

Abstract
Absent, small or homeotic 2‐like protein (ASH2L) is a core component of a multimeric histone methyltransferase complex that is involved in the maintenance of active transcription, participating in several cancers, however the biological function and molecular mechanism of ASH2L in endometrial cancer (ECa) are largely unknown. Endometrial cancer is a common malignant tumor in women and the incidence of this cancer is on the rise. Estrogen‐ERα signaling, as an oncogenic pathway, plays a crucial role in endometrial carcinogenesis. Therefore, further exploration of the molecular mechanisms around ERα‐mediated gene transcription in ECa would be helpful to the understanding of tumor development and to finding a new therapeutic target for ECa. Here, our study demonstrated that ASH2L was highly expressed in ECa samples, and higher expression of ASH2L was positively correlated with a poor prognosis. Moreover, we identified that ASH2L associated with ERα and that knockdown of ASH2L resulted in decreased expression of a subset of the estrogen‐induced target genes, including paired box 2 (PAX2), an oncogenic gene in ECa. ASH2L was recruited to cis‐regulatory elements in PAX2, thereby altering histone H3K4me3 and H3K27me3 levels, to enhance ERα‐mediated transactivation. Finally, depletion of ASH2L suppressed endometrial cancer cell proliferation and migration. Our findings suggest that ASH2L participates in the promotion of ECa progression, if not totally at least partially, via upregulation of PAX2 transcription.
Keywords: ASH2L, co‐regulator, endometrial cancer, histone methylation, transcription
Expression of ASH2L was positively correlated with a poor prognosis in endometrial cancer. ASH2L enhanced ERα‐mediated transactivation and knockdown of ASH2L resulted in decreased expression of PAX2 by altering histone H3K4me3 and H3K27me3 levels and recruitment of ERα. Depletion of ASH2L suppressed endometrial cancer cell proliferation and migration.
![]()
Abbreviations
- E2
17β‐estradiol
- ECa
endometrial cancer
- EtOH
ethanol
- NOD‐SCID
nonobese diabetic‐severe combined immunodeficiency disease
1. INTRODUCTION
Endometrial cancer (ECa) is a reproductive malignancy with increased morbidity and mortality. Clinically, approximately 80% of ECa are estrogen‐dependent type I endometrioid adenocarcinomas, accompanied by hyperlipidemia, anovulation, and other hyperestrogenic risk factors such as obesity. 1 , 2 , 3 It has been reported that adipose tissue has the ability to synthesize estrogen, which continuously activates the ERα signaling pathway, promoting excessive proliferation of endometrium and even causing cancer. 4 Estrogen and selective estrogen‐receptor modulators (SERMs) are considered to be involved in endometrial carcinogenesis via their functions in the regulation of gene transcription. Therefore, clarification of the molecular mechanisms underlying the function of estrogen/SERMs and the ERα signaling pathway in endometrial carcinogenesis is crucial.
ERα is a member of a steroid hormone receptor superfamily. In the presence of estrogen, ERα enters the nucleus and binds to cis‐regulatory elements; in the nucleus it recruits a series of co‐regulators to modulate transcription of ERα target genes. It has been demonstrated that ERα co‐regulators are involved in the modulation of ERα action to promote cell proliferation, invasion, and metastasis in breast cancer. 5 , 6 Therefore, the identification of novel co‐regulators of ERα would be helpful for understanding the development of ERα‐related cancers and in finding new therapeutic targets. However, the cofactors associated with ERα in endometrial cancer are largely unknown.
ASH2L is a core subunit of a multimeric histone methyltransferase complex involved in triggering histone H3 lysine 4 (H3K4) trimethylation, thereby mediating active gene transcription. Loss of ASH2L protein leads to a global reduction in H3K4 trimethylation. 7 , 8 ASH2L contains an amino‐terminal plant homeodomain (PHD) finger and winged‐helix (WH) motifs, suggesting that ASH2L has the potential to recognize DNA and histones. 9 , 10
ASH2L, acting as a cofactor of several transcription factors, participates in the regulation of gene transcription. ASH2L enhances Tbx1, Pax7, or Mef2‐mediated transcriptional activity to be involved in embryogenesis and stem cell differentiation by increasing histone H3K4me3 levels. 11 , 12 , 13 ASH2L specifically promotes Ap2δ‐mediated gene transcription. 14 ASH2L interacts with MK1 to promote TNFα‐induced proinflammatory transcription. 15 In addition, it has been demonstrated that ASH2L interacts with MYC to enhance MYC‐mediated gene transcription by controlling demethylation of H3K27 and acetylation of H3K27, cooperating with CBP/p300. 16 ASH2L participates in the crosstalk between H2B ubiquitination and H3K4 methylation to modulate gene transcription. 17
It has been shown that ASH2L plays an important role in the progression of multiple tumors. ASH2L upregulates GATA3‐induced transcription of ESR1 in breast cancer cells. 18 Conversely, ASH2L is recruited to the promoter region of apoptosis‐related genes mediated by p53, thereby co‐activating p53 function to promote cell apoptosis in colorectal cancer. 19 ASH2L protein is highly expressed in cervical cancer, and ASH2L depletion inhibits HeLa cell proliferation. 18 However, the molecular mechanisms underlying the biological function of ASH2L in endometrial cancer progression are still elusive.
In this study, we identified that ASH2L is highly expressed in endometrial cancer, and that higher expression of ASH2L is positively correlated with a poor prognosis in ECa. We demonstrated that ASH2L associates with ERα and enhances ERα‐induced transactivation. Depletion of ASH2L led to a decrease in transcription of ERα‐regulated genes, including PAX2. Moreover, we provide evidence to show that ASH2L is recruited to the cis‐regulatory elements at the ERα target gene PAX2, thereby modulating histone H3K4me3 and H3K27me3 levels. Furthermore, knockdown of ASH2L significantly suppressed cell proliferation and migration in endometrial cancer. Therefore, ASH2L participates in promotion of endometrial cancer progression via modulation of PAX2 transcription, providing a potential target for endometrial cancer therapy.
2. MATERIALS AND METHODS
2.1. Plasmids and cell cultures
Expression plasmids of human ASH2L (#15548) and MLL1 (#20873) were purchased from the ADDGENE company. A series of truncated mutants of ASH2L was cloned into te pcDNA3.1 vector containing a FLAG‐tag. Plasmid WDR5 (CAT#: RC200162) was purchased from OriGene Technologies. Expression plasmids for ERα, ERα‐AF1, and ERα‐AF2 were kindly provided by Dr. Shigeaki Kato. 20
A detailed description of cell culture is provided in the Supporting Information.
2.2. Antibodies
Antibodies used in this study: anti‐ASH2L (A300‐107A; Bethyl Laboratories), anti‐ASH2L (12331‐1‐AP; Proteintech Group), anti‐FLAG (4110‐FG; GNI), anti‐ERα (D8H8) (#8664; Cell Signaling Technology), anti‐ERα (F10) (sc‐8002; Santa Cruz Biotechnology), anti‐MLL1 (A300‐37A; Bethyl Laboratories), anti‐WDR5 (A302‐429A; Bethyl Laboratories), anti‐PAX2 (TA327502S; OriGene Technologies), anti‐Cyclin D1 (60186‐1‐lg; Proteintech Group), anti‐GAPDH (AC033; ABclonal Technology), anti‐Ki67 (sc‐15402; Santa Cruz Biotechnology), anti‐trimethyl H3‐K27 (07‐449; Millipore), anti‐trimethyl H3‐K4 (05‐745R; Millipore).
