

Abstract
Formalin‐fixed paraffin‐embedded (FFPE) tissues used for routine pathological diagnosis are valuable for cancer genomic analysis; however, the association between mutation status derived from these specimens and prognosis in pancreatic ductal adenocarcinoma (PDAC) remains unclear. We analyzed 50 cancer‐related gene mutations including driver genes in PDAC, using next‐generation sequencing (NGS) to clarify the association between gene mutations and prognosis. DNA was extracted from FFPE tissues obtained from 74 patients with untreated resectable PDAC who underwent surgery at our institution between 2013 and 2018. Fifty of the 74 patients with DNA extracts from FFPE samples suitable for NGS were analyzed. The prevalence of driver gene mutations was as follows: 84% for KRAS, 62% for TP53, 32% for SMAD4, and 18% for CDKN2A. There were no cases of single SMAD4 mutations; its rate of coincidence with KRAS or TP53 mutations was 30% and 2%, respectively. The combination of KRAS and SMAD4 mutations resulted in significantly shorter relapse‐free survival (RFS; median survival time [MST], 12.3 vs. 28.9 months, P = .014) and overall survival (OS; MST, 22.3 months vs. not reached, P = .048). On multivariate analysis, the combination of KRAS and SMAD4 mutations was an independent prognostic factor for RFS (hazard ratio [HR] 4.218; 95% confidence interval [CI], 1.77‐10.08; P = .001) and OS (HR 6.730; 95% CI, 1.93‐23.43; P = .003). The combination of KRAS and SMAD4 mutations in DNA obtained from FFPE tissues is an independent poor prognostic factor in PDAC.
Keywords: KRAS, next‐generation sequencing, pancreatic ductal adenocarcinoma, prognosis, SMAD4
Next‐generation sequencing (NGS) assay targeted 50 cancer‐related genes using DNA extracted from 74 formalin‐fixed paraffin‐embedded (FFPE) tissues of patients with pancreatic ductal adenocarcinoma; mutations were detected in 94% of the 50 (70%) analyzed cases. First, the detection of KRAS and SMAD4 mutations in the resected specimen implied an increase in tumor recurrence with a poor chance of survival. Second, DNA extracted from FFPE tissue was feasible for analysis by NGS to arrive at an accurate prognosis.
![]()
Abbreviations
- CA19‐9
carbohydrate antigen 19‐9
- CHPv2
Cancer Hotspot Panel version 2
- CI
confidence interval
- FFPE
formalin‐fixed paraffin embedded
- HR
hazard ratio
- IPMC
intraductal papillary mucinous carcinoma
- MST
median survival time
- NCCN
National Comprehensive Cancer Network
- NGS
next‐generation sequencing
- OS
overall survival
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
- R
residual tumor
- RFS
relapse‐free survival
1. INTRODUCTION
Despite progress made in health care, PDAC retains a poor record of prognosis. 1 In the United States, the estimated number of newly diagnosed PDAC patients in 2019 is 56 770. Pancreatic ductal adenocarcinoma is the fourth leading cause of cancer‐related death, with a mortality rate of 10% in 2019. 2 Due to aggressive tumor behavior and late clinical detection, the 5‐year survival rate of patients with PDAC is 10%. 2 Although surgical resection remains the only curative treatment for patients without distant metastasis of PDAC, only 20%‐30% of patients are eligible for resection at the time of diagnosis. 2 , 3 In addition, most patients who undergo curative pancreatectomy experience recurrence. Although the prognosis for patients with PDAC has improved due to multimodal treatment, 4 no effective molecularly targeted drug has been established. Recently, gene mutations have been reported to be effective biomarkers for the determination of therapeutic strategies in various cancers, 5 , 6 and some druggable gene mutations have been reported to improve survival rates. 7 , 8 In advanced PDAC patients with distant metastasis, germline BRCA mutations have been reported to be druggable genes 9 ; however, biomarkers have not yet been established for therapeutic targets or precision medicine in resected PDAC.
