


Abstract
The ability to block gene expression in bacteria with the catalytically inactive mutant of Cas9, known as dCas9, is quickly becoming a standard methodology to probe gene function, perform high-throughput screens, and engineer cells for desired purposes. Yet, we still lack a good understanding of the design rules that determine on-target activity for dCas9. Taking advantage of high-throughput screening data, we fit a model to predict the ability of dCas9 to block the RNA polymerase based on the target sequence, and validate its performance on independently generated datasets. We further design a novel genome wide guide RNA library for E. coli MG1655, EcoWG1, using our model to choose guides with high activity while avoiding guides which might be toxic or have off-target effects. A screen performed using the EcoWG1 library during growth in rich medium improved upon previously published screens, demonstrating that very good performances can be attained using only a small number of well designed guides. Being able to design effective, smaller libraries will help make CRISPRi screens even easier to perform and more cost-effective. Our model and materials are available to the community through crispr.pasteur.fr and Addgene.
INTRODUCTION
In bacteria, the catalytically dead variant of Cas9 (dCas9) can bind to DNA strongly enough to block transcription initiation and transcription elongation (1,2). Guide RNAs can be easily reprogrammed to direct dCas9 to any position of interest with a protospacer adjacent motif (PAM), which in the case of the widely used S. pyogenes Cas9 is a simple 5′-NGG-3′ downstream of the target (3–5). While directing dCas9 to either strand of DNA effectively blocks transcription initiation, binding of the guide RNA to the non-template strand (coding strand) is necessary to efficiently block the running RNA polymerase (RNAP) (1,2). This technique to block gene expression is known as CRISPR interference (CRISPRi) and has already been used in a wide range of bacterial species (6,7). High-throughput CRISPRi screens have led to the better characterisation of essential genes, the understanding drugs’ mode of action and the identification of bacteriophage host factors (8–11). Libraries of up to ∼105 guide RNAs can be easily constructed through on-chip oligonucleotide synthesis (12). The guide RNA sequences direct dCas9 binding and are used in the library context as barcodes to measure the abundance of each sgRNA in a mixed culture through next-generation sequencing. While CRISPRi screens are akin to transposon-based high throughput methods such as Tn-seq or TraDIS (13), or to the study of deletion strain libraries such as the KEIO collection (14), they present several notable advantages. The expression of dCas9 can be inducible, enabling the study of essential genes which cannot be deleted and are lost in transposon based methods. The repression level of the target gene can be fine-tuned by playing with the level of complementarity between the guide and the target (2,15). The ability to rationally design the guide library allows targeting any desired set of genes, including small ones that might be missed by transposon insertion screens. Finally, CRISPRi enables to perform whole genome screens with a relatively small library size compared to the high density of transposon insertions required to achieve comparable results (8,9).
In a recent study, we performed a pooled genome-wide screen with ∼92 000 different guide RNAs targeting random positions along the chromosome of E. coli MG1655 (12). This screen revealed important design rules for conducting dCas9 mediated knockdowns in E. coli. In particular, optimizing the expression level of dCas9 is important to avoid a mysterious toxicity phenomenon that occurs when using guide RNAs that share specific five bases seed sequences (referred to as ‘bad seeds’). This observation led us to construct E. coli strain LC-E75, a MG1655 derivative carrying dCas9 under the control of a Ptet promoter integrated at the phage 186 attB site (12). In this strain, the ribosome binding site of dCas9 was optimized to enable strong on-target repression while limiting toxicity and off-target effects. While using strain LC-E75 improved the consistency of the results as compared to a strain where dCas9 expression was not optimized, we could still observe an important variability in the effect of guide RNAs that target within the same essential genes (Figure 1A).
Figure 1.

A linear model trained on screening data predicts guide activity. (A) High variability in the effect of guides (log2FC) targeting the essential gene secA. The log2FC of guides is plotted along the position on the chromosome of E. coli MG1655 (NC_000913.3). (B) A linear (L1) model was trained to predict the activity of guides based on the target sequence. The sequence logo reflects the coefficient of each base in the model, drawn using logomaker (29). Positive values indicate a positive effect of the base on dCas9 activity. Note that the GG of the PAM are not fitted by the model and are displayed with an arbitrary size for ease of reading. Positions 15–20 refer to the last six bases of the target sequence. Positions +1 to +16 refer to positions after the PAM. (C) The activity of 32 guides targeting lacZ was measured in a Miller assay. The log10 of the repression fold is plotted versus the predicted guide activity. (D, E) The activity of 33 guides targeting sfGFP was measured through FACS-seq by Hawkins et al. (16). The measured guide activity is plotted against the activity predicted by the model. The R2 and Pearson R values are indicated on the plots.
