously shown there is no loss in predictive power by screening C-terminal libraries versus internal ZF libraries. Nevertheless, it is important to note that internal ZFs in ZF-TFs are under the influence of both an N and C-terminal ZF and their BRPs.
In addition to predicting functional units, we have also shown that BRP substitutions can reduce or ablate overall function in a set of natural TFs (Supplementary Figure S9) and provide categories of BRPs that would allow for a first approximation of the consequence of BRP mutations (Supplemental Tables S10 and S11). What's more, mutations at these positions have already been shown to have disease-related consequences. Mutations at BRP position 9 are enriched in cancer samples (31). Interestingly the most common change is from Arg at position 9 to Ile. Our data would predict that when an Ile is present at position 9 only four possible BRPs are likely to be functional and even in these cases, a mutation that changes an Arg to an Ile would modify the protein's mode which could have severe consequences on function. In fact, over 71% of these mutations found in cancer samples transition a mode 1 ZF pair out of mode (Supplemental Table S17). Therefore, our data would suggest that that these mutations severely reduce function by disrupting the natural adjacent finger geometry.
Finally, the impact of our investigation of adjacent finger geometry is not limited to predictions of function, but has also underscored the impact that ZF plasticity could have on the utility of the domain and its prevalent use in complex eukaryotes. The evolutionary expansion of the ZF-TFs has been of great interest in recent years. While the driving forces behind this expansion have been the focus of multiple articles, few mechanistic explanations have been offered for how the ZF domain would enable this expansion. Certainly, the modularity of the domain enables the addition and subtraction of ZFs through duplication of the domain. However, modularity is more complicated than simple duplication as our work demonstrates that duplication events could easily result in nonfunctional ZF pairs as their function depends on whether the new BRs are compatible. With this in mind, the screens reported here demonstrate that the most common BRPs of mode 1 enable more flexibility in base recognition than all other modes. If duplication were to result in the presentation of the new ZF in an alternative mode, our results suggest that the amino acid strategy of the helix would have a lower probability to functionally engage the DNA. Not surprisingly, common model organisms that use the ZF as their most common DBD also utilize mode 1 with the highest frequency. In addition, the increased function and helical diversity enabled by mode 1 increases the likelihood that a mutation at one of the core helical residues will still bind DNA, potentially allowing a transition in specificity. The ability to sample new targets, while the second allele of the TF maintains the normal function of the protein, could offer additional evolutionary advantages provided by mode 1 ZFs, and limited in other geometries. Ultimately, our data support a model where the helical plasticity enabled by the mode 1 BRPs simplifies specificity transitions when evolution would favor them and provide advantages for the duplication of the domain.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank current and former members of the Noyes lab for the thoughtful suggestions on the work and manuscript. We thank T.M.C. for her valued insight, support, and feedback on the manuscript. We would also like to thank the NYU Langone Genome Technology Center for their help in determining the best approach for sequencing our results. In addition, we would like to acknowledge HPC support from a Compute Canada Resource Allocation and the NVIDIA academic GPU grant program.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [R01GM118851, R01GM133936 to A.L.M., C.C.V, D.O.G., D.M.I., J.M.S., M.G., M.B.N.]. Funding for open access charge: NIH [R01GM118851].
Conflict of interest statement. None declared.
