


Abstract
During infection of a host, Pseudomonas aeruginosa orchestrates global gene expression to adapt to the host environment and counter the immune attacks. P. aeruginosa harbours hundreds of regulatory genes that play essential roles in controlling gene expression. However, their contributions to the bacterial pathogenesis remain largely unknown. In this study, we analysed the transcriptomic profile of P. aeruginosa cells isolated from lungs of infected mice and examined the roles of upregulated regulatory genes in bacterial virulence. Mutation of a novel regulatory gene pvrA (PA2957) attenuated the bacterial virulence in an acute pneumonia model. Chromatin immunoprecipitation (ChIP)-Seq and genetic analyses revealed that PvrA directly regulates genes involved in phosphatidylcholine utilization and fatty acid catabolism. Mutation of the pvrA resulted in defective bacterial growth when phosphatidylcholine or palmitic acid was used as the sole carbon source. We further demonstrated that palmitoyl coenzyme A is a ligand for the PvrA, enhancing the binding affinity of PvrA to its target promoters. An arginine residue at position 136 was found to be essential for PvrA to bind palmitoyl coenzyme A. Overall, our results revealed a novel regulatory pathway that controls genes involved in phosphatidylcholine and fatty acid utilization and contributes to the bacterial virulence.
INTRODUCTION
The survival and propagation of bacteria depend on their abilities to promptly respond and adapt to the changing environment. Upon encountering host environments, pathogenic bacteria face tremendous challenges, including temperature shift, nutrient availability, host defence mechanisms, competition from commensal microorganisms, etc. (1,2). Bacteria utilize various mechanisms of signal sensing and transduction to alter gene expression, achieving physiological adaptation. Regulatory proteins play critical roles in these gene regulations. By binding to specific DNA sequences, regulatory proteins directly influence the transcription of mRNAs and small regulatory RNAs (3,4).
Pseudomonas aeruginosa is a versatile opportunistic pathogen that causes various acute and chronic infections in human (5,6). In cystic fibrosis (CF) patients, P. aeruginosa colonizes in the respiratory tract, leading to deterioration of lung functions (7,8). The bacterium harbours an arsenal of virulence factors, enabling its colonization, dissemination and persistence. For instance, P. aeruginosa utilizes type III secretion system (T3SS) to directly inject effector proteins into host cells, leading to cell malfunction or death (9,10). In a murine acute pneumonia model, it has been demonstrated that the effector proteins are preferentially injected into neutrophils and timely expression of the T3SS is critical for the bacterial pathogenesis (10–12).
Meanwhile, the ability to acquire and efficiently utilize nutrients from host is crucial for bacterial infections. Human lungs are coated with surfactant that is mainly composed of proteins and lipids, with ∼80% of the surfactant lipid being phosphatidylcholine (PC) (13). Previous studies demonstrated that PC is one of the major nutrient sources for P. aeruginosa during lung infection, supporting the high-cell-density growth of the bacteria (14). PC is cleaved by lipases and phospholipase C (PlcH) secreted by the P. aeruginosa, resulting in phosphorylcholine, glycerol and long chain fatty acids (mainly palmitic and oleic acids), which are further metabolized by the choline (bet), glycerol (glp) and β–oxidation pathways, respectively (15).
In Escherichia coli, the enzymes involved in fatty acids degradation have been well characterized, including a fatty acyl-CoA synthetase (FadD), an acyl-CoA dehydrogenase (FadE), a 3-ketoacyl-CoA thiolase (FadA), and an enoyl-CoA hydratase and 3-hydroxyacyl-CoA dehydrogenase (FadB) (16,17). In enteric bacteria, a GntR family transcriptional regulator, FadR, coordinately regulates genes involved in the degradation and biosynthesis of fatty acids in response to long-chain acyl-CoAs (18–20). In addition, FadR is involved in the regulation of virulence factors in pathogenic bacteria, such as Vibrio cholera, Salmonella and enterohemorrhage E. coli (EHEC) (21–24). P. aeruginosa harbours six fadD and five fadBA homologs, namely fadD1, 2, 3, 4, 5, 6 and fadBA1,2, 3, 4, 5, respectively. Among those genes, fadD1, 2, 4 and fadBA1, 4, 5 play major roles in the bacterial fatty acids degradation and fitness in a murine lung infection model (14,25–27). In P. aeruginosa, the regulator PsrA directly represses the expression of fadBA5 operon and long-chain fatty acids derepress the expression (28,29). However, the regulatory mechanisms for other fatty acid degradation genes remain elusive.
Here in this study, we investigated the transcriptome profile of P. aeruginosa in a murine acute pneumonia model. A regulatory gene pvrA (PA2957, Pseudomonas virulence regulator A) was found to contribute to the bacterial virulence. Results from further analysis demonstrated that PvrA coordinately regulates the genes involved in PC and long-chain fatty acid catabolism.
MATERIALS AND METHODS
Ethic statements
All animal studies were performed in compliance with the National and Nankai University guidelines regarding the use of animals in research. The animal experiment protocol was approved by the animal care and use committee of the College of Life Sciences of Nankai University with the permission number NK-04-2012.
Bacterial strains, plasmids and culture media
The bacterial strains and plasmids used in this study are listed in Supplementary Table S1. Bacterial cells were grown in Luria–Bertani (LB) broth that is composed of 10 g/l tryptone, 5 g/l yeast extract, 5 g/l NaCl, pH 7.4. For solid LB medium, 15 g/l agar was added. Medium with sole carbon source was prepared in the 1× M9 medium (49.4 mM Na2HPO4, 24.0 mM KH2PO4, 9 mM NaCl, 19 mM NH4Cl, 0.5 mM MgSO4) with 0.2% (w/v) Brij-58, supplemented with 0.1% (w/v) glucose or 0.4% (w/v) PC, choline, glycerol or palmitic acid, resulting in Glu-M9, PC-M9, Ch-M9, Gl-M9 or FA-M9 (15,25,26).
Animal experiment
Mouse acute pneumonia was performed as previously described (30). Pseudomonas aeruginosa strains were grown in LB at 37°C overnight and subcultured into fresh LB medium to an OD600 of 1.0. The bacterial cells were then washed with the phosphate-buffered saline (PBS) and adjusted to 1 × 109 CFU/ml for PAO1 or 2 × 108 CFU/ml for PA14. Each 6–8 weeks old female BALB/c mouse was anesthetized with an intraperitoneal injection of 100 μl 7.5% chloral hydrate. Then 20 μl bacterial suspension was intranasally inoculated into each mouse. 12 hours post infection (hpi), the mice were sacrificed by CO2. Lungs were isolated and homogenized in 1% proteose peptone (Solarbio, Beijing, China). The bacterial loads were determined by plating assay.
Transcriptome sequencing and data analysis
Wild type PAO1 was cultured in LB broth at 37°C and harvested at log phase (OD600 of 0.8–1.0), then subjected to RNA purification. Bronchi alveolar lavage fluid (BALF) was collected as previously described (31). Briefly, 6 hpi, the mice were sacrificed by inhalation of CO2. 2 ml PBS containing 0.05 mM EDTA was injected into lungs through trachea by a vein detained needle (BD, Angiocath). Bacteria in the BALF were harvested by centrifugation. Total RNA was isolated using the Trizol Reagent (Thermo Fisher Scientific, USA) and a Direct-zol RNA Miniprep kit (ZYMO, USA). Eukaryotic RNA was then removed using an MICROB Enrich kit (Ambion, USA). The quantity and integrity were determined using a NanoDrop spectrophotometer (Thermo Scientific) and a Bioanalyzer 2100 system (Agilent). For mRNA sequencing, a Ribo-Zero rRNA Removal Kit (Illumina, San Diego, CA, USA) was used to selectively remove rRNA. Random oligonucleotides and SuperScript III were used to synthesize the first strand cDNA. The second strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase treatment. After adenylation of the 3′ ends of the DNA fragments, Illumina PE adapter oligonucleotides were ligated to prepare for hybridization. To select cDNA fragments ∼300 bp in length, the library fragments were purified using the AMPure XP system (Beckman Coulter, Beverly, CA, USA). DNA fragments with adaptor molecules ligated on both ends were then selectively enriched using Illumina PCR Primer Cocktail in a 15 cycle PCR reaction. The products were purified with the AMPure XP system and quantified using the Agilent high sensitivity DNA assay on a Bioanalyzer 2100 system (Agilent). The DNA library was then sequenced on a NextSeq 500 platform (Illumina) by Shanghai Personal Biotechnology Cp. Ltd.
The sequence reads were mapped onto PAO1 reference genome (NC_002516.2) using a CLC genomics Workbench 8.0 (CLC Bio-Qiagen, Aarhus, Denmark). The count data of expression values were then analyzed using a DESeq package of R/Bioconductor. Differentially expressed genes were identified by performing a negative binomial test using the DESeq software, with the cut-off fold-change larger than 2. The raw sequence reads were normalized by dividing with size factors, then transformed to log2(N + 1).
Reverse transcription and quantitative RT PCR
The cDNA was synthesized from 1 μg total RNA using random primers and PrimeScript Reverse Transcriptase (TaKaRa, Dalian, China). Specific Primers (Supplementary Table S1) were used for quantitative RT PCR. 1 ng of cDNA was mixed with 4 pmol of forward and reverse primers and SYBR Premix Ex Taq™ II (TaKaRa) in a total reaction volume of 20 μl. No-template and no-reverse transcription controls were carried out for each experiment. An initial denaturation step at 98°C for 5 min was followed by 40× repeated cycles of denaturation for 5 s at 98°C, primer annealing for 15 s at 55°C and elongation for 15 s at 72°C. The signals were read every cycle. Melting curves were generated by a final denaturation step for 10 s at 98°C and recorded within the range of 65–99°C. A slope of 3.3 was adjusted. The results were analysed using a CFX Connect Real-Time system (Bio-Rad, USA). RNA from three biological replicates were analysed and three technical replicates were performed. Additional details are included in the ‘MIQE checklist’ (Supplementary Table S2) in compliance with the MIQE guidelines (Minimum Information for Publication of Quantitative real-time PCR Experiments) (32).
