


Abstract
We identified over 1000 instances of water-nucleobase stacking contacts in a variety of RNA molecules from a non-redundant set of crystal structures with resolution ≤3.0 Å. Such contacts may be of either the lone pair-π (lp–π) or the OH–π type, in nature. The distribution of the distances of the water oxygen from the nucleobase plane peaks at 3.5 Å for A, G and C, and approximately at 3.1–3.2 Å for U. Quantum mechanics (QM) calculations confirm, as expected, that the optimal energy is reached at a shorter distance for the lp–π interaction as compared to the OH–π one (3.0 versus 3.5 Å). The preference of each nucleobase for either type of interaction closely correlates with its electrostatic potential map. Furthermore, QM calculations show that for all the nucleobases a favorable interaction, of either the lp–π or the OH–π type, can be established at virtually any position of the water molecule above the nucleobase skeleton, which is consistent with the uniform projection of the OW atoms over the nucleobases ring we observed in the experimental occurrences. Finally, molecular dynamics simulations of a model system for the characterization of water-nucleobase stacking contacts confirm the stability of these interactions also under dynamic conditions.
INTRODUCTION
‘Water molecules constitute an integral part of helical and non-helical structures of nucleic acids’ (1). They are well known to stabilize the three dimensional structure of DNA and RNA and to assist drug binding and catalytic reactions, based on their ability to form hydrogen bonds with the electronegative atoms of both the nucleobases and the phosphate-2′-(deoxy)ribose backbone (1–15). Further, studies describing the hydration patterns around RNA canonical and non-canonical base pairs confirmed that base pair hydration mostly involves H-bonding between water molecules and the bases (2,16,17). In addition, a water molecule can be involved in interactions with the π cloud of the aromatic ring of a nucleobase. The chemical nature of such interactions can be: either (i) of the OH–π type, when a hydrogen atom of a water gives an ‘aromatic H-bond’ with the π cloud of the base, or (ii) of the lone pair-π (lp–π) type, when the interaction occurs between a lone pair of the water oxygen and the π cloud of the base.
A number of studies in the last two decades have been experimentally and theoretically characterizing the OH–π and lp–π interactions of water with model organic compounds, also mimicking nucleobases and side chains of aromatic amino acids (18–23). A correlation between the ‘molecular electronegativity’ of the aromatic molecule and the type of established interaction has been proposed (24). The main conclusion being that the OH–π interaction is more favorable in case of molecules providing a negative electrostatic potential on the aromatic ring, such as benzene (20,25), while lp–π interactions are preferred in case of electron deficient neutral rings, such as hexafluorobenzene, and of protonated rings (26–28). In this context, Wheeler and coworkers questioned that the effects of substituents on the stacking of aromatic rings arise from the induced π-polarization and suggested instead that they arise from direct interactions between the substituent and the other ring (29–32). In proteins, OH–π and lp–π contacts have been shown to represent energetically favorable interactions, with the OH–π contacts especially being a weaker but nonetheless important class of interactions (28,33,34).
In RNA, Egli et al. reported examples of both OH–π and lp–π interactions in the crystal structure of a 28-mer ribosomal frameshifting RNA pseudoknot from beet western yellow virus (35,36). In the absence of experimental knowledge of the H-atom positions (obviously missing from the crystal structure), the water-nucleobase contacts were assigned to a specific type of interaction based on the distance between the water oxygen (Ow) and the aromatic plane of the nucleobase, and on the possible involvement of water in H-bonds with surrounding acceptor/donor atoms. They were classified as ‘water-nucleobase stacking interactions’, since they involve the π cloud of the nucleobases and represent a surrogate of the base-base stacking, missing for the involved nucleobases. Later on, Egli and Sarkhel performed a survey of lp–π interactions in structures of different molecules, including proteins and nucleic acids, complemented by quantum mechanics calculations (37). Based on their calculations, they showed that distances for the lp–π interactions are shorter than for the OH–π ones (ranges of 2.7–3.1 and 3.2–3.5 Å, respectively). Further, they showed that the lp–π interaction between water and uracil is comparable in energy to the OH–π interaction between water and a neutral cytosine, due to relatively positive polarization of uracil, and that it is even more stable for a protonated cytosine, as for protonated aromatic rings in general. On these bases, they concluded that lp–π interactions are expected to be more frequent in nucleic acids, also due to the absence in their backbone of H-bond donors (except for RNA 2′-OH ribose) to engage the lone pairs of the water oxygen, and called for a systematic search of the structures in the Protein Data Bank, to retrieve other examples of lp–π interactions (37).
Although the role of water-π interactions in the context of biomolecular structures is thus widely recognized, a comprehensive structural and energetic characterization is still missing for RNA molecules. As the space of RNA architectures is known to be vast and largely uncharacterized, the relevance of such weak interactions cannot be overlooked, especially for such molecules. They can indeed be part, together with the ribose-base stacking interactions (38–40), of a variety of strategies that RNA molecules employ to achieve the stability of their overall fold and of specific structural motifs (10,38,41–43).
In the attempt to fill this gap, in this contribution we extended the work of Egli to the analysis of a comprehensive non-redundant set of RNA high-resolution crystal structures (44), using the same approach we used to characterize ribose-nucleobase stacking interactions in RNAs (38). First, we systematically searched the dataset for water-nucleobase stacking contacts, and we identified over 1000 water-nucleobase stacking contacts in 293 structures, representing a variety of RNA functional molecules. We found that the nucleobase usage in them is biased toward uracil and guanine, and that in the detected interactions the oxygen atoms are not centered on the nucleobases but spread all over their surface. Based on the analysis of the structural context, we classified them as OH–π or lp–π interactions, when possible. Further, we classified bridging waters as those waters being simultaneously involved in water-nucleobase stacking interactions and in classical H-bonds with other RNA atoms.
Then, we complemented the structural analyses with quantum mechanics calculations, which have been proved to give insights on the structural stability and energetics of H-bonding and π–π interactions between nucleobases (12,23,45–61). In addition, quantum mechanics calculations are particularly useful to study non-covalent interactions observed in experimental structures, including the interaction of water with the π ring of the nucleobases (23,37). Herein, we performed a state-of-the-art characterization of the OH–π and lp–π interaction energies for each of the four canonical nucleobases (A, U, G, C), for the electron rich benzene and electron deficient hexafluorobenzene rings, used as references and for the N3-protonated cytosine (hereon C+ or ‘protonated cytosine’), for which at least one water-nucleobase stacking interaction has been structurally characterized (36). We calculated potential energy curves for the OH–π and lp–π interactions involving all the above aromatic systems, from which we derived optimal interaction distances. We found out that the OH–π interaction is energetically more favorable for all the nucleobases but for uracil and protonated cytosine, for which the lp–π one is preferred. This can be satisfactorily explained based on their calculated electrostatic potential maps. Finally, to test the stability of water-nucleobase stacking contacts under dynamic conditions we also performed molecular dynamics (MD) simulations of the 28-mer RNA pseudoknot system, whose water-base stacking interactions were characterized by Egli and coworkers (35,36)
MATERIALS AND METHODS
Structural dataset
Our dataset consisted of the 630 crystal structures from the non-redundant 3D structures dataset for RNA by Leontis and Zirbel (62), version 1.89 (dated: 5 December 2014), having a resolution ≤3.0 Å and including at least one crystallization water.
Identification of water–nucleobase contacts
Water-nucleobase stacking contacts were identified using the setup shown in the Figure 1. The nucleobases of all the instances in the dataset were oriented in a Cartesian frame as follows. The geometric center of the heterocycle skeleton was selected to be the origin of the frame. The x-axis was set as the line passing through the origin and the N3 atom for pyrimidines, or the line passing through the origin and the middle point of the N1–C2 bond for purines. The y-axis formed a 90° angle with the x-axis, with the C6 atom of purines and the C4 atom of pyrimidines lying in the xy-plane at positive y values. A right-handed frame was formed by building the z-axis vector as the cross product of the vectors along the x-axis and y-axis.
Figure 1.
