

Abstract
KRAS mutation is frequently seen in a subtype of ovarian cancer categorized as type 1. The KRAS‐MAPK pathway, which is closely involved in type 1 cancer progression, is under the regulation of receptor tyrosine kinases (RTKs). AXL, one of the RTKs, has been reported to be overexpressed in ovarian cancer and contributes to the poor prognosis. However, there is no useful target‐based agent against such gene profiles. We examined the combined effect of the dual RAF/MEK inhibitor CH5126766 and AXL inhibitor R428 on the growth of ovarian cancer HEY‐T30 and OVCAR‐5 cell lines, both of which bear KRAS mutation and express AXL at a high level, using the WST‐8 assay and the colony formation assay. The synergistic effect of the combination was evaluated by the combination index. The apoptotic cells were analyzed by flow cytometry. The expression of apoptotic proteins and the phosphorylation of MAPK and AKT pathway proteins were investigated by western blotting. We found that CH5126766 and R428 suppressed the phosphorylation of ERK and AKT, respectively, and their combination synergistically inhibited the growth of both cell lines with enhancement of apoptosis accompanied by the Bim upregulation. Combined treatment with CH5126766 and R428 is expected as the novel therapeutic option for KRAS‐mutated ovarian cancer with high expression of AXL.
Keywords: AXL inhibitor, combined treatment, dual RAF/MEK inhibitor, KRAS, ovarian cancer
The combined treatment with a dual RAF/MEK inhibitor CH5126766 and AXL inhibitor R428 synergistically inhibited the growth of ovarian cancer HEY‐T30 and OVCAR‐5 cell lines, both of which bear KRAS mutation and express AXL at a high level, accompanied by inducing apoptosis.

1. INTRODUCTION
Ovarian cancer shows a wide variety of pathological characteristics, due to the diversity of gene profiles and mechanism of carcinogenesis. 1 Based on recent studies, ovarian cancer is histologically categorized into 2 broad subtypes, type 1 and 2. 2 , 3 , 4 Type 1 cancer, including low‐grade serous adenocarcinoma, endometrioid adenocarcinoma, mucinous adenocarcinoma, and clear cell carcinoma, is thought to evolve in a stepwise fashion from benign ovarian cystic lesions through a precancerous condition referred to as a borderline malignant tumor, as the consequence of the accumulation of gene mutations. KRAS mutation is the most common, especially in low‐grade serous and mucinous adenocarcinoma. Frequency of KRAS mutation in these type 1 cancers varies among reports, approximately 30%‐50% in low‐grade serous, 5 , 6 50%‐60% in mucinous, 7 , 8 10% in endometrioid, 7 , 8 and 4%‐20% in clear cell carcinoma. 8 , 9 High‐grade serous adenocarcinoma, classified as type 2, is thought to emerge de novo from normal epithelial cells of the fallopian tube due to genome instability caused by p53 mutation, and rarely bear KRAS mutation. 10 As the sensitivity to conventional chemotherapy is rather poor in type 1 compared with that in type 2, 11 a novel therapeutic strategy that is effective against type 1 cancer is needed. However, current conventional chemotherapy does not provide different methods that consider the histological types based on differences in gene profiles.
The RAS‐RAF‐MEK‐ERK pathway, a part of the MAPK signaling cascades, plays a pivotal role in cell growth, and aberrant regulation of this pathway is closely involved in cancer progression. KRAS mutation is the most common among members of this pathway and regarded as the driver oncogene in some cancers. As type 1 ovarian cancer bears KRAS mutation at a high frequency, the RAS‐MAPK pathway would be a key factor in the development of ovarian cancer and so an essential therapeutic target. To date, some clinical studies on low‐grade serous ovarian cancer using MEK inhibitors have been carried out. 6 , 12
The MAPK pathways, and many other pathways regulating cell growth and cancer development, are under the control of receptor tyrosine kinases (RTKs). Receptor tyrosine kinases are transmembrane receptors that transfer extracellular signals into cells. In humans, 58 RTKs classified into 20 families have been identified. 13 Aberrant regulation of RTKs causes excessive activation of their downstream signal cascades, resulting in uncontrolled cell growth. In addition, RTK signaling mediates chemosensitivity and drug resistance in anticancer treatment through interaction with other RTKs. 14 Treatment strategies targeting some RTKs such as epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), human epidermal growth factor receptor 2 (ErbB2/HER2), and Kit have been developed and are already widely applied in clinical settings. AXL, originally cloned from patients with chronic myelogenous leukemia, is one of the mammalian RTKs and belongs to the TAM receptor family. AXL is expressed in a wide range of human cells and tissues and regulates cell survival and growth, cell adhesion and migration, and inflammatory cytokine release. 15 AXL overexpression has been reported in various malignancies including ovarian cancer, 16 and a correlation with poor prognosis has also been reported. 17 , 18 , 19 AXL also dimerizes with other RTKs, such as EGFR, and activates its downstream pathway through reciprocal phosphorylation, resulting in further cancer progression and therapeutic resistance. 20 , 21
In one of the latest epidemiologic studies by Köbel et al, 22 the incidence of low‐grade serous, mucinous, endometrioid, and clear cell carcinoma was reported to be 3.4%, 3.4%, 11.3%, and 12.2%, respectively. Combined with the percentage of KRAS mutation described above, and the report that over 70% of ovarian cancer patients show AXL overexpression, 16 it is inferred that there is a certain percentage of patients bearing KRAS mutation with high AXL expression.