2.3. siRNA transfection and lentiviral infection
Control siRNA (siCtrl) and siRNA duplexes against the gene encoding ASH2L (siASH2L) were transfected into Ishikawa or HEC‐1A cells. The sequences for 3 independent siRNAs (#1, #2 and #3) specially targeting ASH2L are listed in Supporting Information Table S1. For lentiviral infection, control shRNA lentivirus (shCtrl) and 3 shRNAs against ASH2L lentivirus (shASH2L#1, shASH2L, shASH2L#3) targeting the same sequences as same for siASH2L#1, #2, and #3 were generated by the Shanghai GeneChem Company.
2.4. Co‐immunoprecipitation (Co‐IP), GST pull‐down, western blotting, immunofluorescence assay, and luciferase reporter assay
Detailed descriptions of these procedures are included in Supporting Information.
2.5. RNA isolation, reverse transcription, and quantitative real‐time PCR (qPCR)
Total RNA was extracted using Trizol reagent (Invitrogen). Next, 1 μg of RNA was reverse transcribed into cDNA, performed using a PrimeScript™ RT‐PCR Kit (TaKaRa). Using SYBR Premix Ex Taq (TaKaRa), cDNAs were quantified by real‐time qPCR on a LightCycler 96 instrument (Roche Life Science). Gene expression levels were calculated relative to ribosome 18S rRNA. Primers used to detect mRNA amplification are listed in Table S2.
2.6. Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed in accordance with previously described protocols. 21 Cells were harvested for ChIP after treatment with 10−8M E2 for 12 h. Immunoprecipitation of sonicated chromatin solutions was conducted by incubation with anti‐ERα, anti‐ASH2L, anti‐MLL1, anti‐H3K27me3 or anti‐H3K4me3 at 4°C overnight. Protein A agarose/Salmon Sperm was added for 4 h. DNA fragments were extracted with phenol‐chloroform and precipitated in ethanol. Purified DNA was analyzed using qPCR. Results are shown as the percentage of input chromatin. Each set of results was repeated in at least 3 independent experiments. Primer sequences for estrogen response elements (EREs) of PAX2 are listed in Table S3.
2.7. MTS assay, colony formation assay, and transwell assay
For MTS analysis, cells containing shASH2L or shCtrl were plated at a density of 3000 cells per well in 96‐well plates and incubated with 10−8M E2 or EtOH for 0‐7 d. The absorbance in each well was detected at 490 nm wavelength using a microplate reader on d 0, d 1, d 2, d 4, and d 7. For colony formation assay, 1 × 104 cells carrying shASH2L or shCtrl were incubated with 10−8M E2 or EtOH for 7 d. Cells were then fixed and stained with Coomassie blue dye. For transwell experiments, 3 × 104 Ishikawa cells were incubated with 10−8M E2 or EtOH in transwell chambers for 24‐36 h.
2.8. Xenograft tumor growth
Ishikawa cells carrying shASH2L (1.0 × 107 cells/mouse) were injected into the right axillary subcutaneous of 5‐wk‐old female NOD/SCID mice (Vital River Laboratories), and the other side injected with shCtrl cells as the control. Tumor diameter was measured every week using electronic caliper. Tumor volume (mm3) was calculated as volume = (short diameter)2 × (long diameter)/2. 22 Four wk later, tumor‐bearing mice were sacrificed following the guidelines for the humane treatment of animals. All animal experiments complied with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. All procedures of animal experiments were carried out under the supervision and guidelines of the China Medical University of Animal Care Center in compliance with ethical regulations approved by the Animal Ethics Committee of China Medical University. No animals suffered unnecessarily or were hurt at any stage of an experiment.
2.9. Collection of clinical endometrial tissues and immunohistochemistry
Clinical endometrial carcinoma (for this study we selected an endometrioid adenocarcinoma that showed ERα‐positive expression) fresh tissues, benign endometrial tissues (BET), and specimens carrying endometrial carcinoma samples or BET were provided by the Shengjing Hospital of China Medical University. This study was approved by the Ethics Committee of the Medical Faculty of the China Medical University. Tumor tissue samples from ECa patients were obtained without prior treatment from surgeries. Benign endometrial tissues from patients were obtained from general outpatient surgeries. All samples were obtained after receiving the informed consent from patients. Evaluation of ASH2L expression in endometrial tissue was performed using immunohistochemistry as described previously. 23 Average optical density (AOD, IOD SUM/Area SUM) of the nucleus was used to evaluate ASH2L expression. 24
2.10. Statistics
All statistical analysis was performed using the SPSS statistical (22.0) software program. Student 2‐tailed t test was used to determine statistical relevance between groups (*P < .05, **P < .01, and ***P < .001).
3. RESULTS
3.1. ASH2L is highly expressed in endometrial cancer, and the higher expression of ASH2L is positively correlated with a poor prognosis
It has been reported that ASH2L exerts an oncogenic protein or a tumor suppressor in a series of tumors. However, the biological function of ASH2L on endometrial cancer (ECa) remains to be elucidated. To assess the role of ASH2L in ECa, we analyzed the clinical data from the Cancer Genome Atlas (TCGA) and found that the higher expression of ASH2L is positively correlated with poor prognosis in ECa (Figure 1A). We then turned to detect the expression of ASH2L in clinical ECa (we selected the endometrioid adenocarcinoma in this study) tissues. The results from western blotting demonstrated that the expression of ASH2L was obviously higher in ECa tissues (Figure 1B,C). Immunohistochemistry assay was further performed to examine the expression of ASH2L in 130 clinical samples, including 100 endometrioid adenocarcinoma samples and 30 BET samples. The results showed that the expression of ASH2L in ECa was significantly higher (Figure 1D,E) and the intensity of ASH2L staining was gradually enhanced with increase in the pathology grade (Figure 1F and Table 1) suggesting that ASH2L played an important role in ECa progression.
FIGURE 1.
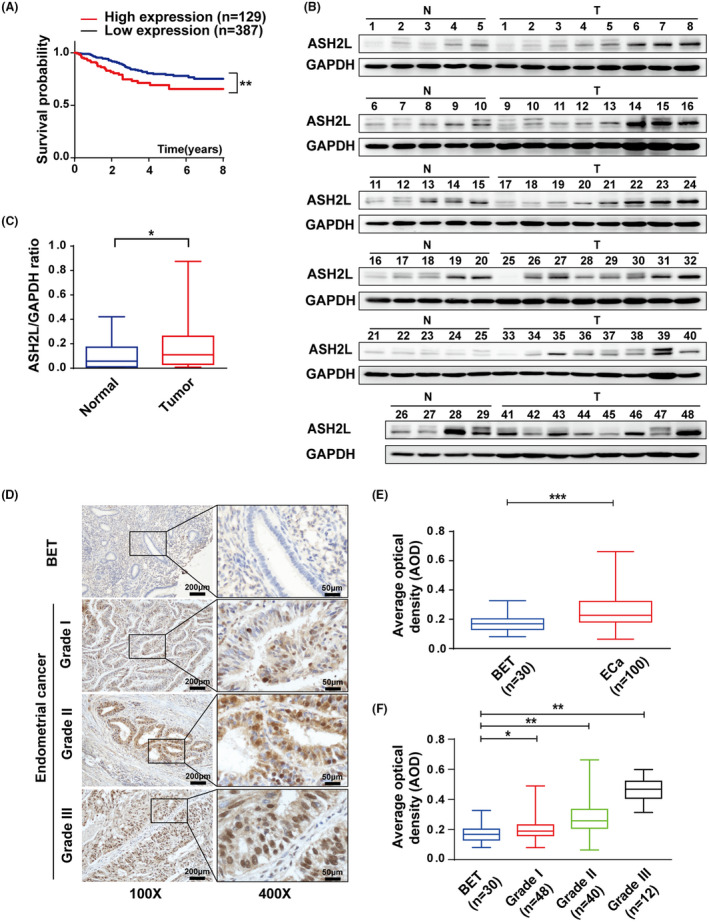
ASH2L is highly expressed in endometrial cancer. A, Kaplan‐Meier analysis for overall survival of 516 endometrial cancer (ECa) patients based on the data from the Cancer Genome Atlas (TCGA). B, Expression of ASH2L in 29 benign endometrial tissues (N) and 48 endometrial cancer (T) detected using western blotting. C, Expression of ASH2L in (B) was quantified using densitometry shown as box and whisker plots. D, Immunohistochemical (IHC) assay in paraffin sections of BET and ECa (histological grades I/II/III) in ×100 magnification (scale bars, 200 μm) and ×400 magnification (scale bars, 50 μm). E and F, Average optical density (AOD) of IHC stained by ASH2L antibody in BET and ECa specimens. Data are shown as mean ± SD, *P < .05, **P < .01, ***P < .001
TABLE 1.