Genomic biomarkers for PDAC have been identified by analyzing the association between somatic gene mutations and their clinicopathological variables. 10 Four driver genes, namely, KRAS, TP53, SMAD4, and CDKN2A, have been reported as representative cancer‐related genes in PDAC. Among them, the KRAS gene mutation is most common, and is associated with poor prognosis. 11 , 12 We previously reported that KRAS mutations in cell‐free DNA obtained from serum in the aftermath of resection are associated with poor prognosis. 13 However, we also previously reported that the survival rate of IPMC patients with a genetic heterogeneous primary tumor containing more than 2 types of KRAS mutation, was better than that of IPMC patients with genetic homogenous primary tumors with only 1 type of KRAS mutation. 14 Recently, PDAC tissues have been subjected to genome‐wide sequencing, and whole‐exon sequencing in particular, and approximately 40 associated gene mutations were discovered. 15 , 16 , 17 , 18 Blackford et al 19 utilized NGS using DNA extracts from resected cancer cells purified from cell lines or xenografts. By analyzing DNA extracts from fresh‐frozen samples, Hayashi et al 20 revealed that the number of driver gene mutations could be a prognostic factor; however, certain gene mutations associated with prognosis were not detected. Based on NGS analysis using FFPE samples, we previously reported that multiple PDAC involves multicentric carcinogenesis and intrapancreatic metastasis, which could be distinguished by comparing driver gene mutations; the latter has a dismal prognosis. 21 According to the NCCN Guidelines of 2019, somatic mutational profiling of tumor tissue is recommended for PDAC. 22 In the future, it is expected that genetic tests from resected specimens will be routine for almost all cases of PDAC. Next‐generation sequencing analysis using DNA extracted from fresh‐frozen samples is well documented; however, only a few reports exist regarding DNA extracted from FFPE samples for PDAC. 23 Due to formalin fixation‐induced degradation, the success rate of NGS analysis using DNA extracted from FFPE tissues is lower than that obtained from fresh‐frozen specimens. 24 However, NGS using FFPE samples is valuable, as these are used for pathological diagnosis in routine clinical practice. Therefore, we hypothesized that NGS analysis using FFPE samples could be applied to PDAC, and that this genetic analysis, including that of driver gene mutations, could identify prognostic predictive factors for PDAC and also clarify the rate of druggable gene mutations in resected PDAC. We undertook targeted deep sequencing using NGS to screen 50 cancer‐related genes from surgically resected PDAC specimens at our institution. We aimed to identify the association between these mutations and clinicopathologic variables as well as the prognostic utility of these mutations.
2. MATERIALS AND METHODS
2.1. Patients
In total, 146 PDAC patients who underwent curative resection at our institution between March 2013 and May 2018 were retrospectively analyzed. All patients were histologically diagnosed with ductal adenocarcinoma. Among these 146 patients, 72 were excluded: for 34 of these patients, samples could not be prepared owing to a lack of consent or other reasons, 32 patients had undergone neoadjuvant treatment, 3 had received chemotherapy for remnant pancreatic cancer, and 3 had undergone R2 resection.
The pathological staging of all specimens, including residual tumors (R) was determined according to the 8th edition of the UICC TNM classification. 25 All participants provided written informed consent. The study was approved by the Human Experimentation Committee of Keio University Hospital (Nos. 20120443 and 20170086), and was carried out in accordance with the 1975 Declaration of Helsinki.
2.2. Clinicopathological characteristics
Clinicopathological variables were extracted from medical records. The clinical variables included age, sex, presence of diabetes mellitus, amount of serum CA19‐9, surgical procedure, operation time, blood loss, postoperative complications evaluated according to the Clavien‐Dindo classification, surgery‐related deaths, and adjuvant chemotherapy. Pathological variables included tumor size, location, and differentiation grade, lymphatic, venous, and intrapancreatic nerve infiltration, serosal, retropancreatic tissue, distal bile duct, duodenal, portal vein, arterial, and extra‐pancreatic nerve plexus invasion, and invasion of other organs. 26
In our institution, most patients with borderline resectable PDAC (according to NCCN resectability classification) undergo neoadjuvant treatment. In addition, some patients diagnosed with T3 or T4 disease according to the 7th edition of the UICC TNM classification have been receiving neoadjuvant chemoradiotherapy since 2003. 27 , 28 Surgical resections included pylorus‐preserving or subtotal stomach‐preserving pancreatoduodenectomy, distal pancreatectomy, and total pancreatectomy. D2 lymph node dissections were undertaken in all patients.
After surgical resection of PDAC, each patient underwent standard postoperative follow‐up. S‐1 or gemcitabine was given as adjuvant chemotherapy at the physician’s discretion. After October 2013, S‐1 was primarily given based on the results of interim JASPAC01 analysis. 29 Successfully completed adjuvant treatment was defined as one that was given for 6 months. Recurrence was defined by definitive evidence of recurrence, which was confirmed with radiographic findings, with or without elevated serum CA19‐9 levels. Physical examinations, toxicity assessments, complete blood cell counts, serum chemistry profiles, and chest‐abdominal computed tomography scans were carried out approximately every 4‐6 months for the first 12 months, and every 6 months thereafter. 28
2.3. Preparation and extraction of DNA from FFPE specimens
Resected specimens were immediately fixed in 10% buffered formalin. The fixed specimens were embedded in paraffin within 1 week. The paraffinized sections were stained with H&E and the main tumor lesions were identified by a pathologist. Ten‐micrometer sections of the primary tumor were cut from each block and placed on glass slides. One section of the main tumor was stained with H&E for orientation; this was subsequently confirmed by a faculty pathologist. Tiny fractions of the main tumor lesions were grossly dissected from the 10‐µm sections, and fractions from 3 or 4 sections were collected in sterile tubes. DNA was extracted and purified from these paraffin sections using the QIAamp DNA FFPE Tissue Kit (Qiagen) according to the manufacturer’s protocol. 13 , 14 DNA concentrations were determined using the TaqMan RNase P Detection Reagents Kit (Applied Biosystems) according to the manufacturer’s protocol. 13
2.4. Targeted sequencing of genomic DNA in FFPE
Amplicon libraries were prepared for 10 nm of individual genomic DNA using an Ion AmpliSeq CHPv2 and Ion AmpliSeq Library Kit 2.0 (Thermo Fisher Scientific). Multiplex PCR was carried out for 20 cycles according to the manufacturer’s protocol. The CHPv2 targeted 2790 Catalogue of Somatic Mutations in Cancer mutational hotspot regions in the following 50 cancer‐related genes: ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAS, GNAQ, HNF1A, HRAS, IDH1, IDH2, JAK2, JAK3, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, and VHL.