Both the RAS‐MAPK pathway and AXL are crucial factors in ovarian cancer development, and thus we examined the combined effect of the inhibitors focusing on KRAS mutation and high AXL expression. CH5126766 was developed by our group as a dual inhibitor of both RAF and MEK. 23 By inhibiting both RAF and MEK, CH5126766 was confirmed to suppress the RAS‐MAPK pathway more strongly than a conventional MEK inhibitor. 23 , 24 , 25 R428, which is also named BGB324 and bemcentinib, is a selective small molecule inhibitor of AXL. 26 The antigrowth effect of this drug on various hematological or solid cancers has been reported, and some clinical trials have already been undertaken. 21 , 27 The present paper provides several lines of evidence that combined treatment with CH5126766 and R428 exerts a synergistic antitumor effect against KRAS‐mutated ovarian cancer cells with high AXL expression.
2. MATERIALS AND METHODS
2.1. Cell lines
Human ovarian cancer HEY‐T30 and TOV‐21G cells were purchased from the ATCC. MCAS cells were purchased from the Japanese Collection of Research Bioresources Cell Bank. OVCAR‐5, SKOV3, IGROV‐1, and OVCAR‐3 cells were obtained as cell lines of NCI‐60 from the NCI Developmental Therapeutics Program. HEY‐T30, SKOV3, IGROV‐1, and OVCAR‐3 cells were maintained in RPMI‐1640 (Nissui Pharmaceuticals) containing 10% FBS, 2 mmol/L glutamine, 50 units/mL streptomycin, and 100 µg/mL penicillin G at 37°C in 5% CO2. OVCAR‐5, MCAS, and TOV‐21G cells were maintained in DMEM (Nissui Pharmaceuticals) containing 10% FBS and 4 mM glutamine.
2.2. Reagents and antibodies
AXL inhibitor R428, ERK inhibitor SCH772984, and AKT inhibitor MK2206 were purchased from Selleck Chemicals. The dual RAF/MEK inhibitor CH5126766 was a kind gift from Chugai Pharmaceutical. Inhibitors were dissolved in DMSO. These agents and DMSO were diluted 1:1000 in culture medium.
Primary Abs were as follows: anti‐AXL (#8661), anti‐AKT (#9272), anti‐phosphorylated (p)‐AKT (Ser473, #4060), anti‐ERK (#9102), anti‐p‐ERK (Tyr202/Tyr204, #9101), anti‐MEK (#9122), anti‐p‐MEK (Ser217/221, #9121), anti‐Bim (#2933), and anti‐Bcl‐xL (#2762) were purchased from Cell Signaling Technology. Anti Bcl‐2 (#sc‐7382) was purchased from Santa Cruz Biotechnology. Anti‐GAPDH (#5G4) was purchased from HyTest. Anti‐β‐actin (A5441) was purchased from Sigma‐Aldrich. Human phospho‐RTK array kit (#ARY001B) was purchased from R&D Systems.
2.3. Cell growth assay
Cells were seeded in 96‐well plates (HEY‐T30, 250 cells/well; OVCAR‐5 and OVCAR‐3, 2000 cells/well; SKOV3, 1000 cells/well). Twenty‐four hours after seeding, the culture medium was replaced with fresh medium and indicated concentrations of anticancer agents. Cells were then incubated for 72 hours and cell viabilities were measured using the CCK‐8 assay (Dojindo Laboratories) according to the manufacturer’s instructions.
2.4. Cytotoxicity assay
Cells were seeded in 96‐well plates (HEY‐T30, 3000 cells/well; OVCAR‐5 and OVCAR‐3, 4000 cells/well). Twenty‐four hours after seeding, the culture medium was replaced by that with indicated concentrations of anticancer agents. Cells were then incubated for 24 hours and cytotoxicity indicated by LDH elimination were measured using a Cytotoxicity LDH Assay Kit (Dojindo Laboratories) according to the manufacturer’s instructions.
2.5. Colony formation assay
Cells were seeded in 6‐well plates (HEY‐T30, 200 cells/well; OVCAR‐5, 1000 cells/well). Twenty‐four hours after seeding, the culture medium was replaced with fresh medium and indicated concentrations of anticancer agents. After 72 hours of incubation, the medium was washed out. Cells were then incubated with growth medium until visible colonies were formed (HEY‐T30, 5 days; OVCAR‐5, 7 days after the washout). Colonies were fixed with 10% formaldehyde and then stained with 0.1% crystal violet, and the colony number was counted. The area occupied by colonies on the digital images was measured using ImageJ software. 28
2.6. Combination index
The combination index (CI) values were calculated using the CalcuSyn software program (Biosoft). In accordance with software criteria, synergism is defined as more than the expected additive effect with a CI less than 1.0.
2.7. Detection of apoptosis
HEY‐T30 cells were seeded at 5 × 103 cells/well, and OVCAR‐5 cells were seeded at 4 × 104 cells/well, incubated in 6‐well plates for 24 hours, and treated with CH5126766 and/or R428 at the indicated concentrations. After 24 or 72 hours of incubation, the analysis of DNA content was undertaken with FACSCaliber (Becton Dickinson). The hypodiploid population (sub‐G1) was quantified using the CellQuest software program (Becton Dickinson) to detect apoptosis.