Chromatin immunoprecipitation
ChIP was performed as previously described with minor modifications (33,34). Pseudomonas aeruginosa cells were cross-linked with 1% formaldehyde in LB medium for 10 min at 37°C with continuous shaking. Crosslinking was stopped by the addition of 125 mM glycine. Bacterial pellets were washed twice with complete proteinase inhibitor cocktail (Roche) in a Tris buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl), and then resuspended in 400 μl nuclei lysis buffer (50 mM Tris–HCl (PH8.0), 10 mM EDTA, 1% Triton X-100, 1% SDS, mini-protease inhibitor cocktail (Roche)) for 30 min. The chromatin was sonicated (Diagenode Bioruptor pico) to sizes of 200–500 bp (20-se on, 30-s off, for 15 cycles). The insoluble debris was spun at 14 000 × g for 10 min at 4°C. 2% of the supernatant was saved as input and 100 μl of the supernatant was incubated with 10 μg anti-Flag antibody (MAB 3118) for 16 hours at 4°C. Then 30 μl of protein G magnetic beads (Life Technologies) was added to each IP reaction and incubated for 2 hours at 4°C with rotation. The immunoprecipitates were then processed with a series of washes: low salt wash buffer (150 mM NaCl, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl (pH 8.1)), high salt wash buffer (500 mM NaCl, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl (pH 8.1)), LiCl wash buffer (0.25 M LiCl, 1% NP40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris–HCl (pH 8.1)) and TE buffer (10 mM Tris–HCl pH 8, 1 mM EDTA). The immune complexes were eluted by 400 μl elution buffer (1% SDS, 0.1 M NaHCO3) and incubated at 65°C for 20 min. To reverse the cross-linking, 20 μl of 5 M NaCl was added to the 400 μl elutes and incubated overnight at 65°C. The samples were further incubated for 1 h at 45°C with 20 mg/ml Proteinase K in 30 mM Tris–HCl pH 6.5 and 10 mM EDTA to digest the proteins. The DNA was purified by phenol/chloroform/isoamyl extraction.
ChIP-Seq
ChIP-Seq libraries were prepared and sequenced following ENCODE guidelines (35) by Wuhan IGENEBOOK Biotechnology Co., Ltd. The DNA fragments (250–350 bp) were selected using SPRI beads and amplified by PCR for 15 cycles following repair and adaptor ligation steps. Libraries were validated on the Bioanalyzer 2100 (Agilent) and Qubit fluorometer (Invitrogen, Carlsbad, CA, USA). The ChIP-seq libraries were sequenced using the HiSeq 2000 system (Illumina) for 50 nt single-end sequencing. The primary analysis of the ChIP-seq reads was carried out as previously described (35). Trimmomatic (version 0.38) was used to filter out low-quality reads. Clean reads were mapped to the P. aeruginosa PA14 genome by Bwa (version 0.7.15), allowing up to two mismatches. Samtools (version 1.3.1) was used to remove potential PCR duplicates, and MACS2 software (version 2.1.1.20160309) was used to call peaks by default parameters (bandwidth, 300 bp; model fold, 5, 50; q value, 0.05). Wig files produced by the MACS software were used for data visualization by IGV (version 2.3.91). GO enrichment analysis was performed using the EasyGO gene ontology enrichment analysis tool. The GO term enrichment was calculated using hypergeometric distribution with a P value cut off of 0.01. P values obtained by the Fisher's exact test were adjusted with FDR for multiple comparisons to detect overrepresented GO terms. To reveal potential roles of genes, cluster Profiler in R package was employed to perform KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis.
Electrophoretic mobility shift assay
The electrophoretic mobility shift assay (EMSA) was performed as previously described with minor modifications (30). Briefly, DNA fragments (400 ng) were incubated with 0, 1.5 or 3 μM purified recombinant PvrA protein at 37°C for 10 min in a 20 μl reaction (10 mM Tris–HCl, pH 7.6, 4% glycerol, 1 mM EDTA, 5 mM CaCl2, 100 mM NaCl, 10 mM-β-mercaptoethanol). Samples were loaded onto an 8% native polyacrylamide gel in 0.5× Tris–borate–EDTA (TBE) buffer (0.044 M Tris base, 0.044 M boric acid, 0.001 M EDTA, pH 8.0) that had been pre-run for 1 h, and then run on ice at 100 V for 1.2 h followed by staining in 0.5× TBE containing 0.5 μg/ml ethidium bromide. Bands were visualized with a molecular imager ChemiDoc™ XRS + (Bio-Rad).
Acetyl coenzyme A measurement
Bacterial intracellular acetyl-coenzyme A (Ac-CoA) content was determined using the PicoProbe Ac-CoA assay kit (Abcam) according to manufacturer's instructions. Briefly, PA14 and the pvrA mutant were grown for 10 h in the Glu-M9 or FA-M9 medium. 30 ml of the bacterial cultures were collected by centrifugation at 12 000 × g for 5 min. After deproteinization with perchloric acid, the coenzyme A (CoA) Quencher and Quencher remover were added into each sample to correct the background generated by free CoA and succinyl-coenzyme A (succ-CoA). The sample was then diluted with the reaction mix, and the fluorescence was measured using a Versamax Tunable microplate reader (Molecular Devices) at the following settings: λex 535 nm; λem 587 nm. The acetyl-CoA standard curve was made in the range of 0–100 pM and the correlation coefficient was 0.990 or higher.
Molecular docking
The docking algorithm glide was used to perform all the molecular docking studies. The two co-crystal structures (PDB ID: 3LSJ, 3E7Q) were prepared and then used to build the energy grid. The grids were generated at the centroid of the co-crystallized ligands. Default settings were employed for both the grid generations and docking. Post-minimization was used to optimize the geometry of the poses.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) was conducted using a MicroCal iTC200 (Malvern) with a cell volume of 400 μl. Both PvrA protein at 30 μM and the ligands of palmitoyl-coenzyme A or palmitic acids at 0.3 mM were dialyzed against the same buffer (20 mM Tris–HCl, 100 mM NaCl, pH 7.5) to minimize artefacts due to minor differences of the buffer composition. The raw data was analysed with the software ORIGIN.
Surface plasmon resonance (SPR) assays
The SPR experiments were carried out using a Biacore T200 instrument (GE Healthcare). In the SPR trials, a biotinylated DNA probe of the fadD1 promoter region was immobilized by streptavidin on the chip surface in an immobilization buffer (10 mM sodium acetate buffer pH 4.0 to 5.0). A serial dilution of the purified PvrA protein was passed over the chip surface for 2 min. The running buffer (HBS-EP buffer, 10 mM HEPES, pH 7.4 containing 150 mM NaCl, 3 mM EDTA and 0.005% (v/v) Surfactant P20) was run at a flow rate of 30 μl/min. The SPR data was analysed with the BIAevaluation v.4.1.1 software (BIAcore).
RESULTS
Transcriptome analysis of P. aeruginosa during infection
To explore the global gene expression profile of P. aeruginosa during infection, we infected mice with a wild type strain PAO1 in an acute pneumonia model and collected bacterial cells from BALF for RNA isolation. In our experiments, mice infected with wild type PAO1 usually began to die at 12 h post infection. Previous studies demonstrated substantial recruitment of neutrophils as early as 6 h after intranasal infection by the P. aeruginosa (30,36,37), thus resulting in an intense interaction between the bacteria and host cells. Therefore, BALF was harvested 6 h post infection from 25 mice and pooled together for bacterial cell isolation and subsequent RNA purification. The global gene expression profile was determined by RNA-seq and compared to that of bacteria grown in LB (Supplementary Figure S1).
Consistent with previous reports, the expression of type III secretion system (T3SS) and iron acquisition associated genes was induced within the host lung environment (Supplementary Table S3). In addition, 19 regulatory genes were upregulated at least 2-fold in vivo (Table 1). To confirm the expression levels of these genes, we repeated the infection with PAO1 and performed real time PCR with isolated bacterial RNA. Consistent with the RNA-seq results, all of the tested regulatory genes were indeed upregulated (Table 1). Four of those genes, including exsA, pchR, typA and fis have previously been demonstrated to play important roles in the bacterial virulence (38–43). To examine whether the expression patterns of these regulatory genes are conserved in different strain backgrounds, we infected mice with another widely used strain PA14 which belongs to a different serogroup (37). All of the 19 regulatory genes were up regulated as well in the PA14 during infection (Table 1), ruling out a strain specific phenomenon.
Table 1.
Upregulated regulatory genes of P. aeruginosa during infection
| mRNA level fold change (in vivo versus LB) | |||||
|---|---|---|---|---|---|
| Gene ID | Gene name | PAO1 RNAseq | PAO1 qPCR | PA14 qPCR | Description |
| PA0191 | 2.82 | 1.82 | 2.58 | transcriptional regulator | |
| PA0253 | 2.93 | 2.07 | 3.88 | transcriptional regulator | |
| PA0448 | 2.36 | 2.30 | 4.19 | transcriptional regulator | |
| PA0707 | toxR | 27.97 | 7.49 | 12.97 | transcriptional regulator ToxR |
| PA1179 | phoP | 9.36 | 3.9 | 24.5 | two-component response regulator PhoP |
| PA1713 | exsA | 11.46 | 9.10 | 20.35 | transcriptional regulator ExsA |
| PA2657 | 2.81 | 5.74 | 2.11 | two-component response regulator | |
| PA2798 | 3.31 | 3.21 | 2.10 | two-component response regulator | |
| PA2957 | 3.33 | 2.55 | 4.41 | transcriptional regulator | |
| PA3006 | psrA | 2.63 | 2.58 | 5.26 | transcriptional regulator PsrA |
| PA3604 | erdR | 2.79 | 5.21 | 2.02 | response regulator ErdR |
| PA3616 | recX | 2.92 | 3.11 | 4.71 | recombination regulator RecX |
| PA3932 | 5.44 | 1.57 | 1.80 | transcriptional regulator | |
| PA4381 | 2.39 | 3.83 | 1.73 | two-component response regulator | |
| PA4227 | pchR | 6.09 | 7.33 | 10.32 | transcriptional regulator PchR |
| PA4776 | pmrA | 3.44 | 14.05 | 8.68 | two-component regulator system response regulator PmrA |
| PA4853 | fis | 5.16 | 4.37 | 3.44 | Fis family transcriptional regulator |
| PA5117 | typA | 6.93 | 3.08 | 4.79 | regulatory protein TypA |
| PA5550 | glmR | 2.82 | 3.14 | 4.88 | GlmR transcriptional regulator |
Roles of the regulatory genes in bacterial virulence
Upregulation of the regulatory genes indicated that these genes might be involved in bacterial response to the host environment. To test whether these genes contribute to bacterial virulence, we obtained mutants of each individual gene from the PA14 transposon (Tn) mutant library (44). While for those not available in the Tn mutant library, deletion mutants were generated in the PA14 background, including ΔPA0253, ΔPA0707, ΔPA2957 and ΔPA3616. Mice were infected with each of the mutant strain intranasally. Mutation of PA2957 resulted in significantly lower bacterial colonization compared to the wide type PA14 (Figure 1A, Supplementary Figure S2). To verify the role of PA2957 in virulence, we examined the survival rates in the murine pneumonia model. Infection with wild type PA14 caused death in all the infected mice within 48 hours. However, mutation of the PA2957 delayed the death and resulted in 40% survival (Figure 1B). Complementation with a PA2957 gene driven by its native promoter restored the bacterial colonization capacity and virulence (Figure 1A, B). In addition, mutation of the PA2957 did not affect bacterial growth rate in LB (Supplementary Figure S4), suggesting that the attenuated virulence might be due to reduced virulence gene expression or defective adaptation to the host in vivo environment. Accordingly, we designated PA2957 as pvrA (Pseudomonas virulence regulator A). The PvrA is a TetR-family transcriptional regulator with unknown regulatory targets. Sequence analysis revealed that PvrA is conserved among Pseudomonads (Supplementary Figure S3).