Relationship between ASH2L expression and clinical pathologic features in ECa
| Characteristics | Number of patients (n = 100) | ASH2L expression | P‐values a | |
|---|---|---|---|---|
| Low (n = 49) | High (n = 51) | |||
| Age | ||||
| <55 (y) | 25 | 15 | 10 | .274 |
| ≥55 (y) | 75 | 34 | 41 | |
| FIGO stage | ||||
| I | 77 | 46 | 31 | .003 |
| II | 12 | 2 | 10 | |
| III | 11 | 1 | 10 | |
| Histological grade | ||||
| I | 48 | 33 | 15 | <.001 |
| II | 40 | 16 | 24 | |
| III | 12 | 0 | 12 | |
| Myometrial invasion | ||||
| No | 18 | 13 | 5 | .008 |
| Superficial myometrial invasion | 62 | 32 | 30 | |
| Deep myometrial invasion | 20 | 4 | 16 | |
| Lymph node metastasis | ||||
| No | 94 | 48 | 46 | .175 |
| Yes | 6 | 1 | 5 | |
| PRα status | ||||
| Negative | 16 | 2 | 14 | .007 |
| Positive | 84 | 47 | 37 | |
FIGO, International Federation of Obstetrics and Gynecology.
Χ2 test.
3.2. ASH2L interacts with ERα in ECa‐derived cell lines
ERα has been considered as a crucial factor in endometrial carcinogenesis (especially in endometrioid adenocarcinoma). We therefore turned to ask whether ASH2L associates with ERα in ECa‐derived cell lines. Protein expression of ASH2L or ERα in several ECa‐derived cell lines was detected using western blotting (Figure S1A). Co‐immunoprecipitation (Co‐IP) experiments were performed. The results demonstrated that endogenous ASH2L associates with ERα in ECa‐derived cell lines (Ishikawa cells and HEC‐1A cells) (Figures 2A and S1B). Moreover, Ishikawa cells were transiently transfected with expression plasmids for ERα and ASH2L, and Co‐IP experiments were performed. The results showed that ERα associates with Flag‐tagged ASH2L and the association between them was a little stronger in the presence of E2 (Figure 2B). In addition, GST pull‐down experiments were performed to examine the direct interaction between ASH2L and ERα truncated mutants, including ERα‐AF1 (29‐180aa) and ERα‐AF2 (282‐595aa) as indicated. The results showed that ASH2L mainly binds to ERα‐AF2 (Figure 2C). Moreover, to detect the subcellular distribution of ASH2L and ERα in cells, immunofluorescence experiments were performed in Ishikawa cells and COS7 cells. The data demonstrated that following treatment with E2, endogenous ASH2L and ERα were mainly distributed in the nucleus (Figure 2D). In addition, the expression plasmids encoding FLAG‐tagged ASH2L‐FL (full length) or its truncated mutations as indicated were transfected together with ERα into COS7 cells. The results showed that, in the presence of E2, ASH2L‐FL or ASH2L‐N1/N2/C1 was co‐localized in the nucleus with ERα, however ASH2L‐C2 was distributed in the whole cell, suggesting that the nuclear location sequence of ASH2L was mainly located in the WH domain (Figure 2E,F).
FIGURE 2.
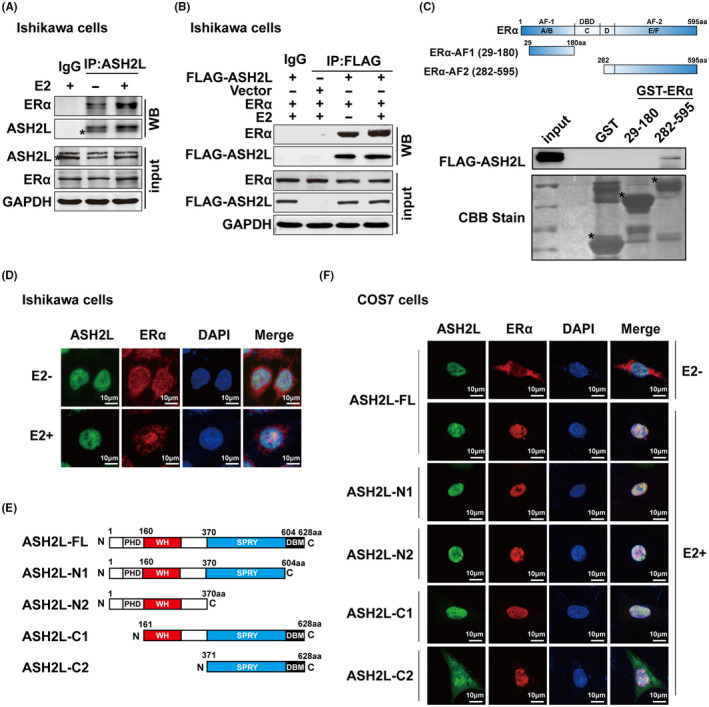
ASH2L interacts with ERα in ECa‐derived cell lines. A, Endogenous interaction between ASH2L and ERα in Ishikawa cells was verified by co‐immunoprecipitation. *Asterisk in (A) on the second band in the ASH2L panels indicates the expression of ASH2L. B, Exogenous association between ASH2L and ERα in Ishikawa cells which are transiently expressing ERα and FLAG‐ASH2L plasmids by Co‐IP. C, Identification of binding domains in ERα for ASH2L interaction. Bound proteins in GST pull‐down experiments were analyzed using immunoblotting using anti‐FLAG antibody and equal loading of GST‐ERα deletion mutants was assessed by Coomassie brilliant blue staining. Schematic representation of GST‐tagged ERα‐AF1 (29‐180) and ERα‐AF2 (282‐595) was shown at the top of (C). *Asterisk placed to indicate the expression position of GST or GST‐ERα truncated mutation proteins. D, Immunofluorescence confocal experiments were used to identify the localizations of endogenous ERα (red) and ASH2L (green). E, Diagram of full‐length (FL) and truncated mutants of ASH2L. F, Fluorescence confocal experiments were used to identify the localizations of exogenous ERα (Red) and ASH2L‐FL or its truncations (green). Nucleus was stained using DAPI (blue). Scale bars, 10 μm
3.3. ASH2L upregulates ERα‐mediated transactivation in ECa cells
To explore the functional domain of ASH2L for enhancement of ERα action, a luciferase reporter assay was performed in Ishikawa cells as indicated. Our results demonstrated that ASH2L‐FL significantly enhanced ERα‐mediated transcriptional activity following the treatment with E2, while the ASH2L truncated mutations had no obvious effects on activity. These results suggest that ASH2L needs to be completely assembled to co‐activate ERα action (Figure 3A). We further examined the effects of ASH2L on the transactivation mediated by ERα or ERα truncated fragments, including ERα‐AF1 with a constitutive function and ERα‐AF2 with a ligand‐dependent function in COS7 cells. The results showed that ASH2L significantly increased ERα‐ or ERα‐AF2‐induced transactivation. However, enhancement of ASH2L on ERα‐AF1 action was not observed (Figure 3B). Moreover, ASH2L deletion inhibited ERα‐induced transcriptional activity by 60%‐70% (Figure 3C). Knockdown efficiency of ASH2L by siRNA against ASH2L (siASH2L) or shASH2L in Ishikawa cells or HEC‐1A cells was confirmed using western blotting (Figure S2).