Here, by using the differences between the effects of guides targeting essential genes as a measure of guide activity, we fit a linear model able to predict dCas9’s ability to block the RNA polymerase. This model was validated on a newly generated dataset of 32 guides blocking the expression of lacZ in E. coli as well as on data generated by Hawkins et al. on a set of guides targeting sfGFP both in E. coli and Bacillus subtilis (16). We further design and test a novel genome wide library for E. coli MG1655 (EcoWG1), in which guides were selected according to our model predictions and rules to avoid bad seeds and off-targets.
MATERIALS AND METHODS
Model building and training
Starting with the 92 000 guides of the Cui et al. dataset, we filtered the data to keep guides targeting the 247 genes targeted by five guides or more and with a median log2FC smaller than –2. The resulting 2765 guides were used to fit our model. We first fitted a simple linear model with L1 regularization using 10-fold cross-validation to select alpha, and using as features the one-hot-encoded primary sequence data of the target and the surrounding 20 nt on each side. L1 models include a regularization term that pushes coefficients to zero and are commonly used in feature selection. The coefficients of this model highlighted the importance of the seed sequence of the guide, of the N in the NGG PAM, and interestingly of the sequence downstream of the PAM (Supplementary Figure S1). We then sought to determine the best sequence range to consider in our model. Starting from the three positions to which the L1 model gives the most weight as the only features (Supplementary Figure S1), we progressively increased the sequence range provided to the model in a stepwise manner using 10-fold cross-validation. The final sequence range we selected includes the last six bases of the guide, the N of the NGG PAM and the following 16 bases (Figure 1B).
Library design
For each guide we computed the number of perfectly matched targets in the genome (ntargets), the number of off-targets with a perfect identity to the seed sequence of x nucleotides (noff_x), the number of off-targets on the non-template strand of genes with a perfect identity to the seed sequence of x nucleotides (noff_x_gene), the number of off-targets in promoter regions with a perfect identity to the seed sequence of x nucleotides (noff_x_prom), the presence or absence of a bad seed sequence (badseed), whether the guide is positioned in the first or second half of the gene (second_half), the score quartile predicted by our model and finally the score itself. The bad seeds considered here are the 30 worst seed sequences identified in strain LC-E18 in our previous study (12). Only off-target positions with an NGG PAM were considered. To account for the poor annotation of transcription initiation sites, and to ensure that our strategy can be applied to genomes where the position of transcription start sites is not available, we used as a proxy for promoters all sequences located with –100 and +20 bases of gene starts.
Using insights from our previous publications, we arbitrarily ranked guides in each gene sequentially by criteria in the following order: ntargets, noff_12, noff_11_gene, noff_9_prom, badseed, score_quartile, second_half, score. Note that only the last criteria is a continuous variable, making it possible to perform this sequential sorting. Finally, we selected the five best guides for each gene. Note that the activity prediction model provided on crispr.pasteur.fr was slightly improved compared to the one used to generate the EcoWG1 library and cannot be used to generate the exact same library.
Library cloning
Guides selected by our sorting strategy were ordered for synthesis (from Twist Bioscience) in the following form: 5′-TAGCTCAGTCCTAGGTATAATACTAGT-(guide sequence)-GTTTTAGAGCTAGAAATAGCAAGTTAA-3′
A two-step PCR protocol was used to amplify the ssDNA oligo pool. First, the complementary strand was synthesized using 2.5 pmol of primer LC297 in a reaction volume of 30 ul, using the KAPA HiFi PCR kit with the following protocol: initial denaturation, 80 sec at 98°C, followed by extension for 120 sec at 72°C. The second step consists of a PCR amplification of the above extension product with the following protocol: initial denaturation, 60 sec at 95°C, followed by 6 cycles of denaturation, annealing and extension, 20 sec at 98°C, 15 sec at 60°C and 20 sec at 72°C respectively, and a final extension for 5 min at 72°C. For this second step, 100 pmol of primers LC296 and LC297 were added to the extension reaction, to a final volume of 50 uL of reaction using the KAPA HiFi PCR kit. The final PCR product was gel purified followed by Gibson assembly with the psgRNA (Addgene #114005) backbone amplified with primers LC293 and LC294 (17). The Gibson Assembly product was electroporated (1.8 kV, 25 uF, 200 Ω) into E. coli MG1655 using 1 mm cuvettes. The cells were recovered for 1 h in a shaking incubator at 37°C, and then spread on 12 cm × 12 cm square agar plates (LB supplemented with 50 ug/ml Kanamycin). Plates were incubated for 4 h at 37°C, after which the cells were pooled together by washing off the cells from the plates. Miniprep extraction was performed on the pooled cells.