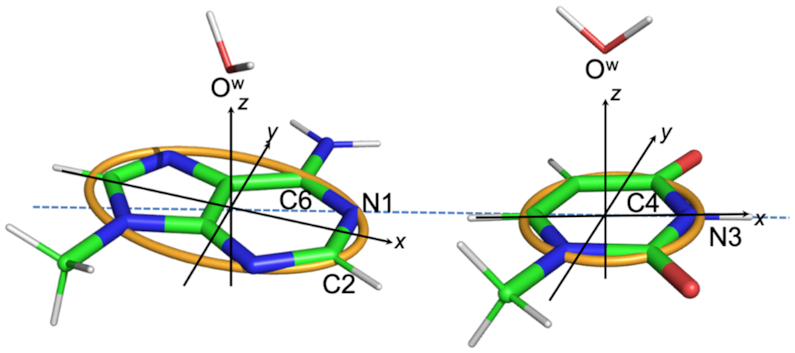
Definition of the reference Cartesian frame on the nucleobases and of the parameters used to define the position of the OW relatively to the nucleobase. The origin is at the geometrical center of the heterocycle skeleton, the x-axis passing through the N3 atom for pyrimidines and through the middle point of N1–C2 bond for purines, the y-axis forms a 90° angle with the x-axis, with the C6 atom of purines and the C4 atom of pyrimidines lying in the xy-plane at positive y values, the z-axis is the cross product of the versors (unit vectors indicating the directions) along the x and y-axes, thus forming a right-handed frame. The orange curve defines a circle of radius 1.5 Å in the xy-plane of pyrimidines, and an ellipse with minor and major axes equal to 1.5 and 2.5 Å in the xy-plane of purines. A water and a nucleobase are considered to be interacting if the projection of OW on the xy-plane is within the orange circle/ellipse, with the vertical distance (along the z axis) in the −4.0 to +4.0 Å range. The instances of OW-nucleobase stacking contacts with specification of their PDB IDs and residues involved are reported in Supplementary Table S1.
Water-base contacts were defined based on two conditions. First, the rise of the OW atom from the nucleobase plane had to fall in the −4.0 to +4.0 Å range. Second, the projection of the OW atom on the nucleobase plane had to follow within a circle of radius 1.5 Å (center at the frame origin) for pyrimidines or within an ellipse with major axis of 2.5 Å and minor axis 1.5 Å (again centered at the origin) for purines. This procedure allowed us to identify a total of 1008 water–base contacts in 293 PDB structures involving all four nucleobases. Of course, choosing cutoff values in a structural search always has some arbitrariness. In the present case, the cutoff values were selected to enforce that the projection of OW on the base plane is within the heterocycle ring. For the vertical distance, consistently with literature we considered a cutoff of ±4.0 Å (38).
Of the 1008 water-nucleobase contacts identified based on the above criteria, a small fraction, 39, involved waters with an occupancy below 1. EDIA (Electron Density for Individual Atoms) values (63), representing a quality control for the placement of water molecules in X-ray structures, were also calculated for the oxygen atoms of 916 waters (belonging to 270 structures) over the total 1008 involved in stacking contacts, using the ProteinsPlus server (64). Such EDIA values, all reported in Supplementary Table S1, are above 0.4 for 77% of the analysed water molecules (>0.8 for 21% of them, Supplementary Figure S3), placing them within medium or well resolved waters. For 92 waters (9% of the total), EDIA values could not be obtained as the electron density maps of the corresponding structures are unfortunately unavailable (see Supplementary Table S1). These numbers are stable to the change of the structures resolution cut-off. When we limited our analysis to the 869 stacked waters in the subset of 236 structures with a resolution ≤2.5 Å, we observed indeed 190 waters (22% of the total) with an EDIA value >0.8, and 668 waters (77% of the total) with an EDIA value above 0.4.
Putative protonated A and C nucleobases were identified based on observed H-bond distances from acceptor atoms for their N1 and N3 atoms, respectively (50). The analysis of the structural motifs in the structures with PDB IDs: 1S72, 3NKB and 4LVZ has been done using the DSSR tool (65). In the structural context analyses, the screening up for H-bond donor/acceptor atoms available for water molecules H-bonded to those involved in the stacking contacts was limited to their first coordination shell.
Quantum mechanics (QM) calculations
For modeling a OH–π interaction, one hydrogen atom of a water molecule was oriented towards the geometrical center of the nucleobase (see Figure 1). For modeling a lp–π interaction, an ideal geometry was considered, with the Ow pointing toward the geometrical center of the nucleobase and the two hydrogens pointing away from the nucleobase, in a plane orthogonal to the plane of the nucleobase (Figure 1) (25,33). Model geometries were obtained analogously for benzene and hexafluorobenzene. The distance between OW atom and the nucleobases centroid was performed at 0.1 Å increments, in the 2.4–6.0 Å range, and at 1.0 Å increments in the 6.0–8.0 Å range, with the water and the nucleobases frozen at the PBE1PBE/TZVP optimized geometries.
Interaction energies (Ebind) were calculated using equation (1):
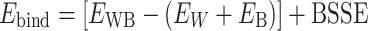 |
(1) |
where EWB is the electronic energy of the water–base complex, EW and EB are the electronic energies of the isolated water and nucleobase fragments forming the complex, and BSSE is the basis set superposition error (66).
The water–base stacking interaction energies were evaluated at the coupled cluster level of theory, with iterative inclusion of single and double excitations and perturbative inclusion of triple excitations (CCSD(T)), which is considered the golden standard in electronic structure calculations including stacking interactions in nucleic acids (67). The domain-based local pair-natural orbital (DLPNO) approximation (68–70), as implemented in the ORCA package (71), was used to accelerate calculations. Tighter than the default ‘TightPNO’ DLPNO settings (TCutPairs = 10−5, TCutPNO = 10−7 and TCutMKN = 10−3) were used (72). The triple and quadruple-ζ correlation consistent basis sets of Dunning augmented with diffuse functions were used in the present work to describe hydrogen, carbon, nitrogen and oxygen atoms (73). The correlation fitting basis sets cc-pVQZ/C developed by Hättig and co-workers (74), necessary for the resolution of identity approximation as a part of DLPNO scheme, were used. All cc-pVQZ/C basis sets were used as implemented in ORCA 4.0 suite of programs (75).
To account for basis set incompleteness effects, we applied the 2-point extrapolation schemes for Hartree-Fock and DLPNO-CCSD(T) correlation energies proposed by Helgaker and co-workers (76–78), see Equations (2 and 3). For two adjacent triple and quadruple-ζ basis sets:
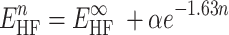 |
(2) |
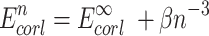 |
(3) |
where n = 3 and 4 for triple and quadruple-ζ basis sets,  are the Hartree-Fock and correlation energies at the complete basis set (CBS) limit, α/β are parameters to be obtained from a system of the two equations. In addition, the standard counterpoise correction (CP) (79) has been applied to CBS extrapolated energies to calculate interaction energies. Final Ebind thus correspond to the DLPNO-CCSD(T)/CBS level of theory. Electrostatic potentials were calculated as previously described (38), and are here mapped on electron density isosurfaces corresponding to a value of 0.0004 atomic units, scaled between −30 and +30 kcal/mol.
are the Hartree-Fock and correlation energies at the complete basis set (CBS) limit, α/β are parameters to be obtained from a system of the two equations. In addition, the standard counterpoise correction (CP) (79) has been applied to CBS extrapolated energies to calculate interaction energies. Final Ebind thus correspond to the DLPNO-CCSD(T)/CBS level of theory. Electrostatic potentials were calculated as previously described (38), and are here mapped on electron density isosurfaces corresponding to a value of 0.0004 atomic units, scaled between −30 and +30 kcal/mol.
Potential energy interaction maps were built by scanning the grid positions located on the lines connecting the nucleobase centroid to each atom of the ring (see Supplementary Figure S1). On all the grid points the distance between OW and the nucleobase plane was fixed to the ideal values of 3.0 and 3.5 Å for the lp–π and OH–π geometries. Energies of the water-base stacking interaction were calculated for all the scanned grid single points at the DLPNO-CCSD(T)/CBS level.