2.8. Western blot assays
Cells treated with agents were lysed with RIPA buffer supplemented with protease inhibitors (#25955; Nacalai Tesque) and phosphatase inhibitors (#07574; Nacalai Tesque). An equal amount of protein was electrophoresed on a Mini‐PROTEAN TGX gel (Bio‐Rad Laboratories) and then transferred onto a PVDF membrane (Immobilon‐P transfer membrane; Merck‐Millipore). Membranes were incubated with specific primary Ab overnight at 4°C. Anti‐rabbit or anti‐mouse IgG and HRP‐linked Ab (GE Healthcare) were used to detect primary Ab binding. The binding was visualized by ECL chemiluminescent detection reagent (Chemi‐Lumi One; Nacalai Tesque, or Immobilon Western Chemiluminescent HRP Substrate; Merck‐Millipore), and imaged and quantified by the ChemiDoc Imaging System (Bio‐Rad). All immunoblots are representative of a minimum of 3 independent experiments.
3. RESULTS
3.1. Combined treatment with CH5126766 and R428 synergistically reduces the growth of ovarian cancer cell lines bearing KRAS mutation with high AXL expression
First, to identify RTKs that markedly influence ovarian cancer progression, we screened the RTK activity of KRAS‐mutated ovarian cancer cell line HEY‐T30 cells by phospho‐RTK array and found that AXL, EGFR, and ROR‐2 were highly phosphorylated (Figure 1A). In addition, a number of reports have shown that high AXL expression is detected in most malignant tumors, including ovarian cancer. 16 As mentioned above, AXL activity causes resistance to inhibitors of other RTKs, including EGFR, 29 , 30 , 31 and we focused on AXL for further investigation. We then examined AXL expression levels of 4 ovarian cancer cell lines harboring KRAS mutation, and 3 cell lines without KRAS mutation, according to the COSMIC database, for comparison (Figure 1B). In KRAS‐mutated cells, HEY‐T30 showed excessively high AXL expression. Three other cell lines with KRAS mutation also showed AXL expression, and OVCAR‐5 showed higher expression among them; HEY‐T30 and OVCAR‐5 were selected for further investigation. In the cells without KRAS mutation, excessively high AXL expression in SKOV3 and no detectable AXL expression was seen in 2 other cell lines, consistent with a previous report. 32
Figure 1.
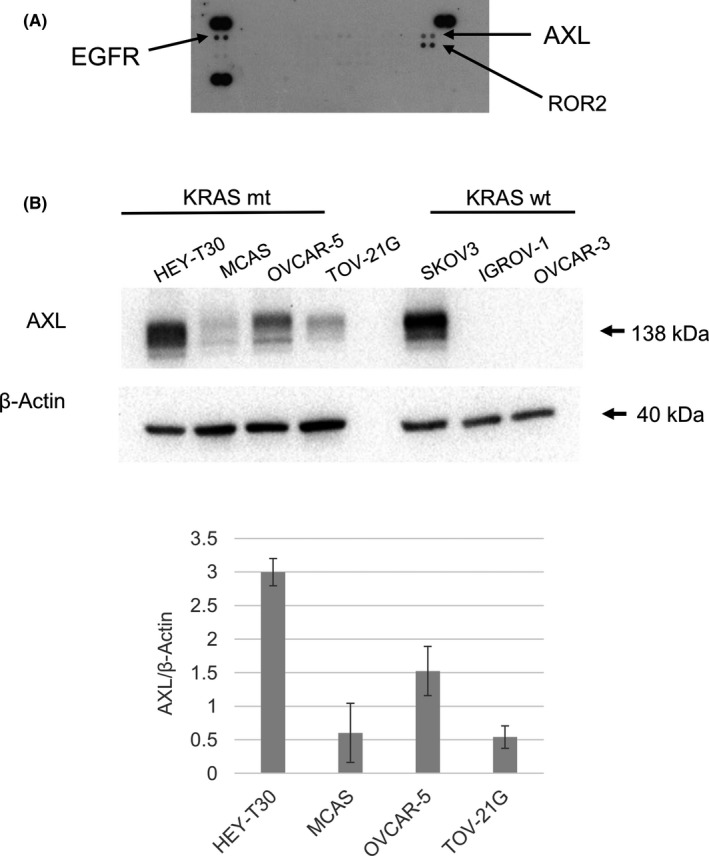
AXL is expressed in KRAS‐mutated ovarian cancer cell lines. A, Phospho‐receptor tyrosine kinase array. KRAS‐mutated ovarian cancer cell line HEY‐T30 cells were evaluated by array analysis. Data are representative of 2 independent experiments. EGFR, epidermal growth factor receptor; ROR2, receptor tyrosine kinase‐like orphan receptor 2. B, Expression levels of AXL were compared among 4 KRAS‐mutated (mt) ovarian cancer cell lines and 3 cell lines without KRAS mutation (wt) by western blot analysis. Cells at 80%‐90% confluency were lysed for analysis. Equal amounts of lysed protein were applied for electrophoresis, and the chemiluminescence signal was quantified. The densitometric analysis data were expressed as relative values compared with loading control (β‐actin) being 1
The individual or combined effects of inhibition of RAS‐MAPK and AXL in these cell lines were examined by cell growth assay. Each single treatment with the dual RAF/MEK inhibitor CH5126766 and AXL kinase inhibitor R428 suppressed the growth of both HEY‐T30 and OVCAR‐5 cells in a dose‐dependent manner, although the dose dependency of single treatment by R428 in OVCAR‐5 cells was lower than in HEY‐T30 cells (Figure 2A). Next, we assessed the effects of combined inhibition of MAPK and AXL on cell growth. The concentration of each drug was set within the range in which each drug alone may exert moderate growth suppression. The cytotoxicity in this concentration range was not observed by LDH assay (Figure S1). More growth suppression with the combination compared with each single treatment was confirmed in both cell lines (Figure 2B). The CI calculated by CalcuSyn at indicated drug concentrations was lower than 1 (0.78 in HEY‐T30 cells and 0.67 in OVCAR‐5 cells), indicating the synergistic effect of this combination (Figure 2B). In addition, this combination also significantly suppressed growth of the colonies in terms of both the number and area compared with each single treatment (Figure 3).
