

Abstract
Immune‐checkpoint inhibitors improve the survival of head and neck squamous cell carcinoma (HNSCC) patients. Although recent studies have demonstrated that the tumor immune microenvironment (TIME) has critical roles in immunotherapy, the precise mechanisms involved are unclear. Therefore, further investigations of TIME are required for the improvement of immunotherapy. The frequency of effector regulatory T‐cells (eTregs) and the expression of immune‐checkpoint molecules (ICM) on eTregs and conventional T‐cells (Tconvs) both in peripheral blood lymphocytes (PBL) and tumor‐infiltrating lymphocytes (TIL) from HNSCC patients were analyzed by flow cytometry and their distributions were evaluated by multi‐color immunofluorescence microscopy. High frequency eTreg infiltration into HNSCC tissues was observed and high expressions of CD25, FOXP3, stimulatory‐ICM (4‐1BB, ICOS, OX40 and GITR) and inhibitory‐ICM (programmed cell death‐1 [PD‐1] and cytotoxic T‐lymphocyte‐associated protein‐4 [CTLA‐4]) were found on invasive eTregs. In contrast, the expression of stimulatory‐ICM on Tconvs was low and the expression of inhibitory‐ICM was high. In addition, ICM‐ligands (programmed cell death‐1 [PD‐L1], galectin‐9 and CEACAM‐1) were frequently expressed on cancer cells. PD‐L1 and galectin‐9 were also expressed on macrophages. PD‐1+ T‐cells interacted with PD‐L1+ cancer cells or PD‐L1+ macrophages. This suggested that in TIL, eTregs are highly activated, but Tconvs are exhausted or inactivated by eTregs and immune‐checkpoint systems, and ICM and eTregs are strongly involved in the creation of an immunosuppressive environment in HNSCC tissues. These suggested eTreg targeting drugs are expected to be a combination partner with immune‐checkpoint inhibitors that will improve immunotherapy of HNSCC.
Keywords: head and neck squamous cell carcinoma, immune checkpoint, immunotherapy, regulatory T‐cell, tumor immune microenvironment
Frequencies of eTregs populations (CD45RA‐FOXP3hi) in CD4+ T‐cells of PBL and TIL from HNSCC patients were compared. High frequency of eTreg infiltration into HNSCC tissues was revealed. In addition, MFI of CD25 and FOXP3 in eTreg of TIL were higher than in PBL, which indicates that eTreg in TIL are not only highly infiltrated into the tissues but also are highly activated.

1. INTRODUCTION
Between 400 000 and 600 000 new cases of head and neck cancers (HNC) are diagnosed worldwide each year, with between 223 000 and 300 000 deaths occurring annually. 1 In Japan, the projected HNC incidence and mortality in 2018 were reported to be 27 600 and 8700, respectively; these figures are gradually increasing every year. 2
The standard therapies for HNC are surgery, radiotherapy, chemotherapy and radio‐chemotherapy. FP treatment (a combination of 5‐fluorouracil [5‐FU] and platinum drugs) or FP+ cetuximab is the standard therapy used for unresectable recurrent and distant metastatic cases. 3 , 4 , 5 , 6 , 7 Recently, the programmed cell death‐1 (PD‐1) antibody medicines nivolumab and pembrolizumab demonstrated significant improvement in survival for recurrent and/or metastatic head and neck squamous cell cancers (HNSCC) in the CheckMate 141 8 and KEYNOTE‐012 studies, 9 respectively, and have been approved worldwide. However, more than 70% of patients are still non–responders for these PD‐1 antibody medicines. Therefore, further development and improvement of immune therapeutics is required.
The tumor suppressive immune microenvironment is related to cancer progression and prognosis. The immune checkpoint system, 10 metabolic conditions, 11 and infiltration of immunosuppressive cells such as Tregs, 12 myeloid derived suppressive cells (MDSC) 13 and M2 macrophages 14 are considered to be important factors for the creation of the tumor suppressive immune microenvironment. Therefore, a comprehensive analysis of the tumor suppressive immune microenvironment constituted by these factors is required to improve the efficacy of cancer immunotherapy. Because Tregs have highly immunosuppressive functions 15 , 16 , 17 and the numbers of Tregs infiltrating into cancer tissues are reported to be correlated with prognosis in many cancers, 18 , 19 , 20 , 21 , 22 including HNC, 23 , 24 the regulation of Tregs is one direction for the development of novel cancer immunotherapies. 12 Tregs can be classified into three fractions by flow cytometric two‐dimensional analysis using CD45RA and forkhead box P3 (FOXP3) staining as follows: resting Treg/naïve Treg (CD45RA+FOXP3lo), activating Treg/effector Treg (CD45RA−FOXP3hi) and non–Treg (CD45RA−FOXP3lo). 17 Using this classification, immunosuppressive activity is observed for effector Tregs (eTreg) and naive Tregs (nTreg) but not non–Tregs. 17 In addition, it was reported that in colorectal cancers (CRC), cancer tissues were infiltrated by CD45RA−FOXP3lo non–Tregs that secreted inflammatory cytokines, and CRC with an abundant infiltration of non–Tregs showed a significantly better prognosis than those with a predominant infiltration of CD45RA−FOXP3hi cells. 25 Because Tregs have been detected as CD4+CD25+ cells or CD4+FOXP3+ cells in many previous reports, it is difficult to determine which Tregs have suppressive activity. Therefore, we attempted to analyze the eTreg fraction by flow cytometric analysis using CD45RA and FOXP3 to specifically detect highly immunosuppressive Tregs. Immune‐checkpoint molecules (ICM) on eTregs were also detected by flow cytometry to estimate the activation and functional status of eTregs. In addition, the distribution of ICM‐expressing cells in cancer tissues was analyzed by multi‐color immunofluorescence microscopy. Thus, we specifically evaluated the immunosuppressive microenvironment in HNSCC tissues to explore new directions for the development of future HNSCC immunotherapy.
2. MATERIALS AND METHODS
2.1. Patients and specimens
Peripheral venous blood samples, draining lymph node (DLN) samples and surgical cancer samples were obtained from 28 patients with HNSCC who underwent primary surgery between January 2017 and April 2019 in the Department of Otorhinolaryngology and Surgical Pathology, Aichi Medical University Hospital. All subjects signed written informed consent that was approved by the Institutional Review Committee (reference number: 2016‐H171). Metastatic DLN were discriminated from non–metastatic DLN by using clinical PET‐CT and enhanced cervical CT status and also distinguished by intraoperative findings, such as size and hardness. Finally, the metastatic DLN were also defined from the pathological confirmation by microscopy. Patient characteristics are presented in Table 1.
Table 1.
Patient list in this study
| Case number | Age | Gender | Tumor site | TNM classification | Previous treatment | ||
|---|---|---|---|---|---|---|---|
| T | N | M | |||||
| 1 | 74 | F | Maxi | 4a | 1 | 0 | NAC |
| 2 | 61 | M | Maxi | 4a | 2b | 0 | NAC |
| 3 | 79 | M | Hypo | 4a | 1 | 0 | — |
| 4 | 78 | M | Hypo | 4a | 1 | 0 | — |
| 5 | 75 | M | Hypo | 4a | 2a | 0 | — |
| 6 | 66 | M | Hypo | 3 | 1 | 0 | — |
| 7 | 55 | M | Hypo | 3 | 2b | 0 | NAC |
| 8 | 84 | M | Hypo | 4 | 2b | 0 | — |
| 9 | 71 | F | Hypo | 4a | 0 | 0 | — |
| 10 | 64 | F | Hypo | 4a | 0 | 0 | CCRT |
| 11 | 68 | M | Lar | 2 | 2b | 0 | NAC |
| 12 | 61 | M | Lar | 2 | 0 | 0 | NAC, RT |
| 13 | 80 | M | Lar | 1a | 0 | 0 | RT |
| 14 | 79 | M | Lar | 4a | 2a | 0 | — |
| 15 | 65 | M | Lar (R) | 2 | 0 | 0 | CCRT |
| 16 | 76 | M | Lar (R) | 1a | 0 | 0 | RT |
| 17 | 68 | M | Lar (R) | 3 | 0 | 0 | NAC, RT |
| 18 | 60 | M | Oro | 4a | 1 | 0 | NAC |
| 19 | 73 | M | Oro | 1 | 1 | 0 | NAC |
| 20 | 73 | M | Oro (M) | 2 | 2a | 0 | NAC |
| 21 | 58 | F | Oral | 2 | 0 | 0 | NAC |
| 22 | 77 | M | Oral | 2 | 0 | 0 | — |
| 23 | 81 | M | Oral | 3 | 2a | 0 | — |
| 24 | 65 | F | Oral | 3 | 1 | 0 | chemo, RT |
| 25 | 76 | F | Oral | 2 | 2b | 0 | — |
| 26 | 47 | M | Oral | 4b | 0 | 0 | NAC |
| 27 | 71 | F | Oral | 2 | 0 | 0 | — |
| 28 | 77 | M | Oral (R) | 2 | 2 | 0 | RT |
Abbreviations: CCRT, chemo + RT; chemo, chemotherapy; hypo, hypopharynx; lar, larynx; M, metastasis; maxi, maxi‐sinus; NAC, neoadjuvant chemotherapy; oro, oropharynx; R, relapse; RT, radiotherapy.