Figure 1.
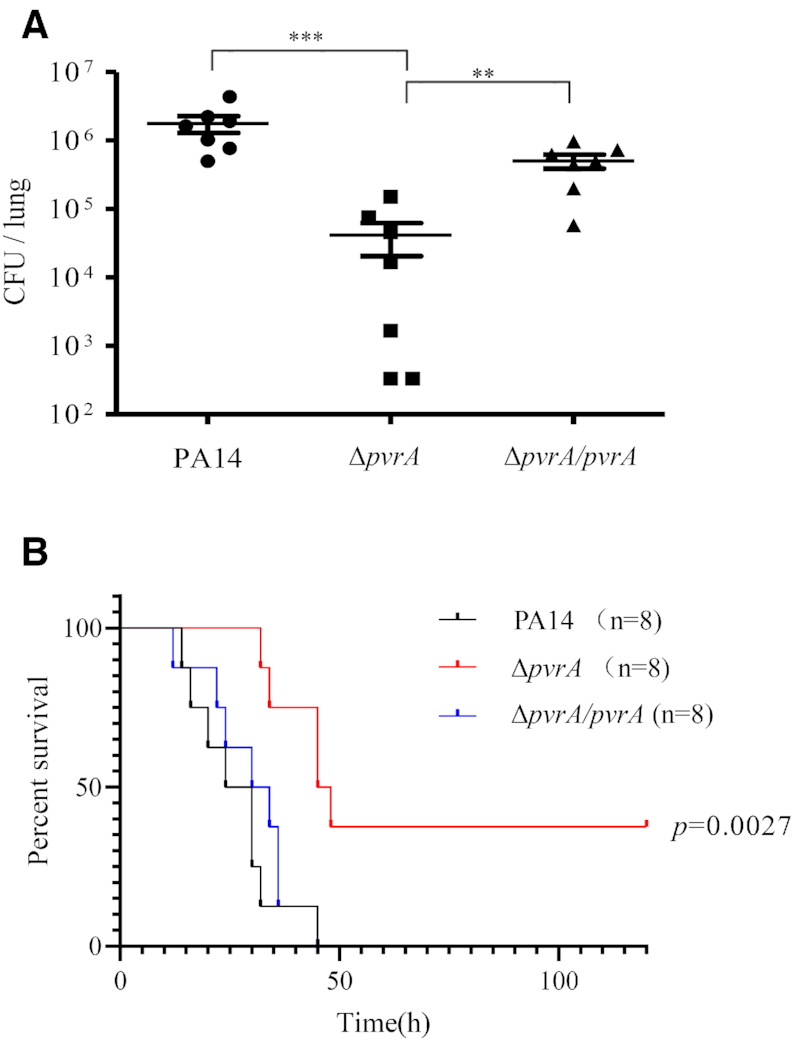
Mutation of pvrA reduces the virulence of PA14. (A). Bacterial colonization in the murine acute pneumonia model. Each mouse was infected intranasally with 4 × 106 CFU of wild type PA14, ΔpvrA or the complemented strain (ΔpvrA/pvrA). 12 hours post infection, mice were sacrificed and bacterial loads in the lungs were determined. Bars represent medians, and error bars represent standard deviations. *P < 0.05; ***P < 0.001 by Student's t-test. (B). Survival rates of infected mice. Mice were infected intranasally with 6 × 106 CFU of wild type PA14, the ΔpvrA mutant or the complemented strain (ΔpvrA/pvrA). The mice were monitored for 5 days. P values were calculated by the log-rank test.
PvrA directly regulates genes related to bacterial virulence and metabolism
To identify genes directly regulated by the PvrA, we overexpressed a C-terminal FLAG tagged pvrA in wild type PA14 and performed a CHIP-seq assay. The DNA binding sequences, loci and enrichment fold were shown in Supplementary Table S4. Of note, the potential regulatory targets of PvrA include genes involved in the utilization of phosphatidylcholine (PC) from the host, such as plcH, fadD1, fadD6 and PA0508 (Table 2) (15,25,26). The plcH encodes phospholipase C that cleaves PC into phosphorylcholine, glycerol and fatty acids (14). Both fadD1 and fadD6 encode the fatty acyl-CoA synthetase and PA0508 is highly homologous to the fadE of E. coli (45,46). In addition, PvrA was also found to bind to the promoter regions of glcB, maeB and aprA (Table 2). The glcB encodes a malate synthase which is a key enzyme in the glyoxylate shunt, while maeB encodes a malic enzyme that influences the malate level in the glyoxylate cycle and controls the initiation of the gluconeogenesis from the glyoxylate shunt (47,48). The glyoxylate shunt serves as an alternative to the tricarboxylic acid cycle, and is essential for acetate and fatty acid metabolism in bacteria (46,49). The aprA encodes an extracellular alkaline metalloproteinase that contributes to bacterial virulence (50,51).
Table 2.
Potential PvrA regulated genes identified via ChIP-seq analysis
| Gene ID of PA14 | Gene ID of PAO1 | Summit in PA14 | Fold enrichment | Gene name | Description |
|---|---|---|---|---|---|
| PA14_06300 | PA0482 | 557225 | 29.27 | glcB | Malate synthase G |
| PA14_13110 | PA3924 | 1124193 | 28.27 | fadD6 | Probable medium-chain acyl-CoA ligase |
| PA14_06640 | PA0508 | 578862 | 18.95 | fadE | Probable acyl-CoA dehydrogenase |
| PA14_21370 | PA3299 | 5478678 | 10.14 | fadD1 | Long-chain-fatty-acid–CoA ligase |
| PA14_53360 | PA0844 | 4729534 | 7.90 | plcH | Hemolytic phospholipase C |
| PA14_66680 | PA5046 | 5956289 | 6.01 | meaB | Malic enzyme |
| PA14_48060 | PA1249 | 4279068 | 5.25 | aprA | Alkaline metalloproteinase |
EMSA was utilized out to confirm the binding of the identified promoter regions by PvrA. Band shifts were observed with the promoter regions of plcH, fadD1, fadD6, PA0508, glcB, maeB and aprA (Figure 2). A MEME analysis on the ChIP-seq data revealed the potential PvrA binding site as 5′-CGGTCA-3′ that was present in the probes used in the EMSAs (Figure 3A). Deletion of the potential binding sequence from the promoter region of fadD1 abolished the retardation by PvrA (Figure 3B), indicating a specific binding by the PvrA.
Figure 2.

Binding of PvrA to the promoter regions of genes involved in PC utilization and fatty acid metabolism. Interaction between PvrA and its target DNA was examined by EMSA. Increasing amount of the purified PvrA protein was incubated with the promoter regions of plcH, fadD1, fadD6, PA0508, aprA, glcB, maeB and pvrA. The mixtures were electrophoresed on an agarose gel and the bands were visualized under UV light following ethidium bromide stain. Data represent results from three independent experiments.
Figure 3.
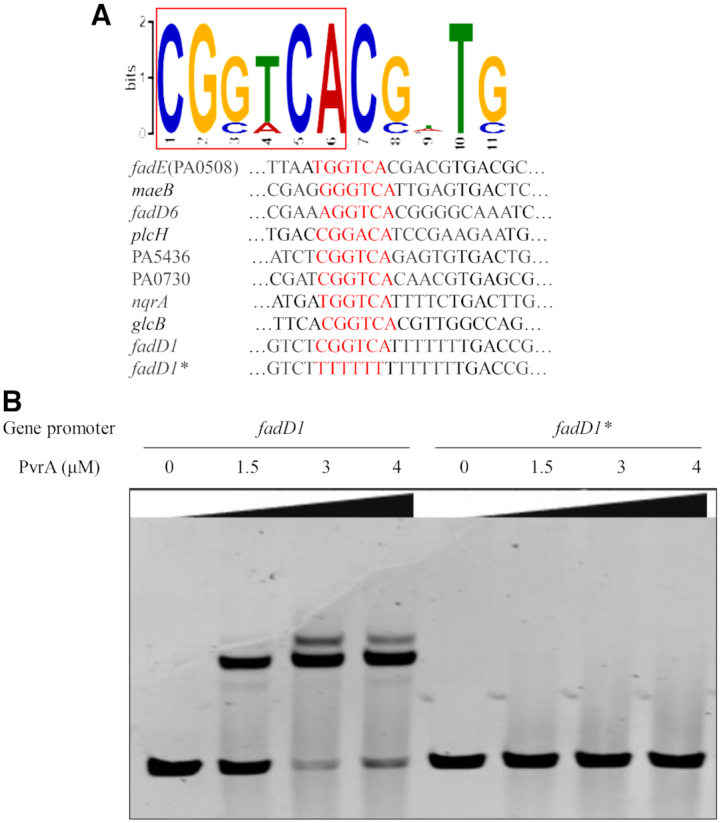
Identification of the potential PvrA binding motif. (A). Potential PvrA binding motif was identified by MEME from the ChIP-seq peak regions. Representative sequences bound by PvrA in the EMSAs are listed below. The conserved sequence is shown in red. (B). In the EMSA, indicated amount of the purified PvrA protein was incubated with the fadD1 promoter region or the same fragment with the predicted binding motif deleted (fadD1*). The bands were visualized by staining with EB.
To examine the role of PvrA in gene regulation during infection, we infected mice with wild type PA14 and a ΔpvrA mutant. In wild type PA14, the expression of pvrA, plcH, fadD1, fadD6, PA0508, aprA, glcB and maeB was upregulated in vivo, however, their expression levels were significantly reduced in the ΔpvrA mutant (Figure 4A, B). Therefore, the above results suggested that PvrA directly controls the expression of plcH, fadD1, fadD6, PA0508, glcB, maeB and aprA.
Figure 4.