Figure 2.
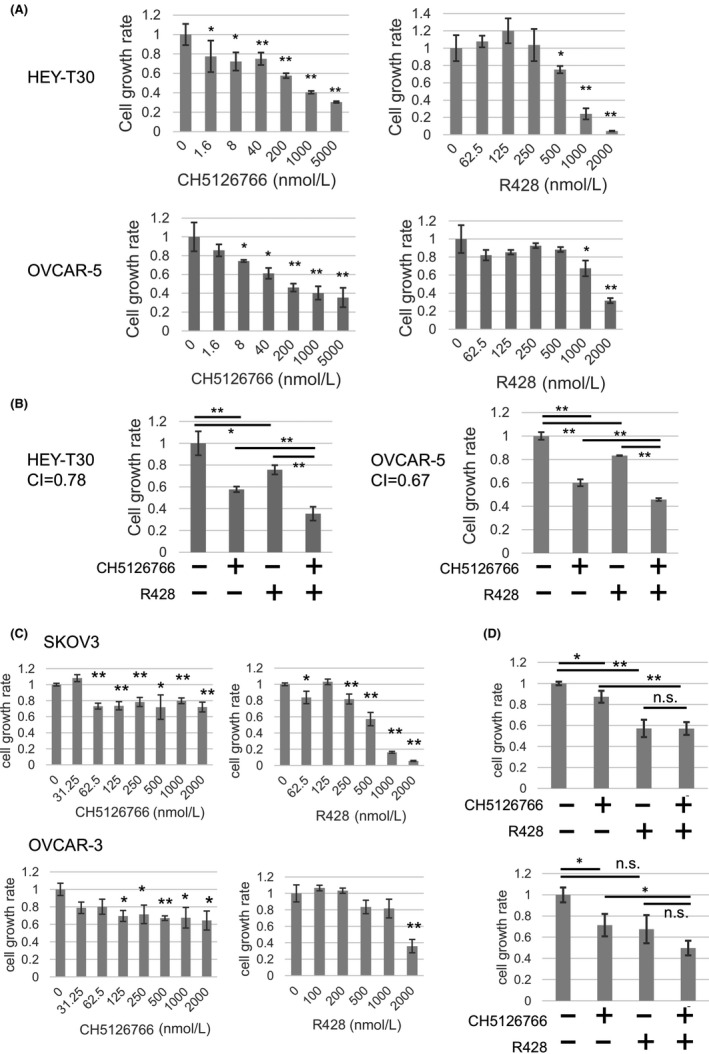
Combined treatment with CH5126766 and R428 synergistically inhibits the growth of both HEY‐T30 and OVCAR‐5 cells. A, HEY‐T30 and OVCAR‐5 cells were treated with CH5126766 or R428 at indicated concentrations. 0 nM indicates the control treated with 0.1% DMSO. B, HEY‐T30 and OVCAR‐5 cells were treated with 0.1% DMSO or CH5126766 and/or R428 for 72 hours. The concentrations of agents applied to each cell line were as follows, and all the following experiments were carried out at the same concentrations: HEY‐T30, 200 nmol/L CH5126766 and/or 500 nmol/L R428; OVCAR‐5 200 nmol/L CH5126766 and/or 1000 nmol/L R428. CI, combination index. C, SKOV3 and OVCAR‐3 cells were treated with CH5126766 or R428 at indicated concentrations. 0 nM indicates the control treated with 0.1% DMSO. D, Evaluation of the combined treatment with 250 nmol/L CH5126766 and/or 500 nmol/L R428. Data represent the mean ± SD of 3 determinations. *P < .05, **P < .01 (vs. control). The cell growth rate of the 0.1% DMSO‐treated cells was regarded as 1. This experiment was carried out at least 3 times for each cell line. n.s., not significant
Figure 3.