Sequencing adapters were joined to the amplification products using unique barcodes (Ion Xpress Barcode Adapters 1‐16 Kit; Thermo Fisher Scientific) and purified using Agencourt AMPure XP Reagent (Beckman Coulter) according to the manufacturer’s protocol. 24
2.5. Statistical analysis
Statistical analysis was undertaken using SPSS statistics version 25.0 (IBM Japan). Clinicopathological variables were compared between patients with and without driver gene mutations. Categorical variables were compared by the χ2 test and continuous variables were compared by the Mann‐Whitney U test. Survival duration was calculated using the Kaplan‐Meier method and comparisons were made between groups using a log‐rank test. A Cox proportional hazards model was used to determine independent prognostic factors among the clinicopathological and genomic variables. Variables with P < .10 as determined by univariate analysis were entered into a forward, stepwise backward multivariate analysis. All statistical tests were 2‐tailed, and P < .05 was considered significant.
3. RESULTS
3.1. Mutations among 50 cancer‐related genes
Seventy‐four FFPE samples were used for DNA extraction, and 3 samples (4.1%) with DNA concentrations less than 0.85 ng/μL were excluded, as recommended by the manufacturer. Library preparation and sequencing by CHPv2 were carried out on 71 samples; however, 21 samples failed due to insufficient amplified DNA. Among the 74 samples, 50 (67.6%) were eligible for NGS analysis.
In total, 125 mutations were detected in 23 genes (Figure 1A). No mutations were detected in 3 (6.0%) cases. Detailed information on the driver gene mutations is shown in Table 1. KRAS was the most commonly mutated gene, with mutations observed in 42 cases (84.0%). This was followed by TP53 in 32 (64.0%), SMAD4 in 16 (32.0%), CDKN2A in 9 (18.0%), PIK3CA in 3 (6.0%), RET in 3 (6.0%), EGFR in 2 (4.0%), ERBB4 in 2 (4.0%), MLH1 in 2 (4.0%), and NOTCH1 in 2 (4.0%) cases. Other variants detected in 1 case included hotspot mutations in APC, ATM, GNAS, HRAS, JAK2, JAK3, NRAS, PTEN, RB1, SMARCB1, SRC, and STK11. No mutations were identified within the hotspot regions of the remaining 27 genes. A single mutation was identified in 4 cases (8.0%), 2 mutations were identified in 22 cases (44.0%), 3 mutations were identified in 9 cases (18.0%), and 4 or more mutations were identified in 12 cases (24.0%).
FIGURE 1.
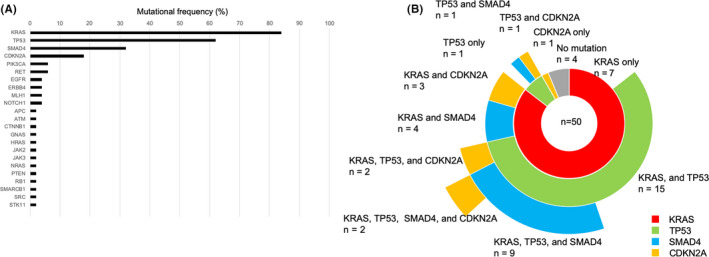
Gene mutation profile of pancreatic ductal adenocarcinoma (PDAC). A, Frequency of somatic mutations in 50 patients with PDAC. B, Combination of driver gene mutations in 50 PDAC patients
TABLE 1.
Details of driver gene mutations in 50 patients with pancreatic ductal adenocarcinoma
| Parameter | KRAS (n = 42) | TP53 (n = 31) | SMAD4 (n = 16) | CDKN2A (n = 9) |
|---|---|---|---|---|
| Mutation, n (%) | ||||
| Missense | 42 (100.0) | 25 (80.6) | 9 (56.3) | 5 (55.6) |
| Nonsense | 0 (0.0) | 4 (12.9) | 4 (25.0) | 3 (33.3) |
| Frameshift insertion | 0 (0.0) | 1 (3.2) | 2 (12.5) | 0 (0.0) |
| Frameshift deletion | 0 (0.0) | 1 (3.2) | 1 (6.3) | 1 (11.1) |
| KRAS mutant subtype, n (%) | ||||
| G12D | 20 (47.6) | |||
| G12V | 15 (35.7) | |||
| G12R | 4 (9.5) | |||
| Q61H | 3 (7.1) | |||
The frequently observed mutation patterns of the driver genes included a single mutation in KRAS, TP53, and CDKN2A genes in 7 (14.0%), 1 (2.0%), and 1 (2.0%) cases, respectively. Mutations were observed in 2 driver genes, KRAS/TP53 in 15 cases (30.0%) and KRAS/SMAD4, KRAS/CDKN2A, TP53/SMAD4, and TP53/CDKN2A in 4 (8.0%), 3 (6.0%), 1 (2.0%), and 1 (2.0%) cases, respectively. There were 9 (18.0%) and 1 (4.0%) cases with 3 mutated driver genes, namely KRAS/TP53/SMAD4 and KRAS/TP53/CDKN2A, and 2 cases (4.0%) with mutations in all 4 driver genes (Figure 1B). We used CHPv2, which includes 10 druggable genes among the 50 target genes. Ten mutations were found in the druggable genes ATM, NOTCH1, PIK3CA, PTEN, and RET; however, these mutations were detected in only 8 cases (16.0%). Detailed information pertaining to the mutation analyses are shown in Table S1.