Molecular dynamics (MD) simulations
All the MD simulations were performed using NAMD (80), with the CHARMM36 all-atom nucleic acids force field for RNA (81) and the TIP3P water model for explicit water environment. The methodology used for the RNA modeling, system setup, equilibration, and simulations was similar to that used in previous studies (57,82,83). The initial coordinates for RNA were taken from the experimental structure with PDB ID: 1L2X and resolution 1.25 Å (36). We notice that the occupancy of OW189, stacked on A24, is 0.50 in the 1L2X structure. All the missing hydrogens were added to the RNA using VMD (84). When modeling the system, the N3 atom of the cytosine at position 8 (C8) was protonated, in agreement with experimental observations (85,86). All the crystal water molecules and ions were kept in their initial positions. The RNA system along with water and ions was energetically minimized for ∼5000 steps by employing the steepest descent (SD) algorithm, while fixing the non-hydrogen atoms. The entire minimized system was then immersed in a water box with dimensions 70 Å × 67 Å × 65 Å and the water molecules within the cut-off distance of 2 Å from the RNA system were deleted. Sufficient number of Na+ and Cl− ions were added to these systems in order to attain a 0.15 M concentration. Then, the system was subjected to a 1000 step SD minimization, followed by 500 ps simulations in a NVT ensemble while having a weak harmonic constraint of 5 kcal/mol Ų on the heavy atoms of RNA. In this step, the system was gradually heated from 100 to 300 K. The equilibrated system was used as an input for running long-time scale MD simulations. Three MD simulations, each 50 ns long, were performed in a NPT ensemble by assigning different velocities to the initial systems. All the restraints applied in the previous steps were removed during these production simulations. The SHAKE algorithm was used to constrain the covalent bonds involving hydrogens, which allowed using a 2 fs time step. Periodic boundary conditions were used during minimization and MD simulations. The particle mesh Ewald summation (PME) method was employed for treating the long-range electrostatic interactions (87). Lennard–Jones (LJ) interactions were truncated at 12 Å, by setting a force smooth switch function from 10 to 12 Å (88). The Nosé–Hoover thermostat (89) and Langevin piston (90) were employed for maintaining temperature and pressure, respectively. The coordinates were saved every 2 ps for further analyses. All the three independent simulations show no structural deformation as indicated by their calculated root-mean-square-displacement (RMSD) trends (see Supplementary Figure S2).
RESULTS AND DISCUSSION
Statistical and structural analysis of water–nucleobase stacking contacts
Occurrences of water-nucleobase stacking contacts were searched in a non-redundant dataset of 630 RNA structures, featuring a resolution of 3.0 Å or better and at least one crystallization water, by using the geometrical criteria discussed in the Methods section and shown in Figure 1. Using this procedure, we identified 1008 instances of water–nucleobase stacking contacts (197, 351, 165, 295 instances for A, G, C and U, respectively). They include three instances involving putative protonated A (A+) and four involving putative protonated C (C+), based on observed H-bond distances from acceptor atoms for their N1 and N3 atoms, respectively (50). At least one instance of water-nucleobase stacking contact has been observed in 293 structures (which correspond to 46.5% of the available PDB structures in the dataset), and as many as 198 instances of water-base stacking contacts have been observed in the large ribosomal subunit of H. marismortui structure (PDB ID: 1S72). The average number of crystallization waters in the above 293 structures is 279, while this number drops to 131 for the ensemble of 337 structures where the water-base stacking contacts were missing.
The identified water-base stacking contacts are located in a variety of RNA molecules, including 31 tRNAs, 7 ribozymes, 17 riboswitches and 3 ribosomes, from H. marismortui, E. coli and T. thermophilus. As exemplary cases, we report in Figure 2 three different functional RNAs, 23S–5S rRNA from H. marismortui, human HDV ribozyme and a chimeric riboswitch (between the S. mutans folT THF aptamer domain and B. subtilis metE expression platform), along with the 198, 5 and 8 occurrences, respectively, of nucleobase-water contacts in them and the structural motifs they are found in. As illustrated in figure, the water-nucleobases stacking contacts are located in a variety of structural elements, mostly other than regular double helices (stems). This is expected, as a water-nucleobase stacking contact can be impaired by a perfect stacking between two neighboring nucleobases. However, in case of small deviations from ideality of a stem, resulting in a less efficient base-to-base stacking, a water-nucleobase contact can be observed (see for instance the case of C46 in the THF riboswitch, Figure 2C).
Figure 2.

Water–nucleobase stacking contacts in three case studies. (A) 3D representation of 23S-5S rRNA from H. marismortui (95) with blue, green, red, and orange spheres representing adenine, guanine, uracil, cytosine bases involved in water–base interactions, respectively (left), and number of occurrences of bases involved in the interactions in different structural motifs (right). (B, C) 3D representation of HDV ribozyme (96) and THF riboswitch (97) highlighting the bases involved in water–base stacking contacts, the same bases are listed along with the structural motifs they are located in.
A prevalent involvement of G and U in forming water-base stacking contacts is observed, representing together 64% of the total instances (35% for G and 29% for U, Figure 3A), while A and C are involved in stacking contacts with water in the 20% and 16% of the total cases. Reducing the cutoff distance of the rise parameter for the identification of the water-base stacking contacts to 3.5 Å and then to 3.0 Å still results in the prevalence of water-nucleobases stacking contacts involving G and U, with water–U interactions representing 45% of the instances with a cutoff of 3.0 Å (see Figure 3B). The above discussion indicates that U is more likely to form stacking contacts with water at shorter distances. Within the analysed range of rise distances, the majority of the instances fall within 3.1 and 3.7 Å, with a peak around 3.5 Å for A, G and C, and around 3.1–3.2 Å for U, which is in line with the above results. The observed predominance of G-water stacking contacts seems to merely reflect the relative abundance of the different nucleobases in the analysed set of structures, where G is indeed the most represented base, with a frequency of 32%. As for U, its frequency is only 19%, therefore it is tempting to suggest that its preferential involvement in such contacts could be related to it being the only nucleobase with tendency to give stacking contacts with water at low distances (as also confirmed by energy calculations, see below). The 1008 nucleobases stacked to water represent altogether the 4.0% of all the nucleobases present in the above set of 293 structures, including those located in regular stems and perfectly stacked to neighboring nucleobases.
Figure 3.

(A) Total number of instances (count and percentage) of water-nucleobase stacking contacts. The distance cutoff between OW and the nucleobase plane (NP) was set at 4.0, 3.5 and 3.0 Å and the corresponding counts and percentages of the nucleobases participating in OW-nucleobase stacking contacts have been reported, (B) Distribution of the vertical distances, i.e. the distances between OW and the nucleobase plane in the recorded stacking interactions, is reported for each nucleobase.
Finally, we also analysed the occurrence of water-nucleobase contacts in a subset of high-resolution structures, by lowering the resolution cutoff from 3.0 Å, to 2.6 Å, to 2.0 Å and to 1.5 Å. As a result of these analyses, we found that the trend in the usage of the different nucleobases was preserved for all the resolution cutoffs (Supplementary Figure S3).
Analysis of the distribution of the OW atoms over the plane of the nucleobases (Figure 4) shows that they are distributed all over the area of the heterocycle rings. For U and G a bias towards the C2 and N7–C8 atoms is observed, while for C and A a quite uniform projection of OW atoms is observed throughout the nucleobase ring.
Figure 4.
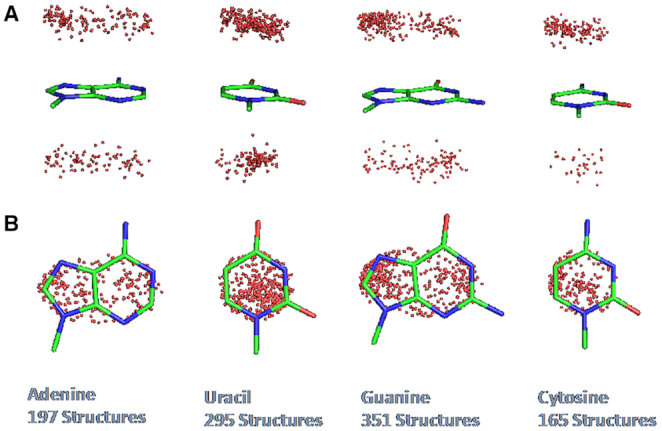
(A) Side view and (B) top view of the projection of OW, shown as a red dot, on the base plane in the observed water-nucleobase stacking contacts.
Analysis of the structural context: bridging vs. dangling waters and lp–π versus OH–π water-nucleobase interactions
We classified waters involved in the stacking contacts with nucleobases in ‘bridging’ and ‘dangling’ ones. Bridging waters are those connecting to each other two regions of the RNA or the RNA to the bound protein in RNA/protein complexes, and might thus have a structural role. This is a consequence of giving a stacking contact with a nucleobase and being at the same time involved in at least one H-bond with: (i) donor/acceptor atoms from the RNA or a bound protein and/or (ii) water molecules which are in turn involved in H-bonds with the RNA/protein. Remaining waters, giving no H-bond to any macromolecular atom, are instead classified as dangling waters, and can be considered as representatives of solvation waters at the RNA surface. We searched therefore for donor/acceptor atoms at H-bonding distance from the waters involved in the stacking contacts and, as a result of this analysis, we could identify 735 bridging and 273 dangling waters, corresponding to 73% and 27% of the total instances, respectively. Analysis of the solvent accessibility of the stacked nucleobases showed that, while those stacked to bridging waters explore a wide range of possible accessibilities, not surprisingly, the nucleobases stacked to the dangling waters are biased towards high accessibility values (Supplementary Figures S4 and S5).