FIGURE 3.
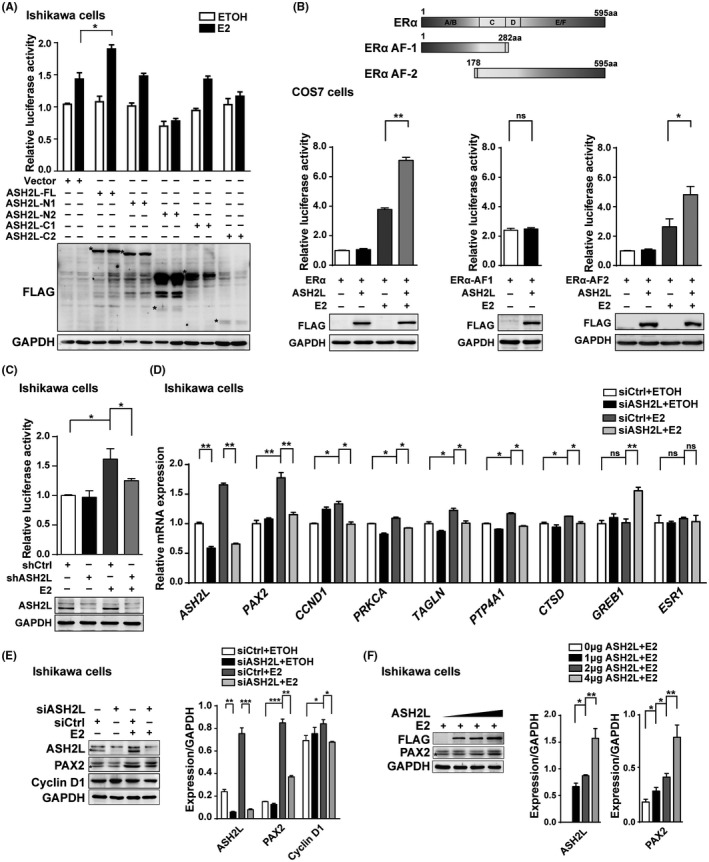
ASH2L upregulates ERα‐mediated transactivation in ECa cells. A, Luciferase reporter assay was performed to verify the effects of ASH2L‐FL or different truncation on the ERα‐mediated transcriptional regulation above; western blotting shows the expression of ASH2L‐FL and its truncation below. B, Schematic representation of ERα, ERα‐AF1, and ERα‐AF2 is shown at the top of the panel. Luciferase reporter assays were performed to detect the effects of ASH2L on the transcriptional activity mediated by ERα or its truncated mutants in COS7 cells. Expression of ASH2L was analyzed using western blotting. C, Transcriptional activity of ERα was detected in Ishikawa cells infected with lentivirus carrying shASH2L or shCtrl. Expression of endogenous ASH2L below. D, Real‐time qPCR assay was performed to detect the effects of ASH2L on activation of several genes regulated by E2‐ERα. ASH2L was knocked down by specific siRNA in Ishikawa cells. E, Western blotting was performed to detect the effects of ASH2L on the expression of PAX2 or Cyclin D1 in Ishikawa cells. F, Western blotting was performed to detect the effects of different doses of ASH2L on the expression of PAX2. Quantification of protein expression is shown as histograms on the right. *Asterisk in the western blotting is placed to indicate the position of labeled proteins. In the histogram, the bars represent the mean ± SD (n = 3), *P < .05, **P < .01, ***P < .001
We further examined the influence of ASH2L on endogenous ERα‐regulated gene transcription in ECa cells using real‐time qPCR. In agreement with the results in the luciferase assay, ASH2L depletion resulted in a significant decrease in mRNA expression of PAX2 and CCND1, which were induced by ERα in Ishikawa cells (Figure 3D). Furthermore, ASH2L depletion decreased the transcription levels of PAX2, CTSD, and PRKCA in HEC‐1A cells (Figure S3A). In addition, depletion of ASH2L suppressed the protein expression of PAX2 or cyclin D1 in Ishikawa or HEC‐1A cells (Figures 3E and S3B,C). Ectopic expression of ASH2L increased the expression of PAX2 in Ishikawa cells in a dose‐dependent manner (Figure 3F). Therefore, our results indicated that ASH2L co‐activates ERα‐mediated transactivation in ECa‐derived cells.
3.4. ASH2L associates with MLL1/WDR5 to enhance ERα‐induced transactivation
ASH2L is a subunit of multimeric histone methyltransferase complexes, which mainly mediate methylation of histone H3 lysine 4 (H3K4) for involvement in active transcription. ASH2L regulates the methyltransferase activity of MLL1 by interacting with other components. 7 , 25 We therefore turned to ask whether ASH2L associated with the core subunits of histone methyltransferase complex to co‐activate ERα action in endometrial cancer cells. Co‐IP experiments were performed to examine the association of ERα with ASH2L, MLL1, or WDR5. The results demonstrated that endogenous ERα interacted with ASH2L, MLL1, or WDR5 with, or without E2. Importantly, knockdown of ASH2L weakened the interaction between MLL1 and ERα, nevertheless there were no obvious effects on the interaction of ERα with WDR5 (Figure 4A). The results suggested that ERα interacts with ASH2L together with MLL1, and WDR5, meanwhile ASH2L is required for the association between MLL1 and ERα. To identify the possible domains in ASH2L/ERα for interaction with MLL1 and WDR5 in the presence of E2, Ishikawa cells were co‐transfected with expression plasmids encoding ASH2L‐FL or its truncated mutants for Co‐IP experiments. The results demonstrated that the interaction between ASH2L and MLL1 or WDR5 did not occur when the SPRY domain was deleted in ASH2L, whereas the interaction of ASH2L with ERα was not influenced by deletion of the SPRY domain in ASH2L, suggesting that the SPRY domain was necessary for association between ASH2L and MLL1/WDR5, but not for ERα (Figure 4B).
FIGURE 4.