2.2. Flow cytometry
Peripheral blood lymphocytes (PBL), non–metastatic DNL lymphocytes (NM‐DLNL), metastatic DLN lymphocytes (M‐DLNL) and tumor infiltrated lymphocytes (TIL) were prepared from HNSCC patients as follows. PBL were isolated from heparinized peripheral blood by Ficoll‐Paque PLUS (GE Healthcare) gradient centrifugation. For NM‐DLNL, M‐DLNL and TIL, lymphnodes and cancer tissues obtained by surgical operation were minced in the cell detachment solution, Accutase (Innovative Cell Technologies), for 15 minutes at room temperature, and were isolated from the extracted cells by Ficoll‐Paque PLUS gradient centrifugation. PBL, NM‐DLNL, M‐DLNL and TIL were incubated with fluorochrome‐conjugated mAbs to CD45RA, CD4 and CD8 for 20 minutes at 4°C, and were washed once with washing buffer (PBS containing 0.2% human albumin and 2 mmol/L EDTA). Next, the cells were incubated with fluorochrome‐conjugated mAbs to CD25 or ICM such as 4‐1BB, inducible T‐cell co‐stimulator (ICOS), glucocorticoid‐induced TNF receptor (GITR), OX40, PD‐1, cytotoxic T‐lymphocyte‐associated protein‐4 (CTLA‐4), T‐cell immunoglobulin and mucin domain‐3 (TIM‐3) or lymphocyte‐activation gene‐3 (LAG‐3) for 20 minutes at 4°C. After washing, the cells were fixed with fixative solution (eBioscience, CA, USA) for 30 minutes at 4°C, and were washed with permeabilized solution twice, followed by incubation with Alexa488 conjugated mAb to FOXP3 for 30 minutes at 4°C. Then the cells were analyzed on a FACSCanto II (BD Biosciences) with the aid of FlowJo software (Tree Star). The monoclonal antibodies used are listed in Table 2.
Table 2.
Antibodies used in this study
| Antibodies | Clones | Manufactures | Catalogue number |
|---|---|---|---|
| Flow cytometry | |||
| CD4‐APC | RPA‐T4 | Biolegend | 300514 |
| CD8‐PerCP | SK1 | BD | B043‐0087‐120 |
| CD25‐PE | M‐A251 | BD | 555432 |
| CD45RA‐APC‐H7 | 5H9 | BD | 561212 |
| FOXP3‐Alexa488 | 236A/E7 | BD | 561181 |
| 4‐1BB‐PE | 4B4‐1 | BD | 555956 |
| ICOS‐PE | DX29 | BD | 557802 |
| OX40‐PE | ACT35 | BD | 555838 |
| GITR‐BV421 | 108‐17 | Biolegend | 371208 |
| PD‐1‐BV421 | EH12.2H7 | Biolegend | 329920 |
| CTLA‐4‐BV421 | BNI3 | BD | 562743 |
| TIM‐3‐PE | 7D3 | BD | 563422 |
| LAG‐3‐BV421 | 11C3C65 | Biolegend | 369314 |
| CCR4‐PE | L291H4 | Biolegend | 359412 |
| Immunohistochemistry | |||
| PD‐L1 | 28‐8 | Abcam | ab205921 |
| Galectin‐9 | DR94A | CST | 54330 |
| CEACAM‐1 | EPR4049 | Abcam | ab108397 |
| CD8 | 1A5 | Biogenex | MU422‐UC |
| CD3 | SP162 | Spring bio | M4622 |
| PD‐1 | NAT105 | Abcam | ab52587 |
| ICOS | SP98 | Spring bio | M3982 |
| GITR | D9I9D | CST | 44689 |
| FOXP3 | 236A/E7 | Abcam | ab20034 |
2.3. Multi‐color immunofluorescence microscopy
Multi‐immunofluorescent staining with 4‐μm‐thick paraffin‐embedded sections was performed using the tyramide signal amplification (TSA) system (PerkinElmer) for the detection of ICM such as PD‐1, ICOS and GITR, ICM ligands such as programmed cell death receptor ligand 1 (PD‐L1), galectin‐9 and carcinoembryonic antigen‐related cell adhesion molecule‐1 (CEACAM‐1), and T‐cell lineage markers such as CD3 and CD8, and FOXP3 in HNSCC tissues. The monoclonal antibodies used are listed in Table 2. De‐paraffinized sections were incubated with antigen retrieval buffer (ab208572; Abcam) for 10 minutes at 110°C in an autoclave. After cooling down to 60°C, the sections were blocked with 2% horse serum for 10 minutes at room temperature. Primary antibody reaction with appropriately diluted mAbs was performed for 60 minutes at room temperature. After washing with PBS for 5 minutes three times, secondary antibody reactions were performed with peroxidase polymer anti–mouse IgG or anti–rabbit IgG (Vector Laboratories) for 30 minutes at room temperature. The sections were washed with PBS for 5 minutes three times, then the stained molecules were visualized with the TSA system following the manufacturer’s recommended procedure. Before progressing to the next molecule staining, the sections were heated with 10 mmol/L Tris‐HCl containing 2 mmol/L EDTA (pH 9.0) for 10 minutes at 95°C to remove the binding antibodies from the first molecule staining step. Second, third and fourth molecule staining steps used the same staining procedure as for the first staining step described above. The multi‐color staining images were captured using a digital pathology slide scanner, Aperio CS2 (Leica Microsystems KK), and analyzed with Aperio ImageScope software. Immunofluorescence analysis was performed using an immuno‐oncologist (SS) and two well‐experienced pathologists (TT and TT). Expression frequencies and the distribution of each molecule on cancer cells or lymphocytes were evaluated for each sample by visual inspection. The expression frequencies were indicated as the average percentage in any three areas of 0.6 mm2.
2.4. Tumor immune microenvironment classification of cancer tissues
The distribution of lymphocytes and CD8+ T‐cells in the tissues was investigated with hematoxylin and eosin (HE) staining and conventional immunohistochemistry with anti–CD8 mAb, and TIME classification was characterized into three types according to previous reports 26 , 27 , 28 : “immune‐desert” in which very few T‐cells infiltrated into cancer tissues; “immune‐excluded” in which T‐cells infiltrated into stromal areas but not into the nest; and “immune‐inflamed” in which T‐cells infiltrated into the nest and stroma (Figure S1).
2.5. Statistical analysis
Correlations between two variables were assessed using Spearman’s rank correlation coefficient (Rs). Differences between two groups were examined with the Student t test.
3. RESULTS
3.1. Flow cytometric analysis of lymphocytes in head and neck squamous cell carcinoma patients: eTregs and Tconvs
3.1.1. Significant infiltration of eTregs into head and neck squamous cell carcinoma tissues
The eTreg population in CD4+ lymphocytes (CD4+CD45RA−FOXP3hi) from HNSCC patients was evaluated (Figure 1). The eTreg population of TIL (n = 24; average 36.63%; SD, 12.53) was approximately nine times higher than that of PBL (n = 28; average, 4.28%; SD; 3.72) (Figure 1C,G). This suggested that eTregs predominantly infiltrated into the HNSCC tissues. The population of CD25+ cells was compared between eTregs, CD4+ Tconvs (CD4+CD45RA−FOXP3−) and CD8+ Tconvs (CD8+CD45RA−). The CD25+ population of eTregs was markedly higher than that of CD4+ and CD8+ Tconvs, both in PBL and TIL, which re–confirmed the significance of CD25 as a marker of Tregs (Figure 1E,F,H).