The resulting psgRNA_EcoWG1 library (addgene #131625) was then introduced into strain LC-E75 by electroporation: 20 ul of LC-E75 electrocompetent cells were mixed with 50 ng of psgRNA-EcoWG1 and electroporated using 1 mm electroporation cuvettes (1.8 kV, 25 uF, 200 Ω). After the pulse, cells were recovered in 980 ul of LB in a shaking incubator at 37°C for 1 h, followed by spreading on 12 cm × 12 cm square agar plates (LB supplemented with 50 ug/ml kanamycin). The plates were incubated for 4 h at 37°C, after which the cells were pooled together by washing off the cells from the plates using LB medium. The pool of cells was aliquoted and stored using a final glycerol concentration of 20% at –80°C. The sequence of the primers used for library construction are listed in Supplementary Table S1.
CRISPR screen
A frozen tube of LC-E75 cells carrying the psgRNA_EcoWG1 library was thawed. The cells were diluted 1:100 into 100 ml of LB medium without antibiotics, and grown in a shaking incubator at 37°C until OD600 reached 0.2. At that point, a 50 ml sample of culture was taken as time-point 0. Simultaneously, 3 ml of culture were added to 12 ml of fresh LB, and the 15 ml were plated on 15, 12 cm × 12 cm square LB agar plates supplemented with 1 uM aTc. The plates were incubated at 37°C for 3 h, after which the cells from five plates were washed off and pooled together as time-point 1 (3H). The remaining plates were incubated at 37°C for an additional 3 h, after which the cells from five plates were washed off and pooled together as time-point 2 (6H). The remaining plates were incubated at 37°C for a further 18 h, after which they were all washed off and pooled together as time-point 3 (24H). A sample from time-point 3 was diluted to an OD600 of 0.2 in a final volume of 1 ml. The volume was then increased by the addition of 4 ml of LB to enable plating on 15, 12 cm × 12 cm square agar plates (LB supplemented with 1 uM aTc). After 6 h of incubation at 37°C for 6 h, cells from five plates were washed off and pooled together as time-point 4 (24+6H). The whole experiment was performed twice.
Illumina sequencing
We used here the customized Illumina sequencing method previously described in (12). First, plasmids were extracted at different time points immediately after collecting the cells by performing a miniprep. Then, a first PCR was performed to amplify the guide RNA using different indexes for each sample (Supplementary Table S2). The product of this PCR was purified by running the sample in a 2% agarose gel (120 V for 65 min) followed by extraction of the band. A second PCR was performed to add both a second index to each sample (Supplementary Table S3) and the Illumina attachment sequences. The PCR products were purified through gel extraction once again using the same protocol, after which ∼200 ng of each sample (measured using a Nanodrop 2000c) were pooled together, followed by a last gel extraction.
The final concentration of the pooled and purified samples was measured using the KAPA Library Quantification Kit (Illumina). Sequencing was then performed using primer LC609 as custom read 1 primer. We also used custom index primers: LC499 was used to read index 1, and LC610 was used to read index 2 (Supplementary Table S4). Sequencing was performed on a NextSeq 500 benchtop sequencer. The first two bases read by primer LC609 are the same for all clusters. To avoid low diversity issues, the first 2 cycles were set as dark cycles thanks to a custom sequencing program which can be provided by Illumina upon request. We then performed an additional 20 normal sequencing cycles to read the guide RNA sequence.
Data processing
More than 107 reads were obtained for each sample, with the exception of one of the repeats of the last time point (24+6H) for which we only obtained 7 × 105 reads. Read counts were normalized by the total number of reads obtained for each sample. Log2 fold change (log2FC) was computed between each time point and the time point 0 of the experiment on the read counts (+1 to avoid computing the log of 0). Guides for which fewer than 20 reads were obtained at time point 0 were removed from the analysis. The log2FC obtained for each time point were very strongly correlated between replicates, with a slightly lower correlation between the two replicates of the last time point due to the lower amount of data obtained for one of them (Pearson-r of 0.98, 0.99, 0.99 and 0.95 for the four time points respectively). As a consequence we used for each guide the average log2FC of the two biological replicates, with the exception of the last time point for which we only consider the replicate for which more than 107 reads were obtained. The read counts for each guide in the library are provided as supplementary data 1, and log2FC as supplementary data 2.