3.2. Patient characteristics
Fifty patients were successfully analyzed by NGS in this study, and their characteristics are shown in Table 2. The median age of the patients was 71.5 (38‐85) years; 32 patients were male and 18 female. Pancreaticoduodenectomy, distal pancreatectomy, and total pancreatectomy were carried out in 27, 20, and 3cases, respectively. R0 resection was undertaken in 29 cases (58%), and R1 resection in 21 cases. Adjuvant chemotherapy was given in 42 cases and successfully completed in 29 cases.
TABLE 2.
Characteristics of patients with pancreatic ductal adenocarcinoma (n = 50)
| Parameter | n = 50 |
|---|---|
| Age, y; median (range) | 71.5 (38‐85) |
| Sex, n (%) | |
| Male | 32 (64.0) |
| Female | 18 (36.0) |
| Preoperative CA19‐9, IU/mL; median (range) | 100 (1‐2392) |
| Neoadjuvant treatment, n (%) | 0 (0.0) |
| Induction of adjuvant treatment, n (%) | 42 (84.0) |
| Successfully completed adjuvant treatment, n (%) | 29 (58.0) |
| Regimen of adjuvant treatment, n (%) | |
| S‐1 | 26 (52.0) |
| Gemcitabine | 1 (2.0) |
| Gemcitabine + S‐1 | 2 (4.0) |
| Surgical procedure, n (%) | |
| Pancreaticoduodenectomy | 27 (54.0) |
| Distal pancreatectomy | 20 (40.0) |
| Total pancreatectomy | 3 (6.0) |
| Severe complications, n (%) | 7 (14.0) |
| Surgery‐related deaths, n (%) | 0 (0.0) |
| Tumor location, n (%) | |
| Head | 28 (56.0) |
| Body or tail | 22 (44.0) |
| Tumor size, mm; median (range) | 29.5 (12‐65) |
| Residual tumor status, n (%) a | |
| R0 | 29 (58.0) |
| R1 | 21 (42.0) |
| R2 | 0 (0.0) |
| Pathological T status, n (%) a | |
| pT1 | 7 (14.0) |
| pT2 | 35 (70.0) |
| pT3 | 8 (16.0) |
| pT4 | 0 (0.0) |
| Pathological N status, n (%) a | |
| pN0 | 10 (20.0) |
| pN1 | 21 (42.0) |
| pN2 | 19 (38.0) |
| Pathological stage, n (%) a | |
| IA | 4 (8.0) |
| IB | 6 (12.0) |
| IIA | 0 (0.0) |
| IIB | 20 (40.0) |
| III | 18 (36.0) |
| IV | 2 (4.0) |
CA19‐9, carbohydrate antigen 19‐9.
Based on UICC TNM classification (8th edition).
3.3. Survival analysis
At the time of analysis, 26 (52.0%) patients had recurrence and 14 (28.0%) patients had died. The median time from the date of surgery to the last follow‐up was 18.7 months (range, 5.1‐50.0 months) for all patients. The median time of RFS was 18.2 months, but that for OS was not reached.
Patients with SMAD4 mutations had significantly poorer RFS than those with WT SMAD4 (MST, 12.3 vs. 23.4 months, respectively; 1‐ and 3‐year RFS, 54.7%, 13.0% vs. 69.0%, 33.2%, respectively, P = .034; Figure 2A). Patients with SMAD4 mutations had poorer OS than those with WT SMAD4 (MST, 22.3 months vs. not reached, respectively; 1‐ and 3‐year OS, 87.1%, 30.5% vs. 93.8%, 70.9%, respectively, P = .063; Figure 2B). The SMAD4 WT group had a significantly higher amount of venous infiltration (P = .044) than the SMAD4 mutation group. There was no significant difference in the other parameters such as age, sex, diabetes mellitus, tumor location, resectability, clinical stage, tumor diameter, R0 resection, pathological TNM, and pathological stage between the 2 groups (Table S2).
FIGURE 2.