Further, since standard crystallographic techniques cannot locate hydrogen atoms, to possibly distinguish between lp–π and OH–π water-nucleobase interactions, we searched for potential H-bond acceptors from RNA, proteins or other water molecules in proximity of OW, allowing to establish a likely H-bond network defining the position of the hydrogen atoms bound to OW (see Supplementary Figure S6, and Supplementary Table S2). Over the 1008 water-nucleobase contacts we found, 349 occurrences (34.5% of the total) can have the two hydrogens on OW involved in H-bonds (see Table 1) with: (i) two RNA/protein atoms (288), (ii) one RNA/protein atom and one oxygen from another water molecule (52), (iii) two oxygens from other waters (9). In case of water molecules, the acceptor nature of their oxygen was verified by checking that it was in turn at H-bond distance from two RNA/protein acceptor atoms (see SI for details). In all the above 349 occurrences, no H atom on OW is available for an OH–π interaction with a nucleobase and therefore they may be assigned as lp–π interactions. Within this analysis, 82 contacts of the 198 shown in Figure 2 for 23S–5S rRNA from H. marismortui and 6 of out of 9 contacts found in the chimeric riboswitch could be assigned as lp−π interactions.
Table 1.
Total number of OW-nucleobase contacts and numbers of instances with Ow hydrogens involved or not in H-bonds with surrounding RNA, protein or water acceptor atoms
| All occurrences | Occurrences where Ow is at H-bond distance from 2 acceptor atoms (putative lp–π) | Occurrences where Ow is at H-bond distance from 1 acceptor atom | |||||
|---|---|---|---|---|---|---|---|
| Nucleobase | # Ow-nucleobase contacts | Both from RNA/protein | 1 from RNA/protein, 1 from a water | Both from waters | From RNA/protein or a water | Occurrences where Ow is at H-bond distance from no acceptor atom | Number of waters with EDIA ≥ 0.8/0.4a |
| A | 197 | 65 | 11 | 2 | 28 | 91 | 41/136 |
| U | 295 | 68 | 16 | 3 | 66 | 142 | 59/206 |
| G | 351 | 113 | 16 | 4 | 63 | 155 | 65/241 |
| C | 165 | 42 | 9 | 0 | 32 | 82 | 31/123 |
| All | 1008 | 288 | 52 | 9 | 189 | 470 | 196/706 |
| % | - | 28 | 5 | 1 | 19 | 47 | 21/77b |
aValues above 0.8 mark well supported, values in range 0.4 to 0.8 mark medium supported and values below 0.4 mark badly supported atoms.
bCalculated relatively to the 916 waters from the 270 structures having an electron density map available.
In the remaining 659 water-nucleobase contacts, 189 occurrences have only one acceptor atom at H-bond distance from OW, 227 of them have no acceptor atom at H-bond distance from OW, and 243 have a water molecule at H-bond distance from OW (independently from the presence of another water/protein/RNA acceptor), but it was impossible to uniquely assign the H-bonds network around these water molecules based on the analysis of their first coordination shell. In these 659 occurrences, corresponding to 65.5% of the total, at least one hydrogen atom is in principle available for an OH–π interaction with a nucleobase, and thus it is impossible to assign them as lp–π or a OH–π interaction. In some cases, two heavy donor atoms from neighboring residues are found at H-bond distance from OW, presumably engaging in H-bonds both of its lone pairs. This would imply the formation of OH–π stacking interactions with the nucleobase (two examples of such putative OH–π interactions are shown in Supplementary Figure S7). However, clearly the H-bond network hypothesis remains uncertain for them, as the positions of the hydrogens possibly involving the OW lone pairs in H-bonding are missing from the structures.
Model systems and energy calculations
Potential energy curves
Potential energies of ideal lp–π and OH–π water-nucleobase interactions have been calculated for the four canonical nucleobases and, considering its presence in a water-base stacking contact in the 28-mer RNA pseudoknot system characterized by Egli and coworkers (35,36), for protonated cytosine. For the OH–π interaction, one hydrogen atom bound to Ow was oriented toward the centroid of the nucleobase. For modeling a pure lp–π interaction, an ideal geometry was considered, with the Ow pointing towards the centroid of the nucleobase and the two hydrogens pointing away and forming with Ow a plane orthogonal to the plane of the nucleobase (25,33). Model geometries were obtained analogously for benzene and hexafluorobenzene, having an extreme electron rich and electron deficient π cloud, respectively, and used therefore as reference systems. The distance between Ow and the aromatic ring was varied between 2.4 and 8.0 Å for both the lp–π and OH–π interactions (see Materials and Methods for details). The resulting potential energy curves (PECs) are reported in Figure 5A.
Figure 5.

(A) Potential Energy Curves for all the nucleobases (A, U, G, C), and for benzene and hexafluorobenzene, with the water oriented to give rise to pure lp–π (left) and OH–π (right) interactions. Interaction energies in kcal/mol (y-axis) are reported versus the OW-ring distances, varying between 2.4 and 8.0 Å (x-axis). (B) Electrostatic potentials of nucleobases, water, benzene and hexafluorobenzene. Electrostatic potentials were mapped on electron density isosurfaces corresponding to a value of 0.0004 atomic units, and are scaled between –30 and 30 kcal/mol.
Among the neutral systems, benzene and hexafluorobenzene represent the extremes of the obtained plots, with their PECs bracketing the PECs of the four nucleobases (Figure 5). For benzene the OH–π interaction is favored, with the minimum at ≈3.3 Å, while the PEC for the lp–π interaction, matching the one previously obtained by Ran and Hobza (25), is repulsive at every OW-benzene distance. As already suggested, these results can be explained in terms of attractive/repulsive electrostatic interaction between the differently oriented water molecule and the negative electrostatic potential above the aromatic ring of benzene (Figure 5B) (25,91). In contrast, for hexafluorobenzene the lp–π interaction is favored over the OH–π one, which only shows a shallow minimum around 3.3 Å. As already reported (26,27,92), this can be explained considering the positive electrostatic potential above the aromatic ring of hexafluorobenzene, due to the electron-withdrawing fluorine substituents.
As for the neutral nucleobases, the PECs show well defined energy minima for both the OH–π and lp–π geometries. However, the OH–π interaction is clearly favored over the the lp–π interaction for A and C (by 0.90 and 1.15 kcal/mol), it is slightly favored for G (by 0.23 kcal/mol), while for U the lp–π interaction is clearly favored over the OH–π interaction (by 1.96 kcal/mol), with the OH–π PEC nearly overlapping the replusive PEC of hexafluorobenzene. In all the cases, the OH–π and lp–π PECs have the minimum at approximately 3.4–3.5 and 3.0–3.1 Å, supporting Egli's suggestion that the OW-base distance can be considered as one of the parameters defining the type of interaction in crystallographic structures (86). The single point energy values calculated at all the vertical distances for each nucleobase are reported in Supplementary Tables S3–S7. As expected, for the protonated cytosine the lp–π interaction is strongly favored (energy minimum of −8.6 kcal/mol) and the OH–π interaction is strongly disfavored (energy minimum of +2.1 kcal/mol) at every distance (see Supplementary Figures S8–S10).
Interaction energies for different projections of Ow on the nucleobase plane
From our structural analysis (see Figure 4) it is clear that, in the experimental occurrences of water–nucleobase stacking contacts, the water oxygen is not always projected towards the centroid of the nucleobase. Instead, its projection is quite spread to cover more or less all the aromatic ring. Therefore, in order to evaluate whether and how the energetics of the stacking interactions is affected by the OW position relatively to the nucleobase centroid, we calculated the water–nucleobase interaction energy at different geometries corresponding to the water sliding horizontally in a plane parallel to the nucleobase plane (see Methods for details). The distance between OW and the nucleobase plane was set to the ideal values of 3.0 and 3.5 Å for the lp–π and OH–π geometries, respectively. On a quantitative ground, the average interaction energies for A, U, G and C are −2.20 ± 0.34, −0.40 ± 0.23, −1.50 ± 0.52 and −1.15 ± 0.38 kcal/mol, respectively, for the OH–π interaction and 1.01±0.72, −1.92±0.59, −1.37±0.72 and −1.17±0.59 kcal/mol, for the lp–π interaction.
Analysis of the contour maps of Figure 6 indicates that sliding the water molecule along a given direction has, usually, opposite effects on the OH–π and lp–π interactions and that the two energy profiles for each base are quite complementary. Overall, for U, the lp–π interaction is favored over the OH–π one at any sliding position of the water over the nucleobase plane, in agreement with its prevalently positive electrostatic potential. For C, instead, energies for the two interactions are comparable, with the lp–π one favored around N1 and the OH–π one favored around N3 (Figure 6), where the electrostatic potential is remarkably negative (see Figure 5).