Miller assays
We designed 32 sgRNAs to target randomly chosen positions on the coding (non-template) strand of LacZ. The only constraints for the choice were: (i) the target should not be located in the first 100 bp of lacZ. Such a target might interfere with transcription initiation rather than transcription elongation. (ii) The target should not be located within the last 100 bp of lacZ to limit the risks of generating a truncated version of LacZ that is still active. (iii) The guide RNA should not have off-targets in the genome of E. coli MG1655. We consider an off-target if there is a perfect match of 9nt or more between the seed region of the guide and either strand of a promoter region (defined as –200 to +50 relative to gene start). We also consider an off-target if there is a perfect match of 11nt between the seed of the guide and the coding strand of any gene of E. coli MG1655.
The 32 guides were cloned in plasmid psgRNA. This arrayed library as well as a control psgRNA carrying a non-target guide RNA were introduced into strain ACE1. ACE1 was built by integrating dCas9 at the primary 186 attB site using plasmid pLC143. Plasmid pLC143 is a derivative of plasmid pOSIP-KO-RBS2-dCas9 described in (12), which carries the weak RBS controlling the expression of dCas9 in strain LC-E75. The difference between ACE1 and LC-E75 is that the later also carries a mCherry reporter integrated at the lambda attB site. Overnight cultures were diluted to an initial OD600 = 0.003 in 1 ml of LB + kanamycin 50 ug/ml in a 96 well deep-well plate (Masterblock 96-well, 2 ml, V-bottom plates by Greiner Bio-one). The cultures were grown in an orbital plate shaker (450 rpm) in the presence of 1 mM IPTG and 1 uM aTc, for 6 h at 37°C. The OD600 was recorded using a plate reader, after which 20 ul of each culture was mixed with 180 ul of Miller assay buffer in a 96-well plate. The Miller assay buffer should be prepared fresh before every use, and is composed of 150 ul TZ8 (100 mM Tris–HCl pH 8, 10 mM KCl and 1 mM MgSO4), 40 ul ONPG (stock at 4 mg/ml in water), 1.9 ul of 2-mercaptoethanol and 0.9 ul of polymyxin B sulphate (stock at 20 mg/ml in water). The 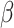 -galactosidase (
-galactosidase (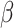 ) activity was measured as the slope of OD420 versus time normalized by the OD600 of the culture before preparing the Miller reaction. Guide activity was estimated as:
) activity was measured as the slope of OD420 versus time normalized by the OD600 of the culture before preparing the Miller reaction. Guide activity was estimated as: 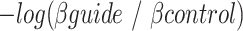 where
where 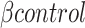 is the
is the 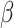 -galactosidase activity of the control non-targeting guide. Guide sequences and results are available as supplementary data 3.
-galactosidase activity of the control non-targeting guide. Guide sequences and results are available as supplementary data 3.
RESULTS
A linear model captures the importance of positions within and downstream the target sequence
We previously performed a genome-wide CRISPRi screen in E. coli MG1655 (12). Using this dataset, we take advantage of the differences in the efficiency of guides that target essential or fitness genes to investigate the sequence determinants of dCas9 activity. During growth of the library in a pool, guide RNAs that block the expression of essential or fitness genes are depleted from the library. The fold-change of each guide is a factor of how strongly the guide blocks the RNA polymerase and the fitness defect produced when the target gene, or operon, is silenced. To take this gene effect into account, we computed as a measure of guide activity the difference between the log2 transformed fold change (log2FC) of a guide and the median log2FC of guides targeting the same gene. Out of the 92 000 guides of the Cui et al. dataset, our filtered dataset contains the 2765 guides targeting the 247 genes with a median log2FC smaller than –2. Models trained to predict Cas9 cleavage activity either in eukaryotic cells or bacteria have no predictive power on this dataset (Spearman R of 0.07 for Doench et al. (18), 0.01 for Moreno-Mateos et al. (19), 0.07 for Guo et al. (20)). We thus investigated the sequence requirements to effectively silence genes with dCas9 in E. coli.