Figure 1.
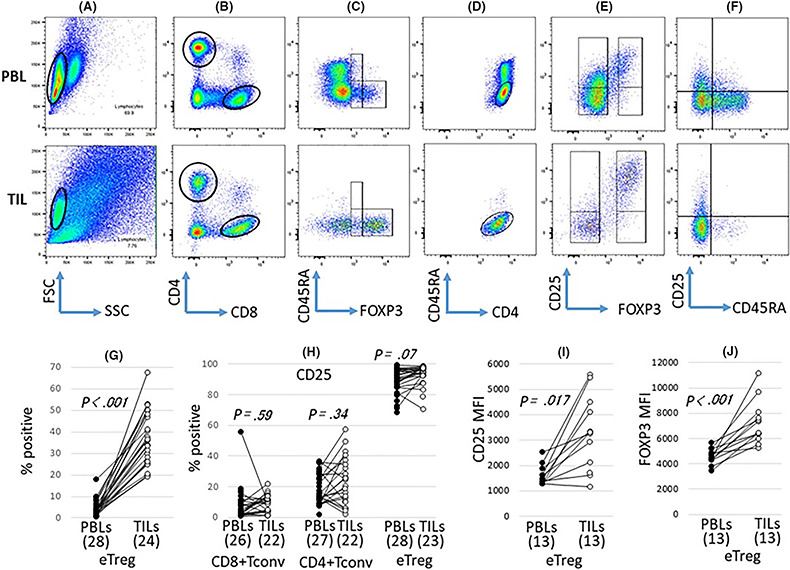
Significant infiltration of eTregs into head and neck squamous cell carcinoma (HNSCC) tissues. Peripheral blood lymphocytes (PBL) and tumor‐infiltrating lymphocytes (TIL) from patients with HNSCC were stained with mAb to CD4, CD8, CD45RA, CD25 and FOXP3. The frequency of eTregs and CD25 expression on eTregs and Tconvs was analyzed by flow cytometry. A representative analysis strategy is shown for case 23 (A–F). The lymphocytes from PBL and TIL were gated in the cytograms (A) and separated by CD4 and CD8 (B). Then, CD4‐positive cells were separated by CD45RA and FOXP3 (C). The cells were gated on CD45RA+/FOXP3lo, CD45RA−/FOXP3lo and CD45RA−/FOXP3high, and CD45RA−/FOXP3high cells were determined to be eTregs (C). The CD4‐positive cells gated in (B) were gated on CD45RA−/CD4+ (D) and CD25 expression was analyzed in the FOXP3 negative and positive populations (E). CD8‐positive cells gated in (B) were separated by CD45RA and CD25, and CD25 expression was analyzed (F). eTreg frequencies (G) and the mean fluorescence intensity (MFI) of eTregs (J) were compared between PBL and TIL. CD25 frequencies in each fraction (H) and the MFI of eTregs (I) were compared between PBL and TIL
3.1.2. High activation of eTregs with high expression of immune‐checkpoint molecules, CD25 and FOXP3 in tumor‐infiltrating lymphocytes
Expressions of ICM in eTregs and Tconvs were evaluated (Figures 2 and 3). Positive populations of stimulatory molecules such as 4‐1BB, ICOS, OX40 and GITR in eTregs were markedly higher in TIL than PBL. Although significant differences were not observed in eTregs when the CD25+ population was compared between PBL and TIL (Figure 1H), the mean fluorescence intensity (MFI) in eTregs was higher in TIL than PBL (Figure 1I). In addition, the MFI of FOXP3 in eTregs was also higher in TIL than PBL (Figure 1J). These findings indicate that eTregs infiltrating into HNSCC tissues were highly activated.
Figure 2.
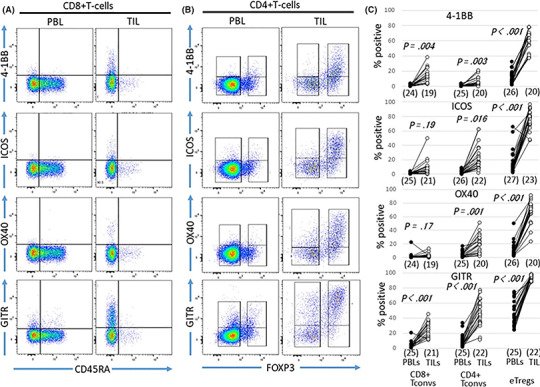
Expression of stimulatory immune‐checkpoint molecules (ICM) on eTregs and Tconvs in peripheral blood lymphocytes (PBL) and tumor‐infiltrating lymphocytes (TIL) from head and neck squamous cell carcinoma (HNSCC) patients. Expression of stimulatory ICM in PBL and TIL on CD8+ Tconvs (A), CD4+ Tconvs and eTregs (B) is shown for case 23. Frequencies of stimulatory ICM in each fraction were compared between PBL and TIL (C)
Figure 3.
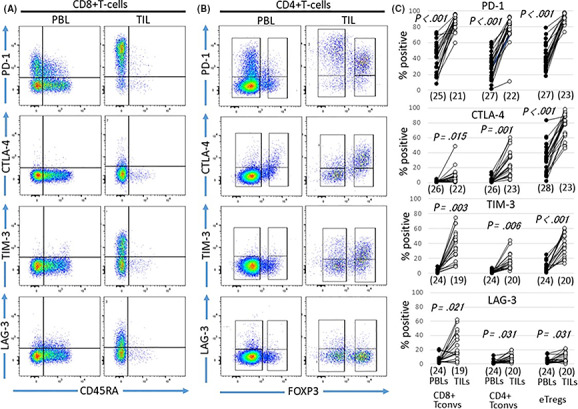
Expression of inhibitory immune‐checkpoint molecules (ICM) on eTregs and Tconvs in peripheral blood lymphocytes (PBL) and tumor‐infiltrating lymphocytes (TIL) from head and neck squamous cell carcinoma (HNSCC) patients. Expression of stimulatory ICM in PBL and TIL on CD8+ Tconvs (A), CD4+ Tconvs and eTregs (B) is shown for case 23. Frequencies of stimulatory ICM in each fraction were compared between PBL and TIL (C)
3.1.3. Various immune‐checkpoint molecules’ expression level on Tconvs
The positive population of stimulatory molecules in Tconvs was also higher in TIL than PBL but was lower than that in eTregs (Figure 2). Differences in positive populations of the stimulatory molecules were observed between CD4+ Tconvs and CD8+ Tconvs. The 4‐1BB+ population was higher in CD8+ Tconvs than CD4+ Tconvs (Figure 2). In contrast, the positive populations of ICOS, OX40 or GITR were higher in CD4+ Tconvs than CD8+ Tconvs (Figure 2). The populations of inhibitory molecules, PD‐1, CTLA‐4 and TIM‐3, but not LAG‐3, in eTregs were higher in TIL than PBL, and in Tconvs they were also higher in TIL compared with PBL (Figure 3). The positive populations of each inhibitory molecule were compared among three subsets: eTregs, CD4+ Tconvs and CD8+ Tconvs. PD‐1 was very high in all three subsets, CTLA‐4 was higher in eTregs than in CD4+ Tconvs and CD8+ Tconvs, and TIM‐3 and LAG‐3 were higher in CD8+ Tconvs than in CD4+ Tconvs and eTregs (Figure 3).
3.1.4. Association among cell populations in tumor‐infiltrating lymphocytes
Each ICM‐positive population of T cells was compared with the eTreg population (Figure S2). A significant positive correlation was found between the eTreg population and positive populations of PD‐1 and TIM‐3 in CD8+ Tconvs but not between the eTreg population and any ICM‐positive populations in CD4+ Tconvs. In addition, we evaluated the correlation between each ICM population in CD4+ Tconvs, CD8+Tconvs and eTregs (Tables S1‐S3). A significant correlation was observed between 12 pairs, 6 pairs and 7 pairs of CD4+ Tconvs, CD8+Tconvs and eTregs, respectively. This suggested that ICM expression in HNSCC tissues was closely modulated, especially in CD4+Tconvs. A CTLA‐4‐positive population in CD4+ Tconvs significantly correlated with positive populations of stimulatory ICM, such as 4‐1BB, ICOS, OX40 and GITR, which are related to cell growth. However, these correlations were not observed for eTregs. PD‐1‐positive populations in CD4+ Tconvs and CD8+ Tconvs were not correlated with any ICM‐positive populations. However, PD‐1‐positive populations were correlated with 4‐1BB and GITR in eTregs, and the expression of 4‐1BB, which is a marker for T‐cell activation and proliferation, on PD‐1‐positive T cell subsets in TIL was investigated. The eTregs highly expressed 4‐1BB compared with the Tconvs (Figure S3).