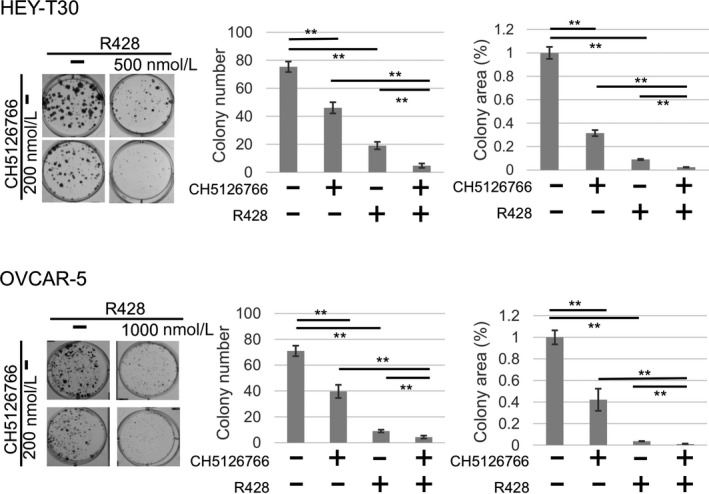
Colony formation assay. HEY‐T30 and OVCAR‐5 cells were treated with 0.1% DMSO or CH5126766 and/or R428 at the indicated concentrations for 72 h. Cells were washed out and incubated with growth medium for 5 (HEY‐T30) or 7 (OVCAR‐5) additional days. Data on the colony area are presented as the percentage compared with the control (DMSO). This experiment was carried out at least 3 times for each cell line. *P < .05, **P < .01
In addition, to confirm whether the combination effect was specific in the cells with KRAS mutation and high AXL expression, we tested the single or combined effect of CH5126766 and R428 in the cell lines without KRAS mutation (Figure 2C,D). In SKOV3 (KRAS wt/AXL high) cells, single treatment with CH5126766 exerted a marginal inhibitory effect that did not show dose‐dependency, reflecting KRAS wt. In contrast, R428 inhibited the growth of SKOV3 cells, reflecting high expression of AXL. Furthermore, no synergistic suppression was shown. In OVCAR‐3 (KRAS wt/AXL low) cells, CH5126766 showed a marginal inhibitory effect as well as in SKOV3, reflecting KRAS wt. R428 exerted growth inhibition in OVCAR‐3 cells, with less dose‐dependency than in SKOV3 or HEY‐T30 (excessively high AXL expression). Significant synergistic growth suppression in combination treatment was not shown. From these results, the combination effect of these 2 drugs was considered as a specific event in cell lines with KRAS mutation and high AXL expression.
3.2. Combined treatment with CH5126766 and R428 enhances apoptosis with induction of proapoptotic protein Bim
We then searched for the mechanism of the combined treatment reducing cell growth in HEY‐T30 cells, by flow cytometry. At 24 hours, each single treatment did not cause detectable apoptosis, as shown in the sub‐G1 population; however, a minor level of apoptosis was induced by combination treatment with statistical significance. However, in a later phase (72 hours after treatment), each single drug caused a mild level of apoptosis and the apoptotic change was more potent when CH5126766 and R428 were combined. The induction of apoptosis at 72 hours was also seen in OVCAR‐5 cells (Figure 4).
Figure 4.
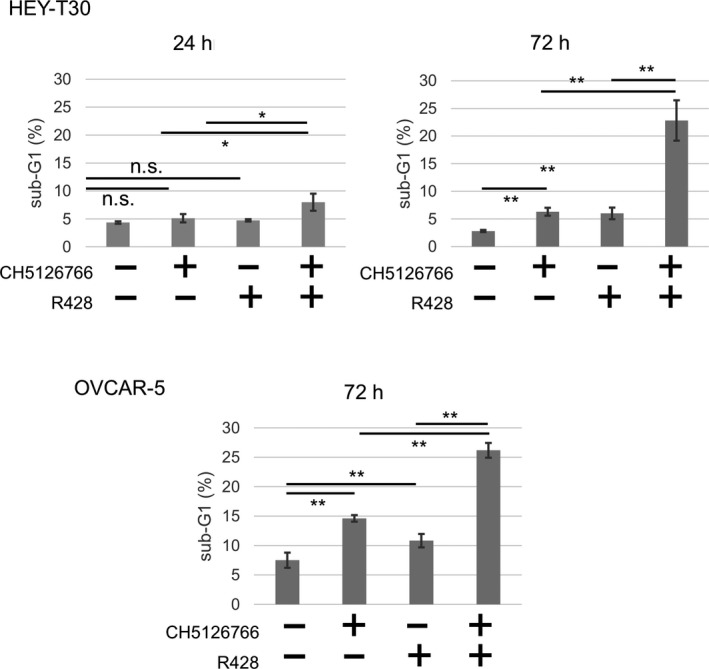
Combined treatment with CH5126766 and R428 enhances apoptosis compared with each treatment alone. HEY‐T30 and OVCAR‐5 cells were treated with 0.1% DMSO or CH5126766 and/or R428 at the indicated concentrations for 24 or 72 h, and the level of apoptosis (sub‐G1 population) was analyzed. Data represent the mean ± SD of 3 determinations. *P < .05, **P < .01. n.s., not significant
Western blot analysis showed that the proapoptotic protein Bim was upregulated by combined treatment at approximately 24 hours (OVCAR‐5) and 72 hours (HEY‐T30) after treatment (Figure 5). CH5126766 treatment alone weakly induced Bim, and it was markedly enhanced by the combination in both cell lines. Other apoptosis‐related proteins, including Bcl‐xL and Bcl‐2, did not show clear changes with either single or combined treatment in any cell line at 72 hours (Figure 5).
Figure 5.