We first sought to investigate the importance of the primary sequence of the target. We determined the best possible sequence range to consider in our model using L1 regression and stepwise feature selection (see Materials and Methods). A simple L1 regression model using as features the last six bases of the guide, the N of the NGG PAM and the following 16 bases was able to predict guide activity with a Pearson R of 36.7 ± 5.4% (10-fold cross-validation average and standard deviation) (Figure 1B). More complex models did not yield any substantial improvement. The most important positions to determine the silencing activity of dCas9 are the last base of the guide where a ‘T or C’ are favored, the ‘N’ of the PAM where ‘C’ is favored, and the base immediately after the PAM where a ‘G’ should be avoided and a ‘C’ favored. Interestingly, bases downstream of the PAM also play a role in dCas9 activity. To effectively block the RNAP, the guide RNA needs to bind the non-template strand. In this orientation, the RNAP will arrive on the PAM side of the target. The sequence downstream of the PAM thus corresponds to the sequence read by the RNA polymerase while bumping into dCas9. Published RNAP immunoprecipitation and sequencing data shows that transcription is interrupted 16 nucleotides before the PAM (1), matching the number of nucleotides selected as features in our model.
Model validation on independent datasets
We then sought to obtain independent experimental validation that our model is able to predict how well guide RNAs can direct dCas9 to block transcription elongation. To this end, we measured how well 32 guides targeting the coding strand of lacZ could block its expression. Residual β-galactosidase activity was measured after 6 h of dCas9 induction through a Miller assay (21). We observed a Pearson correlation coefficient of 0.66 (P-value = 3 × 10−5) between the activity predicted by our model and the guide activity inferred from the Miller assay (Figure 1C). The better performance of our model on this validation dataset than on our training dataset can likely be explained by the fact that a Miller assay is a much cleaner measurement of dCas9 activity than what can be estimated from the log2FC of guides in a pooled CRISPR screen. This result suggests that despite the noisy nature of activity measurements used to fit our model, we were able to capture biologically relevant features that are predictive of dCas9’s ability to block the RNAP. To evaluate the ability of our model to predict on-target activity of guide RNAs not just in E. coli but also in other species we took advantage of a recently published dataset in which the activity of 33 guides targeting sfGFP was measured through FACS-seq in both E. coli and B. subtilis (16). In this dataset the activity of the guides in E. coli correlates with their activity in B. subtilis, indicating that sequence features that determine guide activity are at least partly shared between species (Supplementary Figure S2). Our model was able to predict the activity of these guides with a Pearson R of 0.63 (P-value = 1 × 10−4) and 0.53 (P-value = 2 × 10−3) in E. coli and B. subtilis respectively (Figure 1D, E).
Design rules for guide RNA design
We further established guide RNA design rules to select the best possible guides for targeting a gene of interest. Our rules attempt to select guides whose predicted activity falls in the top quartile while avoiding off-targets and toxic seed sequences. We specifically avoid off-targets with 11 nucleotides of perfect identity or more between the seed sequence and the non-template strand of a gene, as well as off-targets with nine nucleotides of perfect identity or more to promoter regions in either orientation. Only off-targets with an NGG PAM motif are considered. These design choices were made based on off-target effects observed in our previous dataset (12).
Benchmarking the EcoWG1 library
These design rules were used to design the EcoWG1 library, containing ∼20 000 guides targeting the non-template strand of every open reading frame and annotated small RNAs in the chromosome of E. coli MG1655 with five guides per gene. This library was cloned in plasmid psgRNA and introduced into strain LC-E75 by electroporation. The resulting strain library was plated on LB agar with 1 uM aTc for up to 24H, followed by resuspension in LB and replating with aTc for an additional 6H (Figure 2A). The psgRNA plasmid was extracted at different time points (3H, 6H, 24H, 24+6H), the library sequenced and the number of reads for each guide used to compute log2FC (see Methods). The average number of generations performed was estimated to be ∼6 at 3H, ∼11 at 6H, ∼14 at 24H and ∼25 at 24+6H. Gene scores were estimated as the median log2FC of guides targeting the gene.
Figure 2.

Performance of the EcoWG1 library. (A) Experimental setup for the screen performed with the genome-wide EcoWG1 library in strain LC-E75 (MG1655 with dCas9 controlled by a Ptet promoter integrated at the 186 attB site). (B) Distribution of the depletion scores of non-essential and essential genes at the different time points of the experiment. The gene depletion score is computed as the median log2FC of the guides targeting the gene. (C) AUC of gene essentiality prediction using increasing numbers of randomly picked guides per gene in each dataset. Three random draws are shown for each dataset.
To evaluate the quality of the EcoWG1 library, we first assessed how well this novel dataset could be used to predict gene essentiality in E. coli using a recent TraDIS dataset as ground truth (22). Looking at the distribution of gene scores, one can observe how guides targeting essential genes are depleted during the course of the experiment. It is quite interesting to note a bimodal distribution at intermediary time points, where some essential genes show strong depletion scores already at early time points, while the effect of other essential genes can only be seen at later time points (Figure 2B).