Figure 6.

Contour diagrams showing the interaction energies for the lp–π and OH–π interactions between water and the A/G/C/U bases. Energy values are color coded, from red (−3 kcal/mol) to blue (+1.5 kcal/mol). Singe point energies were calculated for both the types of interaction on a grid made of 4 points for each line connecting the nucleobase centroid to each atom of the ring (See Methods and Supplementary Figure S1). Water-nucleobase distances were frozen at 3.0 Å and 3.5 Å for the lp–π and the OH–π interaction, respectively.
Regarding A, the OH–π interaction is clearly favored as compared to the lp–π one for stacking over the six-membered ring and the N7 atom, featuring a neutral to negative electrostatic potential, while the stability of the two interactions become comparable for stacking over two other atoms of the five-membered ring, C8 and N9 (where the lp–π is even favored), possibly due to the positive electrostatic potential around them.
Finally, for G the two interactions exhibit a comparable stability around the central region of the nucleobase, but are favored one over the other at the ring extremes. Specifically, the OH–π interaction prevails and the lp–π interaction is clearly unstable around the N7 atom on the five-membered ring, where the electrostatic potential is negative, while the lp–π interaction becomes favored around two atoms of the six-membered ring, N1 and C2, where the electrostatic potential is neutral to positive.
As a concluding remark, favorable energies for either of the two interactions, lp–π or OH–π, are observed all over the surface of each of the four nucleobases, consistently with the quite uniform projection of OW atoms over their rings, which we observed in the instances of water-nucleobase stacking contacts in experimental RNA structures (see Figure 4).
Molecular dynamics simulations
In order to check the stability of the water-nucleobase stacking contacts in a complete RNA structure and under dynamic conditions, we performed MD simulations on the 28-mer ribosomal RNA pseudoknot from beet western yellow virus (PDB ID: 1L2X, resolution 1.25 Å). The crystallographic structure of this RNA presents four instances of water-nucleobase stacking contacts, according to Egli and coworkers, who classified them as possible lp–π or OH–π interactions, based on the distance of the OW from the nucleobase ring centroid and considerations on their structural context (86). In particular, the OW71-C8 and OW73-A20 contacts were classified as lp–π interactions, while OH–π interactions were proposed to be formed between OW175 and A9, and between A24 and two water molecules, OW120 (stacked on top of the five membered ring) and OW189 (stacked on top of the six membered ring). Within our structural analysis, waters involved in the OW71-C8 and OW189/OW120-A24 interactions can be classified as bridging waters, and thus we discuss them below. Conversely, we classified the water molecules involved in the OW175-A9 and OW73-A20 interactions as dangling (solvation) waters, and we discuss them in the Supporting Information. As very similar results were obtained from the three independent trajectories we performed (see Supplementary Figures S11–S14), for the sake of simplicity only one of them is presented and discussed below.
The OW71-C8 interaction involves a protonated cytosine, as shown by experimental evidence (93,94). Therefore, in our MD setup a protonated C8 (at the N3 position) was used. From the pseudoknot crystal structure two H-bonds can be deduced, involving the two hydrogens on OW71, one with A9 (via the O1P atom) and one with another surrounding water molecule (OW68). The absence of hydrogen atoms available for an OH–π interaction confirms this stacking contact as a lp–π interaction. In addition, at H-bonding distance from OW71 there is also the N4 atom of C11, possibly engaging a lone pair on OW71 in a H-bond with one of its hydrogens (see Figure 7).
Figure 7.

Dynamic behavior of the lp–π OW71-C8 interaction in the ribosomal frameshifting RNA pseudoknot from beet western yellow virus (PDB ID: 1L2X). (A) Superimposition of 500 frames from the MD trajectory. For the sake of clarity, sugars and nucleobases for residue 9 and 10 are not shown, (B) H-bonds of OW71 with surrounding RNA/water atoms are shown in the frame extracted at 18.5 ns, (C) Time evolution of all the relevant distances (between: OW71 and the nucleobase plane, OW71 and OW68 or water molecules exchanging with it, OW71 and C11(N4), and OW71 and A9(O1P)) are shown. The color code of the plots is the same used in (B). The distance average and standard deviation values are also given in the relative plots.
All the above distances are plotted versus the MD simulation time in Figure 7. The OW71-C8 plot (reporting the distance between OW71 and the centroid of the C8 nucleobase) shows that the water–nucleobase contact is maintained during the simulation, for an average distance of 2.96 ± 0.14 Å matching the crystal distance of 2.93 Å. In addition, the interaction can be classified as lp–π, as it is OW71 and not the OW71-hydrogens to point towards the C8 base plane during the whole simulation. This is consistent with the positive charge delocalized over C8, resulting in a strongly positive electrostatic potential above the nucleobase plane. No surrounding water molecule replaces the initial water molecule during the simulation, which is indicative of a very stable interaction also under dynamic conditions.
The H-bonds between OW71 and the neighboring A9(O1P) and C11(N4) are also maintained during the simulation. The average H-bond distance between OW71 and A9(O1P) is 2.80±0.19 Å, which compares with the value of 2.92 Å in the crystal structure, while the distance between OW71 and C11(N4) is on average 3.03 ± 0.18 Å, which compares with the experimental value of 2.95 Å.
As for the interaction of OW71 with OW68, after 5.2 ns the water molecule representing OW68 in the initial simulation box starts exchanging with surrounding water molecules. Nevertheless, a water molecule in the OW68 site or slightly displaced (examples from 500 equidistant frames are shown in Figure 7A) maintains the H-bond with OW71 all throughout the simulation (precisely in 99% of the frames). The average distance of the OW71–OW68 H-bond is 2.91 ± 0.18 Å (comparing with a distance of 2.80 Å in the crystal structure).
The interaction of A24 with OW189, stacked on its 6-membered ring, and with OW120, stacked on its 5-membered ring, has been hypothesized to be of the OH–π type by Egli and coworkers. However, we notice that, based on the network of H-bond donors and acceptors in proximity of OW189 in the crystal structure, OW189 is expected to have no hydrogen available for an OH–π interaction with A24. In any case, this is clearly an unstable interaction, considering that the occupancy of such a water in the crystal structure is only 0.5.
All the above distances are plotted versus the MD simulation time in Figure 8. Both the stacking contacts exhibit a quite dynamic behavior, with OW189 and OW120 not staying on site and being both exchanged by other water molecules from the bulk solvent. The stacking contact of OW120 on the 5-membered ring is however preferentially maintained. It is indeed observed in 92% of the frames, featuring an average distance from the nucleobase ring of 3.31±0.29 Å, which compares with the experimental value of 3.44 Å between OW120 and A24. It is noteworthy that, during the dynamics, OW120 (or the water molecule exchanging with it) gets engaged in 2 H-bonds, with two acceptor atoms from the RNA backbone: OP2(A25), with an average distance 2.82 ± 0.03 Å, and O2′(C8), with an average distance of 3.03 ± 0.31 Å. This implies that no hydrogen on OW120 is available for an OH–π interaction with A24, and the only possible interaction with the nucleobase must be of the lp- π type. In the crystal structure the distances between OW120 and OP2(A25)/O2′(C8) are well above the thresholds for the H-bond definition (3.82 Å/4.41 Å). Therefore, this is clearly the result of a rearrangement of the RNA backbone around OW120 under dynamic conditions. Considering that both the OH–π and the lp–π interactions are energetically favored over the adenine 5-membered ring (see Figure 6), our results suggests that the OH–π interaction can convert to a similarly stable lp–π interaction under dynamic conditions, due to the additional stability deriving from two classical H-bonds involving the water molecule and ‘bridging’ two RNA regions.
Figure 8.

(A) Top: H-bonds of OW1 (or OW120) with surrounding RNA/water atoms and the stacking interaction with the nucleobase are shown as dashed lines in the frame extracted at 26.4 ns. Bottom: Time evolution of the relevant distances discussed in the text. The color code of the plots is the same used for the dashed lines in the top panel. The distance average and standard deviation values are also given in the relative plots. (B) Top: H-bonds of OW1 (or OW120) and OW2 (or OW189) with surrounding RNA/water atoms and the stacking interactions with the nucleobase are shown in the frame extracted at 14.5 ns. Bottom: Time evolution of the relevant distances discussed in the text. The color code of the plots is the same used for the dashed lines in the top panel. The distance average and standard deviation values are also given in the relative plots. The OW2-A24 plot shows some discontinuities as frames for which the OW2-A24 distance is larger than the threshold of 4.0 Å (that we used to define water-nucleobase stacking interactions) were not considered.