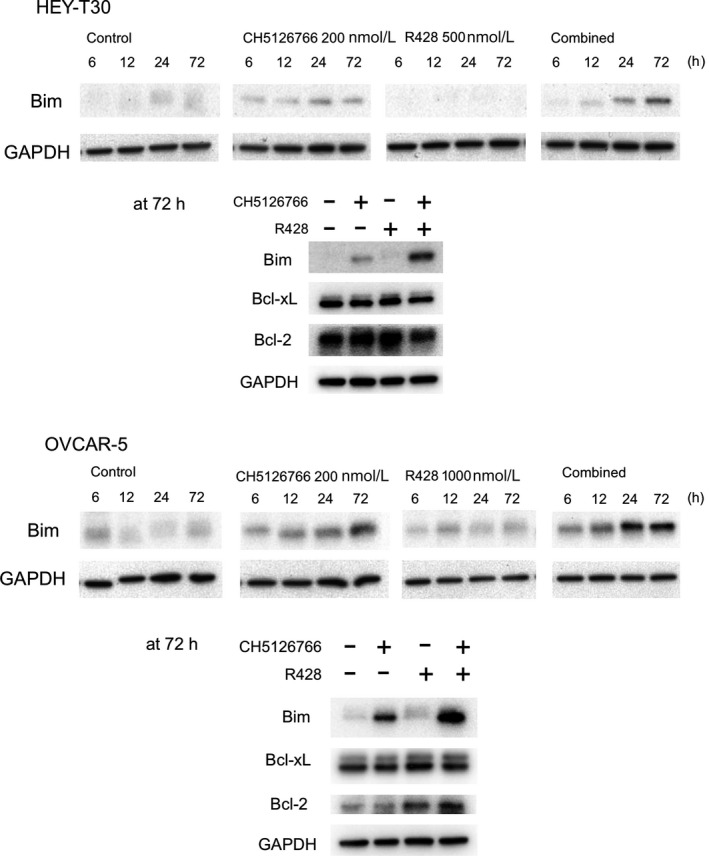
Combined treatment with CH5126766 and R428 induces apoptosis accompanied by Bim induction in both HEY‐T30 and OVCAR‐5 cells. Western blotting of Bim, Bcl‐xL, and Bcl‐2 was undertaken with the lysate treated for 6, 12, 24, and 72 h with or without CH5126766 and/or R428 at the indicated concentrations. GAPDH is shown as a loading control. In cases where a single loading control is shown, a single membrane was run, stripped, and reprobed for the second and further proteins
3.3. Combined inhibition of AXL by R428 and RAF/MEK by CH5126766 cooperatively suppresses phosphorylation of AKT and ERK
We evaluated the phosphorylation status of ERK and AKT, which indicates the activity of MAPK and PI3K‐AKT pathways, respectively, both of which are major upstream regulators of Bim. At 6 hours after treatment, CH5126766 downregulated the phosphorylation of ERK accompanied by the suppression of phosphorylated MEK, and R428 downregulated the phosphorylation of AKT in both cell lines (Figure 6). CH5126766 alone did not affect the AKT status but the downregulation of p‐AKT was augmented by the combination (Figure 6). Combined treatment augmented the downregulation of p‐ERK in OVCAR‐5 cells, whereas in HEY‐T30 cells CH5126766 alone strongly suppressed p‐ERK and the additional effect by R428 was not confirmed in combination.
Figure 6.
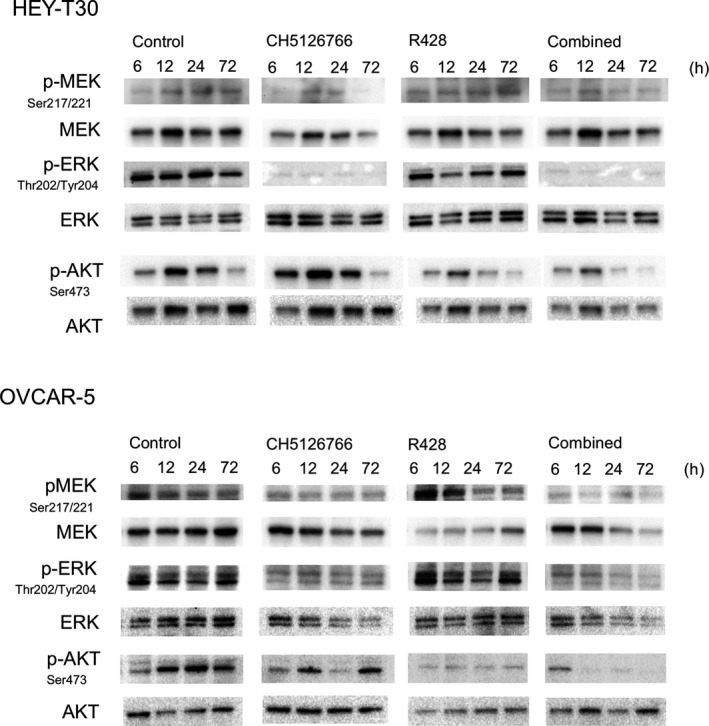
Combined treatment with CH5126766 and R428 cooperatively suppresses phosphorylation of AKT and ERK. Western blotting for phosphorylated (p)‐MEK (Ser217/221), MEK, p‐AKT (Ser473), AKT, p‐ERK (Thr202/Tyr204), and ERK was carried out with lysate treated for 6, 12, 24, and 72 h with or without CH5126766 and/or R428 at the indicated concentrations
4. DISCUSSION
This is the first report verifying the synergistic effect of the dual RAF/MEK inhibitor CH5126766 and AXL kinase inhibitor R428 in ovarian cancer cell lines bearing both KRAS mutation and high AXL expression. We found that R428 exerts a significant growth inhibitory effect in ovarian cancer cells with high AXL expression, and CH5126766 exerts a significant growth inhibitory effect in ovarian cancer cells with KRAS mutation. From this result, it is considered that the efficacy of the combined treatment might be determined by the expression level of AXL and KRAS status; thus, synergistic growth suppression effect was the specific feature of the cell lines that combine KRAS mutation and high AXL expression.