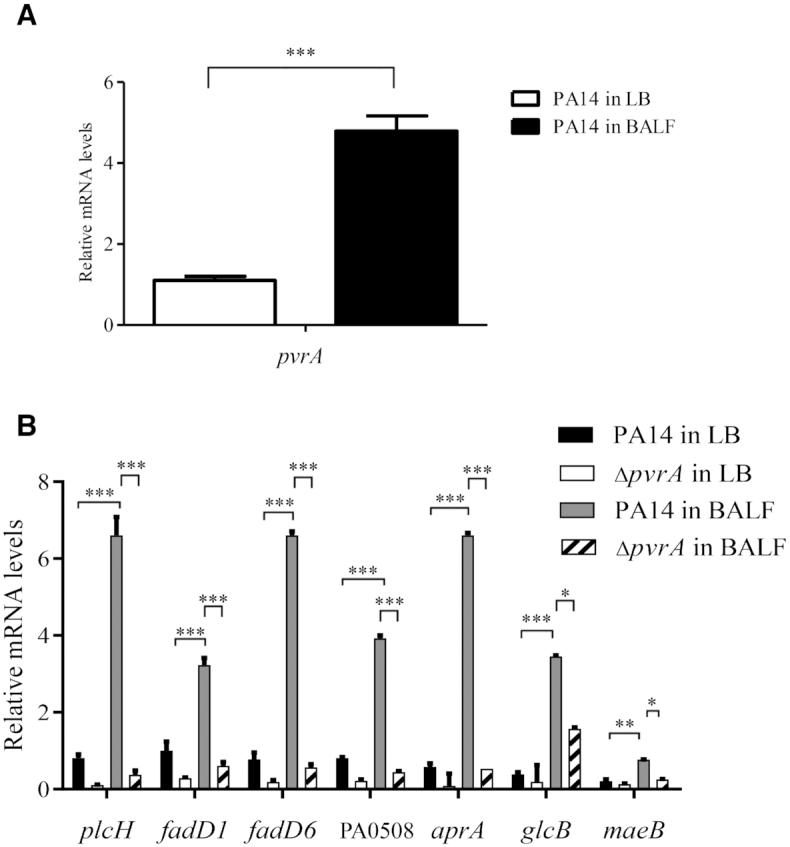
Role of PvrA in gene regulation during infection. Wild type PA14 and the ΔpvrA mutant were grown in LB to an OD600 of 1.0. A portion of the bacteria were subjected to RNA isolation. The remaining bacteria were washed and resuspended in PBS. Mice were infected with the bacteria intranasally. The bacteria were isolated from BALF 6 hpi. The mRNA levels of indicated genes were determined by real time PCR with rpsL (encoding the 16S rRNA) as the internal control. (A). Relative mRNA levels of pvrA of PA14 in BALF and LB medium. (B). Relative mRNA levels of plcH, fadD1, fadD6, PA0508, aprA, glcB, maeB in BALF or LB. *P < 0.05; **P < 0.01; ***P < 0.001 by Student's t-test. Data represent the mean ± standard deviation from three samples.
PvrA controls bacterial utilization of exogenous PC and palmitic acid
Since PlcH, FadD1, FadD6 and PA0508 all are involved in the utilization of PC, we examined the role of PvrA in bacterial utilization of PC. Wild type PA14 and the ΔpvrA mutant were grown in M9 medium with glucose or PC as the sole carbon source (designated as Glu-M9 and PC-M9, respectively). In Glu-M9, the two strains grew at the same rate (Figure 5A), however, a growth retardation was observed for the ΔpvrA mutant in PC-M9 (Figure 5B). We then examined the expression levels of the genes involved in PC utilization. In Glu-M9, the mRNA level of plcH was similar between wild type PA14 and the ΔpvrA mutant, whereas the mRNA levels of fadD1, fadD6 and PA0508 were approximately 2–3 folds lower in the ΔpvrA mutant. Compared to the cells grown in Glu-M9, growth in PC-M9 induced the expression of plcH, fadD1 and PA0508 in wild type PA14 by 18.5-, 5- and 9-fold respectively, whereas the induction of the corresponding genes in the ΔpvrA mutant was 12-, 3.5- and 2-fold, thus enlarging the differences in their expression levels between the two strains (Figure 5C). As for fadD6, growth in PC-M9 induced the similar fold of increase in the expression in both strains, thus the expression level difference remained at 2-fold (Figure 5C). We then examined the roles of PvrA regulated genes in the bacterial utilization of PC. A ΔplcH mutant grew similarly as the wild type strain in PC-M9 in the first 20 h. Then the OD540 started to drop and maintained at a low level after 25 h (Supplementary Figure S5A). However, deletion of fadD1, fadD6 and PA0508 individually or simultaneous deletion of fadD1, fadD6 (ΔfadD1ΔfadD6) or fadD1, fadD6 and PA0508 (ΔfadD1ΔfadD6ΔPA0508) did not affect the bacterial growth in PC-M9 (Supplementary Figure S5A). As a control, all the tested strains grew similarly in Glu-M9 (Supplementary Figure S5B). This result is consistent with the role of PlcH in the cleavage of PC, which is required for the bacterial utilization of PC (14).
Figure 5.
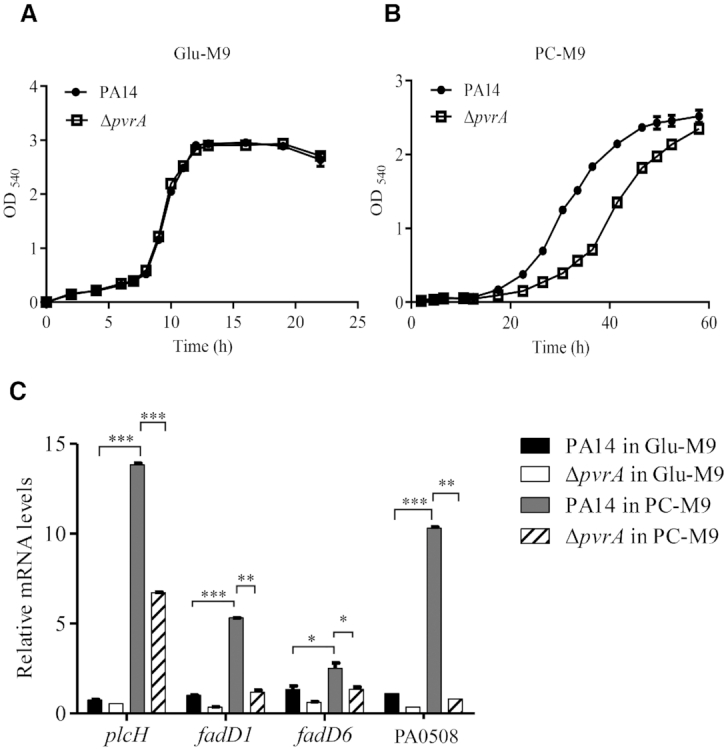
PvrA contributes to the bacterial utilization of PC. Same amount of wild type PA14 and the ΔpvrA mutant were inoculated in Glu-M9 (A) and PC-M9 (B). The bacterial growth was monitored by measuring OD540. (C) PA14 and the ΔpvrA mutant were inoculated into Glu-M9 or PC-M9 and grown to an OD600 of 1.0. The relative mRNA levels of plcH, fadD1, fadD6 and PA0508 were determined by real time PCR. rpsL was used as the internal control. *P < 0.05; **P < 0.01; ***P < 0.001 by Student's t-test. Data represent the mean ± standard deviation from three samples.
The exogenous PC is cleaved by P. aeruginosa into choline, glycerol and long chain fatty acids before catabolized by various pathways (15). We therefore examined the utilization of the individual carbon source by the ΔpvrA mutant. A growth defect was observed in the ΔpvrA mutant when palmitic acid was the sole carbon source (Figure 6A), whereas the mutant grew normally when choline or glycerol was the sole carbon source (Figure 6B, C). Acetyl-CoA is a key β-oxidation intermediate in the fatty acids catabolism. The ΔpvrA mutant produced less acetyl-CoA than the wide type PA14 when grown in the palmitic acid-M9 medium (FA-M9), whereas no difference was observed when the two strains were grown in the Glu-M9 medium (Figure 6D). In addition, when palmitic acid was the sole carbon source (FA-M9), mutation of pvrA significantly reduced the expression of plcH, fadD1, fadD6, PA0508 and glcB (Figure 6E). However, when glycerol or choline was the sole carbon source, the expression levels of those genes were similar between the wide type PA14 and the ΔpvrA mutant, except that fadD6 and PA0508 were downregulated in the ΔpvrA mutant in Gly-M9 and Cho-M9, respectively (Figure 6E). These results suggest an important role of PvrA in the bacterial utilization of palmitic acid.
Figure 6.

Growth analyses and genes expression of PA14 and the ΔpvrA mutant on palmitic acid, glycerine and choline. Same amount of wild type PA14 and the ΔpvrA mutants were inoculated in FA-M9 (A), Gly-M9 (B) or Cho-M9 (C). The bacterial growth was monitored by measuring OD540. (D). PA14, the ΔpvrA mutant and the complemented strain were grown in Gly-M9 or FA-M9 for 10 h. The intracellular acetyl-CoA concentration was determined and normalized to the corresponding total protein concentration. (E). PA14 and the ΔpvrA mutant were grown in FA-M9, Gly-M9 or Cho-M9 for 10 h. The relative mRNA levels of fadD1, fadD6, PA0508, plcH, glcB were determined by real time PCR. *P < 0.05; **P < 0.01; ***P < 0.001 by Student's t-test. Data represent the mean ± standard deviation from three samples.
To examine the roles of the PvrA regulated genes in the bacterial utilization of palmitic acid, we monitored the growth of the strains with mutations in those genes in FA-M9. Deletion of fadD1, fadD6, PA0508 individually, or simultaneous deletion of fadD1 and fadD6 did not affect the bacterial growth (Supplementary Figure S6). Triple deletion of fadD1, fadD6 and PA0508 (ΔfadD1ΔfadD6ΔPA0508) resulted in a slight growth defect (Supplementary Figure S6). These results suggest that the other fatty acyl-CoA synthetases (FadD2, FadD3, FadD4, FadD5) and acyl-CoA dehydrogenases (PA0506 and PA0507) might support the growth of the triple mutant in FA-M9.