We further compared the performance of the EcoWG1 library to the data we obtained previously with the Cui 2018 library (∼9 guides per gene on average but with large fluctuations from gene to gene, grown over 17 generations) and data generated by Wang and colleagues with a library of ∼60 000 guides (∼15 guides per gene grown over 15 generations) (8). The comparison was performed on a subset of genes for which all datasets carried at least five guides per gene. Receiver operating characteristic (ROC) curves were computed for each dataset using gene scores and the area under the curve (AUC) used to compare their performance (Supplementary Figure S3). The EcoWG1 library enabled the prediction of gene essentiality with AUCs of 85.4% (3H), 97.7% (6H), 97.7% (24H) and 98.9% (24+6H), while our previous dataset and that of Wang both obtained AUCs of 97.3%. Note that both the Cui 2018 and the Wang 2018 libraries have more guides per gene than the EcoWG1 library, showing that the effect measured with a few well designed guides can be as reliable as the effect measured from a larger number of randomly designed guides. We wondered how small the number of guides designed with our rules could be and still provide reliable results. AUCs of gene essentiality predictions were computed using one to five randomly picked guides in each gene (Figure 2C). Our results demonstrate the better performance of EcoWG1 compared to previous libraries. This is especially evident when only a single, randomly picked, guide per gene is considered. Using a single guide designed following our rules, one can obtain an AUC almost on a par with the results of previous libraries, which used 9–15 guides per gene. A caveat of these comparisons is that the AUCs of gene essentiality predictions is not only a factor of the library design but also of how the experiments were conducted. In particular, experiments in which more generations were performed will tend to separate essential genes from non-essential genes more as the log2FC increases over time. Nonetheless, when considering a single guide per gene, the EcoWG1 library already outperformed the other libraries with as little as ∼11 generations (6H).
DISCUSSION
We provide a novel model to predict the ability of guide RNAs to direct dCas9 to block the RNAP elongation in E. coli when binding to the non-template strand (coding strand). This model highlights the importance of bases surrounding the PAM sequence, bases in the seed sequence and to a lower degree bases up to 16nt downstream of the PAM. Most of the models built so far to predict the on-target activity of Cas9 have focused on genome editing in Eukaryotes and were shown to frequently be in disagreement (23). However, they tend to agree on certain key features. The ideal PAM motif seems to be CGGH in several independent studies, consistently with the prediction of our model (19,24). Conversely, previous models consistently identified that a G as the last base of the guide increases genome editing efficiency, while our model indicates that Y (C or T) is better at that position to block the RNAP. This discrepancy could come from the fact that stable binding of dCas9 to the target is essential to block the RNAP, while stable binding after cleavage by Cas9 likely hinders genome editing by blocking access of the DNA repair machinery to the cleavage site (25). Accordingly, it was shown that displacing Cas9 after cleavage with the RNAP can enhance genome editing efficiency in mammalian cells (25).
The importance of bases as far as 16nt downstream of the PAM that we identify here is however not a common feature of previous activity prediction models. Bases downstream of the PAM are the ones encountered by the RNAP when colliding with dCas9. Differences in the primary sequence could affect the ability of the RNAP to kick out dCas9. Another non-exclusive hypothesis is that there might be a direct interaction between this region and dCas9 affecting repression strength. A recent study employing single molecule approaches identified a direct interaction between Cas9 and bases ∼14nt downstream of the PAM which seem important for Cas9 binding (26). While nothing is known about the impact of the primary DNA sequence on this interaction, it is possible that different sequences could interact more or less strongly with Cas9 which could also explain the importance of bases downstream of the PAM in our model.
Models that predict guide activity for CRISPRi have previously been proposed for mammalian cells (27,28), where efficient repression by dCas9 relies on a protein fusion between dCas9 and the chromatin modifier domain KRAB. The mode of action of this dCas9-KRAB fusion is very different from the transcriptional repression investigated here, which uses dCas9 as a roadblock to the RNA polymerase. In mammalian systems, the position of the target relative to the transcription start site and canonical nucleosome-occupied regions are the most important feature (28). Accordingly these models are irrelevant to bacteria.