Our results showed that CH5126766 and R428 clearly downregulated p‐ERK and p‐AKT, respectively, followed by the induction of apoptosis accompanied by the Bim upregulation. As we showed in Figure 5, no significant change in Bim expression by the single treatment of R428 was shown; however, the combination treatment enhanced the Bim induction compared to the single treatment of CH5126766. In addition, we showed in Figure 3 that R428 treatment, with further long time course, strongly suppress cancer cell growth. According to previous report, 33 , 34 AKT and ERK regulate the function of Bim through different mechanisms. Active ERK facilitates the ubiquitination and subsequent degradation of Bim, whereas active AKT inhibits Bim, exerting its apoptotic function by promoting 14‐3‐3 proteins to bind Bim and suppress its activity. Based on the theory above, we hypothesized the following mechanism of reaction sequence: suppression of AKT phosphorylation by R428 might not cause significant quantitative change of Bim. At the early stage of the combined treatment (less than 72 hours), Bim degradation was suppressed by CH5126766, then the apoptotic function was enhanced by R428, followed by cell growth suppression gradually exerted at a later stage. The quantitative change in Bim protein by R428 was too subtle to be detected by the western blot but, with a longer time course, it was able to suppress colony formation. Both PI3K‐AKT and RAF‐MEK‐ERK pathways are the main regulators of the downstream proapoptotic factor Bim; 33 , 34 the simultaneous inhibition of both pathways can efficiently induce Bim expression, through different modes of action.
In melanoma with BRAF mutation, AXL overexpression has been reported to attenuate the sensitivity to inhibitors of BRAF/MEK, and cooperative antitumor effects with AXL inhibition were indicated in vitro and in vivo. 35 , 36 Indeed, a clinical trial evaluating the efficacy of R428 combined with the MEK inhibitor trametinib and BRAF inhibitor dabrafenib is already underway (NCT02872259). There are marked expectations for this combined strategy for clinical use in ovarian cancer patients who bear KRAS mutation and high AXL expression.
In this study, we showed one model of combined molecular‐targeting treatment based on the genetic profile. It is also speculated that the inhibitors of other molecules in intracellular signaling pathways could also show such effects. We examined the efficacy of ERK inhibitor SCH772984 and AKT inhibitor MK2206 in HEY‐T30 cells and observed that SCH772984 exerted significant growth inhibition, whereas the efficacy of MK2206 was limited (Figure S2). These data suggest that RAF/MEK inhibitor could be replaced by ERK inhibitor, but AXL inhibitor could not be replaced by AKT inhibitor. As RTK signals are known to be divided in multiple intracellular pathways, it could not be completely inhibited by inhibiting a single downstream molecule.
Though the group of ovarian cancer patients bearing KRAS mutation and high AXL expression is small in size, the group should not be ignored because of urgent need for novel treatment. The combined inhibition of the RAS‐MAPK pathway and AXL could be a promising treatment strategy for this group of patients.
As it is widely accepted, ovarian cancer shows wide variety in activity of genes due to thier mutation and/or functional disruption of thier upstream regulator, which is one of the reasons why conventional chemotherapy has shown limited effect. Therefore, applying specific combinations of molecular targeting drugs according to individual gene profile could improve the prognosis of patients with ovarian cancer.
CONFLICT OF INTEREST
T. Sakai received a grant from Chugai Pharmaceutical Co., Ltd. (outside of submitted work). All others declare no conflict of interest.
Supporting information
Fig S1
Fig S2
ACKNOWLEDGMENTS
This work was supported by the Research Fund of the Kyoto Prefectural University of Medicine. We thank Professor Seiji Yano and Dr Hiromichi Ebi (Division of Medical Oncology, Cancer Research Institute, Kanazawa University) for helpful information and advice on our experiments.