3.1.5. Correlations in immune populations between peripheral blood lymphocytes and tumor‐infiltrating lymphocytes
The eTreg population in CD4+ T‐cells was compared between PBL and TIL, but a significant correlation was not found. In addition, we compared ICM populations in each subset of T‐cells, including CD8+ Tconvs and CD4+ Tconvs. We found significant positive correlations for GITR, PD‐1 and CTLA‐4 in CD4+ Tconvs and CTLA‐4 in eTregs (Table S4). These ICM on CD4+ Tconvs or eTregs in PBL might be biomarkers for treatment with anti–PD‐1 and/or anti–CTLA‐4.
3.1.6. eTreg population and immune‐checkpoint molecule expression in draining lymph node
The eTreg population in the DLN was evaluated. When the eTreg population in CD4+ T‐cells was compared among PBL, NM‐DLNL, M‐DLNL and TIL, that in M‐DLNL (n = 16, average; 15.11%, SD; 10.48) was significantly higher than in NM‐DLNL (n = 16, average; 5.48%, SD; 2.69) and PBL (n = 28; average, 4.28%; SD; 3.72) but lower than in TIL (n = 24; average 36.63%; SD, 12.53) (Figure S4). We examined that eTreg populations in effector memory CD4+ T‐cells (TEM) and central memory CD4+ T‐cells (TCM) (Figure S5). When ratios of % eTreg in TEM/% eTreg in TCM in collection sites of each sample were compared, the ratios were higher in TIL than NM‐DLNL and M‐DLNL. This indicated that eTreg populations of TIL are biased towards the TEM subset rather than NM‐DLNL and M‐DLNL. Because TCM generally have a high growth capacity and longer life compared with TEM, we believe DLNL are an important source of eTregs in TIL.
3.1.7. CCR4 expression in T‐cell subsets in comparison with CTLA‐4
It is known that CCR4 and CTLA‐4 are the target molecules for Tregs. 29 , 30 These molecule expressions in three subsets, CD4+ Tconvs, CD8+ Tconvs and eTregs, were compared (Figure S6). Both CCR4 and CTLA‐4 were preferentially expressed on eTregs, both in PBL and TIL and were expressed on also CD4+ Tconvs and CD8+ Tconvs both in PBL and TIL (Figure S6). Selectivity for the expression of CTLA‐4 on eTregs both in PBL and TIL was higher than that of CCR4 (Figure S6). Positive populations of CCR4 in each subset were higher than that of CTLA‐4 (Figure S6A‐c,d and B‐a,b). Almost cells of eTregs in TIL were double positive for CCR4 and CTLA‐4 (Figure S6A‐g). In contrast, these molecules’ expression in TIL was reciprocal in CD4+ and CD8+ Tconvs (Figure S6A‐f, B‐d).
3.2. Immunohistochemical analysis of the microenvironment in head and neck squamous cell carcinoma tissues
3.2.1. Tumor immune microenvironment classification of head and neck squamous cell carcinoma tissues and association with clinical features
Among 25 cases of HNSCC, 4 (16%) were classified as “immune‐inflamed,” 18 (72%) as “immune‐excluded” and 3 (12%) as “immune‐desert” based on TIME classification (Table 3). The representative staining examples are shown in Figure S1.
Table 3.
Immunohistochemical analysis of tumor immune microenvironment (TIME) in head and neck squamous cell carcinoma tissues
| Case number | TIME classification | Immunohistochemistry | T‐cell infiltration (cell number) | PD‐1 expression on T‐cells | Treg infiltration | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PD‐L1 | Galectin‐9 |
CEA CAM1 |
Stroma | Nest | Stroma | Nest | Cell number | Frequency in CD4+ T cells | ||||
| Stroma | Nest | Stroma | Nest | |||||||||
| 1 | Excluded | – | Focal | – | ++ | +w | 50% | 90% | + | ‐~+w | 30% | 70% |
| 2 | Excluded | – | – | – | +++ | +w | 80% | 80% | +~++ | –~+w | 30% | nd |
| 3 | Excluded | >50% | 30% | – | ++ | +w | 50% | 50% | + | –~+w | 30% | 10% |
| 4 | Excluded | >50% | Focal | 30% | ++ | +w | 50% | 90% | + | – | 50% | – |
| 5 | Excluded | – | – | Focal | +~++ | +w | 50% | 90% | +w~+ | – | nd | nd |
| 6 | Excluded | 20% | – | Focal | +w~++ | +w | nd | nd | +w~+ | –~+w | nd | nd |
| 7 | Excluded | – | – | 10% | ++ | ‐ | 50% | nd | + | – | 40% | nd |
| 9 | Excluded | 5% | – | – | +~++ | +w~+ | 30% | 70% | + | +w | 50% | 50% |
| 10 | Desert | – | 10% | – | ‐~+w | ‐ | 80% | nd | –~+w | – | 50% | 10% |
| 11 | Inflamed | >50% | >50% | >50% | ++ | ++ | 90% | 90% | +w~+ | +w~+ | 20% | 20% |
| 12 | Excluded | – | 5% | – | +‐++ | +w | nd | nd | + | –~+w | nd | nd |
| 13 | Excluded | >50% | – | – | + | +w~+ | nd | nd | +w~+ | +w | nd | nd |
| 14 | Excluded | <5% | 20% | 10% | +~++ | ‐~+w | 30% | 70% | + | – | 60% | – |
| 15 | Excluded | >50% | 30% | – | +w~++ | +w~+ | 30% | 70% | + | –~+w | 60% | 30% |
| 17 | Desert | – | – | – | + | +w | 50% | 90% | + | –~+w | 50% | 50% |
| 18 | Excluded | – | focal | – | +w~+ | +w~+ | 50% | 70% | +w | +w | nd | nd |
| 20 | Desert | – | – | – | ‐~+ | ‐ | 50% | nd | +w | – | 30% | – |
| 21 | Excluded | 10% | focal | – | +~++ | +w~+ | 50% | 90% | + | +w | 40% | 50% |
| 22 | Excluded | >50% | 30% | – | +++ | ‐ | 80% | nd | +~++ | – | nd | nd |
| 23 | Excluded | – | 10% | focal | +w~++ | +w | 50% | 90% | + | – | nd | nd |
| 24 | Inflamed | >50% | >50% | – | +++ | ++ | 70% | 90% | +~++ | + | nd | nd |
| 25 | Inflamed | >50% | 10% | – | +++ | +~++ | 80% | 80% | +~++ | + | 50% | 50% |
| 26 | Excluded | 30% | – | – | + | ‐ | 50% | nd | +w | – | 50% | – |
| 27 | Inflamed | >50% | >50% | 10% | ++~+++ | +~++ | 80% | 80% | +~++ | + | 60% | 50% |
| 28 | Excluded | >50% | 20% | – | ++~+++ | +w | 30% | 70% | +~++ | –~+w | 40% | 40% |
–, <10 cells/0.6 mm2; +w, 10 ~ 100 cells/0.6 mm2; +, 100 ~ 500 cells/mm2; ++, 500‐1000 cells/0.6 mm2; +++, >1000 cells/0.6 mm2; nd, not determined.
Table S5 shows the association between TIME classification and clinical features (recurrence‐free duration after surgery and survival duration after diagnosis), virus infections (HPV and EBV) and the Brickman index. However, we could not evaluate the associations of these factors with immune responses in this study because the number of cases was too small to analyze. Further study with a higher number of cases is required to determine whether TIME classification is associated with the clinical features.