The CRISPR-ERA tool proposes design rules for bacteria, putting arbitrary weight on two simple features, the presence of extreme GC contents (25%,75%) and the position relative to the TSS (0, +500 bp) (27). In our dataset, no guides had a GC content lower than 25% and only 100 guides had a GC content higher than 75%, making it hard to properly investigate the impact of extreme GC content. We could still see that high GC content guides showed a slight but significant reduction in activity (median = –0.35, Mann–Whitney U P < 0.001). Nonetheless, this feature did not substantially improve the performance of the model and was not included here, but might be included in future models. In bacteria, dCas9 binding within or shortly after the promoter sequence can block the initiation of transcription, which might on average lead to a slightly stronger repression than guides binding further along the gene and which block transcription elongation (1,2,12). While the distance to the TSS was initially proposed as a factor contributing to repression efficiency when blocking transcription elongation in bacteria (1), this was not corroborated by a previous analysis of our dataset (12). The vast majority of guides used here are expected to interfere with transcription elongation and not transcription initiation, and the distance to the TSS was therefore not selected as a feature of our model.
The sequence requirements to efficiently block transcription initiation are likely somewhat different than those we identified here. In particular, it is well documented that guides in both orientations can effectively silence genes when binding the promoter region (1,2). The sequence downstream of the PAM might be less important in this context. Likewise, we do not investigate here the ability of dCas9 to block transcription elongation when targeting the template strand. While binding of dCas9 in this orientation leads to much weaker repression on average, a wide range of repression strengths can still be observed in this orientation which would be interesting to characterize in future work. The reasons why dCas9 is able to block the RNAP much more frequently when binding in one orientation than the other remains to be understood and such analysis could help shed some light on this process.
In a previous study, we provided evidence that dCas9 saturates the target sequences when expressed to a sufficient level, and that repression strength is then controlled predominantly by the rate at which the RNAP can kick-out dCas9 from the DNA and pass through (15). In a non-saturating regime, the repression strength is also influenced by the number of active dCas9:sgRNA complexes present in the cell, which could itself be impacted by the guide RNA sequence, through its expression rate, satability or folding. Possible effects of the guide sequence on target search time could also become apparent in a non-saturating regime. As a consequence, the sequence features that determine strong repression by dCas9 might partly change depending on the expression level of dCas9 and the number of copies of the target in the cell. Further work will be required to investigate these effects. Along the same lines, the reversibility of gene silencing by dCas9 is a useful property of this tool, and it would be interesting to understand whether the features identified by our model can predict the dynamics of de-repression once the inducer of dCas9 is removed.
Finally, our activity prediction model is available through a user friendly interface at crispr.pasteur.fr. An updated version of the CRISPR browser is also made available9. This genome browser can be used to conveniently analyze the results of CRISPR screens and compare them to published data. We hope the EcoWG1 library (addgene #131625) and the tools we provide will be a useful resource for the community.
DATA AVAILABILITY
The code and data used to perform the analyses are available here: https://gitlab.pasteur.fr/dbikard/ecowg1
Illumina reads were deposited on SRA: PRJNA592950 fastq files can be found with the accession number SAMN13443046.
A custom genome browser is available at crispr.pasteur.fr to visualize the data.
Supplementary Material
Notes
Present address: Lun Cui, Department of Bioengineering, School of Pharmaceutical Engineering & Life Science, School of Nursing, Changzhou University, Changzhou, Jiangsu Province, China
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council (ERC) under the Europe Union's Horizon 2020 research and innovation program [677823]; French Government's Investissement d’Avenir program; Laboratoire d’Excellence ‘Integrative Biology of Emerging Infectious Diseases’ [ANR-10-LABX-62-IBEID]; Calvo-Villamañán is supported by the Ecole Doctorale FIRE – Programme Bettencourt and a Veroniki-Holding scholarship. Funding for open access charge: European Research Council.
Conflict of interest statement. None declared.