The stacking contact of OW189 with the 6-membered ring is instead maintained in only 59% of the frames, featuring an average distance from the nucleobase ring of 3.61 ± 1.17 Å, which compares with the experimental value of 3.78 Å between OW189 and A24. This is consistent with the occupancy 0.50 of OW189 in the crystallographic structure (the occupancy of OW120 being instead 1). OW189 uses its hydrogens to H-bond to the N7 atom of A25, for an average distance of 2.99 ± 0.27 Å versus the crystallographic distance of 2.76 Å, and to OW120 itself, for an average distance of 2.96 ± 0.24 Å versus the crystallographic distance of 2.49 Å, being left with no hydrogen for an OH–π interaction with the ring of A24.
Figures 8 shows typical snapshots during the dynamics where either a single water molecule or two water molecules are stacked over A24, with both their hydrogens engaged in H-bonds with neighboring acceptor atoms.
CONCLUSIONS
We have found 1008 instances of water-nucleobase stacking contacts in 293 non-redundant PDB structures, representing a variety of RNA molecules. Based on their structural context, we could assign approximately a third of them as candidate lp–π interactions while the assignment remains uncertain for the remaining ones. The distance of the OW atom from the nucleobase is distributed in the 3–4 Å range, with a peak approximately at 3.5 Å for A, G and C, and approximately at 3.1–3.2 Å for U. Further, 73% of the waters involved in the stacking contacts are usually connected through H-bonds to other regions of the RNA molecule, which suggests that they might have a structural role. QM calculations indicated that the water-nucleobase stacking energy is not negligible, with values of −2.36 kcal/mol for the OH–π interaction for A and of −2.39 kcal/mol for the lp–π interaction for U. Expectedly, the lp–π interaction is clearly stronger (≈−8.6 kcal/mol) in case of a protonated nucleobase, such as C+. Furthermore, by calculating the interaction energy of a water molecule sliding over the nucleobases plane, we have shown that a favorable interaction, of either the lp–π or the OH–π type, can be established at virtually any position of the surface of the four nucleobases. This is consistent with the quite uniform projection of the OW atoms over the nucleobases ring, which we observed in the RNA experimental structures. The geometrical plasticity of the water-nucleobase stacking interactions, in terms of vertical rise from the nucleobase plane and of horizontal slide over the nucleobase skeleton, allows the stacked water molecules to adapt their position to optimize additional and more geometrically stringent interactions, such as H-bonds, with neighboring atoms. MD simulations carried out for a case study, a 28-mer RNA pseudoknot, showed that such interactions, when involving bridging waters, are maintained under dynamic conditions, although they can possibly switch from one to the other type of interaction.
In light of these results, we believe the water-nucleobase stacking contacts to add themselves to the constellation of weak interactions that, together with the well accepted strong ones (i.e. H-bonds and base-base stacking), helped RNA set up a variety of strategies for achieving structural complexity (41).
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
L.C. and M.C. acknowledge King Abdullah University of Science and Technology (KAUST) for support and the KAUST Supercomputing Laboratory for providing computational resources of the supercomputer Shaheen II; R.O. thanks MIUR-FFABR (Fondo per il Finanziamento Attività Base di Ricerca) for funding. Funding for open access charge: King Abdullah University of Science and Technology.
Conflict of interest statement. None declared.
REFERENCES
- 1. Westhof E. Water - an integral-part of nucleic-acid structure. Annu. Rev. Biophys. Biol. 1988; 17:125–144. [DOI] [PubMed] [Google Scholar]
- 2. Auffinger P., Westhof E.. Hydration of RNA base pairs. J. Biomol. Struct. Dyn. 1998; 16:693–707. [DOI] [PubMed] [Google Scholar]
- 3. Auffinger P., Westhof E.. Water and ion binding around RNA and DNA (C,G) oligomers. J. Mol. Biol. 2000; 300:1113–1131. [DOI] [PubMed] [Google Scholar]
- 4. Ball P. Water as an active constituent in cell biology. Chem. Rev. 2008; 108:74–108. [DOI] [PubMed] [Google Scholar]
- 5. Bielecki L., Kulinski T., Adamiak R.W.. Structure and dynamics of adenosine loops in RNA bulge duplexes. RNA hydration at the bulge site. Nato Asi. 3 High Tech. 1999; 70:73–87. [Google Scholar]
- 6. Charette M., Gray M.W.. Pseudouridine in RNA: what, where, how, and why. IUBMB Life. 2000; 49:341–351. [DOI] [PubMed] [Google Scholar]
- 7. Newby M.I., Greenbaum N.L.. Investigation of Overhauser effects between pseudouridine and water protons in RNA helices. Proc. Natl Acad. Sci. U.S.A. 2002; 99:12697–12702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schneider B., Patel K., Berman H.M.. Hydration of the phosphate group in double-helical DNA. Biophys. J. 1998; 75:2422–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sorin E.J., Rhee Y.M., Pande V.S.. Does water play a structural role in the folding of small nucleic acids. Biophys. J. 2005; 88:2516–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Westhof E. Westhof E. Water and Biological Macromolecules. 1993; Boca Raton: CRC Press; 226–243. [Google Scholar]
- 11. Westhof E., Fritsch V.. RNA folding: beyond Watson-Crick pairs. Structure. 2000; 8:R55–R65. [DOI] [PubMed] [Google Scholar]
- 12. Chawla M., Abdel-Azeim S., Oliva R., Cavallo L.. Higher order structural effects stabilizing the reverse Watson-Crick Guanine-Cytosine base pair in functional RNAs. Nucleic Acids Res. 2014; 42:714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rhodes M.M., Reblova K., Sponer J., Walter N.G.. Trapped water molecules are essential to structural dynamics and function of a ribozyme. Proc. Natl Acad. Sci. U.S.A. 2006; 103:13380–13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Walter N.G. Ribozyme catalysis revisited: is water involved. Mol. Cell. 2007; 28:923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ward W.L., Plakos K., DeRose V.J.. Nucleic acid catalysis: metals, nucleobases, and other cofactors. Chem. Rev. 2014; 114:4318–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Auffinger P., Hashem Y.. Nucleic acid solvation: from outside to insight. Curr. Opin. Struct. Biol. 2007; 17:325–333. [DOI] [PubMed] [Google Scholar]
- 17. Auffinger P., Westhof E.. RNA solvation: a molecular dynamics simulation perspective. Biopolymers. 2000; 56:266–274. [DOI] [PubMed] [Google Scholar]
- 18. Althagbi H.I., Bernstein D.R., Crombie W.C., Lane J.R., McQuiston D.K., Oosterwijk M.A., Saunders G.C., Zou W.Y.. The crystal structures of 1-(4-halo-2,3,5,6-tetrafluorophenyl)-3-benzylimidazolium bromides: the relative importance of anion-pi, lone pair-pi, pi-pi stacking and halogen bonding interactions. J. Fluor. Chem. 2018; 206:61–71. [Google Scholar]
- 19. Duran-Solares G., Fugarolas-Gomez W., Ortiz-Pastrana N., Lopez-Sandoval H., Villasenor-Granados T.O., Flores-Parra A., Altmann P.J., Barba-Behrens N.. Lone pair center dot center dot center dot pi interactions on the stabilization of intra and intermolecular arrangements of coordination compounds with 2-methyl imidazole and benzimidazole derivatives. J. Coord. Chem. 2018; 71:1935–1958. [Google Scholar]
- 20. Durec M., Marek R., Kozelka J.. Water-Tryptophan Interactions: Lone-pair…pi or O-H…pi? Molecular dynamics simulations of beta-Galactosidase suggest that both modes can Co-exist. Chem. Eur. J. 2018; 24:5849–5859. [DOI] [PubMed] [Google Scholar]
- 21. Garau C., Quinonero D., Frontera A., Ballester P., Costa A., Deya P.M.. Anion-pi interactions: must the aromatic ring be electron deficient. New J. Chem. 2003; 27:211–214. [Google Scholar]
- 22. Kim D., Tarakeshwar P., Kim K.S.. Theoretical investigations of anion-pi interactions: The role of anions and the nature of pi systems. J. Phys. Chem. A. 2004; 108:1250–1258. [Google Scholar]