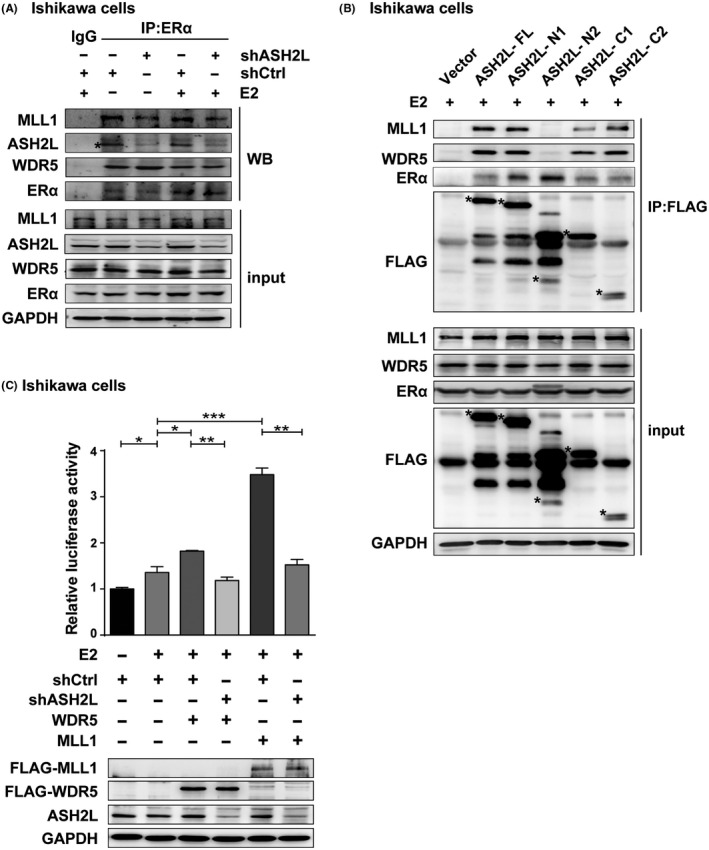
ASH2L associates with MLL1/WDR5 to enhance ERα‐induced transactivation. A, Co‐IP was performed to detect the interactions between endogenous ERα and ASH2L/MLL1/WDR5 in shCtrl and shASH2L Ishikawa cells. B, Interaction domains of ASH2L with MLL1/WDR5, or ERα in Ishikawa cells was tested by Co‐IP. C, Luciferase assay shows the effects induced by deletion of ASH2L on the ERα‐mediated transactivation, which is upregulated by WDR5 or MLL1 in Ishikawa cells. Expression of WDR5 and MLL1 was detected using western blotting. *Asterisks placed to indicate the position of expression of FLAG‐ASH2L truncated mutants. Data are shown as mean ± SD (n = 3), *P < .05, **P < .01, ***P < .001
Having shown that ASH2L is required for association between MLL1 and ERα, we then turned to perform luciferase assays to examine the influence of ASH2L depletion on the effect of MLL1 or WDR5 (MLL1/WDR5) on ERα action. The results showed that the function of MLL1/WDR5 in enhancement of ERα‐mediated transcriptional activity was clearly inhibited by ASH2L depletion (Figure 4C). These results suggested that ASH2L associates with MLL1/WDR5 to upregulate ERα action, and that ASH2L may act as an indispensable adaptor and function with MLL1/WDR5 to module ERα‐induced transactivation.
3.5. ASH2L facilitates the recruitment of MLL1 to the cis‐regulatory elements of ERα‐regulated gene in endometrial cancer cells
Our data demonstrated that ASH2L is required for endogenous ERα‐regulated gene activation. To examine whether ASH2L or ERα was recruited to the cis‐regulatory elements of ERα‐regulated gene upon E2 induction. Chromatin immunoprecipitation (ChIP) assays were performed in Ishikawa cells. Having established that PAX2 as a putative ERα‐regulated gene 26 , 27 was downregulated by depletion of ASH2L in endometrial cancer cells, we further searched for potential ERα‐binding sites in the promoter region of PAX2 based on bioinformatics analysis. We named the sites PAX2‐ERE I/II/III/IV (Figure 5A). ChIP assay results demonstrated that ASH2L or ERα was recruited to PAX2‐ERE II/III in the presence of E2 (Figure 5B). To test whether ASH2L influenced the histone modification levels or MLL1 recruitment to PAX2‐ERE II/III regions, ChIP assays were further performed in Ishikawa cells following knockdown of ASH2L and with or without E2; antibodies used are as indicated. The results showed that ASH2L deletion reduced the recruitment of ERα or MLL1, and resulted in a reduction in H3K4me3 levels at PAX2‐ERE II/III regions following treatment with E2 (Figure 5C). In addition, interestingly, we observed that H3K27me3 levels were increased by knockdown of ASH2L (Figure 5C). To further address the interplay between genomic recruitment of ERα to PAX2 and to histones H3K4me3 and H3K27me3, we used publicly available ChIP‐seq data. 28 , 29 All ChIP‐seq data were downloaded from Gene Expression Omnibus (GEO) DataSets (https://www.ncbi.nlm.nih.gov/gds). ERα and H3K4me3 ChIP‐seq data were obtained from the GSE120756 dataset; H3K27me3 data were obtained from the GSE23701 dataset. The results showed that enrichment of ERα regions over the whole PAX2 gene was accompanied by an enhancement in H3K4me3 levels, but a reduction in H3K27me3 levels as indicated (Figure 5D,E). Together these results suggested that ASH2L or ERα is recruited to PAX2‐ERE II/III regions, which are cis‐regulatory elements of PAX2 in endometrial cancer. In addition, ASH2L facilitated the recruitment of MLL1 or ERα to the PAX2‐ERE II/III region, thereby controlling H3K4me3 and H3K27me3 levels to enhance the transactivation induced by ERα.
FIGURE 5.

ASH2L facilitates the recruitment of MLL1 to the cis‐regulatory elements of the ERα‐regulated gene in endometrial cancer cells. A, Schematic diagram of the searched estrogen response elements (EREs) at cis‐regulatory elements of PAX2. B, ChIP assay with indicated antibodies shows the recruitment of ASH2L and ERα at different EREs. C, ChIP assay shows the recruitment of indicated protein and histone modification at EREII/III. DNA fragments were amplified using qPCR with the primers indicated in (A). D and E, ChIP‐Seq profiles and the corresponding peaks of genomic regions around PAX2. Error bars represent mean ± SD (n = 3), *P < .05, **P < .01, ***P < .001
3.6. ASH2L depletion inhibits cell proliferation and migration in endometrial cancer
To investigate the physiological function of ASH2L in the progression of endometrial cancer, MTS assay experiments were performed to study the effect of ASH2L on cell proliferation in ECa‐derived cells. The results in Ishikawa cells shown in the line charts indicated that cell proliferation promoted by E2 was inhibited by deletion of ASH2L (shASH2L) (Figures 6A and S4A,B). The same experiments were performed in HEC‐1A cells (Figure S4C‐E). Consistently, the results from cell colony formation experiments showed that the number of cell clones formed by Ishikawa cells or HEC‐1A cells was significantly reduced by ASH2L depletion (Figures 6B,C and S4F,G). To further confirm the effect of ASH2L on cell growth/proliferation in Ishikawa cells, colony formation assays were further performed using 2 additional shRNAs against ASH2L (shASH2L #1 and #3). The results demonstrated that ASH2L depletion by different loci of shASH2L significantly inhibited cell growth/proliferation in Ishikawa cells (Figure S5A‐D). To know whether the function of ASH2L on cell proliferation is related to ERα in ECa cells, cell proliferation experiments were performed with depletion of ERα. The results showed that proliferation induced by overexpression of ASH2L was significantly reduced by knockdown of ERα (Figure 6D,E). Efficiency of siRNA against ERα or expression of ASH2L was detected using western blotting (Figure 6F). These data suggest that ASH2L is involved in the promotion of ECa‐derived cell proliferation, and the promotion of cell proliferation induced by ASH2L is at least partially related to ERα.
FIGURE 6.