REFERENCES
- 1. Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A.. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013; 152:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bikard D., Jiang W., Samai P., Hochschild A., Zhang F., Marraffini L.A.. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013; 41:7429–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mojica F.J.M., Díez-Villaseñor C., García-Martínez J., Almendros C.. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009; 155:733–740. [DOI] [PubMed] [Google Scholar]
- 4. Jiang W., Bikard D., Cox D., Zhang F., Marraffini L.A.. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013; 31:233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sternberg S.H., Redding S., Jinek M., Greene E.C., Doudna J.A.. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014; 507:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramachandran G., Bikard D.. Editing the microbiome the CRISPR way. Philos. Trans. R. Soc. B: Biol. Sci. 2019; 374:20180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peters J.M., Koo B.-M., Patino R., Heussler G.E., Hearne C.C., Qu J., Inclan Y.F., Hawkins J.S., Lu C.H.S., Silvis M.R. et al.. Enabling genetic analysis of diverse bacteria with Mobile-CRISPRi. Nat. Microbiol. 2019; 4:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang T., Guan C., Guo J., Liu B., Wu Y., Xie Z., Zhang C., Xing X.-H.. Pooled CRISPR interference screening enables genome-scale functional genomics study in bacteria with superior performance. Nat. Commun. 2018; 9:2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rousset F., Cui L., Siouve E., Becavin C., Depardieu F., Bikard D.. Genome-wide CRISPR-dCas9 screens in E. coli identify essential genes and phage host factors. PLos Genet. 2018; 14:e1007749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peters J.M., Colavin A., Shi H., Czarny T.L., Larson M.H., Wong S., Hawkins J.S., Lu C.H.S., Koo B.-M., Marta E. et al.. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell. 2016; 165:1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu X., Gallay C., Kjos M., Domenech A., Slager J., van Kessel S.P., Knoops K., Sorg R.A., Zhang J.-R., Veening J.-W.. High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol. Syst. Biol. 2017; 13:931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cui L., Vigouroux A., Rousset F., Varet H., Khanna V., Bikard D.. A CRISPRi screen in E. coli reveals sequence-specific toxicity of dCas9. Nat. Commun. 2018; 9:1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van Opijnen T., Camilli A.. Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nature reviews. Microbiology. 2013; 11:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H.. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006; 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vigouroux A., Oldewurtel E., Cui L., Bikard D., van Teeffelen S.. Tuning dCas9’s ability to block transcription enables robust, noiseless knockdown of bacterial genes. Mol. Syst. Biol. 2018; 14:e7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hawkins J.S., Silvis M.R., Koo B.-M., Peters J.M., Jost M., Hearne C.C., Weissman J.S., Todor H., Gross C.A.. Modulated efficacy CRISPRi reveals evolutionary conservation of essential gene expression-fitness relationships in bacteria. 2019; bioRxiv doi:15 October 2019, pre-print: not peer reviewed 10.1101/805333. [DOI] [PMC free article] [PubMed]
- 17. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A. 3rd, Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 18. Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. et al.. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016; 34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moreno-Mateos M.A., Vejnar C.E., Beaudoin J.-D., Fernandez J.P., Mis E.K., Khokha M.K., Giraldez A.J.. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods. 2015; 12:982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo J., Wang T., Guan C., Liu B., Luo C., Xie Z., Zhang C., Xing X.-H.. Improved sgRNA design in bacteria via genome-wide activity profiling. Nucleic Acids Res. 2018; 46:7052–7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Low K.B. Experiments in molecular genetics. Q. Rev. Biol. 1974; 49:151–151. [Google Scholar]
- 22. Goodall E.C.A., Robinson A., Johnston I.G., Jabbari S., Turner K.A., Cunningham A.F., Lund P.A., Cole J.A., Henderson I.R.. The essential genome of Escherichia coli K-12. mBio. 2018; 9:e02096-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.-B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J. et al.. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016; 17:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doench J.G., Hartenian E., Graham D.B., Tothova Z., Hegde M., Smith I., Sullender M., Ebert B.L., Xavier R.J., Root D.E.. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014; 32:1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Clarke R., Heler R., MacDougall M.S., Yeo N.C., Chavez A., Regan M., Hanakahi L., Church G.M., Marraffini L.A., Merrill B.J.. Enhanced bacterial immunity and mammalian genome editing via RNA-polymerase-mediated dislodging of Cas9 from double-strand DNA breaks. Mol. Cell. 2018; 71:42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Q., Wen F., Zhang S., Jin J., Bi L., Lu Y., Li M., Xi X.-G., Huang X., Shen B. et al.. The post-PAM interaction of RNA-guided spCas9 with DNA dictates its target binding and dissociation. Sci. Adv. 2019; 5:eaaw9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu H., Wei Z., Dominguez A., Li Y., Wang X., Qi L.S.. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics. 2015; 31:3676–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Horlbeck M.A., Gilbert L.A., Villalta J.E., Adamson B., Pak R.A., Chen Y., Fields A.P., Park C.Y., Corn J.E., Kampmann M. et al.. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife. 2016; 5:e19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tareen A., Kinney J.B.. Logomaker: beautiful sequence logos in Python. Bioinformatics. 2020; 36:2272–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code and data used to perform the analyses are available here: https://gitlab.pasteur.fr/dbikard/ecowg1
Illumina reads were deposited on SRA: PRJNA592950 fastq files can be found with the accession number SAMN13443046.
A custom genome browser is available at crispr.pasteur.fr to visualize the data.