REFERENCES
- 1. Weirauch M.T., Yang A., Albu M., Cote A.G., Montenegro-Montero A., Drewe P., Najafabadi H.S., Lambert S.A., Mann I., Cook K. et al.. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014; 158:1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Emerson R.O., Thomas J.H.. Adaptive evolution in zinc finger transcription factors. PLoS Genet. 2009; 5:e1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schmitges F.W., Radovani E., Najafabadi H.S., Barazandeh M., Campitelli L.F., Yin Y., Jolma A., Zhong G., Guo H., Kanagalingam T. et al.. Multiparameter functional diversity of human C2H2 zinc finger proteins. Genome Res. 2016; 26:1742–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pavletich N.P., Pabo C.O.. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science. 1991; 252:809–817. [DOI] [PubMed] [Google Scholar]
- 5. Wolfe S.A., Nekludova L., Pabo C.O.. DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct. 2000; 29:183–212. [DOI] [PubMed] [Google Scholar]
- 6. Liu H., Chang L.H., Sun Y., Lu X., Stubbs L.. Deep vertebrate roots for mammalian zinc finger transcription factor subfamilies. Genome Biol. Evol. 2014; 6:510–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Najafabadi H.S., Mnaimneh S., Schmitges F.W., Garton M., Lam K.N., Yang A., Albu M., Weirauch M.T., Radovani E., Kim P.M. et al.. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat. Biotechnol. 2015; 33:555–562. [DOI] [PubMed] [Google Scholar]
- 8. Enuameh M.S., Asriyan Y., Richards A., Christensen R.G., Hall V.L., Kazemian M., Zhu C., Pham H., Cheng Q., Blatti C. et al.. Global analysis of Drosophila Cys(2)-His(2) zinc finger proteins reveals a multitude of novel recognition motifs and binding determinants. Genome Res. 2013; 23:928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Persikov A.V., Rowland E.F., Oakes B.L., Singh M., Noyes M.B.. Deep sequencing of large library selections allows computational discovery of diverse sets of zinc fingers that bind common targets. Nucleic Acids Res. 2014; 42:1497–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Persikov A.V., Wetzel J.L., Rowland E.F., Oakes B.L., Xu D.J., Singh M., Noyes M.B.. A systematic survey of the Cys2His2 zinc finger DNA-binding landscape. Nucleic Acids Res. 2015; 43:1965–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Najafabadi H.S., Garton M., Weirauch M.T., Mnaimneh S., Yang A., Kim P.M., Hughes T.R.. Non-base-contacting residues enable kaleidoscopic evolution of metazoan C2H2 zinc finger DNA binding. Genome Biol. 2017; 18:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T.. The human transcription factors. Cell. 2018; 175:598–599. [DOI] [PubMed] [Google Scholar]
- 13. Grey C., Baudat F., de Massy B.. PRDM9, a driver of the genetic map. PLos Genet. 2018; 14:e1007479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baker Z., Schumer M., Haba Y., Bashkirova L., Holland C., Rosenthal G.G., Przeworski M.. Repeated losses of PRDM9-directed recombination despite the conservation of PRDM9 across vertebrates. Elife. 2017; 6:e24133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Imbeault M., Helleboid P.Y., Trono D.. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature. 2017; 543:550–554. [DOI] [PubMed] [Google Scholar]
- 16. Barazandeh M., Lambert S.A., Albu M., Hughes T.R.. Comparison of ChIP-Seq data and a reference motif set for human KRAB C2H2 zinc finger proteins. G3 (Bethesda). 2018; 8:219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klug A. The discovery of zinc fingers and their development for practical applications in gene regulation and genome manipulation. Q Rev. Biophys. 2010; 43:1–21. [DOI] [PubMed] [Google Scholar]
- 18. Ramirez C.L., Foley J.E., Wright D.A., Muller-Lerch F., Rahman S.H., Cornu T.I., Winfrey R.J., Sander J.D., Fu F., Townsend J.A. et al.. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat. Methods. 2008; 5:374–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sander J.D., Dahlborg E.J., Goodwin M.J., Cade L., Zhang F., Cifuentes D., Curtin S.J., Blackburn J.S., Thibodeau-Beganny S., Qi Y. et al.. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat. Methods. 2011; 8:67–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rebar E.J., Greisman H.A., Pabo C.O.. Phage display methods for selecting zinc finger proteins with novel DNA-binding specificities. Methods Enzymol. 1996; 267:129–149. [DOI] [PubMed] [Google Scholar]
- 21. Jolma A., Yan J., Whitington T., Toivonen J., Nitta K.R., Rastas P., Morgunova E., Enge M., Taipale M., Wei G. et al.. DNA-binding specificities of human transcription factors. Cell. 2013; 152:327–339. [DOI] [PubMed] [Google Scholar]