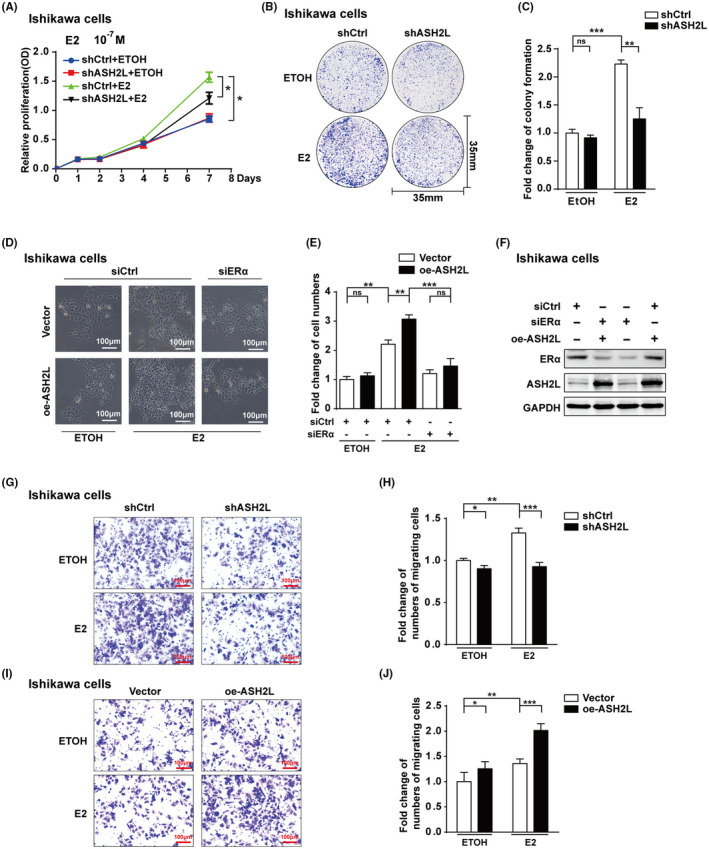
ASH2L depletion inhibits the endometrial cancer cell growth/proliferation and migration. A‐F, Effect of ASH2L on cell growth/proliferation in Ishikawa cells. A, MTS assay was performed in Ishikawa cells carrying shCtrl or shASH2L (sequence from siASH2L #2). B and C, Colony formation experiments in Ishikawa cells with shCtrl and shASH2L. D and E, Promotion function of ectopic expression of ASH2L on cell proliferation was reduced by ERα depletion in Ishikawa cells. Scale bars, 100 μm. F, Expression of ASH2L and ERα in (D) was detected by western blotting. G‐J, Effect of ASH2L on cell migration in Ishikawa cells. oe‐ASH2L stands for overexpression of ASH2L. Data are shown as mean ± SD (n = 3), *P < .05, **P < .01, ***P < .001
To examine whether ASH2L exerts a biological function on the migration of ECa cells, transwell experiments were performed. Ishikawa cells with stable knockdown (shASH2L) or overexpression of ASH2L (oe‐ASH2L) were respectively placed in a Corning chamber for 36 or 24h (Figure 6G‐J). To further confirm the effect of ASH2L on cell migration in Ishikawa cells, transwell experiments were further performed with shASH2L #1 and #3 (Figure S5E‐H). Collectively, these results demonstrated that ASH2L depletion inhibited cell migration; consistently, ectopic expression of ASH2L promoted cell migration in Ishikawa cells.
We then further conducted subcutaneous xenograft tumor experiments in mice. Ishikawa cells (1 × 107 cells/mice) infected with lentivirus carrying shASH2L or shCtrl were injected separately into right (shASH2L) or left flanks (shCtrl) of 5‐wk‐old female NOD/SCID mice (n = 10) for 4 wk. The results showed that the growth rate of xenograft tumors formed by shASH2L Ishikawa cells was significantly slower than those from shCtrl cells (Figure 7A). Both size and mass of xenograft tumors formed by shASH2L Ishikawa cells were significantly smaller than those of shCtrl cells (Figure 7B,C). qPCR assay or western blotting was performed to confirm that the RNA or protein levels of ASH2L in the xenograft tumors from shASH2L Ishikawa cells were significantly lower than those from the control group. Expression of PAX2 was also suppressed in tissues formed by shASH2L Ishikawa cells (Figure 7D‐F). In addition, immunohistochemistry results showed that protein expression of ASH2L was reduced and accompanied by a reduction in Ki67 in the xenograft tumor tissues carrying shASH2L (Figure 7G‐H). Taken together, these results suggest that ASH2L depletion significantly decreased the growth rate of the xenograft tumor.
FIGURE 7.
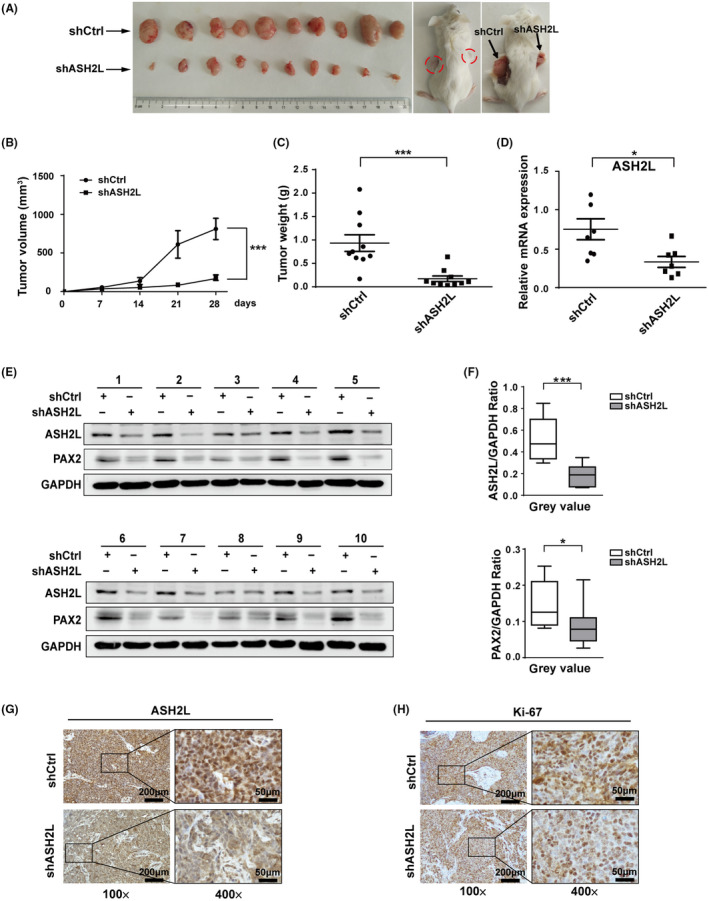
Knockdown of ASH2L inhibits tumor growth in mice xenograft tumor. A, Pictures show xenograft tumors formed by Ishikawa cells in mice. B and C, Tumor volume (B) and weight (C) of xenografts in each group were shown. D, RNA levels of ASH2L in each group were detected by qPCR. E and F, Protein levels of ASH2L and PAX2 in every xenograft tumor tissue were detected by western blotting. G and H, Expression of ASH2L and proliferation‐related protein Ki67 in the xenograft tumor tissues were examined by immunohistochemistry. Data are shown as mean ± SD. *P < .05, ***P < .001
4. DISCUSSION
As an essential subunit of a multimeric histone methyltransferase complex, ASH2L participates in regulation of multiple transcriptional factors to exert various biological functions. However, the role of ASH2L in the regulation of ERα‐induced transcriptional activity and its biological function in endometrial cancer (ECa) are still largely unknown. In this study, we provide evidence to demonstrate that ASH2L plays an oncogenic role in ECa and participates in enhancement of the transcription of ERα target genes, including PAX2.