Palmitoyl-coenzyme A is a ligand of PvrA
In the bacterial utilization of fatty acids, the fatty acids are converted into fatty acyl-CoA first in cytosol and then catabolized through the β-oxidation pathway (15). In E. coli, the long chain fatty acyl-CoA is a ligand for FadR, influencing its regulatory function (18–20). Therefore, we suspected that palmitoyl-coenzyme A might serve as a ligand for PvrA. A molecular docking analysis revealed a possible interaction between palmitoyl-coenzyme A and PvrA (Figure 7A). The R136 forms three hydrogen bonds with the oxygen atoms in the oxygen-phosphorus bonds of the palmitoyl-coenzyme A, thus might serve as a critical residue for the interaction (Figure 7B). Indeed, ITC assays revealed active interaction between PvrA and palmitoyl-coenzyme A but not with palmitic acid (Figure 7C, D). Replacement of the R136 with an alanine (R136A mutation) diminished the interaction between PvrA and palmitoyl-coenzyme A (Figure 7E, F).
Figure 7.

Interaction between PvrA and palmitoyl-coenzyme A. (A) Model structure of the interaction between palmitoyl-coenzyme A and PvrA by the MetaPocket program. (B) Binding modes of palmitoyl-coenzyme A in the PvrA binding pocket. Dotted lines represent hydrogen-bonds between palmitoyl-coenzyme A and the arginine residue at the 136 position of PvrA. (C–F) Direct binding of palmitoyl-coenzyme A to the soluble PvrA was measured by isothermal titration microcalorimetry (ITC). Isothermal titration microcalorimetry of palmitoyl-coenzyme A (C) or palmitic acid (D) (0.3 mM) binding to 0.03 mM PvrA protein at 25°C. Palmitoyl-coenzyme A exhibited binding affinity of KD = 3.10 × 10−5 M to PvrA; No detectable binding of palmitic acid to PvrA was exhibited. (E) ITC analysis revealed the affinity between PvrA R136A mutant and palmitoyl-coenzyme A was KD = 2.62 × 10−4 M. (F). Interaction between the buffer and palmitoyl-coenzyme A was analysed as negative control. Affinity and molar ratio are indicated. ND: No detectable binding.
To determine whether palmitoyl-coenzyme A affects the DNA binding ability of PvrA, we performed EMSA assay with the promoter region of fadD1. Supplementation of 0.25 μM palmitoyl-coenzyme A to 3 μM PvrA protein resulted in undetectable free DNA probe and an obvious super shift band (Figure 8A, lane 6), whereas in the absence of palmitoyl-coenzyme A or in the presence of the same molar concentration of palmitic acid, free probe remained and a lighter super shift was observed with the same amount of PvrA (Figure 8A, lanes 10). Meanwhile, the R136A mutation abolished the effect of palmitoyl-coenzyme A on the probe binding (Figure 8B).
Figure 8.

Palmitoyl-coenzyme A enhances the binding of PvrA to the target DNA fragment. The binding of wild type PvrA (A) or the mutated PvrA (R136A) (B) to the fadD1 promoter was examined by EMSA. The fadD1 promoter fragment was incubated with increasing concentrations of PvrA or the mutated PvrA (R136A) with or without the indicated concentrations of palmitoyl-coenzyme A. The DNA bands were visualized by EB staining. The free probes are indicated by arrowheads and the super shifts are indicated by arrows.
To confirm the role of palmitoyl-coenzyme A in enhancing the affinity of PvrA to the fadD1 promoter region, we performed a surface plasmon resonance (SPR) assay. Consistent with the EMSA results, the presence of palmitoyl-coenzyme A increased the binding affinity of the PvrA to the DNA probe by approximately 3-fold, as determined by reduction of the equilibrium dissociation constant (KD) from 1.493 × 10−8 M to 4.716E × 10−9 M (Figure 9A, B). The R136A mutation did not affect the affinity of PvrA to the probe (KD = 1.261 × 10−8 M), however, the presence of palmitoyl-coenzyme A did not increase its affinity (KD = 1.596 × 10−8 M) (Figure 9C, D).
Figure 9.
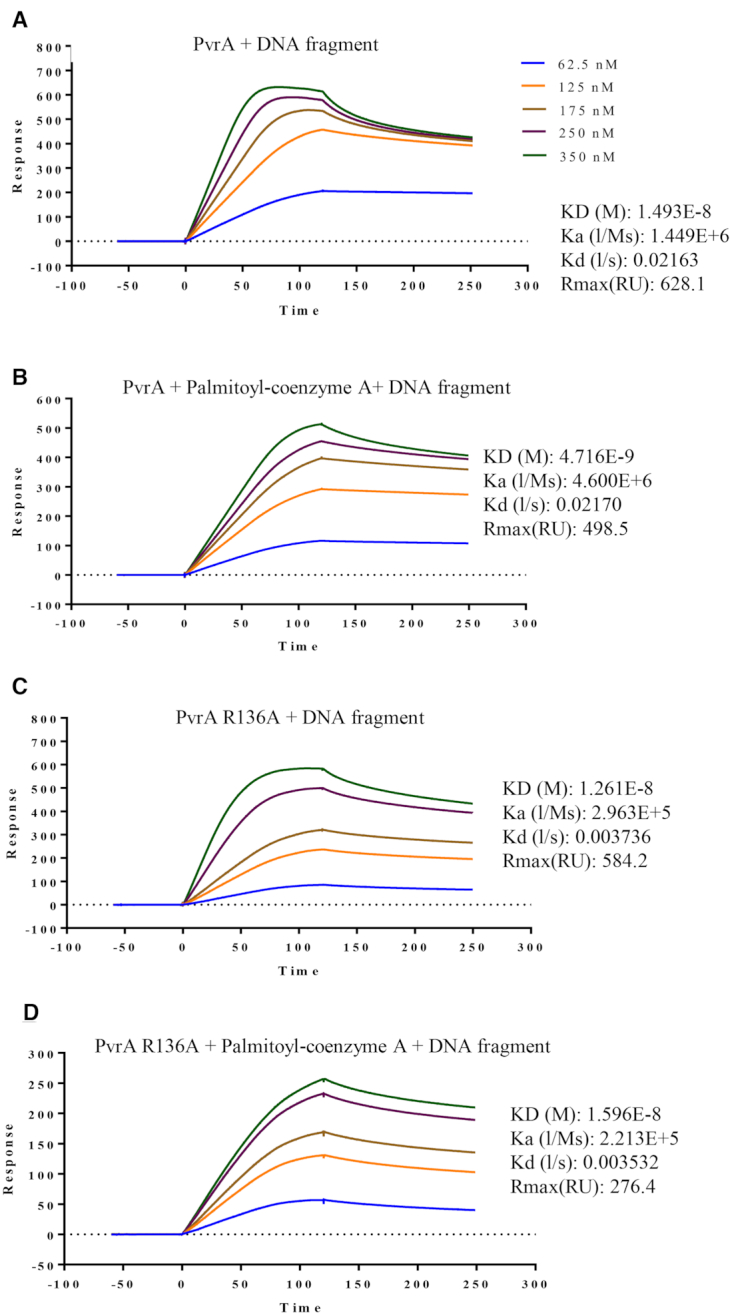
Surface Plasmon Resonance (SPR) analyses of binding of PvrA to the fadD1 promoter fragment. The PvrA or PvrA(R136A) protein at the indicated concentrations were injected over the immobilized DNA fragment of the fadD1 promoter region (30 bp). (A, B) The binding affinities of PvrA to the DNA probe in the absence (A) or presence (B) of palmitoyl-coenzyme A. (C, D) The binding affinities of PvrA(R136A) to the DNA probe in the absence (C) or presence (D) of palmitoyl-coenzyme A. ka, association rate constant; kd, dissociation rate constant; KD, equilibrium dissociation constant. The concentrations of the proteins and values of ka, kd and KD are indicated in the inset to the figures.
In addition, complementation of the ΔpvrA mutant with the R136A allele restored neither the induction of fadD1, fadD6, PA0508 and plcH by palmitic acid (Figure 10A) nor the bacterial virulence in the murine acute pneumonia model (Figure 10B). In combination, these results suggest that palmitoyl-coenzyme A is the likely ligand for PvrA, activating the plcH and fatty acid utilization genes.
Figure 10.
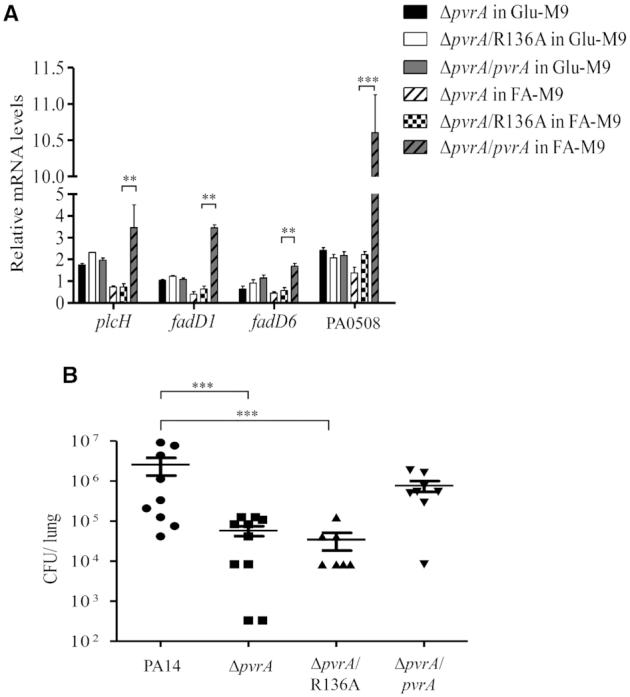
Role of the R136 in the function of PvrA. (A) The ΔpvrA mutant and the mutant carrying a wild type pvrA or the R136A mutant allele were grown in Glu-M9 and FA-M9. The relative mRNA levels of plcH, fadD1, fadD6 and PA0508 were determined by real time PCR. rpsL was used as the internal control. (B) Mice were infected intranasally with 4 × 106 CFU of the indicated strains. 12 hpi, the mice were sacrificed and bacterial loads in the lungs were determined by plating. Data represents the mean ± standard deviation from three samples. **P < 0.01, ***P < 0.001 by Student's t-test.
Roles of PvrA regulated genes in the bacterial virulence
To evaluate the contributions of the PvrA regulated genes in the bacterial virulence, we infected mice with strains deficient in the individual genes of plcH, fadD1, fadD6 and PA0508. No attenuation was observed in those mutants (Figure 11). Next, we constructed double, triple and quadruple mutants. No significant difference in the bacterial load was observed between wild type PA14 and the strains of ΔfadD1ΔfadD6, ΔfadD1ΔPA0508 and ΔfadD1ΔfadD6ΔPA0508. However, the ΔfadD1ΔfadD6ΔPA0508ΔplcH mutant displayed ∼50-fold reduction of bacterial loads in the lungs of infected mice (Figure 11). These results indicated that PvrA might contribute to bacterial virulence by coordinately regulating genes involved in the PC and fatty acid utilization within the host lung environment.