Umemura S, Sowa Y, Iizumi Y, Kitawaki J, Sakai T. Synergistic effect of the inhibitors of RAF/MEK and AXL on KRAS‐mutated ovarian cancer cells with high AXL expression. Cancer Sci. 2020;111:2052–2061. 10.1111/cas.14414
REFERENCES
- 1. Prat J. Ovarian carcinomas: five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch. 2012;460:237‐249. [DOI] [PubMed] [Google Scholar]
- 2. Koshiyama M, Matsumura N, Konishi I. Recent concepts of ovarian carcinogenesis: type I and type II. Biomed Res Int. 2014;2014:934261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shih IM, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004;164:1511‐1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kurman RJ, Shih IM. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol. 2010;34:433‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singer G, Kurman RJ, Chang HW, Cho SK, Shih IM. Diverse tumorigenic pathways in ovarian serous carcinoma. Am J Pathol. 2002;160:1223‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Farley J, Brady WE, Vathipadiekal V et al. Selumetinib in women with recurrent low‐grade serous carcinoma of the ovary or peritoneum: an open‐label, single‐arm, phase 2 study. Lancet Oncol. 2013;14:134‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gemignani ML, Schlaerth AC, Bogomolniy F et al. Role of KRAS and BRAF gene mutations in mucinous ovarian carcinoma. Gynecol Oncol. 2003;90:378‐381. [DOI] [PubMed] [Google Scholar]
- 8. Auner V, Kriegshäuser G, Tong D et al. KRAS mutation analysis in ovarian samples using a high sensitivity biochip assay. BMC Cancer. 2009;9:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Singer G, Oldt R, Cohen Y et al. Mutations in BRAF and KRAS characterize the development of low‐grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95:484‐486. [DOI] [PubMed] [Google Scholar]
- 10. Kurman RJ, Shih IM. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer–shifting the paradigm. Hum Pathol. 2011;42:918‐931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gurung A, Hung T, Morin J, Gilks CB. Molecular abnormalities in ovarian carcinoma: clinical, morphological and therapeutic correlates. Histopathology. 2013;62:59‐70. [DOI] [PubMed] [Google Scholar]
- 12. Miller CR, Oliver KE, Farley JH. MEK1/2 inhibitors in the treatment of gynecologic malignancies. Gynecol Oncol. 2014;133:128‐137. [DOI] [PubMed] [Google Scholar]
- 13. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu AM, Huang PH. Receptor tyrosine kinase coactivation networks in cancer. Cancer Res. 2010;70:3857‐3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008;100:35‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun W, Fujimoto J, Tamaya T. Coexpression of Gas6/Axl in human ovarian cancers. Oncology. 2004;66:450‐457. [DOI] [PubMed] [Google Scholar]
- 17. Verma A, Warner SL, Vankayalapati H, Bearss DJ, Sharma S. Targeting Axl and Mer kinases in cancer. Mol Cancer Ther. 2011;10:1763‐1773. [DOI] [PubMed] [Google Scholar]
- 18. Willis S, Villalobos VM, Gevaert O et al. Single gene prognostic biomarkers in ovarian cancer: a meta‐analysis. PLoS ONE. 2016;11:e0149183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen PX, Li QY, Yang Z. Axl and prostasin are biomarkers for prognosis of ovarian adenocarcinoma. Ann Diagn Pathol. 2013;17:425‐429. [DOI] [PubMed] [Google Scholar]
- 20. Elkabets M, Pazarentzos E, Juric D et al. AXL mediates resistance to PI3Kα inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell. 2015;27:533‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scaltriti M, Elkabets M, Baselga J. Molecular pathways: AXL, a membrane receptor mediator of resistance to therapy. Clin Cancer Res. 2016;22:1313‐1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Köbel M, Kalloger SE, Huntsman DG et al. Differences in tumor type in low‐stage versus high‐stage ovarian carcinomas. Int J Gynecol Pathol. 2010;29:203‐211. [DOI] [PubMed] [Google Scholar]
- 23. Ishii N, Harada N, Joseph EW et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res. 2013;73:4050‐4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lito P, Saborowski A, Yue J et al. Disruption of CRAF‐mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25:697‐710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wada M, Horinaka M, Yamazaki T, Katoh N, Sakai T. The dual RAF/MEK inhibitor CH5126766/RO5126766 may be a potential therapy for RAS‐mutated tumor cells. PLoS ONE. 2014;9:e113217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holland SJ, Pan A, Franci C et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010;70:1544‐1554. [DOI] [PubMed] [Google Scholar]
- 27. Feneyrolles C, Spenlinhauer A, Guiet L et al. Axl kinase as a key target for oncology: focus on small molecule inhibitors. Mol Cancer Ther. 2014;13:2141‐2148. [DOI] [PubMed] [Google Scholar]
- 28. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schoumacher M, Burbridge M. Key roles of AXL and MER receptor tyrosine kinases in resistance to multiple anticancer therapies. Curr Oncol Rep. 2017;19:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Z, Lee JC, Lin L et al. Activation of the AXL kinase causes resistance to EGFR‐targeted therapy in lung cancer. Nat Genet. 2012;44:852‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taniguchi H, Yamada T, Wang R et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun. 2019;10:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rankin EB, Fuh KC, Taylor TE et al. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010;70:7570‐7579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome‐dependent degradation of the BH3‐only protein, Bim. J Biol Chem. 2003;278:18811‐18816. [DOI] [PubMed] [Google Scholar]
- 34. Qi XJ, Wildey GM, Howe PH. Evidence that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL apoptotic function. J Biol Chem. 2006;281:813‐823. [DOI] [PubMed] [Google Scholar]
- 35. Boshuizen J, Koopman LA, Krijgsman O et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody‐drug conjugate and BRAF/MEK inhibitors. Nat Med. 2018;24:203‐212. [DOI] [PubMed] [Google Scholar]
- 36. Zuo Q, Liu J, Huang L et al. AXL/AKT axis mediated‐resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene. 2018;37:3275‐3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2