In the presence of estrogen, ERα enters the nucleus with recruitment of a series of cofactors to induce the target gene transcription. The estrogen/ERα signaling pathway plays a crucial role in carcinogenesis and tumor progression of type I ECa and ERα‐positive breast cancer, although the molecular mechanism of modulation of ERα signaling in ECa has not been clearly elucidated. Dozens of ERα cofactors have been identified to be important for breast cancer progression, however a description of ERα cofactors in ECa remains elusive. It has been mentioned that SRC‐3 as an ERα cofactor is shared between breast cancer and ECa, and it is still unknown whether endometrial cancer‐specific ERα cofactors exist. 27 A previous study described the crosstalk between the glucocorticoid receptor (GR) and ERα in ECa, demonstrating that GR genomic binding sites are reprogrammed in ECa cells following treatment with E2 and dexamethasone (Dex), in which more GR were recruited to the sites normally bound by ERα to promote ECa progression. 30 Therefore, modulation of ERα‐induced transcriptional activation would be crucial for ECa. In this study, our results demonstrated that ASH2L enhanced ERα‐induced transcriptional activity in ECa‐derived cell lines, but had no influence on ESR1 gene transcription. It has been reported previously that, in breast cancer cells, ASH2L associates with transcription factor GATA3 to increase ESR1 gene transcription. Therefore, these results suggested that ASH2L is essential for ECa and breast cancer cell growth in a different ERα action modulation manner.
A comparison of the ERα binding sites in breast cancer and ECa demonstrated that only a small fraction of the binding sites was shared between endometrial cancer cells (ECC‐1) and breast cancer cells (T‐47D). 31 This finding indicates that there are significant differences in ERα‐mediated gene transcription that might be caused by different ERα partners in these tumors. 27 , 32 Here, we demonstrated that ASH2L as a partner of ERα upregulates the transcription of endogenous ERα‐regulated genes, including PAX2, a member of the PAX transcription factor family. ChIP assay results demonstrated that, in ECa‐derived cell lines, ASH2L or ERα is recruited to the ERE II/III regions of PAX2. PAX2 is induced by tamoxifen/E2‐ERα to exert oncogenic function in ECa. 26 , 33 In this study, in an analysis of the sequence at the promoter region of PAX2, we searched several half EREs located at transcriptional regulatory sequences of PAX2 and found that the binding sites of transcription factors such as AP‐1, C/EBPβ, 34 SP1 or NF‐κB 26 , 35 , 36 were nearby the half EREs of PAX2. Whether these transcription factors could form heterodimers with ERα to induce ERα‐regulated genes in ECa still need to be elucidated further.
We have shown that ASH2L associates with ERα together with the core subunits of the ASH2L–KMT2 complex. Moreover, ASH2L depletion reduces the association between ERα and MLL1, but not other subunits. Our results suggest that ERα interacts with ASH2L, and that ASH2L facilitates the association between ERα and MLL1. Binding experiments demonstrated that ASH2L interacts directly with ERα, and that the ASH2L SPRY domain is required for association of ASH2L with MLL1 and WDR5, but not with ERα. Therefore, it appears that ASH2L is an adaptor for the association between ERα and the ASH2L–KMT2 complex.
ChIP experiments have further demonstrated that ASH2L depletion reduced the recruitment of MLL1 or ERα to the ERE II/III regions of PAX2, and resulted in a reduction in histone H3K4me3 levels and an enhancement in H3K27me3 levels. These results suggested that ERα, together with ASH2L, control the modification of both H3K4me3 and H3K27me3. Trimethylation of histone H3K27 as a marker for transcriptional repression is mainly catalyzed by EZH2, which is the core subunit of Polycomb repressive complex 2 (PRC2). Demethylation of H3K27me3 is carried out at least by KDM6A (also known as UTX) or KDM6B, which plays a crucial role in cancer pathogenesis. 37 , 38 KDM6A has been identified in the ASH2L–KMT2 complex, 39 , 40 suggesting that KDM6A could be involved in the KMT2 complex to control histone modification in the context of transcription. In addition, KDM6A has been reported to associate with ERα and functions in ERα‐mediated transcription to be involved in breast cancer progression. 38 Taken together, these findings implied that cooperation between the ASH2L–KMT2 complex, KDM6A, and ERα controls histone H3K4me3 and H3K27me3 on the cis‐regulatory elements of ERα‐regulated genes.
In summary, our studies demonstrated that ASH2L associates with ERα together with the ASH2L–KMT2 complex, thereby regulating histone H3K4me3 and H3K27me3 levels to maintain active transcription on ERα target genes. We provide evidence that ASH2L acts as a novel co‐activator of ERα to promote cell proliferation and migration of endometrial cancer, providing potential targets that could be exploited in the treatment of endometrial cancer (Figure 8).
FIGURE 8.
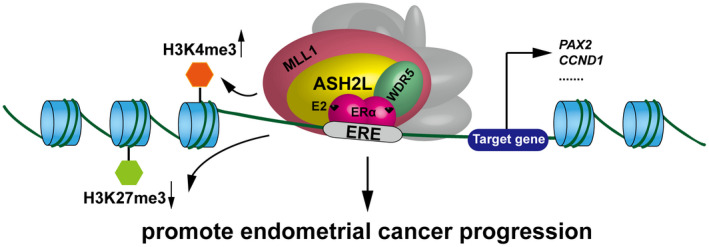
Schematic representation for ASH2L acting as a co‐activator of ERα to be involved in promotion of endometrial cancer progression
DISCLOSURE
The authors declare that they have no conflicts of interest.
Supporting information
Tables S1‐S3
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Yunlong Huo and Dr. Hongyan Zhang for excellent technique assistance. We thank Dr. Xin Zhou for kindly providing the clinical endometrial cancer samples. We thank Dr. Shigeaki Kato (Soma Central Hospital, Fukusima, Japan) for providing ERα and pERE‐tk‐luc plasmids and giving important comments for the draft of this paper. This study was supported by the National Natural Science Foundation of China (31871286 for Yue Zhao, 81872015 for Chunyu Wang, 31701102 for Shengli Wang, 81702800 for Renlong Zou, and 81502438 for Tingting Zhou, 81902889 for Wensu Liu).