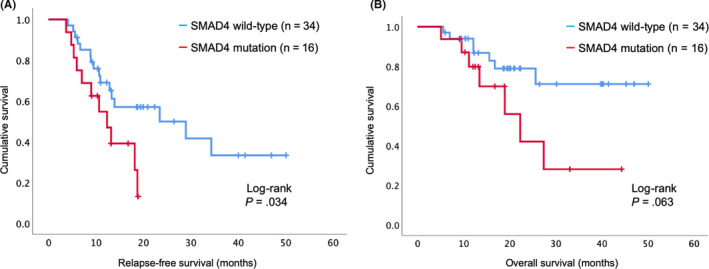
Survival curve using the Kaplan‐Meier method for all patients with pancreatic ductal adenocarcinoma (PDAC) (n = 50). A, Relapse‐free survival curve according to SMAD4 gene mutation status. B, Overall survival curve according to SMAD4 gene mutation status
There were no single‐mutation cases of SMAD4, and 15 (93.8%) of all cases of SMAD4 mutations were coincident with KRAS mutations. Patients with a combination of KRAS and SMAD4 mutations had significantly poorer RFS than those with WT KRAS or SMAD4 (MST, 12.3 vs. 28.9 months; 1‐ and 3‐year RFS, 51.4%, 11.4% vs. 69.9%, 33.8%, P = .014; Figure 3A). Patients with a combination of KRAS and SMAD4 mutations also had significantly poorer OS than those with WT KRAS or SMAD4 (MST, 22.3 months vs. not reached, respectively; 1‐ and 3‐year OS, 78.3%, 27.4% vs. 94.2%, 71.4%, respectively, P = .048; Figure 3B).
FIGURE 3.
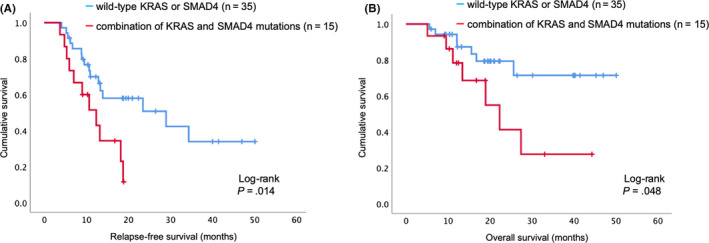
Survival curve generated by the Kaplan‐Meier method for patients with pancreatic ductal adenocarcinoma. A, Relapse‐free survival curve based on the combined KRAS and SMAD4 gene mutation status. B, Overall survival curve based on the combined KRAS and SMAD4 gene mutation status
On univariate Cox regression analysis, SMAD4 mutation, a combination of KRAS and SMAD4 mutations, lymphatic and intrapancreatic nerve infiltration, serosal, extrapancreatic nerve plexus invasion, pathological T status, pathological N status, and successfully completed adjuvant treatment were identified as significant predictive factors for RFS (Table 3). Likewise, for OS, a combination of KRAS and SMAD4 mutations, portal vein invasion, pathological T status, and successfully completed adjuvant treatment were significant predictive factors. On multivariate Cox regression analysis, a combination of KRAS and SMAD4 mutations (HR = 4.218; 95% CI, 1.77‐10.08; P = .001) and successfully completed adjuvant treatment (HR = 0.226; 95% CI, 0.10‐0.52; P < .001) were independent predictive factors for RFS (Table 3), whereas a combination of KRAS and SMAD4 mutations (HR = 6.730; 95% CI, 1.93‐23.43; P = .003) and successfully completed adjuvant treatment (HR = 0.068; 95% CI, 0.02‐0.27; P < .001) were identified as independent predictive factors for OS (Table 3).
TABLE 3.
Univariate and multivariate Cox regression analysis of prognostic factors for relapse‐free survival and overall survival in patients with pancreatic ductal adenocarcinoma
| Relapse‐free survival | Overall survival | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Univariate | Multivariate | Univariate | Multivariate | |||||||
| HR | P value | HR | 95% CI | P value | HR | P value | HR | 95% CI | P value | |
| KRAS mutation | 0.847 | .763 | 0.525 | .336 | ||||||
| KRAS mutant subtype | 0.861 | .464 | 1.016 | .958 | ||||||
| TP53 mutation | 1.154 | .720 | 1.245 | .716 | ||||||
| SMAD4 mutation | 2.337 | .040 | 2.611 | .074 | ||||||
| CDKN2A mutation | 1.157 | .770 | 2.496 | .131 | ||||||
| Combination of KRAS and SMAD4 mutations | 2.639 | .019 | 4.218 | 1.765‐10.079 | .001 | 2.767 | .057 | 6.730 | 1.934‐23.426 | .003 |
| Preoperative serum CA19‐9 | 1.000 | .214 | 1.001 | .146 | ||||||
| Tumor size ≥20 mm | 1.703 | .387 | 2.176 | .454 | ||||||
| Tumor differentiation grade | 0.670 | .313 | 1.182 | .785 | ||||||
| Lymphatic infiltration ≥2 | 2.786 | .061 | 2.020 | .359 | ||||||
| Venous infiltration ≥2 | 1.388 | .548 | 2.375 | .405 | ||||||
| Intrapancreatic nerve infiltration ≥2 | 2.928 | .080 | 29.19 | .230 | ||||||
| Serosal invasion | 2.116 | .059 | 1.146 | .814 | ||||||
| Retro pancreatic tissue invasion | 1.332 | .601 | 26.98 | .349 | ||||||
| Portal vein invasion | 1.895 | .100 | 3.213 | .052 | ||||||
| Arterial invasion | 0.572 | .449 | 0.653 | .684 | ||||||
| Extrapancreatic nerve plexus invasion | 2.428 | .061 | 1.547 | .577 | ||||||
| Pathological T status | 2.107 | .041 | .092 | 2.339 | .078 | .332 | ||||
| Pathological N status | 1.713 | .061 | .561 | 1.275 | .501 | .863 | ||||
| Pathological stage | 1.189 | .259 | 1.163 | .474 | ||||||
| Residual tumor status | 1.640 | .208 | 1.137 | .824 | ||||||
| Severe complications | 0.433 | .560 | 0.592 | .614 | ||||||
| Successfully completed adjuvant treatment | 0.295 | .002 | 0.226 | 0.098‐0.521 | <.001 | 0.228 | .013 | 0.068 | 0.017‐0.274 | <.001 |
Abbreviations: CA19‐9, carbohydrate antigen 19‐9; CI, confidence interval; HR, hazard ratio.