3.2.2. Immune‐checkpoint molecules’ ligand expression in the nest and stroma
Immune‐checkpoint molecule ligand expression in the tissues was investigated by immunohistochemistry. The PD‐L1‐positive percentage on the nest in 25 cases of HNSCC was 60% (15/25), which is comparable to previous reports of HNSCC, 8 NSCLC 31 and gastric cancer, 32 and higher than that of renal cancer, 33 urothelial cancer 34 and melanoma. 35 All of the 4 “immune‐inflamed” cases were positive for PD‐L1 on cancer cells at the positive rate of over 50%. In contrast, there were 6 in 18 “immune‐excluded” cases and none in the 3 “immune‐desert” cases (Table 3). Galectin‐9 also tended to be expressed in “immune‐inflamed” cases more than in “immune‐excluded” and “immune‐desert” cases (Table 3). CEACAM‐1 was expressed in 5 cases at the positive rate of over 10% (Table 3). Representative expressions of these molecules are shown in Figure 4A. In the stroma, PD‐L1 and/or galectin‐9 were expressed in a population of macrophages (Figure 4B) in all cases. These PD‐L1 and/or galectin‐9‐expressing macrophages were distributed along the periphery of the cancer nest (Figure 4C) in “immune‐excluded” cases. In “immune‐inflamed” cases, PD‐L1‐positive macrophages were markedly infiltrated into the nests (Figure 4D). CEACAM‐1 was not expressed on macrophages but was widely expressed on neutrophils (data not shown).
Figure 4.
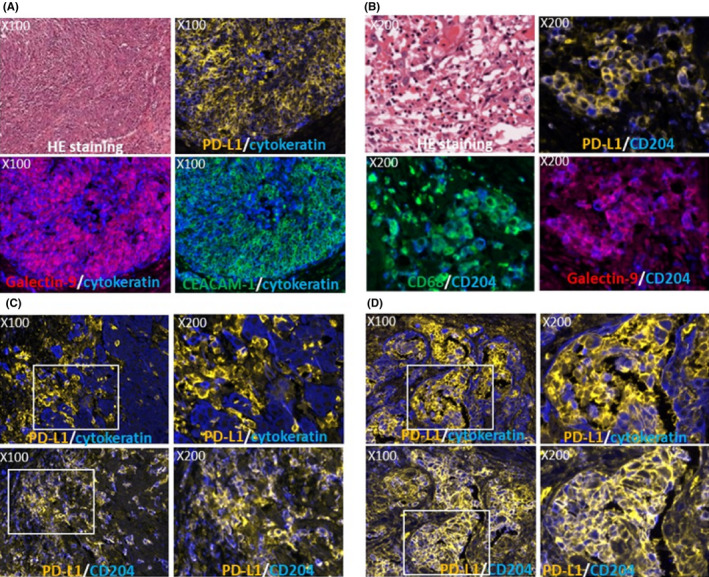
Expression of immune‐checkpoint molecule (ICM) ligands in head and neck squamous cell carcinoma (HNSCC) tissues. ICM ligands expression in the nest of case 11 is shown (A). Representative double immunostaining images with programmed cell death‐1 (PD‐1) in orange and cytokeratin in blue (upper right), galectin 9 in red and cytokeratin in blue (lower left) and CECAM‐1 in green and cytokeratin in blue (lower right) are shown in the same area with HE images (upper left). ICM ligand expression in the stroma is shown (B). Representative double immunostaining images with PD‐L1 in orange and CD204 in blue (upper right), CD68 in green and CD204 in blue (lower left) and galectin‐9 in red and CD204 in blue (lower right) are shown in the same area with the HE images (upper left). Distribution of programmed cell death receptor ligand 1 (PD‐L1) (orange) expressing macrophages along the periphery of the nest (cytokeratin in blue) was observed (C, upper panel) in an “immune‐excluded” case (case 14). In an “immune‐inflamed” case (case 24), high infiltration of PD‐L1 (orange)‐expressing macrophages into the nest (cytokeratin in blue) were observed (D, upper panel). These macrophages were positive for CD204 (C and D, lower panel). Upper right and lower right images of (C) and (D) indicate the expanded images of the white square left of the images. Magnifications are indicated in each image
3.2.3. Frequency and distribution of programmed cell death‐1 expressing T‐cells in the tissues
Programmed cell death‐1 expression on T‐cells in the tissues was examined by double immunostaining of CD3 and PD‐1 (Table 3, Figure 5A). Most of the T‐cells in TIL were PD‐1‐positive by immunohistochemistry, consistent with the results by flow cytometry. PD‐1+ T‐cell frequency in the tissues was higher in “inflamed cases” than “excluded cases.” PD‐1+ T‐cell frequency in the nest was higher than the stroma in 10 of 12 “immune‐excluded” cases, although it was almost equal between the nest and stroma in “immune‐iinflamed” cases (Table 3, Figure 5B). Interaction of PD‐1+ T‐cells and PD‐L1+ cancer cells and/or macrophages was observed (Figure 5C).
Figure 5.
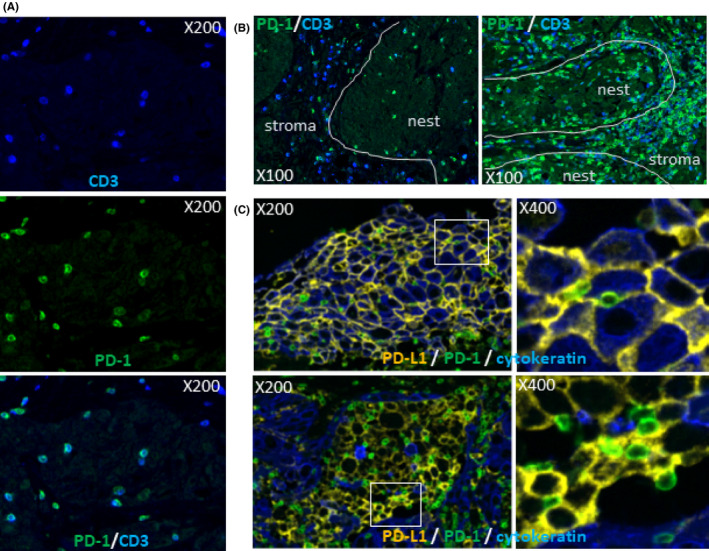
Programmed cell death‐1 (PD‐1) expression on T‐cells and interaction with programmed cell death receptor ligand 1 (PD‐L1) expressing cells in head and neck squamous cell carcinoma (HNSCC) tissues. HNSCC tissues were double stained with PD‐1 and CD3 and representative PD‐1 expression on T‐cells in HNSCC tissue is shown in (A) (upper; CD3 in blue, middle; PD‐1 in green, lower; merge). PD‐1 expressing T‐cells were preferentially distributed in the nest of the “immune‐excluded” case [case 15 (B, left)] and were equally distributed both in the nest and stroma [case 27 (B, right)]. Interactions of PD‐L1 (orange) with cancer cells (cytokeratin+ in blue) (upper images) or macrophages (cytokeratin) with PD‐1 (green) on lymphocytes are shown for case 24. Upper right and lower right images indicate the expanded images of the white square left of the images. White lines indicate the border between the nest and stroma. Magnifications are indicated in each image
3.2.4. Frequency and distribution of Tregs in the tissues
The frequency of FOXP3 expression in CD4+ T‐cells was evaluated by triple immunostaining of CD3, CD8 and FOXP3 (Figure 6A) using 14 HNSCC tissues. In this evaluation, CD3+CD8− T‐cells were measured as CD4+ T‐cells because the staining intensity of commercially available CD4 antibody is weak and CD4 is expressed on monocytes and macrophages. The results are shown in Table 3. The average and SD were 43.5% and 12.2, respectively, which are approximately 1.2 times higher than the results from flow cytometry (CD45RA−FOXP3hi). Any differences in the frequency of FOXP3 expression in CD4+ T‐cells were not found between “immune‐inflamed” cases and “immune‐excluded cases,” or the stroma and the nest. Tregs (CD3+CD8−FOXP3+) distribution in the tissues was evaluated using the same triple staining. Infiltration of Tregs was restricted in the stroma and many of them were distributed along the periphery of the nests in “immune‐excluded” cases, while they were also observed in the nest as equal as the stroma in “immune‐inflamed” cases (Figure 6B). ICOS and GITR was preferentially expressed on FOXP3+ cells (Figure 6C), consistent with the results from flow cytometry. The ICOS and/or GITR‐positive cells were more than 50% positive for FOXP3. However, the expression frequency of PD‐1 on FOXP3+ cells (Figure 6C) was lower than the results from flow cytometry. The sensitivity of immunofluorescence for the detection of PD‐1 is thought to be lower than for flow cytometry. The expression of other ICM (PD‐L2, LSECtin, CTLA‐4, TIM‐3, LAG‐3, OX40 and 4‐1BB) and CD25 in the tissues was not investigated in this study because we could not find satisfactory antibodies for paraffin‐embedded sections, although we tried several commercially available antibodies.