Figure 11.
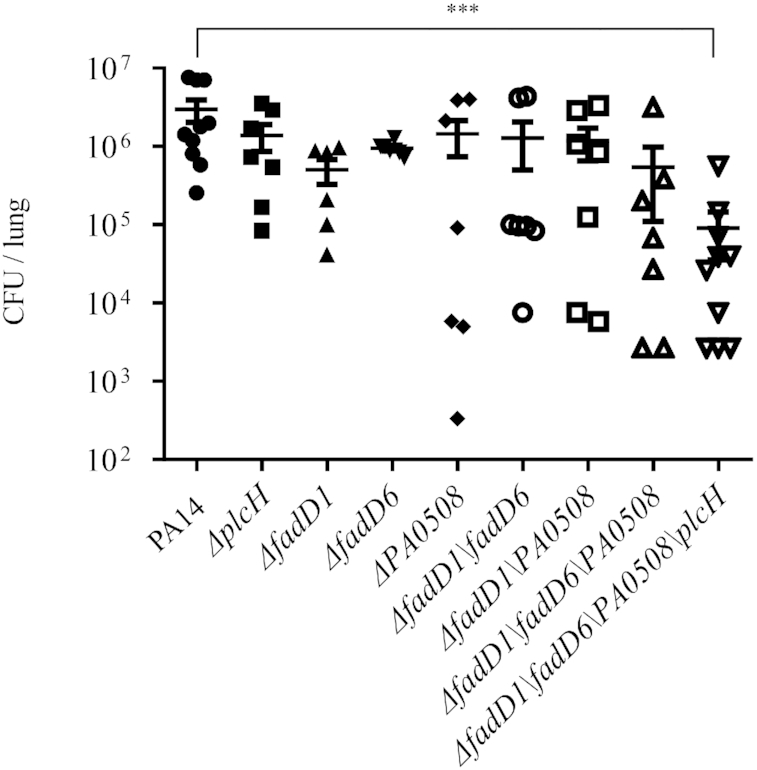
Roles of the PvrA regulated genes in the bacterial virulence. The indicated strains were grown in LB to an OD600 of 1.0. Mice were infected intranasally with 4 × 106 CFU of each strain. The mice were sacrificed 12 hpi and the bacterial loads in the lungs were determined by plating. Bars represent medians, and error bars represent standard deviations. ***P < 0.001 by Student's t-test.
DISCUSSION
In this study, we analysed the transcriptome of P. aeruginosa in a murine acute pneumonia model and focused on upregulated regulatory genes. PvrA was found to be upregulated during infection and contributes to the bacterial virulence. PvrA is a TetR family protein with a helix-turn-helix (HTH) motif at the N-terminus. We found that PvrA controls the expression of plcH, fadD1, fadD6, PA0508 that are required for the bacterial utilization of PC and long chain fatty acids.
Besides an essential role in the utilization of PC, the PlcH of P. aeruginosa is also involved in the acquisition of nutrients from erythrocytes (52), contributing to the bacterial virulence in a mouse burn model (53) and systemic infection in a leukopenic mouse model (52). The expression of plcH is regulated by GbdR via a positive feedback mechanism (54,55). PC is degraded by the PlcH, generating choline, which is further degraded into glycine betaine and dimethylglycine (56). GbdR is activated by glycine betaine and dimethylglycine, upregulating the expression of plcH (54). When environmental inorganic phosphate concentration is low, PhoB can also activate the expression of plcH (57). Under hypoxic conditions, the plcH is repressed by Anr (58). Besides, the H-NS family of proteins, MvaT and MvaU, repress the expression of plcH (59). In our study, we found that mutation of pvrA reduced the expression level of plcH. These results suggest that plcH is regulated by multiple pathways, indicating potential pleiotropic physiological functions under various environmental conditions. In PA14, plcH was up regulated in BALF collected from the infected mice (Figure 4B). However, plcH was downregulated in PAO1 in BALF as shown in the RNA-seq results (Supplementary Table S3). It might be possible that the role of PlcH in PC utilization in vivo is different between PA14 and PAO1. Another possible cause of the different expression patterns of plcH might be due to the difference in virulence between PA14 and PAO1. In several infection models, PA14 is more virulent than PAO1 (60). Thus in our experiment, PA14 might cause more tissue damages than PAO1, leading to a different host inflammatory response and local environment, which might result in different regulation of plcH. Further studies are needed to elucidate the regulatory pathway of plcH in response to host in vivo environment during the infection.
In enteric bacteria, a GntR family regulator FadR activates the expression of long chain fatty acids (LCFAs) synthesis genes and represses those involved in LCFAs catabolism. The activity of FadR is regulated by long acyl-CoA molecules that are the products of the first reaction of β-oxidation. Interaction with long acyl-CoA molecules abolishes the binding of FadR to its target DNA, resulting in derepression of LCFAs catabolism genes and downregulation of LCFAs synthesis genes (61). In Vibrio cholera, FadR is also a GntR family regulator (62), whereas the FadR in Sulfolobus acidocaldarius, Thermus thermophiles, Bacillus subtilis and Bacillus halodurans belong to the TetR family regulators (63,64). In P. aeruginosa, no fadR homolog has been identified so far. Here we found that a TetR/AcrR family regulator PvrA directly regulates fadD1, fadD6, PA0508, however, no direct binding was identified between PvrA and key genes involved in the LCFAs synthesis. Whether PvrA regulates the LCFAs synthesis genes requires further studies. Amino acid sequence analysis performed by I-TASSER (Iterative Threading ASSEmbly Refinement, zhang.bioinformatics.ku.edu/I-TASSER) (65,66) revealed that the structure of PvrA is similar to that of DesT (data not shown), a regulator controlling the homeostasis of saturated and unsaturated fatty acids in P. aeruginosa (67). Previous studies demonstrated that palmitoyl-coenzyme A is a ligand of DesT (68,69). Since LCFAs metabolism has been shown to play important roles in bacterial virulence, it is necessary to fully understand the regulatory mechanism in P. aeruginosa.
In the EMSA results, at the concentration of 3 μM, the affinity of R136A PvrA for the DNA probe was decreased by the presence of 0.25 μM palmitoylcoenzyme A (Figure 8). However, the SPR results demonstrated that the affinity of R136 PvrA for the DNA was not affected by the palmitoylcoenzyme A (Figure 9). We suspected that the difference might be due to the differences in the sensitivities and detection limits between the two assay methods. In the SPR assay, the protein concentrations were 62.5–350 nM, which were much lower than those used in the EMSA. Thus the affinity of R136A PvrA at 3 μM for the DNA probe was out of the range of the SPR assay. In addition, the difference of the reaction buffers might also contribute to the difference.
In this study, we examined the roles of the PvrA regulated genes in the bacterial utilization of PC, fatty acids and virulence in a murine acute pneumonia model. Mutation of plcH resulted in growth defect when PC was the sole carbon source (PC-M9, Supplementary Figure S5A), indicating an essential role of PlcH in the utilization of PC (15). Fatty acyl-CoA synthetase (FadD) and acyl-CoA dehydrogenase (FadE) are the key enzymes in the bacterial utilization of fatty acids. P. aeruginosa harbours six fadD and three fadE homologs, namely fadD1-6 and PA0506, PA0507 and PA0508, respectively (14,25–27). Previous studies revealed that deletion of the individual fadD homologous gene did not affect the bacterial utilization of fatty acids (26). Simultaneous deletion of fadD1 and fadD2 reduced the bacterial growth when fatty acids were the sole carbon source, however, deletion of fadD6 in the ΔfadD1ΔfadD2 mutant did not further reduce the bacterial growth (26). These results indicate that FadD1 and FadD2 play major roles in the bacterial utilization of fatty acids (26). In this study, we found that PvrA directly controls the expression fadD1, fadD6 and PA0508. Consistent with previous reports, deletion of fadD1 or fadD6 did not affect the bacterial growth in FA-M9. Deletion of PA0508 did not affect the bacterial growth in FA-M9 either, presumably due to the existence of the redundant acyl-CoA dehydrogenases, e.g. PA0506 and PA0507. Simultaneous deletion of fadD1, fadD6 and PA0508 (ΔfadD1ΔfadD6ΔPA0508) did not affect the bacterial growth in PC-M9 (Supplementary Figure S5A), indicating that the strain can at least utilize glycerol and choline derived from PC to support its growth. In FA-M9, theΔfadD1ΔfadD2ΔPA0508 mutant displayed a slight growth defect (Supplementary Figure S6), indicating that the other Fatty acyl-CoA synthetases and acyl-CoA dehydrogenases can support the bacterial utilization of palmitic acid in vitro.
In the acute pneumonia model, the bacterial load of the ΔplcH mutant was similar to the wild type PA14 (Figure 11), indicating that carbon sources other than PC could support the growth of the bacteria in lung. For example, fatty acids derived from phosphoethanolamine or released from dead cells can be utilized by P. aeruginosa (70–73). No significant attenuation was found in the ΔfadD1ΔfadD6ΔPA0508 mutant (Figure 11). We suspected that the glycerol and choline derived from PC and other carbon sources might be sufficient to support the bacterial growth. However, the quadruple mutant (ΔplcHΔfadD1ΔfadD6ΔPA0508) displayed attenuated virulence in the acute pneumonia model (Figure 11). Since PlcH plays a critical role in the bacterial utilization of PC, it is likely that the quadruple mutant cannot utilize the PC in vivo. In addition, deletion of fadD1, fadD6 and PA0508 might impair the bacterial utilization of fatty acids in vivo from sources other than PC, presumably due to the higher enzyme affinity of FadD1, FadD6 and PA0508 to adapt to the special fatty acids environment in the lung than the other enzymes involved in the fatty acids utilization. Thus, the combination of the defective utilization of PC and fatty acids in the lung might result in the attenuated virulence of the ΔplcHΔfadD1ΔfadD6ΔPA0508 mutant. PvrA also regulates the malate synthase gene glcB. Previous studies demonstrated that GlcB plays a key role in bacterial utilization of fatty acids and mutation of glcB reduced the virulence of P. aeruginosa (46,49,74). In combination, these results indicated that PvrA might contribute to the bacterial virulence by controlling the genes involved in PC and fatty acids utilization.
Currently, we are taking efforts to identify additional genes regulated by the PvrA to fully understand the role of PvrA in the bacterial metabolism and virulence. In our ChIP-seq results, PvrA was found to bind to the promoter region of exsA, which encodes the master regulator of the type III secretion system (T3SS) (Supplementary Table S4). Several T3SS genes were found to be upregulated in the BALF (Supplementary Table S3). Further studies are needed to test whether PvrA regulates the T3SS in response to a host signal, e.g. palmitoylcoenzyme A from the utilization of host fatty acids.