- 22. Genomes Project, C. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al.. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Noyes M.B., Meng X., Wakabayashi A., Sinha S., Brodsky M.H., Wolfe S.A.. A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Res. 2008; 36:2547–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bulyk M.L., Huang X., Choo Y., Church G.M.. Exploring the DNA-binding specificities of zinc fingers with DNA microarrays. Proc. Natl Acad. Sci. U.S.A. 2001; 98:7158–7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barrera L.A., Vedenko A., Kurland J.V., Rogers J.M., Gisselbrecht S.S., Rossin E.J., Woodard J., Mariani L., Kock K.H., Inukai S. et al.. Survey of variation in human transcription factors reveals prevalent DNA binding changes. Science. 2016; 351:1450–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Badis G., Berger M.F., Philippakis A.A., Talukder S., Gehrke A.R., Jaeger S.A., Chan E.T., Metzler G., Vedenko A., Chen X. et al.. Diversity and complexity in DNA recognition by transcription factors. Science. 2009; 324:1720–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yin Y., Morgunova E., Jolma A., Kaasinen E., Sahu B., Khund-Sayeed S., Das P.K., Kivioja T., Dave K., Zhong F. et al.. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017; 356:eaaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jolma A., Kivioja T., Toivonen J., Cheng L., Wei G., Enge M., Taipale M., Vaquerizas J.M., Yan J., Sillanpaa M.J. et al.. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010; 20:861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zuo Z., Roy B., Chang Y.K., Granas D., Stormo G.D.. Measuring quantitative effects of methylation on transcription factor-DNA binding affinity. Sci. Adv. 2017; 3:eaao1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garton M., Najafabadi H.S., Schmitges F.W., Radovani E., Hughes T.R., Kim P.M.. A structural approach reveals how neighbouring C2H2 zinc fingers influence DNA binding specificity. Nucleic Acids Res. 2015; 43:9147–9157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Munro D., Ghersi D., Singh M.. Two critical positions in zinc finger domains are heavily mutated in three human cancer types. PLoS Comput. Biol. 2018; 14:e1006290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller J.C., Pabo C.O.. Rearrangement of side-chains in a Zif268 mutant highlights the complexities of zinc finger-DNA recognition. J. Mol. Biol. 2001; 313:309–315. [DOI] [PubMed] [Google Scholar]
- 33. Elrod-Erickson M., Rould M.A., Nekludova L., Pabo C.O.. Zif268 protein-DNA complex refined at 1.6 A: a model system for understanding zinc finger-DNA interactions. Structure. 1996; 4:1171–1180. [DOI] [PubMed] [Google Scholar]
- 34. Noyes M.B. Analysis of specific protein-DNA interactions by bacterial one-hybrid assay. Methods Mol. Biol. 2012; 786:79–95. [DOI] [PubMed] [Google Scholar]
- 35. Oakes B.L., Xia D.F., Rowland E.F., Xu D.J., Ankoudinova I., Borchardt J.S., Zhang L., Li P., Miller J.C., Rebar E.J. et al.. Multi-reporter selection for the design of active and more specific zinc-finger nucleases for genome editing. Nat. Commun. 2016; 7:10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meng X., Brodsky M.H., Wolfe S.A.. A bacterial one-hybrid system for determining the DNA-binding specificity of transcription factors. Nat. Biotechnol. 2005; 23:988–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ichikawa D.M., Corbi-Verge C., Shen M.J., Snider J., Wong V., Stagljar I., Kim P.M., Noyes M.B.. A multireporter bacterial 2-Hybrid assay for the high-throughput and dynamic assay of PDZ domain-peptide interactions. ACS Synth. Biol. 2019; 8:918–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hagberg A.A., Schult D.A., Swart P.J.. 2008; Pasadena, CA: Proceedings of the 7th Python in Science Conference (SciPy 2008) Exploring Network Structure, Dynamics, and Function using NetworkX. [Google Scholar]
- 39. Pedgregosa F., Varoquaux G., Gamfort A., Michel V., Thirion B.. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 2011; 12:2825–2830. [Google Scholar]
- 40. Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J. et al.. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 2020; 17:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Narasimhan K., Lambert S.A., Yang A.W., Riddell J., Mnaimneh S., Zheng H., Albu M., Najafabadi H.S., Reece-Hoyes J.S., Fuxman Bass J.I. et al.. Mapping and analysis of Caenorhabditis elegans transcription factor sequence specificities. Elife. 2015; 4:e06967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Case D.A., Cheatham T.E. 3rd, Darden T., Gohlke H., Luo R., Merz K.M. Jr, Onufriev A., Simmerling C., Wang B., Woods R.J.. The Amber biomolecular simulation programs. J. Comput. Chem. 2005; 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peters M.B., Yang Y., Wang B., Fusti-Molnar L., Weaver M.N., Merz K.M. Jr. Structural survey of zinc containing proteins and the development of the zinc AMBER force field (ZAFF). J. Chem. Theory Comput. 2010; 6:2935–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vriend G. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 1990; 8:52–56. [DOI] [PubMed] [Google Scholar]