Figure 6.
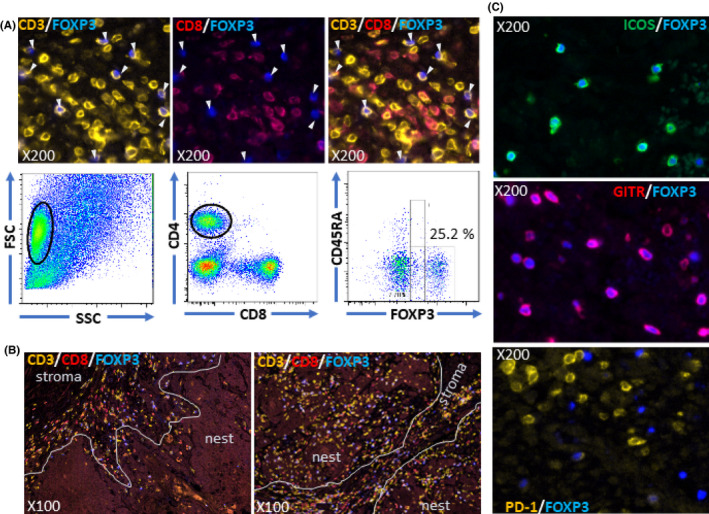
Analysis of Tregs infiltrated into head and neck squamous cell carcinoma (HNSCC) tissues. HNSCC tissues were triple stained with CD3 in orange, CD8 in red and FOXP3 in blue. representative staining images are shown for case 11 (A). The upper left image shows staining with CD3 and FOXP3. All FOXP3+ cells were positive for CD3. The upper middle image shows staining with CD8 and FOXP3. All FOXP3+ cells were negative for CD8. The upper right image shows merging of the left and middle. White arrows indicate FOXP3+ cells. Flow cytometric results of eTreg frequency in CD4+ population from the same case (case 11) are shown in the lower panel. CD3+CD8‐FOXP3+ cells were distributed along the periphery of the nests in the “immune‐excluded” case (case 14, left of B) and were infiltrated both into the nest and stroma in the “immune‐inflamed” case (case 27, right of B). ICOS (C, upper) or GITR (C, middle) were preferentially expressed on FOXP3+ cells but not programmed cell death‐1 (PD‐1) (C, lower). Magnifications are indicated in each image
4. DISCUSSION
We investigated TIME in HNSCC tissues by focusing on the infiltration of eTregs and the expression of ICM.
The eTreg population was evaluated by flow cytometric two‐dimensional analysis of CD4+ fractions using CD45RA and FOXP3 to specifically detect high immunosuppressive fractions. Predominant eTreg infiltration into cancer tissues was elucidated and the infiltrated eTregs were highly activated with high suppressive functions based on their high expression levels of CD25, FOXP3 and ICM. The eTreg frequency in TIL from HNSCC in this study (n = 24, average; 36.6%) was higher than that of melanoma (n = 7 average; 4.65%), 36 gastric cancer (n = 35 average; 9.37%‐36.0%), 37 colon cancer (n = 35 average; close to 18%) 25 and non–small cell lung cancer (n = 20, average; 9.8%). 38 In contrast, there were few CD45RA−FOXP3lo non–Tregs in TIL from HNSCC compared with these tumors. The relevance of eTregs to create an immuno‐suppressive microenvironment was considered higher in HNSCC than in these other tumors.
Immune‐checkpoint molecules on eTregs are related to their function and activation, suggesting that ICM are important targets of novel immunotherapy for HNSCC based on regulation of eTregs. For example, CTLA‐4 is involved in the exhaustion of T‐cells but is also an effector molecule of eTregs that sequesters CD28 from B7 molecules such as CD80 and CD86 expressed on antigen presenting cells. 39 Transforming growth factor (TGF)‐β and the ectonucleotidases CD39 and CD73 are important effector molecules of eTregs. Although we did not investigate the expressions of these molecules, it was previously reported that LAP (membrane‐bound active form of TGF‐β) and CD39 were highly expressed by FOXP3+ Tregs and that CD39+FOXP3+ Tregs simultaneously express CTLA‐4 and TIM‐3. 40 CTLA‐4 was highly expressed on the eTregs of TIL in HNSCC patients, whereas a lower expression of CTLA‐4 was observed on Tconvs, especially CD8+ Tconvs. Interestingly, a CTLA‐4‐positive population in CD4+ Tconvs was significantly correlated with positive populations of stimulatory ICM. However, these correlations were not observed for eTregs. This suggests that CTLA‐4 in CD4+ Tconvs is expressed upon growth stimulation in HNSCC tissues; in contrast, CTLA‐4 in eTregs is constitutively expressed, in accord with a recent study. 41 This indicates that CTLA‐4 functions as an effector molecule of eTregs rather than the exhaustion of Tconvs in HNSCC tissues.
In addition, PD‐L1/PD‐1 interactions augment Treg proliferation and maintain Treg activation status dependent on the lipid phosphatase, phosphatase and tensin homolog (PTEN), 42 and are critical for the extrathymic differentiation of peripherally‐induced Treg cells in vivo. 43 These previous reports and our findings about the expression and distribution of PD‐L1 and PD‐1 in the tissues indicate that the exhaustion of Tconvs and the activation and functional enhancement of eTregs simultaneously occur in HNSCC tissues through PD‐L1/PD‐1 interactions, which deeply suppress the cancer immunity. This suggested that anti–PD‐1 antibody medicines function not only as an immune‐checkpoint blocker but also as an eTreg regulator in HNSCC. However, it was recently reported that PD‐L1/PD‐1 interaction inhibits eTreg activation and PD‐1 blockade causes rapid cancer progression, referred to as hyperprogressive disease. 44 Other recent reports also suggest that PD‐1 expressed on Tregs is related to suppression or exhaustion of Tregs. 45 , 46 For further discussion, we investigated the association among ICM‐positive populations in TIL. PD‐1‐positive populations were correlated with 4‐1BB and GITR in eTregs but not in CD4+ Tconvs and CD8+ Tconvs, which may indicate that PD‐1 expression in eTregs is activation dependent and independent both in CD4+ Tconvs and CD8+ Tconvs. In addition, the PD‐1+ eTregs highly expressed 4‐1BB, which is a marker for T‐cell activation and proliferation, rather than the Tconvs. Therefore, PD‐1 expression may represent exhaustion in Tconvs but activation in eTregs. This suggests that PD‐1+ eTregs are not exhausted and supports the results of a recent study, 47 showing that the expression of genes related to proliferation and suppressive function were positive for PD‐1 in Tregs. However, our discussion is based only on the results of ICM expression by flow cytometric analysis. Therefore, further investigations with functional analysis should be performed using eTregs. Thus, the roles of PD‐1 on eTregs are still unclear. We are continuing to investigate the roles of PD‐1 on eTregs because this might provide an important basis for the development of novel cancer treatments using ICM inhibitors.