4. DISCUSSION
Among the 4 driver genes, SMAD4 mutations were recognized as a prognostic factor for RFS. Furthermore, the combination of KRAS and SMAD4 mutations was found to be an independent prognostic factor for RFS and OS. DNA extracted from FFPE tissues was suitable for analysis by NGS. Druggable gene mutations were detected in 16% of resected FFPE samples of PDAC.
In the current study, 68% of FFPE samples were successfully sequenced. Nakagaki et al 24 reported that all fresh‐frozen specimens were successfully sequenced, but 40% of FFPE specimens were eligible for NGS analyses. The report by Nakagaki et al used samples that were up to 10 years old, although details on the proportions of sample age are unknown; the quality of those samples might have been poorer than those used in the current study, which only included samples up to 5 years of age. However, in the current study, the success rate of analysis decreased when the time to analysis exceeded 3 years (data not shown). The Guidelines for Validation of Next‐Generation Sequencing‐Based Oncology Panels recommend that NGS analysis using DNA extracted from older FFPE blocks (eg older than 3 years) could increase background noise due to deamination. 30 Hence, DNA was extracted from newer samples as per the recommendation. However, this introduces the potential limitation of a shortened follow‐up period. Establishing a biobank for FFPE specimens might not be necessary, as for fresh‐frozen specimens, because FFPE is routinely used for pathological diagnosis and thus, genomic testing can be carried out retrospectively using resected specimens. Using NGS with FFPE should therefore aid in the introduction of genomic testing in daily clinical practice. The current study indicated that DNA extracts from FFPE tissues are eligible for NGS analysis and elicit an accurate prognosis.
In the current study, the mutation rates in the four driver genes were similar to those described in previous reports. 17 , 18 , 20 Among these genes, SMAD4 mutations were found to be an independent prognostic factor for RFS. Furthermore, the combination of KRAS and SMAD4 mutations was an independent prognostic factor for both RFS and OS. However, other mutations, including those in KRAS alone, as well as those in TP53 and CDKN2A, did not stratify cases by RFS or OS. Although KRAS mutations in PDAC occur too frequently to be considered appropriate prognostic factors, previous reports mention that the KRAS mutant subtype G12V or G12D is associated with poor prognosis 31 , 32 ; however, which exact subtype is associated with a worse prognosis is controversial. In the current study, differences in the KRAS mutation subtype had no impact on prognosis.
The transforming growth factor‐β/SMAD4 signaling pathway suppresses tumors that induce cell cycle arrest and apoptosis. 33 , 34 Loss of heterozygosity or the homozygous deletion of SMAD4 was first reported in PDAC, 35 but has since been identified in various types of cancer. 36 , 37 In PDAC, the decreased expression of SMAD4 identified by the immunolabeling of resected specimens has been reported to be associated with poor prognosis. 14 , 38 , 39 , 40 In NGS, SMAD4 mutations are considered a poor prognostic factor in the analysis of DNA extracts from resected PDAC cells purified from cell lines or xenografts, 21 whereas they were not a prognostic factor in the analysis of DNA extracts from FFPE or fresh‐frozen specimens. To our knowledge, the current study is the first to report SMAD4 mutations as a prognostic factor for PDAC in NGS analysis using DNA extracts from FFPE. Wilentz et al reported that SMAD4 mutations could be detected at a rate of 30% in high‐grade PanIN, a precancerous lesion of pancreatic cancer. However, there were no SMAD4 mutations in low‐grade PanIN (non‐PanIN and PanIN 1). 41 In a mouse model, KRAS mutations alone could slow the progression of PanIN to cancer; however, combining KRAS mutations and SMAD4 deletions was found to cause the rapid progression of tumors. 42 In the examination of autopsy cases, SMAD4 gene mutations were found to be a predictor of multiple distant metastases. 43 These previous reports suggest that SMAD4 mutations could lead to cancer progression and, therefore, poor prognosis; this supports the results of the current study. The combination of KRAS and SMAD4 could potentially be used as a prognostic indicator for the selection of appropriate adjuvant treatment regimens, such as multidrug combination therapy or extension of the treatment period.