Zeng K, Wu Y, Wang C, et al. ASH2L is involved in promotion of endometrial cancer progression via upregulation of PAX2 transcription. Cancer Sci. 2020;111:2062–2077. 10.1111/cas.14413
REFERENCES
- 1. Lauby‐Secretan B, Scoccianti C, Loomis D, et al. Body fatness and cancer‐viewpoint of the IARC Working Group. N Engl J Med. 2016;375:794‐798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Raglan O, Kalliala I, Markozannes G, et al. Risk factors for endometrial cancer: an umbrella review of the literature. Int J Cancer. 2018. [DOI] [PubMed] [Google Scholar]
- 3. Morice P, Leary A, Creutzberg C, Abu‐Rustum N, Darai E. Endometrial cancer. Lancet. 2016;387:1094‐1108. [DOI] [PubMed] [Google Scholar]
- 4. Hemsell DL, Grodin JM, Brenner PF, Siiteri PK, MacDonald PC. Plasma precursors of estrogen. II. Correlation of the extent of conversion of plasma androstenedione to estrone with age. J Clin Endocrinol Metab. 1974;38:476‐479. [DOI] [PubMed] [Google Scholar]
- 5. Liang J, Shang Y. Estrogen and cancer. Annu Rev Physiol. 2013;75:225‐240. [DOI] [PubMed] [Google Scholar]
- 6. Winuthayanon W, Lierz SL, Delarosa KC, et al. Juxtacrine activity of estrogen receptor alpha in uterine stromal cells is necessary for estrogen‐induced epithelial cell proliferation. Sci Rep. 2017;7:8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mittal A, Hobor F, Zhang Y, et al. The structure of the RbBP5 beta‐propeller domain reveals a surface with potential nucleic acid binding sites. Nucleic Acids Res. 2018;46:3802‐3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Steward MM, Lee JS, O'Donovan A, Wyatt M, Bernstein BE, Shilatifard A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol. 2006;13:852‐854. [DOI] [PubMed] [Google Scholar]
- 9. Callebaut I, Mornon JP. The PWAPA cassette: Intimate association of a PHD‐like finger and a winged‐helix domain in proteins included in histone‐modifying complexes. Biochimie. 2012;94:2006‐2012. [DOI] [PubMed] [Google Scholar]
- 10. Chen Y, Wan B, Wang KC, et al. Crystal structure of the N‐terminal region of human Ash2L shows a winged‐helix motif involved in DNA binding. EMBO Rep. 2011;12:797‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang NC, Sincennes M‐C, Chevalier FP, et al. The dystrophin glycoprotein complex regulates the epigenetic activation of muscle stem cell commitment. Cell Stem Cell. 2018;22:755‐768. e756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jung Y, Hsieh LS, Lee AM, et al. An epigenetic mechanism mediates developmental nicotine effects on neuronal structure and behavior. Nat Neurosci. 2016;19:905‐914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stoller JZ, Huang LI, Tan CC, et alAsh2l interacts with Tbx1 and is required during early embryogenesis. Exp Biol Med (Maywood). 2010;235:569‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tan CC, Sindhu KV, Li S, et al. Transcription factor Ap2delta associates with Ash2l and ALR, a trithorax family histone methyltransferase, to activate Hoxc8 transcription. Proc Natl Acad Sci U S A. 2008;105:7472‐7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song M, Fang F, Dai X, Yu L, Fang M, Xu Y. MKL1 is an epigenetic mediator of TNF‐alpha‐induced proinflammatory transcription in macrophages by interacting with ASH2. FEBS Lett. 2017;591:934‐945. [DOI] [PubMed] [Google Scholar]
- 16. Ullius A, Luscher‐Firzlaff J, Costa IG, et al. The interaction of MYC with the trithorax protein ASH2L promotes gene transcription by regulating H3K27 modification. Nucleic Acids Res. 2014;42:6901‐6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu L, Lee S, Zhou BO, et al. ASH2L regulates ubiquitylation signaling to MLL: trans‐regulation of H3 K4 methylation in higher eukaryotes. Mol Cell. 2013;49:1108‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luscher‐Firzlaff J, Gawlista I, Vervoorts J, et al. The human trithorax protein hASH2 functions as an oncoprotein. Cancer Res. 2008;68:749‐758. [DOI] [PubMed] [Google Scholar]
- 19. Mungamuri SK, Wang S, Manfredi JJ, Gu W, Aaronson SA. Ash2L enables P53‐dependent apoptosis by favoring stable transcription pre‐initiation complex formation on its pro‐apoptotic target promoters. Oncogene. 2015;34:2461‐2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tateishi Y, Kawabe Y‐I, Chiba T, et al. Ligand‐dependent switching of ubiquitin‐proteasome pathways for estrogen receptor. EMBO J. 2004;23:4813‐4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao Y, Takeyama K‐I, Sawatsubashi S, et alCorepressive action of CBP on androgen receptor transactivation in pericentric heterochromatin in a Drosophila experimental model system. Mol Cell Biol. 2009;29:1017‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang X, Kyo S, Nakamura M, et al. Imatinib sensitizes endometrial cancer cells to cisplatin by targeting CD117‐positive growth‐competent cells. Cancer Lett. 2014;345:106‐114. [DOI] [PubMed] [Google Scholar]
- 23. Wang C, Sun H, Zou R, et alMDC1 functionally identified as an androgen receptor co‐activator participates in suppression of prostate cancer. Nucleic Acids Res. 2015;43:4893‐4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu S, Wu F, Gu S, et al. Gene silencing via PDA/ERK2‐siRNA‐mediated electrospun fibers for peritendinous antiadhesion. Adv Sci (Weinh). 2019;6:1801217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Y, Han J, Zhang Y, et alStructural basis for activity regulation of MLL family methyltransferases. Nature. 2016;530:447‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu H, Chen Y, Liang J, et al. Hypomethylation‐linked activation of PAX2 mediates tamoxifen‐stimulated endometrial carcinogenesis. Nature. 2005;438:981‐987. [DOI] [PubMed] [Google Scholar]
- 27. Rodriguez AC, Blanchard Z, Maurer KA, Gertz J. Estrogen signaling in endometrial cancer: a key oncogenic pathway with several open questions. Horm Cancer. 2019;10:51‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kong SL, Li G, Loh SL, Sung WK, Liu ET. Cellular reprogramming by the conjoint action of ERalpha, FOXA1, and GATA3 to a ligand‐inducible growth state. Mol Syst Biol. 2011;7:526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang LU, Ozark PA, Smith ER, et al. TET2 coactivates gene expression through demethylation of enhancers. Sci Adv. 2018;4:eaau6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vahrenkamp JM, Yang CH, Rodriguez AC, et al. Clinical and Genomic Crosstalk between Glucocorticoid Receptor and Estrogen Receptor alpha In Endometrial Cancer. Cell Rep. 2018;22:2995‐3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gertz J, Savic D, Varley K, et alDistinct properties of cell‐type‐specific and shared transcription factor binding sites. Mol Cell. 2013;52:25‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shang Y. Molecular mechanisms of oestrogen and SERMs in endometrial carcinogenesis. Nat Rev Cancer. 2006;6:360‐368. [DOI] [PubMed] [Google Scholar]
- 33. Kahraman K, Kiremitci S, Taskin S, Kankaya D, Sertcelik A, Ortac F. Expression pattern of PAX2 in hyperplastic and malignant endometrium. Arch Gynecol Obstet. 2012;286:173‐178. [DOI] [PubMed] [Google Scholar]
- 34. Rotinen M, Celay J, Alonso MM, Arrazola A, Encio I, Villar J. Estradiol induces type 8 17beta‐hydroxysteroid dehydrogenase expression: crosstalk between estrogen receptor alpha and C/EBPbeta. J Endocrinol. 2009;200:85‐92. [DOI] [PubMed] [Google Scholar]
- 35. Dong J, Tsai‐Morris CH, Dufau ML. A novel estradiol/estrogen receptor alpha‐dependent transcriptional mechanism controls expression of the human prolactin receptor. J Biol Chem. 2006;281:18825‐18836. [DOI] [PubMed] [Google Scholar]
- 36. Cheung E, Acevedo ML, Cole PA, Kraus WL. Altered pharmacology and distinct coactivator usage for estrogen receptor‐dependent transcription through activating protein‐1. Proc Natl Acad Sci U S A. 2005;102:559‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morgan MA, Shilatifard A. Chromatin signatures of cancer. Genes Dev. 2015;29:238‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim J‐H, Sharma A, Dhar SS, et al. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014;74:1705‐1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015;15:334‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee MG, Villa R, Trojer P, et alDemethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447‐450. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1‐S3
Supplementary Material