The eTreg population in HNSCC tissues was correlated with the PD‐1‐positive population in CD8+ Tconvs. This suggest that eTreg additively suppress cancer immunity together with PD‐1 expressing on CD8+ Tconvs. Combination therapy of ICM inhibitors and eTreg targeting agents is considered to be synergistic for depression of cancer‐immune suppression. It is reported that a refractory HNSCC patient was successfully treated with anti–PD‐1, nivolumab in combination with anti–CTLA‐4, ipilimumab. 48 Although the action mechanism of ipilimumab is not fully understood, the Tregs targeting mechanism might be important. Ipilimumab target Tregs in melanoma tissue by cell‐mediated cytotoxicity. 30 However, combination therapy with nivolumab and ipilimumab has limitations for use in all patients due to severe adverse events such as colitis, pneumonitis and hepatilis. 49 , 50 Because severe adverse events are limited to skin rash in mogamulizumab treatment, 30 , 51 mogamulizumab is considered manageable for the adverse events rather than ipilimumab. Expression of CCR4 and CTLA‐4 on PBL or TIL from HNSCC patients was compared. Almost all eTregs in TIL were double positive for CCR4 and CTLA‐4, whereas these molecules’ expression was reciprocal in Tconvs. This indicates that population in the Tconv targeted by mogamulizumab is different from that of ipilimumab, although both antibodies target the same population of eTregs. In PBL, the expression level of CCR4 on eTregs was higher than CTLA‐4, and CCR4 was highly expressed on the subpopulation of Tconvs, which was almost negative for CTLA‐4. Thus, the cells targeted by mogamulizumab were different to those of ipilimumab. These findings are interesting for the development of combination therapy of mogamulizumab and anti–PD‐1, such as nivolumab and pembrolizumab.
In addition to CTLA‐4 and PD‐1, the other ICM, 4‐1BB, 52 ICOS, 53 OX40, 54 GITR, 55 TIM‐3 56 and LAG‐3, 57 which are associated with the activation and suppressive functions of eTregs, were highly expressed on eTregs, except for LAG‐3 in HNSCC tissues. In addition, ligands of TIM‐3, galectin‐9 and CEACAM‐1 were frequently expressed in HNSCC tissues. Although other ligands’ expression could not be investigated, many kinds of ICM were considered to be involved in the activation of eTregs in HNSCC so that novel immunotherapy based on eTreg regulation by medicines targeting ICM would be expected. Many antibody medicines to ICM which enhance activation and inhibit exhaustion of Tconvs are under development. In developing ICM antibody medicines, it is necessary to comprehensively examine the effects on both Tconvs and Tregs.
The eTreg population in NM‐DLNL is equal to PBL, and significantly lower than in M‐DLNL and TIL. This indicates that the immune‐response functions in non–metastatic DLN in HNSCC patients remain normal and can fight against the cancer cells. Indeed, for head and neck cancer surgical treatment, neck dissection has been widely used and accepted as the most appropriate surgical treatment. Initially, radical neck dissection that removed almost all NM‐DLN and also M‐DLN was accepted, but functional neck dissection was approved as a safer operation. 58 However, during this type of operation the surgeon could not detect which DLN were metastatic or non–metastatic. From our results, it is considered that the NM‐DLN should be preserved in the primary surgical operation for the adjuvant immunotherapy and/or the immunotherapy after recurrence. Further studies on TIME in the DLN and the cancer tissues are necessary for improvement of HNSCC immunotherapy and also for novel surgical oncological treatment. Interestingly, the eTreg population in TCM was higher in DLNL than TIL. Because TCM generally has high growth capacity and a longer life than TEM, it is considered that DLNL are important as the source of eTregs in TIL and, therefore, eTregs in DLNL are more important as a target for eTreg targeting therapy.
In this study, ICM expressions and eTreg frequency in TIL were investigated by both flow cytometry and immunohistochemistry. Expression frequency of ICOS and GITR on T‐cells or Tregs, and FOXP3 expression frequency in CD4+ T‐cells by immunohistochemistry were comparable to those by flow cytometry. However, PD‐1 expressing on eTregs could not be clearly detected by immunohistochemistry due to the low sensitivity. Improvement of the sensitivity is required. Because the distribution of T‐cell subsets and ICM in the tissues can be investigated by immunohistochemistry, investigations by flow cytometry and immunohistochemistry are important for a comprehensive understanding of TIME. However, the sample numbers in this study were too small for clinicopathological analysis of TIME in HNSCC. Further studies with larger samples are necessary.
In conclusion, our results demonstrated that ICM function in the immune‐checkpoint system but also have important roles for the elicitation of eTreg activation and functions, which might create a tumor suppressive immune microenvironment in HNSCC tissues. Many combination immunotherapies with immune‐checkpoint inhibitors are being investigated in clinical trials for HNSCC. 59 It is important to choose drug combinations that effectively inhibit eTreg functions and immune‐checkpoint systems to improve HNSCC immunotherapy.
DISCLOSURE
Dr Ueda has received consulting remuneration from Terumo and joint research funding from Kyowa Kirin, and belongs to an endowed chair founded by Chugai Pharmaceutical, Ono Pharmaceutical, Rikaken and Medical & Biological Laboratories. Dr Tsuzuki has received consulting remuneration from Chugai Pharmaceutical and honoraria from AstraZeneca plc. Dr Suzuki has received consulting remuneration from Medical & Biological Laboratories. All the other authors declare no competing interests.
Supporting information
Fig S1‐S6
Table S1‐S5
ACKNOWLEDGMENTS
We thank Ms Kyoko Okumura for excellent secretarial assistance. This work was supported by JSPS KAKENHI Grant Numbers C18K07277 and C17K11412, AMED Grant Number JP17ck0106159, the Chubu Regional Consortium for Advanced Medicine (C‐CAM) Sead‐A24 and a research grant from Aichi Medical University.
Suzuki S, Ogawa T, Sano R, et al. Immune‐checkpoint molecules on regulatory T‐cells as a potential therapeutic target in head and neck squamous cell cancers. Cancer Sci. 2020;111:1943–1957. 10.1111/cas.14422
Susumu Suzuki and Tetsuya Ogawa contributed equally to this study.
REFERENCES
- 1. Chaturvedi AK, Anderson WF, Lortet‐Tieulent J, et al. Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers. J Clin Oncol. 2013;31:4550‐4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Center for Cancer Control and Information Services, National Cancer Centre . Projected Cancer Statistics, 2018. https://ganjoho.jp/en/public/statistics/short_pred.html [Google Scholar]
- 3. Gibson MK, Li Y, Murphy B, et al. Randomized phase III evaluation of cisplatin plus fluorouracil versus cisplatin plus paclitaxel in advanced head and neck cancer (E1395): an intergroup trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2005;23:3562‐3567. [DOI] [PubMed] [Google Scholar]
- 4. Forastiere AA, Goepfert H, Maor M, et al. Concurrent chemotherapy and radiotherapy for organ preservation in advanced laryngeal cancer. N Engl J Med. 2003;349:2091‐2098. [DOI] [PubMed] [Google Scholar]
- 5. Adelstein DJ, Li Y, Adams GL, et al. An intergroup phase III comparison of standard radiation therapy and two schedules of concurrent chemoradiotherapy in patients with unresectable squamous cell head and neck cancer. J Clin Oncol. 2003;21:92‐98. [DOI] [PubMed] [Google Scholar]
- 6. Cooper JS, Pajak TF, Forastiere AA, et al. Postoperative concurrent radiotherapy and chemotherapy for high‐risk squamous‐cell carcinoma of the head and neck 9501/Intergroup. N Engl J Med. 2008;359:1116‐1127. [DOI] [PubMed] [Google Scholar]
- 7. Vermorken JB, Mesia R, Rivera F, et al. Platinum‐based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116‐1127. [DOI] [PubMed] [Google Scholar]
- 8. Harrington KJ, Ferris RL, Blumenschein G Jr, et al. Nivolumab versus standard, single‐agent therapy of investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck (CheckMate 141): health‐related quality‐of‐life results from a randomised, phase 3 trial. Lancet Oncol. 2017;18:1104‐1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mehra R, Seiwert TY, Gupta S, et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: pooled analyses after long‐term follow‐up in KEYNOTE‐012. Br J Cancer. 2018;119:153‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56‐61. [DOI] [PubMed] [Google Scholar]
- 11. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16:553‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shitara K, Nishikawa H. Regulatory T cells: a potential target in cancer immunotherapy. Ann N Y Acad Sci. 2018;1417:104‐115. [DOI] [PubMed] [Google Scholar]