Previously, PvrA was shown to be required for the swarming motility of P. aeruginosa (42). In our ChIP-seq assay, genes related to the type IV pili were differentially influenced by the PvrA. However, the twitching motilities were similar between wild type PA14 and the pvrA mutant (42). Further studies are needed to understand the mechanism of PvrA mediated influence on the swarming motility.
Previously, Damron et al. performed a dual-RNA-seq to analyse the transcriptomes of P. aeruginosa and the host in an acute murine pneumonia model 16 hpi (75). Recently, Cornforth et al. compared P. aeruginosa transcriptome during infections of human soft-tissue wounds and CF lungs with that of in vitro laboratory cultures (61). Genes related to iron acquisition were found to be up regulated during those infections, which is consistent with our results, suggesting important roles of these genes during infection. However, genes involved in PC utilization were not up regulated in those studies. We suspect that the difference might be due to different infection stages and sites, e.g. 6 and 16 hpi in the acute pneumonia model as well as mouse lungs versus human soft tissues and CF sputa. Deep analysis of these data might reveal genes essential for P. aeruginosa pathogenesis in different infection scenarios.
Based on our results, we summarize a regulatory network mediated by the PvrA as shown in Figure 12. Since genes regulated by the PvrA play roles in both nutrient utilization and suppression of host immune responses, PvrA might be one of the hubs of the regulatory network that orchestrate gene expression during host infections.
Figure 12.

Proposed model of PvrA mediated regulatory pathways. During infection, PC is degraded by lipases and PlcH, resulting in glycerol, choline and fatty acids. Glycine betaine (GB) and dimethylglycine (DMG) derived from choline are sensed by GbdR, which directly upregulates plcH (57–59). The fatty acids were converted into fatty acyl-CoA. PvrA binds to the fatty acyl-CoA and activates the expression of plcH, fadD1, fadD6, PA0508, aprA, maeB, and glcB. FadD1, FadD6 and PA0508 are key enzymes in the β-oxidation pathway through which a long-chain acyl-CoA molecule is broken down to acetyl-CoA molecules. The las quorum sensing system regulates the expression of aprA (76,77). AprA degrades self flagellin and host complement component C2 to evade recognition by the immune system (50,51). MaeB and GlcB are key enzymes in the glyoxylate shunt which plays an important role in utilizing fatty acids in bacteria (61).
DATA AVAILABILITY
The RNA-seq data have been deposited in the NCBI Short Read Archive (SRA) database with an accession number SRP136746.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Project of China [2017YFE0125600]; National Science Foundation of China [31900115, 31670130, 81670766, 31870130, 31600110]. Funding for open access charge: National Key Research and Development Project of China [2017YFE0125600].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lyczak J.B., Cannon C.L., Pier G.B.. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect. 2000; 2:1051–1060. [DOI] [PubMed] [Google Scholar]
- 2. Skaar E.P., Madhani H.D.. The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 2010; 6:e1000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balasubramanian D., Schneper L., Kumari H., Mathee K.. A dynamic and intricate regulatory network determines Pseudomonas aeruginosa virulence. Nucleic Acids Res. 2013; 41:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang H., Shao X., Xie Y., Wang T., Zhang Y., Wang X., Deng X.. An integrated genomic regulatory network of virulence-related transcriptional factors in Pseudomonas aeruginosa. Nat. Commun. 2019; 10:2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Winsor G.L., Griffiths E.J., Raymond L., Dhillon B.K., Shay J.A., Brinkman F.S.L.. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 2016; 44:D646–D653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mesaros N., Nordmann P., Plesiat P., Roussel-Delvallez M., Van-Eldere J., Glupczynski Y., Malfroot A.. Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. Clin. Microbiol. Infect. 2010; 13:560–578. [DOI] [PubMed] [Google Scholar]
- 7. Smith J.J., Travis S.M., Greenberg E.P., Welsh M.J.. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996; 85:229–236. [DOI] [PubMed] [Google Scholar]
- 8. Goldman M.J., Anderson G.M., Stolzenberg E.D., Kari U.P., Zasloff M., Wilson J.M.. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell. 1997; 88:553–560. [DOI] [PubMed] [Google Scholar]
- 9. Hauser A.R. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 2009; 7:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diaz M.H., Hauser A.R.. Pseudomonas aeruginosa cytotoxin ExoU is injected into phagocytic cells during acute pneumonia. Infect. Immun. 2010; 78:1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Finckbarbançon V., Goranson J., Zhu L., Sawa T., Wienerkronish J.P., Fleiszig S.M., Wu C., Mendemueller L., Frank D.W.. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol. Microbiol. 1997; 25:547. [DOI] [PubMed] [Google Scholar]
- 12. Sun Y., Karmakar M., Taylor P.R., Rietsch A., Pearlman E.. ExoS and ExoT ADP-ribosyltransferase activities mediate Pseudomonas aeruginosa keratitis by promoting neutrophil apoptosis and bacterial survival. J. Immunol. 2012; 188:1884–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bernhard W., Wang J.-Y., Tschernig T., Tummler B., Hedrich H.J., von der Hardt H.. Lung surfactant in a cystic fibrosis animal model: increased alveolar phospholipid pool size without altered composition and surface tension function in cftrm1HGU/m1HGU mice. Thorax. 1997; 52:723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Son M.S. Jr, Matthews W.J., Kang Y., Nguyen D.T., Hoang T.T.. In vivo evidence of Pseudomonas aeruginosa nutrient acquisition and pathogenesis in the lungs of cystic fibrosis patients. Infection and Immunity. 2007; 75:5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun Z., Kang Y., Norris M.H., Troyer R.M., Son M.S., Schweizer H.P., Dow S.W., Hoang T.T.. Blocking phosphatidylcholine utilization in Pseudomonas aeruginosa, via mutagenesis of fatty acid, glycerol and choline degradation pathways, confirms the importance of this nutrient source in vivo. PLoS One. 2014; 9:e103778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fujita Y., Matsuoka H., Hirooka K.. Regulation of fatty acid metabolism in bacteria. Mol. Microbiol. 2007; 66:829–839. [DOI] [PubMed] [Google Scholar]
- 17. Magnuson K., Jackowski S., Rock C.O., Cronan J.E. Jr. Regulation of fatty acid biosynthesis in Escherichia coli. Microbiol. Rev. 1993; 57:522–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cronan J.E., Jr In vivo evidence that acyl coenzyme A regulates DNA binding by the Escherichia coli FadR global transcription factor. J. Bacteriol. 1997; 179:1819–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Raman N., Black P.N., DiRusso C.C.. Characterization of the fatty acid-responsive transcription factor FadR. Biochemical and genetic analyses of the native conformation and functional domains. J. Biol. Chem. 1997; 272:30645–30650. [DOI] [PubMed] [Google Scholar]
- 20. van Aalten D.M., DiRusso C.C., Knudsen J., Wierenga R.K.. Crystal structure of FadR, a fatty acid-responsive transcription factor with a novel acyl coenzyme A-binding fold. EMBO J. 2000; 19:5167–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chatterjee A., Dutta P.K., Chowdhury R.. Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect. Immun. 2007; 75:1946–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lowden M.J., Skorupski K., Pellegrini M., Chiorazzo M.G., Taylor R.K., Jon Kull F.. Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:2860–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spector M.P., DiRusso C.C., Pallen M.J., Garcia del Portillo F., Dougan G., Finlay B.B.. The medium-/long-chain fatty acyl-CoA dehydrogenase (fadF) gene of Salmonella typhimurium is a phase 1 starvation-stress response (SSR) locus. Microbiology. 1999; 145:15–31. [DOI] [PubMed] [Google Scholar]
- 24. Pifer R., Russell R.M., Kumar A., Curtis M.M., Sperandio V.. Redox, amino acid, and fatty acid metabolism intersect with bacterial virulence in the gut. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E10712–E10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kang Y., Zarzycki-Siek J., Walton C.B., Norris M.H., Hoang T.T.. Multiple FadD acyl-CoA synthetases contribute to differential fatty acid degradation and virulence in Pseudomonas aeruginosa. PLoS One. 2010; 5:e13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zarzycki-Siek J., Norris M.H., Kang Y., Sun Z., Bluhm A.P., McMillan I.A., Hoang T.T.. Elucidating the Pseudomonas aeruginosa fatty acid degradation pathway: identification of additional fatty acyl-CoA synthetase homologues. PLoS One. 2013; 8:e64554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Son M.S., Nguyen D.T., Kang Y., Hoang T.T.. Engineering of FRT–lacZ fusion constructs: induction of the Pseudomonas aeruginosa fadBA1 operon by medium and long chain-length fatty acids. Plasmid. 2008; 59:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kang Y., Nguyen D.T., Son M.S., Hoang T.T.. The Pseudomonas aeruginosa PsrA responds to long-chain fatty acid signals to regulate the fadBA5 β-oxidation operon. Microbiology. 2008; 154:1584–1598. [DOI] [PubMed] [Google Scholar]
- 29. Kang Y., Lunin V.V., Skarina T., Savchenko A., Schurr M.J., Hoang T.T.. The long-chain fatty acid sensor, PsrA, modulates the expression of rpoS and the type III secretion exsCEBA operon in Pseudomonas aeruginosa. Mol. Microbiol. 2010; 73:120–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sun Z., Shi J., Liu C., Jin Y., Li K., Chen R., Jin S., Wu W.. PrtR homeostasis contributes to Pseudomonas aeruginosa pathogenesis and resistance against ciprofloxacin. Infect. Immun. 2014; 82:1638–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu W., Huang J., Duan B., Traficante D.C., Hong H., Risech M., Lory S., Priebe G.P.. Th17-stimulating protein vaccines confer protection against Pseudomonas aeruginosa Pneumonia. Am J Respir Crit Care Med. 2012; 186:420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L. et al.. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009; 55:611–622. [DOI] [PubMed] [Google Scholar]