Similar to previous reports, druggable gene mutations were detected in only 16% of cases in the current study. 18 , 20 Cancer Hotspot Panel version 2 does not contain BRCA mutations, which have been reported to be effective targets for treatment with a poly (adenosine diphosphate‐ribose) polymerase inhibitor. 9 In addition, no mutations were detected in 27 genes of CHPv2 in the current study. Therefore, the development of a specific multigene panel might be needed for PDAC.
The current study had some limitations. First, CHPv2 could only detect 50 cancer‐related genes, and could not identify the presence or absence of other related and targetable mutations. Second, the current study had a retrospective design; thus, the results should be verified in more cases, and in a prospective study. Finally, approximately 30% of NGS analyses were excluded due to unsuccessful sequencing.
In conclusion, based on NGS analysis using the FFPE tissue of resected PDAC, the current study revealed that the combination of KRAS and SMAD4 mutations is an independent poor prognostic factor for recurrence and survival. Further prospective investigations based on this analysis are needed in the future.
CONFLICT OF INTEREST
MS has received research funds from Taiho Pharmaceutical, Shionogi, and Eisai. YK has received scholarship endowments from Taiho Pharmaceutical, Chugai Pharmaceutical, Tsumura & Co., Otsuka Pharmaceutical, AsahiKASEI, Eisai, Otsuka Pharmaceutical Factory, Takeda Pharmaceutical, Medicon, Pfizer Japan, Ono Pharmaceutical, Shionogi & Co., and Daiichi Sankyo, and received lecture fees/honoraria from AsahiKASEI, Ono Pharmaceutical, Ethicon, Taiho Pharmaceutical, and Chugai Pharmaceutical, and is an officer/advisor for medical corporations Keiyoukai Keiai Clinic, Hirata Clinic, and Ageo Central General Hospital. The other authors declare that they have no conflicts of interest.
Supporting information
Table S1
Table S2
ACKNOWLEDGEMENTS
This work was supported by JSPS KAKENHI Grant Number 16H06279 (PAGS). We would like to thank Kazumasa Fukuda, a staff member of the Department of Surgery at Keio University School of Medicine, for her help in preparing this manuscript. In addition, we would like to thank Editage for English language editing.
Yokose T, Kitago M, Matsuda S, et al. Combination of KRAS and SMAD4 mutations in formalin‐fixed paraffin‐embedded tissues as a biomarker for pancreatic cancer. Cancer Sci. 2020;111:2174–2182. 10.1111/cas.14425
REFERENCES
- 1. Sant M, Allemani C, Santaquilani M, Knijn A, Marchesi F, Capocaccia R. Survival of cancer patients diagnosed in 1995–1999. Results and commentary. Eur J Cancer. 2009;45:931‐991. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7‐34. [DOI] [PubMed] [Google Scholar]
- 3. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893‐2917. [DOI] [PubMed] [Google Scholar]
- 4. Cloyd JM, Katz MH, Prakash L, et al. Preoperative Therapy and Pancreatoduodenectomy for Pancreatic Ductal Adenocarcinoma: a 25‐Year Single‐Institution Experience. J Gastrointest Surg. 2017;21:164‐174. [DOI] [PubMed] [Google Scholar]
- 5. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non–small‐cell lung cancer with mutated EGFR. N Engl JMed. 2010;362:2380‐2388. [DOI] [PubMed] [Google Scholar]
- 6. Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N Engl JMed. 2013;368:2385‐2394. [DOI] [PubMed] [Google Scholar]
- 7. Kris MG, Johnson BE, Berry LD, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311:1998‐2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA‐Mutated, Hormone Receptor‐Positive Advanced Breast Cancer. N Engl J Med. 2019;380:1929‐1940. [DOI] [PubMed] [Google Scholar]
- 9. Golan T, Hammel P, Reni M, et al. Maintenance Olaparib for Germline BRCA‐Mutated Metastatic Pancreatic Cancer. N Engl J Med. 2019;381:317‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hidalgo M, Von Hoff DD. Translational therapeutic opportunities in ductal adenocarcinoma of the pancreas. Clin Cancer Res. 2012;18:4249‐4256. [DOI] [PubMed] [Google Scholar]
- 11. Shin SH, Kim SC, Hong SM, et al. Genetic alterations of K‐ras, p53, c‐erbB‐2, and DPC4 in pancreatic ductal adenocarcinoma and their correlation with patient survival. Pancreas. 2013;42:216‐222. [DOI] [PubMed] [Google Scholar]
- 12. Schlitter AM, Segler A, Steiger K, et al. Molecular, morphological and survival analysis of 177 resected pancreatic ductal adenocarcinomas (PDACs): Identification of prognostic subtypes. Sci Rep. 2017;7:41064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakano Y, Kitago M, Matsuda S, et al. KRAS mutations in cell‐free DNA from preoperative and postoperative sera as a pancreatic cancer marker: a retrospective study. Br J Cancer. 2018;118:662‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kitago M, Ueda K, Aiura K, et al. Comparison of K‐ras point mutation distributions in intraductal papillary‐mucinous tumors and ductal adenocarcinoma of the pancreas. Int J Cancer. 2004;110:177‐182. [DOI] [PubMed] [Google Scholar]