- 13. Marvel D, Gabrilovich DI. Myeloid‐derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125:3356‐3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aras S, Zaidi MR. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. 2017;117:1583‐1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sakaguchi S, Sakaguchi N, Asano M, et al. Immunologic self‐tolerance maintained by activated T cells expressing IL‐2 receptor alpha‐chains (CD25). Breakdown of a single mechanism of self‐tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151‐1164. [PubMed] [Google Scholar]
- 16. Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211‐5218. [PubMed] [Google Scholar]
- 17. Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899‐911. [DOI] [PubMed] [Google Scholar]
- 18. Tao H, Mimura Y, Aoe K, et al. Prognostic potential of FOXP3 expression in non–small cell lung cancer cells combined with tumor‐infiltrating regulatory T cells. Lung Cancer. 2012;75:95‐101. [DOI] [PubMed] [Google Scholar]
- 19. Merlo A, Casalini P, Carcangiu ML, et al. FOXP3 expression and overall survival in breast cancer. J Clin Oncol. 2009;27:1746‐1752. [DOI] [PubMed] [Google Scholar]
- 20. Wolf D, Wolf AM, Rumpold H, et al. The expression of the regulatory T cell‐specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin Cancer Res. 2005;11:8326‐8331. [DOI] [PubMed] [Google Scholar]
- 21. Jensen HK, Donskov F, Nordsmark M, et al. Increased intratumoral FOXP3‐positive regulatory immune cells during interleukin‐2 treatment in metastatic renal cell carcinoma. Clin Cancer Res. 2009;15:1052‐1058. [DOI] [PubMed] [Google Scholar]
- 22. Gerber AL, Münst A, Schlapbach C, et al. High expression of FOXP3 in primary melanoma is associated with tumour progression. Br J Dermatol. 2014;170:103‐109. [DOI] [PubMed] [Google Scholar]
- 23. Watanabe Y, Katou F, Ohtani H, et al. Tumor‐infiltrating lymphocytes, particularly the balance between CD8(+) T cells and CCR4(+) regulatory T cells, affect the survival of patients with oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109:744‐752. [DOI] [PubMed] [Google Scholar]
- 24. Lu J, Chen XM, Huang HR, et al. Detailed analysis of inflammatory cell infiltration and the prognostic impact on nasopharyngeal carcinoma. Head Neck. 2018;40:1245‐1253. [DOI] [PubMed] [Google Scholar]
- 25. Saito T, Nishikawa H, Wada H, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016;22:679‐684. [DOI] [PubMed] [Google Scholar]
- 26. Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tauriello DVF, Palomo‐Ponce S, Stork D, et al. TGF‐β drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538‐543. [DOI] [PubMed] [Google Scholar]
- 28. Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res. 2016;22:1865‐1874. [DOI] [PubMed] [Google Scholar]
- 29. Kurose K, Ohue Y, Wada H, et al. Phase Ia Study of FoxP3+ CD4 treg depletion by infusion of a humanized anti–CCR4 antibody, KW‐0761, in cancer patients. Clin Cancer Res. 2015;21:4327‐4336. [DOI] [PubMed] [Google Scholar]
- 30. Romano E, Kusio‐Kobialka M, Foukas PG, et al. Ipilimumab‐dependent cell‐mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. 2015;12:6140‐6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first‐line treatment for advanced non–small‐cell lung cancer (CheckMate 012): results of an open‐label, phase 1, multicohort study. Lancet Oncol. 2017;18:31‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fuchs CS, Doi T, Jang RW, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 Clinical KEYNOTE‐059 Trial. JAMA Oncol. 2018;4:e180013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus Ipilimumab versus Sunitinib in advanced renal‐cell carcinoma. N Engl J Med. 2018;378:1277‐1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sharma P, Callahan MK, Bono P, et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open‐label, two‐stage, multi‐arm, phase 1/2 trial. Lancet Oncol. 2016;17:1590‐1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006‐2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fujii H, Arakawa A, Kitoh A, et al. Perturbations of both nonregulatory and regulatory FOXP3+ T cells in patients with malignant melanoma. Br J Dermatol. 2011;164:1052‐1060. [DOI] [PubMed] [Google Scholar]
- 37. Nagase H, Takeoka T, Urakawa S, et al. ICOS+ Foxp3+ TILs in gastric cancer are prognostic markers and effector regulatory T cells associated with Helicobacter pylori. Int J Cancer. 2017;140:686‐695. [DOI] [PubMed] [Google Scholar]
- 38. Kurose K, Ohue Y, Sato E, et al. Increase in activated Treg in TIL in lung cancer and in vitro depletion of Treg by ADCC using an antihuman CCR4 mAb (KM2760). J Thorac Oncol. 2015;10:74‐83. [DOI] [PubMed] [Google Scholar]
- 39. Maeda Y, Nishikawa H, Sugiyama D, et al. Detection of self‐reactive CD8⁺ T cells with an anergic phenotype in healthy individuals. Science. 2014;346:1536‐1540. [DOI] [PubMed] [Google Scholar]
- 40. Jie HB, Gildener‐Leapman N, Li J, et al. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer. 2013;12:2629‐2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tanaka A, Kibayashi T, Tanemura A, et al. Differential control of human Treg and effector T cells in tumor immunity by Fc‐engineered anti–CTLA‐4 antibody. Proc Natl Acad Sci U S A. 2019;116:609‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sharma MD, Shinde R, McGaha TL, et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci Adv. 2015;1:e1500845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen X, Fosco D, Kline DE, et al. PD‐1 regulates extrathymic regulatory T‐cell differentiation. Eur J Immunol. 2014;44:2603‐2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kamada T, Togashi Y, Tay C, et al. PD‐1+ regulatory T cells amplified by PD‐1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A. 2019;116:9999‐10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang B, Chikuma S, Hori S, et al. Nonoverlapping roles of PD‐1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Fagarasan S, Honjo T. Proc Natl Acad Sci U S A. 2016;113:8490‐8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mandal R, Senbabaoglu Y, Desrichard A, et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight. 2016;1:e89829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matoba T, Imai M, Ohkura N, et al. Regulatory T cells expressing abundant CTLA‐4 on the cell surface with a proliferative gene profile are key features of human head and neck cancer. Int J Cancer. 2019;144:2811‐2822. [DOI] [PubMed] [Google Scholar]
- 48. Schwab KS, Kristiansen G, Schild HH, et al. Successful treatment of refractory squamous cell cancer of the head and neck with Nivolumab and Ipilimumab. Case Rep Oncol. 2018;4:17‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bowyer S, Prithviraj P, Lorigan P, et al. Efficacy and toxicity of treatment with the anti–CTLA‐4 antibody ipilimumab in patients with metastatic melanoma after prior anti–PD‐1 therapy. Br J Cancer. 2016;114:1084‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Larkin J, Chiarion‐Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ogura M, Ishida T, Hatake K, et al. Multicenter phase II study of mogamulizumab (KW‐0761), a defucosylated anti–cc chemokine receptor 4 antibody, in patients with relapsed peripheral T‐cell lymphoma and cutaneous T‐cell lymphoma. J Clin Oncol. 2014;32:1157‐1163. [DOI] [PubMed] [Google Scholar]
- 52. Zheng G, Wang B, Chen A. The 4–1BB costimulation augments the proliferation of CD4+CD25+ regulatory T cells. J Immunol. 2004;173:2428‐2434. [DOI] [PubMed] [Google Scholar]
- 53. Ito T, Hanabuchi S, Wang YH, et al. Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity. 2008;28:870‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marabelle A, Kohrt H, Sagiv‐Barfi I, et al. Depleting tumor‐specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. 2013;123:2447‐2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. van Olffen RW, Koning N, van Gisbergen KP, et al. GITR triggering induces expansion of both effector and regulatory CD4+ T cells in vivo. J Immunol. 2009;182:7490‐7500. [DOI] [PubMed] [Google Scholar]
- 56. Gautron AS, Dominguez‐Villar M, de Marcken M, et al. Enhanced suppressor function of TIM‐3+ FoxP3+ regulatory T cells. Eur J Immunol. 2014;44:2703‐2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang CT, Workman CJ, Flies D, et al. Role of LAG‐3 in regulatory T cells. Immunity. 2004;21:503‐513. [DOI] [PubMed] [Google Scholar]
- 58. Bocca E, Pignataro O, Oldini C, et al. Functional neck dissection: an evaluation and review of 843 cases. Laryngoscope. 1984;94:942‐945. [DOI] [PubMed] [Google Scholar]
- 59. Economopoulou P, Kotsantis I, Psyrri A. The promise of immunotherapy in head and neck squamous cell carcinoma: combinatorial immunotherapy approaches. ESMO Open. 2017;1:e000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S6
Table S1‐S5