- 33. Wilbanks E.G., Larsen D.J., Neches R.Y., Yao A.I., Wu C.Y., Kjolby R.A.S., Facciotti M.T.. A workflow for genome-wide mapping of archaeal transcription factors with ChIP-seq. Nucleic Acids Res. 2012; 40:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bush M.J., Chandra G., Bibb M.J., Findlay K.C., Buttner M.J.. Genome-wide chromatin immunoprecipitation sequencing analysis shows that whib is a transcription factor that cocontrols its regulon with whia to initiate developmental cell division in streptomyces. Mbio. 2016; 7:e00523-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P. et al.. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012; 22:1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rangel S.M., Diaz M.H., Knoten C.A., Zhang A., Hauser A.R.. The role of ExoS in dissemination of Pseudomonas aeruginosa during pneumonia. PLoS Pathog. 2016; 11:e1004945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Priebe G.P., Walsh R.L., Cederroth T.A., Kamei A., Coutinhosledge Y.S., Goldberg J.B., Pier G.B.. IL-17 is a critical component of vaccine-induced protection against lung infection by lipopolysaccharide-heterologous strains of Pseudomonas aeruginosa. J. Immunol. 2008; 181:4965–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sawa T., Yahr T.L., Ohara M., Kurahashi K., Gropper M.A., Wienerkronish J.P., Frank D.W.. Active and passive immunization with the Pseudomonas V antigenprotects against type III intoxication and lung injury. Nat. Med. 1999; 5:392. [DOI] [PubMed] [Google Scholar]
- 39. Smith R.S., Wolfgang M.C., Lory S.. An adenylate cyclase-controlled signaling network regulates Pseudomonas aeruginosa virulence in a mouse model of acute pneumonia. Infect. Immun. 2004; 72:1677–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Michel L., González N., Jagdeep S., Nguyenngoc T., Reimmann C.. PchR-box recognition by the AraC-type regulator PchR of Pseudomonas aeruginosa requires the siderophore pyochelin as an effector. Mol. Microbiol. 2005; 58:495–509. [DOI] [PubMed] [Google Scholar]
- 41. Neidig A., Yeung A.T., Rosay T., Tettmann B., Strempel N., Rueger M., Lesouhaitier O., Overhage J.. TypA is involved in virulence, antimicrobial resistance and biofilm formation in Pseudomonas aeruginosa. BMC Microbiol. 2013; 13:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yeung A.T.Y., Torfs E.C.W., Jamshidi F., Bains M., Wiegand I., Hancock R.E.W., Overhage J.. Swarming of Pseudomonas aeruginosa is controlled by a broad spectrum of transcriptional regulators, including MetR. J. Bacteriol. 2009; 191:5592–5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Deng X., Li M., Pan X., Zheng R., Liu C., Chen F., Liu X., Cheng Z., Jin S., Wu W.. Fis regulates type III secretion system by influencing the transcription of exsA in Pseudomonas aeruginosa Strain PA14. Front. Microbiol. 2017; 8:669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liberati N.T., Urbach J.M., Miyata S., Lee D.G., Drenkard E., Wu G., Villanueva J., Wei T., Ausubel F.M.. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:2833–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wells G., Palethorpe S., Pesci E.C.. PsrA controls the synthesis of the Pseudomonas aeruginosa quinolone signal via repression of the FadE homolog, PA0506. PLoS One. 2017; 12:e0189331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miller R.M., Tomaras A.P., Barker A.P., Voelker D.R., Chan E.D., Vasil A.I., Vasil M.L.. Pseudomonas aeruginosa twitching motility-mediated chemotaxis towards phospholipids and fatty acids: specificity and metabolic requirements. J. Bacteriol. 2008; 190:4038–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chang G.G., Tong L.. Structure and function of malic enzymes, a new class of oxidative decarboxylases. Biochemistry. 2003; 42:12721–12733. [DOI] [PubMed] [Google Scholar]
- 48. Crousilles A., Dolan S.K., Brear P., Chirgadze D.Y., Welch M.. Gluconeogenic precursor availability regulates flux through the glyoxylate shunt in Pseudomonas aeruginosa. J. Biol. Chem. 2018; 293:14260–14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maloy S.R., Bohlander M., Nunn W.D.. Elevated levels of glyoxylate shunt enzymes in Escherichia coli strains constitutive for fatty acid degradation. J. Bacteriol. 1980; 143:720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Laarman A.J., Bardoel B.W., Ruyken M., Fernie J., Milder F.J., van Strijp J.A., Rooijakkers S.H.. Pseudomonas aeruginosa alkaline protease blocks complement activation via the classical and lectin pathways. J. Immunol. 2012; 188:386. [DOI] [PubMed] [Google Scholar]
- 51. Casilag F., Lorenz A., Krüger J., Klawonn F., Weiss S., Häussler S.. The LasB elastase of Pseudomonas aeruginosa acts in concert with alkaline protease AprA to prevent flagellin-mediated immune recognition. Infect. Immun. 2015; 84:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kida Y., Shimizu T., Kuwano K.. Cooperation between LepA and PlcH contributes to the in vivo virulence and growth of Pseudomonas aeruginosa in mice. Infect. Immun. 2011; 79:211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ostroff R.M., Wretlind B., Vasil M.L.. Mutations in the hemolytic-phospholipase C operon result in decreased virulence of Pseudomonas aeruginosa PAO1 grown under phosphate-limiting conditions. Infect. Immun. 1989; 57:1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wargo M.J., Ho T.C., Gross M.J., Whittaker L.A., Hogan D.A.. GbdR regulates Pseudomonas aeruginosa plcH and pchP transcription in response to choline catabolites. Infect. Immun. 2009; 77:1103–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hampel K.J., Labauve A.E., Meadows J.A., Fitzsimmons L.F., Nock A.M., Wargo M.J.. Characterization of the GbdR regulon in Pseudomonas aeruginosa. J. Bacteriol. 2014; 196:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wargo M.J. Choline catabolism to glycine betaine contributes to Pseudomonas aeruginosa survival during murine lung infection. PLoS One. 2013; 8:e56850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shortridge V.D., Lazdunski A., Vasil M.L.. Osmoprotectants and phosphate regulate expression of phospholipase C in Pseudomonas aeruginosa. Mol. Microbiol. 1992; 6:863–871. [DOI] [PubMed] [Google Scholar]
- 58. Jackson A.A., Daniels E.F., Hammond J.H., Willger S.D., Hogan D.A.. Global regulator Anr represses PlcH phospholipase activity in Pseudomonas aeruginosa when oxygen is limiting. Microbiology. 2014; 160:2215–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Castang S., Mcmanus H.R., Turner K.H., Dove S.L.. H-NS family members function coordinately in an opportunistic pathogen. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:18947–18952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. He J., Baldini R.L., Deziel E., Saucier M., Zhang Q., Liberati N.T., Lee D., Urbach J., Goodman H.M., Rahme L.G.. The broad host range pathogen Pseudomonas aeruginosa strain PA14 carries two pathogenicity islands harboring plant and animal virulence genes. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:2530–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cornforth D.M., Dees J.L., Ibberson C.B., Huse H.K., Mathiesen I.H., Kirketerp-Moller K., Wolcott R.D., Rumbaugh K.P., Bjarnsholt T., Whiteley M.. Pseudomonas aeruginosa transcriptome during human infection. Proc. Natl. Acad. Sci. U.S.A. 2018; 29:201717525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shi W., Kovacikova G., Lin W., Taylor R.K., Skorupski K., Kull F.J.. The 40-residue insertion in Vibrio cholerae FadR facilitates binding of an additional fatty acyl-CoA ligand. Nat. Commun. 2015; 6:6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang K., Sybers D., Maklad H.R., Lemmens L., Lewyllie C., Zhou X., Peeters E.. A TetR-family transcription factor regulates fatty acid metabolism in the archaeal model organism Sulfolobus acidocaldarius. Nat. Commun. 2019; 10:1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yeo H.K., Park Y.W., Lee J.Y.. Structural basis of operator sites recognition and effector binding in the TetR family transcription regulator FadR. Nucleic Acids Res. 2017; 45:4244–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang J., Zhang Y.. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015; 43:W174–W181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y.. The I-TASSER suite: protein structure and function prediction. Nat. Methods. 2015; 12:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang Y.M., Zhu K., Frank M.W., Rock C.O.. A pseudomonas aeruginosa, transcription factor that senses fatty acid structure. Mol. Microbiol. 2007; 66:622–632. [DOI] [PubMed] [Google Scholar]
- 68. Miller D.J., Zhang Y.M., Subramanian C., Rock C.O., White SW.. Structural basis for the transcriptional regulation of membrane lipid homeostasis. Nat. Struct. Mol. Biol. 2010; 17:971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhu K., Choi K.H., Schweizer H.P., Rock C.O., Zhang Y.M.. Two aerobic pathways for the formation of unsaturated fatty acids in pseudomonas aeruginosa. Mol. Microbiol. 2010; 60:260–273. [DOI] [PubMed] [Google Scholar]
- 70. Hurley B.P., Williams N.L., McCormick B.A.. Involvement of phospholipase A2 in Pseudomonas aeruginosa-mediated PMN transepithelial migration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006; 290:L703–L709. [DOI] [PubMed] [Google Scholar]
- 71. Tamang D.L., Pirzai W., Priebe G.P., Traficante D.C., Pier G.B., Falck J.R., Morisseau C., Hammock B.D., McCormick B.A., Gronert K. et al.. Hepoxilin A(3) facilitates neutrophilic breach of lipoxygenase-expressing airway epithelial barriers. J. Immunol. 2012; 189:4960–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dar H.H., Tyurina Y.Y., Mikulska-Ruminska K., Shrivastava I., Ting H.-C., Tyurin V.A., Krieger J., Croix C.M. St, Watkins S., Bayir E. et al.. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J. Clin. Invest. 2018; 128:4639–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kearns D.B., Robinson J., Shimkets L.J.. Pseudomonas aeruginosa exhibits directed twitching motility up phosphatidylethanolamine gradients. J. Bacteriol. 2001; 183:763–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hagins J.M., Scoffield J., Suh SJ., Silo-Suh L.. Malate synthase expression is deregulated in the Pseudomonas aeruginosa cystic fibrosis isolate FRD1. Can J Microbiol. 2011; 57:186–195. [DOI] [PubMed] [Google Scholar]
- 75. Damron F.H., Oglesbysherrouse A.G., Wilks A., Barbier M.. Dual-seq transcriptomics reveals the battle for iron during Pseudomonas aeruginosa acute murine pneumonia. Sci. Rep. 2016; 6:39172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gambello M.J., Kaye S., Iglewski B.H.. LasR of Pseudomonas aeruginosa is a transcriptional activator of the alkaline protease gene (apr) and an enhancer of exotoxin A expression. Infect. Immun. 1993; 61:1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schuster M., Lostroh C.P., Ogi T., Greenberg E.P.. Identification, timing, and signal specificity of Pseudomonas aeruginosa Quorum-controlled genes: a transcriptome analysis. J. Bacteriol. 2003; 185:2066–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data have been deposited in the NCBI Short Read Archive (SRA) database with an accession number SRP136746.