- 15. Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801‐1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cancer Genome Atlas Research Network . integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32:185‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blackford A, Serrano OK, Wolfgang CL, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009;15:4674‐4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hayashi H, Kohno T, Ueno H, et al. Utility of Assessing the Number of Mutated KRAS, CDKN2A, TP53, and SMAD4 Genes Using a Targeted Deep Sequencing Assay as a Prognostic Biomarker for Pancreatic Cancer. Pancreas. 2017;46(3):335‐340. [DOI] [PubMed] [Google Scholar]
- 21. Fujita Y, Matsuda S, Sasaki Y, et al. Pathogenesis of multiple pancreatic cancers involves multicentric carcinogenesis and intrapancreatic metastasis [published online ahead of print December 4, 2019]. Cancer Sci. 2020;111(2):739‐748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tempero MA. NCCN Guidelines Updates: Pancreatic Cancer. J Natl Compr Canc Netw. 2019;17:603‐605. [DOI] [PubMed] [Google Scholar]
- 23. Wright GP, Chesla DW, Chung MH. Using next‐generation sequencing to determine potential molecularly guided therapy options for patients with resectable pancreatic adenocarcinoma. Am J Surg. 2016;211:506‐511. [DOI] [PubMed] [Google Scholar]
- 24. Nakagaki T, Tamura M, Kobashi K, et al. Targeted next‐generation sequencing of 50 cancer‐related genes in Japanese patients with oral squamous cell carcinoma. Tumour Biol. 2018;40:1010428318800180. [DOI] [PubMed] [Google Scholar]
- 25. Brierley JD, Gospodarowicz MK, Wittekind C. UICC: TNM Classification of Malignant Tumours, 8th edn Hoboken, NJ: Wiley Blackwell; 2017. [Google Scholar]
- 26. Japan Pancreas Society . Classification of Pancreatic Carcinoma, 4th English edn. Tokyo: Kanehara; 2017:70‐79. [Google Scholar]
- 27. Nishimura FY, Nishiyama R, Kitago M, et al. Two cases of pathological complete response to neoadjuvant chemoradiation therapy in pancreatic cancer. Keio J Med. 2015;64:26‐31. [DOI] [PubMed] [Google Scholar]
- 28. Endo Y, Kitago M, Aiura K, et al. Efficacy and safety of preoperative 5‐fluorouracil, cisplatin, and mitomycin C in combination with radiotherapy in patients with resectable and borderline resectable pancreatic cancer: a long‐term follow‐up study. World J Surg Oncol. 2019;17:145‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Uesaka K, Fukutomi A, Boku N, et al. Randomized phase III trial of adjuvant chemotherapy with gemcitabine versus S‐1 for patients with resected pancreatic cancer (JASPAC‐01 study) [abstract]. J Clin Oncol. 2013;31:145. [DOI] [PubMed] [Google Scholar]
- 30. Jennings LJ, Arcila ME, Corless C, et al. Guidelines for Validation of Next‐Generation Sequencing‐Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J Mol Diagn. 2017;19:341‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang J, Löhr JM, Nilsson M, et al. Variant Profiling of Candidate Genes in Pancreatic Ductal Adenocarcinoma. Clin Chem. 2015;61:1408‐1416. [DOI] [PubMed] [Google Scholar]
- 32. Bournet B, Muscari F, Buscail C, et al. KRAS G12D mutation subtype is a prognostic factor for advanced pancreatic adenocarcinoma. Clin Transl Gastroenterol. 2016;7:e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shi Y, Massague J. Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell. 2003;113:685‐700. [DOI] [PubMed] [Google Scholar]
- 34. Attisano L, Wrana JL. Signal transduction by the TGF‐beta superfamily. Science. 2002;296:1646‐1647. [DOI] [PubMed] [Google Scholar]
- 35. Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350‐353. [DOI] [PubMed] [Google Scholar]
- 36. Kang YK, Kim WH, Jang JJ. Expression of G1‐S modulators (p53, p16, p27, cyclin D1, Rb) and Smad4/Dpc4 in intrahepatic cholangiocarcinoma. Hum Pathol. 2002;33:877‐883. [DOI] [PubMed] [Google Scholar]
- 37. Royce SG, Alsop K, Haydon A, et al. The role of SMAD4 in early‐onset colorectal cancer. Colorectal Dis. 2010;12:213‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tascilar M, Skinner HG, Rosty C, et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2001;7:4115‐4121. [PubMed] [Google Scholar]
- 39. Biankin AV, Morey AL, Lee CS, et al. DPC4/Smad4 expression and outcome in pancreatic ductal adenocarcinoma. J Clin Oncol. 2002;20:4531‐4542. [DOI] [PubMed] [Google Scholar]
- 40. Oshima M, Okano K, Muraki S, et al. Immunohistochemically detected expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer. Ann Surg. 2013;258:336‐346. [DOI] [PubMed] [Google Scholar]
- 41. Wilentz RE, Iacobuzio‐Donahue CA, Argani P, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Can Res. 2000;60:2002‐2006. [PubMed] [Google Scholar]
- 42. Bardeesy N, Cheng KH, Berger JH, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006;20:3130‐3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Iacobuzio‐Donahue CA, Fu B, Yachida S, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27:1806‐1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2