- 45. Ryckaert J.P., Ciccotti G., Berendsen H.J.C.. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977; 23:327–341. [Google Scholar]
- 46. Toukmaji A., Sagui C., Board J., Darden T.. Efficient particle-mesh Ewald based approach to fixed and induced dipolar interactions. J. Chem. Phys. 2000; 113:10913–10927. [Google Scholar]
- 47. Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R.. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984; 81:3684–3690. [Google Scholar]
- 48. Perez A., Marchan I., Svozil D., Sponer J., Cheatham T.E. 3rd, Laughton C.A., Orozco M.. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys J. 2007; 92:3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roe D.R., Cheatham T.E. 3rd. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013; 9:3084–3095. [DOI] [PubMed] [Google Scholar]
- 50. Weirauch M.T., Cote A., Norel R., Annala M., Zhao Y., Riley T.R., Saez-Rodriguez J., Cokelaer T., Vedenko A., Talukder S. et al.. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 2013; 31:126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weirauch M.T., Hughes T.R.. A catalogue of eukaryotic transcription factor types, their evolutionary origin, and species distribution. Subcell Biochem. 2011; 52:25–73. [DOI] [PubMed] [Google Scholar]
- 52. Gupta A., Christensen R.G., Rayla A.L., Lakshmanan A., Stormo G.D., Wolfe S.A.. An optimized two-finger archive for ZFN-mediated gene targeting. Nat. Methods. 2012; 9:588–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Persikov A.V., Singh M.. De novo prediction of DNA-binding specificities for Cys2His2 zinc finger proteins. Nucleic Acids Res. 2014; 42:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gupta A., Christensen R.G., Bell H.A., Goodwin M., Patel R.Y., Pandey M., Enuameh M.S., Rayla A.L., Zhu C., Thibodeau-Beganny S. et al.. An improved predictive recognition model for Cys(2)-His(2) zinc finger proteins. Nucleic Acids Res. 2014; 42:4800–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu H., Kim P.M., Sprecher E., Trifonov V., Gerstein M.. The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput. Biol. 2007; 3:e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Noyes M.B., Christensen R.G., Wakabayashi A., Stormo G.D., Brodsky M.H., Wolfe S.A.. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell. 2008; 133:1277–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kazemian M., Brodsky M.H., Sinha S.. Genome Surveyor 2.0: cis-regulatory analysis in Drosophila. Nucleic Acids Res. 2011; 39:W79–W85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hemavathy K., Guru S.C., Harris J., Chen J.D., Ip Y.T.. Human Slug is a repressor that localizes to sites of active transcription. Mol. Cell Biol. 2000; 20:5087–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Alleyne T.M., Pena-Castillo L., Badis G., Talukder S., Berger M.F., Gehrke A.R., Philippakis A.A., Bulyk M.L., Morris Q.D., Hughes T.R.. Predicting the binding preference of transcription factors to individual DNA k-mers. Bioinformatics. 2009; 25:1012–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu J., Stormo G.D.. Context-dependent DNA recognition code for C2H2 zinc-finger transcription factors. Bioinformatics. 2008; 24:1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Persikov A.V., Osada R., Singh M.. Predicting DNA recognition by Cys2His2 zinc finger proteins. Bioinformatics. 2009; 25:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. LeCun Y., Bengio Y., Hinton G.. Deep learning. Nature. 2015; 521:436–444. [DOI] [PubMed] [Google Scholar]
- 63. Lambert S.A., Albu M., Hughes T.R., Najafabadi H.S.. Motif comparison based on similarity of binding affinity profiles. Bioinformatics. 2016; 32:3504–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hashimoto H., Wang D., Horton J.R., Zhang X., Corces V.G., Cheng X.. Structural basis for the versatile and methylation-dependent binding of CTCF to DNA. Mol. Cell. 2017; 66:711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Preprocessed sequencing and code are available upon request. The model of specificity can be applied to ZFs using the utility ‘ZFPred’ found at repository: www.gitlab.com/kimlab/zfgeomodes