- 23. Novotny J., Bazzi S., Marek R., Kozelka J.. Lone-pair-pi interactions: analysis of the physical origin and biological implications. Phys. Chem. Chem. Phys. 2016; 18:19472–19481. [DOI] [PubMed] [Google Scholar]
- 24. Reyes A., Fomina L., Rumsh L., Fomine S.. Are water-aromatic complexes always stabilized due to pi-H interactions? LMP2 study. Int. J. Quant. Chem. 2005; 104:335–341. [Google Scholar]
- 25. Ran J., Hobza P.. On the nature of bonding in lone pair center dot center dot center dot pi-Electron Complexes: CCSD(T)/Complete basis set limit calculations. J. Chem. Theo. Comput. 2009; 5:1180–1185. [DOI] [PubMed] [Google Scholar]
- 26. Gallivan J.P., Dougherty D.A.. Can lone pairs bind to a pi system? The water center dot center dot center dot hexafluorobenzene interaction. Org. Lett. 1999; 1:103–105. [DOI] [PubMed] [Google Scholar]
- 27. Danten Y., Tassaing T., Besnard M.. On the nature of the water-hexafluorobenzene interaction. J. Phys. Chem. A. 1999; 103:3530–3534. [Google Scholar]
- 28. Scheiner S., Kar T., Pattanayak J.. Comparison of various types of hydrogen bonds involving aromatic amino acids. J. Am. Chem. Soc. 2002; 124:13257–13264. [DOI] [PubMed] [Google Scholar]
- 29. Wheeler S.E. Understanding substituent effects in noncovalent interactions involving aromatic rings. Acc. Chem. Res. 2013; 46:1029–1038. [DOI] [PubMed] [Google Scholar]
- 30. Wheeler S.E. Local nature of substituent effects in stacking interactions. J. Am. Chem. Soc. 2011; 133:10262–10274. [DOI] [PubMed] [Google Scholar]
- 31. Wheeler S.E., Houk K.N.. Substituent effects in the benzene dimer are due to direct interactions of the substituents with the unsubstituted benzene. J. Am. Chem. Soc. 2008; 130:10854–10855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bloom J.W., Raju R.K., Wheeler S.E.. Physical nature of substituent effects in XH/pi interactions. J. Chem. Theo. Comput. 2012; 8:3167–3174. [DOI] [PubMed] [Google Scholar]
- 33. Jain A., Ramanathan V., Sankararamakrishnan R.. Lone pair center dot center dot center dot pi interactions between water oxygens and aromatic residues: Quantum chemical studies based on high-resolution protein structures and model compounds. Protein Sci. 2009; 18:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steiner T. Hydrogen bonds from water molecules to aromatic acceptors in very high-resolution protein crystal structures. Biophys. Chem. 2002; 95:195–201. [DOI] [PubMed] [Google Scholar]
- 35. Sarkhel S., Rich A., Egli M.. Water-nucleobase “stacking”: H-pi and lone pair-pi interactions in the atomic resolution crystal structure of an RNA pseudoknot. J. Am. Chem. Soc. 2003; 125:8998–8999. [DOI] [PubMed] [Google Scholar]
- 36. Egli M., Minasov G., Su L., Rich A.. Metal ions and flexibility in a viral RNA pseudoknot at atomic resolution. Proc. Natl Acad. Sci. U.S.A. 2002; 99:4302–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Egli M., Sarkhel S.. Lone pair-aromatic interactions: to stabilize or not to stabilize. Acc. Chem. Res. 2007; 40:197–205. [DOI] [PubMed] [Google Scholar]
- 38. Chawla M., Chermak E., Zhang Q.Y., Bujnicki J.M., Oliva R., Cavallo L.. Occurrence and stability of lone pair-pi stacking interactions between ribose and nucleobases in functional RNAs. Nucleic Acids Res. 2017; 45:11019–11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. D’Ascenzo L., Leonarski F., Vicens Q., Auffinger P.. ‘Z-DNA like’ fragments in RNA: a recurring structural motif with implications for folding, RNA/protein recognition and immune response. Nucleic Acids Res. 2016; 44:5944–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. D’Ascenzo L., Leonarski F., Vicens Q., Auffinger P.. Revisiting GNRA and UNCG folds: U-turns versus Z-turns in RNA hairpin loops. RNA. 2017; 23:259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grosjean H., Westhof E.. An integrated, structure- and energy-based view of the genetic code. Nucleic Acids Res. 2016; 44:8020–8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rozov A., Demeshkina N., Khusainov I., Westhof E., Yusupov M., Yusupova G.. Novel base-pairing interactions at the tRNA wobble position crucial for accurate reading of the genetic code. Nat. Commun. 2016; 7:10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Butcher S.E., Pyle A.M.. The molecular interactions that stabilize RNA tertiary Structure: RNA motifs, patterns, and networks. Acc. Chem. Res. 2011; 44:1302–1311. [DOI] [PubMed] [Google Scholar]
- 44. Berman H., Henrick K., Nakamura H., Markley J.L.. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007; 35:D301–D303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chawla M., Credendino R., Oliva R., Cavallo L.. Structural and energetic impact of Non-Natural 7-Deaza-8-Azaadenine and its 7-Substituted derivatives on H-bonding potential with uracil in RNA molecules. J. Phys. Chem. B. 2015; 119:12982–12989. [DOI] [PubMed] [Google Scholar]
- 46. Chawla M., Credendino R., Poater A., Oliva R., Cavallo L.. Structural stability, acidity, and halide selectivity of the fluoride riboswitch recognition site. J. Am. Chem. Soc. 2015; 137:299–306. [DOI] [PubMed] [Google Scholar]
- 47. Chawla M., Oliva R., Bujnicki J.M., Cavallo L.. An atlas of RNA base pairs involving modified nucleobases with optimal geometries and accurate energies. Nucleic Acids Res. 2015; 43:6714–6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chawla M., Poater A., Besalu-Sala P., Kalra K., Oliva R., Cavallo L.. Theoretical characterization of sulfur-to-selenium substitution in an emissive RNA alphabet: impact on H-bonding potential and photophysical properties. Phys. Chem. Chem. Phys. 2018; 20:7676–7685. [DOI] [PubMed] [Google Scholar]
- 49. Chawla M., Poater A., Oliva R., Cavallo L.. Structural and energetic characterization of the emissive RNA alphabet based on the isothiazolo[4,3-d]pyrimidine heterocycle core. Phys. Chem. Chem. Phys. 2016; 18:18045–18053. [DOI] [PubMed] [Google Scholar]
- 50. Chawla M., Sharma P., Halder S., Bhattacharyya D., Mitra A.. Protonation of base pairs in RNA: context analysis and quantum chemical investigations of their geometries and stabilities. J. Phys. Chem. B. 2011; 115:1469–1484. [DOI] [PubMed] [Google Scholar]
- 51. Oliva R., Cavallo L.. Frequency and effect of the binding of Mg2+, Mn2+, and Co2+ ions on the guanine base in Watson-Crick and reverse Watson-Crick base pairs. J. Phys. Chem. B. 2009; 113:15670–15678. [DOI] [PubMed] [Google Scholar]
- 52. Oliva R., Cavallo L., Tramontano A.. Accurate energies of hydrogen bonded nucleic acid base pairs and triplets in tRNA tertiary interactions. Nucleic Acids Res. 2006; 34:865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Oliva R., Tramontano A., Cavallo L.. Mg2+ binding and archaeosine modification stabilize the G15 C48 Levitt base pair in tRNAs. RNA. 2007; 13:1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sharma P., Chawla M., Sharma S., Mitra A.. On the role of Hoogsteen:Hoogsteen interactions in RNA: ab initio investigations of structures and energies. RNA. 2010; 16:942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sharma P., Sharma S., Chawla M., Mitra A.. Modeling the noncovalent interactions at the metabolite binding site in purine riboswitches. J. Mol. Model. 2009; 15:633–649. [DOI] [PubMed] [Google Scholar]
- 56. Spackova N., Cubero E., Sponer J., Orozco M.. Theoretical study of the guanine → 6-thioguanine substitution in duplexes, triplexes, and tetraplexes. J. Am. Chem. Soc. 2004; 126:14642–14650. [DOI] [PubMed] [Google Scholar]
- 57. Sponer J., Bussi G., Krepl M., Banas P., Bottaro S., Cunha R.A., Gil-Ley A., Pinamonti G., Poblete S., Jureacka P. et al.. RNA structural dynamics as captured by molecular simulations: a comprehensive overview. Chem. Rev. 2018; 118:4177–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sponer J., Jurecka P., Hobza P.. Accurate interaction energies of hydrogen-bonded nucleic acid base pairs. J. Am. Chem. Soc. 2004; 126:10142–10151. [DOI] [PubMed] [Google Scholar]
- 59. Sponer J., Leszczynski J., Hobza P.. Nature of nucleic acid-base stacking: nonempirical ab initio and empirical potential characterization of 10 stacked base dimers. Comparison of stacked and H-bonded base pairs. J. Phys. Chem. 1996; 100:5590–5596. [Google Scholar]
- 60. Sponer J., Mladek A., Sponer J.E., Svozil D., Zgarbova M., Banas P., Jurecka P., Otyepka M.. The DNA and RNA sugar-phosphate backbone emerges as the key player. An overview of quantum-chemical, structural biology and simulation studies. Phys. Chem. Chem. Phys. 2012; 14:15257–15277. [DOI] [PubMed] [Google Scholar]
- 61. Sponer J.E., Reblova K., Mokdad A., Sychrovsky V., Leszczynski J., Sponer J.. Leading RNA tertiary interactions: structures, energies, and water insertion of A-minor and P-interactions. A quantum chemical view. J. Phys. Chem. B. 2007; 111:9153–9164. [DOI] [PubMed] [Google Scholar]
- 62. Leontis N.B., Zirbel C.L.. Nonredundant 3D Structure Datasets for RNA Knowledge Extraction and Benchmarking. Nucleic Acids and Molecular Biology. 2012; 27:Berlin, Heidelberg: Springer. [Google Scholar]
- 63. Nittinger E., Schneider N., Lange G., Rarey M.. Evidence of water molecules-a statistical evaluation of water molecules based on electron density. J. Chem. Inf. Model. 2015; 55:771–783. [DOI] [PubMed] [Google Scholar]
- 64. Fahrrolfes R., Bietz S., Flachsenberg F., Meyder A., Nittinger E., Otto T., Volkamer A., Rarey M.. ProteinsPlus: a web portal for structure analysis of macromolecules. Nucleic Acids Res. 2017; 45:W337–W343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lu X.J., Bussemaker H.J., Olson W.K.. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 2015; 43:e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mentel L.M., Baerends E.J.. Can the counterpoise correction for basis set superposition effect be justified. J. Chem. Theo. Comput. 2014; 10:252–267. [DOI] [PubMed] [Google Scholar]
- 67. Kruse H., Mladek A., Gkionis K., Hansen A., Grimme S., Sponer J.. Quantum chemical benchmark study on 46 RNA backbone families using a dinucleotide unit. J. Chem. Theo. Comput. 2015; 11:4972–4991. [DOI] [PubMed] [Google Scholar]
- 68. Riplinger C., Neese F.. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013; 138:034106. [DOI] [PubMed] [Google Scholar]
- 69. Riplinger C., Pinski P., Becker U., Valeev E.F., Neese F.. Sparse maps-A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. J. Chem. Phys. 2016; 144:024109. [DOI] [PubMed] [Google Scholar]
- 70. Riplinger C., Sandhoefer B., Hansen A., Neese F.. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013; 139:134101. [DOI] [PubMed] [Google Scholar]
- 71. Neese F. The ORCA program system. Wires Comput Mol Sci. 2012; 2:73–78. [Google Scholar]
- 72. Liakos D.G., Sparta M., Kesharwani M.K., Martin J.M.L., Neese F.. Exploring the accuracy limits of local pair natural orbital coupled-cluster theory. J. Chem. Theory Comput. 2015; 11:1525–1539. [DOI] [PubMed] [Google Scholar]
- 73. Dunning T.H. Gaussian-basis sets for use in correlated molecular calculations. 1. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989; 90:1007–1023. [Google Scholar]
- 74. Weigend F., Kohn A., Hattig C.. Efficient use of the correlation consistent basis sets in resolution of the identity MP2 calculations. J. Chem. Phys. 2002; 116:3175–3183. [Google Scholar]
- 75. Neese F. Software update: the ORCA program system, version 4.0. Wires Comput. Mol. Sci. 2018; 8:e1327. [Google Scholar]
- 76. Halkier A., Helgaker T., Jørgensen P., Klopper W., Koch H., Olsen J., Wilson A.K.. Basis-set convergence in correlated calculations on Ne, N-2, and H2O. Chem. Phys. Lett. 1998; 286:243–252. [Google Scholar]
- 77. Helgaker T., Klopper W., Koch H., Noga J.. Basis-set convergence of correlated calculations on water. J. Chem. Phys. 1997; 106:9639–9646. [Google Scholar]
- 78. Halkier A., Helgaker T., Jørgensen P., Klopper W., Olsen J.. Basis-set convergence of the energy in molecular Hartree-Fock calculations. Chem. Phys. Lett. 1999; 302:437–446. [Google Scholar]
- 79. Boys S.F., Bernardi F.. Calculation of small molecular interactions by differences of separate total Energies - some procedures with reduced errors. Mol. Phys. 1970; 19:553–566. [Google Scholar]
- 80. Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K.. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005; 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Denning E.J., Priyakumar U.D., Nilsson L., Mackerell A.D.. Impact of 2′-Hydroxyl sampling on the conformational properties of RNA: update of the CHARMM All-Atom additive force field for RNA. J. Comput. Chem. 2011; 32:1929–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gorle S., Pan Y.G., Sun Z.Q., Shlyakhtenko L.S., Harris R.S., Lyubchenko Y.L., Vukovic L.. Computational model and dynamics of monomeric full-length APOBEC3G. ACS Cent. Sci. 2017; 3:1180–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Suresh G., Priyakumar U.D.. DNA-RNA hybrid duplexes with decreasing pyrimidine content in the DNA strand provide structural snapshots for the A- to B-form conformational transition of nucleic acids. Phys. Chem. Chem. Phys. 2014; 16:18148–18155. [DOI] [PubMed] [Google Scholar]
- 84. Humphrey W., Dalke A., Schulten K.. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 85. Pallan P.S., Marshall W.S., Harp J., Jewett F.C., Wawrzak Z., Brown B.A., Rich A., Egli M.. Crystal structure of a luteoviral RNA pseudoknot and model for a minimal ribosomal frameshifting motif. Biochemistry. 2005; 44:11315–11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sarkhel S., Rich A., Egli M.. Water-nucleobase “stacking”: H-pi and lone pair-pi interactions in the atomic resolution crystal structure of an RNA pseudoknot. J. Am. Chem Soc. 2003; 125:8998–8999. [DOI] [PubMed] [Google Scholar]
- 87. Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G.. A smooth particle mesh ewald method. J. Chem. Phys. 1995; 103:8577–8593. [Google Scholar]
- 88. Steinbach P.J., Brooks B.R.. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994; 15:667–683. [Google Scholar]
- 89. Hoover W.G. Canonical dynamics - equilibrium phase-space distributions. Phys. Rev. A. 1985; 31:1695–1697. [DOI] [PubMed] [Google Scholar]
- 90. Feller S.E., Zhang Y.H., Pastor R.W., Brooks B.R.. Constant-pressure molecular-dynamics simulation - the langevin piston method. J. Chem. Phys. 1995; 103:4613–4621. [Google Scholar]
- 91. Prakash M., Samy K.G., Subramanian V.. Benzene-Water (BZW(n) (n = 1-10)) Clusters. J. Phys. Chem. A. 2009; 113:13845–13852. [DOI] [PubMed] [Google Scholar]
- 92. Amicangelo J.C., Irwin D.G., Lee C.J., Romano N.C., Saxton N.L.. Experimental and theoretical characterization of a lone pair-pi complex: water-hexafluorobenzene. J. Phys. Chem. A. 2013; 117:1336–1350. [DOI] [PubMed] [Google Scholar]
- 93. Kim Y.C., Su L., Maas S., O’Neill A., Rich A.. Specific mutations in a viral RNA pseudoknot drastically change ribosomal frameshifting efficiency. Proc. Natl Acad. Sci. U.S.A. 1999; 96:14234–14239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Nixon P.L., Giedroc D.P.. Energetics of a strongly pH dependent RNA tertiary structure in a frameshifting pseudoknot. J. Mol. Biol. 2000; 296:659–671. [DOI] [PubMed] [Google Scholar]
- 95. Klein D.J., Moore P.B., Steitz T.A.. The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J. Mol. Biol. 2004; 340:141–177. [DOI] [PubMed] [Google Scholar]
- 96. Chen J.H., Yajima R., Chadalavada D.M., Chase E., Bevilacqua P.C., Golden B.L.. A 1.9 angstrom crystal structure of the HDV ribozyme precleavage suggests both lewis acid and general acid mechanisms contribute to phosphodiester cleavage. Biochemistry. 2010; 49:6508–6518. [DOI] [PubMed] [Google Scholar]
- 97. Trausch J.J., Batey R.T.. A disconnect between high-affinity binding and efficient regulation by antifolates and purines in the tetrahydrofolate riboswitch. Chem. Biol. 2014; 21:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.